
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanism of the feedback-inhibition resistance in aspartate kinase of Corynebacterium pekinense: from experiment to MD simulations†
Xiaoting Liu‡
 ab,
Caijing Han‡abc,
Li Fangab,
Zhanqing Fanab,
Yanan Wangab,
Xin Gaoab,
Junhua Shiab and
Weihong Min*ab
ab,
Caijing Han‡abc,
Li Fangab,
Zhanqing Fanab,
Yanan Wangab,
Xin Gaoab,
Junhua Shiab and
Weihong Min*ab
aCollege of Food Science and Engineering, National Engineering Laboratory of Wheat and Corn Deep Processing, Jilin Agricultural University, Changchun 130118, Jilin, People's Republic of China. E-mail: minwh2000@jlau.edu.cn; Fax: +86-431-8451-7235; Tel: +86-139-4491-9697
bNational Engineering Laboratory of Wheat and Corn Deep Processing, Changchun 130118, Jilin, China
cSchool of Public Health, Weifang Medical University, Weifang 261042, Shandong, China
First published on 22nd December 2020
Abstract
In microorganisms and plants, aspartate kinase (AK) is the initial committed enzyme of the biosynthesis of the aspartate acid family amino acids and is inhibited by end products. In the paper, we mutated the key allosteric regulatory site A380 around the binding site of the Lys inhibitor in Corynebacterium pekinense AK (CpAK). A single-mutant A380C was obtained with 12.35-fold higher enzyme activity through high-throughput screening. On this basis, T379 as another key allosteric regulatory site was further modified, and the double-mutant T379N/A380C with 22.79-fold higher enzyme activity was obtained. Molecular dynamics (MD) simulations were used to investigate the mechanism of allosteric inhibition by Lys. The results indicated that the binding of Lys with CpAK resulted in conformational changes and a larger distance between the phosphorus atom of ATP and the oxygen atom of Asp, which was detrimental for the catalytic reaction. However, the mutation of allosteric sites opens the “switch” of allosteric regulation and can prevent the conformational transformation. Some key residues such as G168, R203, and D193 play an important role in maintaining the substrate binding with CpAK and further enhance the enzyme activity.
1. Introduction
Aspartate kinase (AK, EC 2.7.2.4), also named aspartokinase, is the first and typical allosteric enzyme responsible for the synthesis of the aspartate acid family amino acids including lysine, threonine, methionine, and isoleucine.1 AK catalyzes phosphorylation of the γ-carboxylate group of aspartate in microorganisms and plants and is regulated by end-products through feedback inhibition.2 For instance, AKIII from Escherichia coli and CaAK from Clostridium acetobutylicum are sensitive to Lys,3,4 while MjAK and TtAK separately found in Methanococcus jannaschii and Thermus thermophilus are both subjected to the feedback inhibition by threonine.5,6 Additionally, AKI from Arabidopsis thaliana is inhibited synergistically by lysine and S-adenosylmethionine,7 whereas CgAK (from Corynebacterium glutamicum) and SyAK (from Synechocystis) are sensitive to Lys plus Thr in a concerted manner.8,9So far, most structurally determined AKs are homo-oligomeric enzymes composed of α subunits, while CgAK, MjAK, TtAK, and PaAK (from Pseudomonas aeruginosa) display a unique α2β2-type heterotetrameric structure which contains equimolar α and β subunits encoded by in-frame overlapping genes. In this α2β2-type AK, the N-terminal portion of the α subunit serves as a catalytic domain, whereas the β subunit and the C-terminal region of the α subunit function as regulatory domains. The catalytic domain is further divided into two lobes, the N-lobe making up the Asp-binding site and the C-lobe offering a nucleotide-binding pocket for ATP. A key feature of the regulatory domain is the conserved ACT domain which is found in a wide variety of allosteric enzymes.10 In recent years, the allosteric regulation of AK has attracted profound attention from scholars. The general allosteric mechanisms of AK have been concluded: the effector binding leads to a conformational transition from a substrate-ATP bound relaxed state (R-state, close conformation) to the inactive tense state (T-state, open conformation), which in turn hinders the catalytic reaction.3,11
To promote the industrial production of the Asp-derived amino acids, engineering of regulation and activity of AK is important. Therefore, feedback-resistant mutations are successfully made in genes encoding for LysC (aspartate kinase).2,9,11 Apart from the enzymological interest, AK is an attractive target for developing new antibiotics and antitubercular drugs because of its absence in human and animals.12 For example, PaAK plays crucial roles in biofilm-associated antibiotic-resistance and infections, which demonstrates its potential application in anti-P. aeruginosa treatment.13,14 Mycobacterium tuberculosis (Mtb) AK has been identified as a potential drug target in Mtb, especially in multidrug-resistant Mtb.12,15
In our previous study, CpAK from Corynebacterium pekinense has been found to be a novel allozyme (Fig. 1) and T379 as well as A380 around the binding site of inhibitor Lys are the conserved allosteric regulatory sites. Elucidation of its allosteric regulatory mechanism is of great significance for the construction of highly efficient microbial strains.16–18 However, up to date, the detailed allosteric mechanism of CpAK is still unclear. In this work, we designed and expressed novel mutants with high-enzyme activity as well as feedback inhibition resistance on the basis of sites 379 and 380 by using site-directed mutagenesis and high-throughput screening techniques. Furthermore, molecular dynamics (MD) simulations and microscale thermophoresis (MST) were applied to study the interaction between AK and substrate or inhibitor, which sheds light on the allosteric regulation.
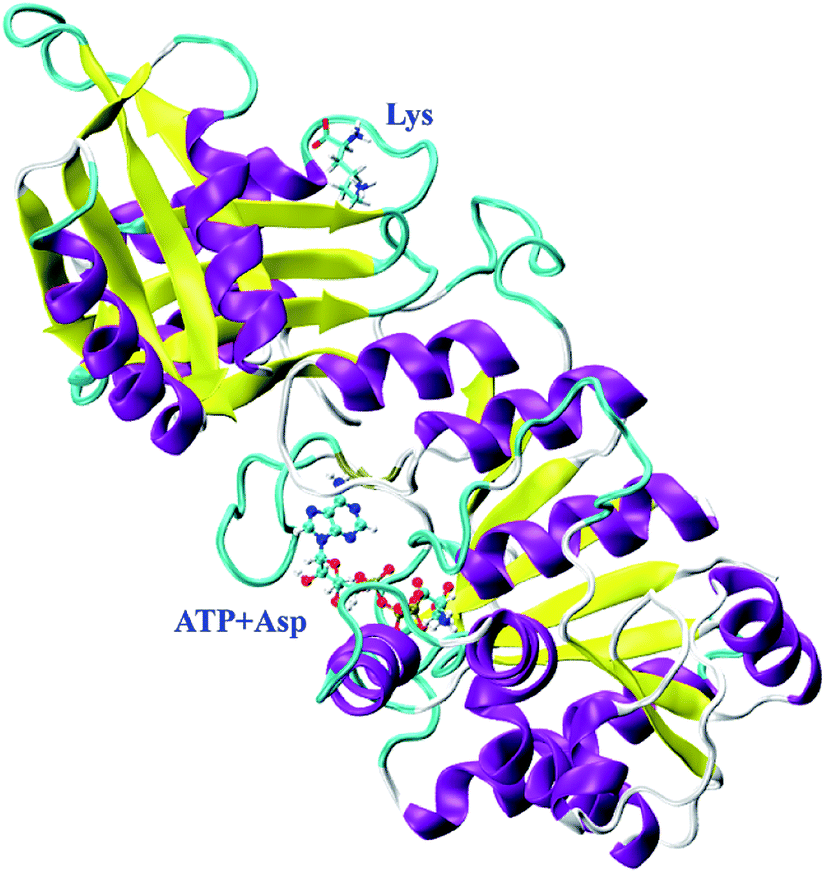 | ||
| Fig. 1 Structure of CpAK. | ||
2. Results and discussions
2.1. Construction and purification of mutant strains
The single mutant A380C and double variant T379N/A380C were obtained by site-directed mutagenesis and high-throughput screening. The recombinant plasmid with about 7000 bp (pET-28a was 5369 bp and the target gene AK was 1266 bp) was used as template to perform PCR (polymerase chain reaction) amplification under the action of mutant primers (Fig. 2A). The target band was also detected at 7000 bp (Fig. 2B), which demonstrated the mutation was successfully induced. AK gene with 1266 bp band was amplified by PCR, applying the mutant strain as the template and the cloning primers as the primers, and then sent to Shanghai Sangong for sequencing (Fig. 2C). The components of the crude enzyme were collected after the purification process, of which the peaks f1 and f2 in Fig. 2D were corresponding with the 2, 3 and 4, 5 bands in Fig. 2E, respectively. There was no obvious band at 48 kDa, implying that the target protein was fully purified by the His Trap column. The stripe 6 (Fig. 2E) corresponding to the peak f3 (Fig. 2D) had an obvious band at 48 kDa, indicating that AK has been purified successfully. The stripe 7 was the results of western blotting, indicating that the purified AK protein was successful. | ||
| Fig. 2 Electrophoretic verification and purification of AK. (A) Agarose gel electrophoresis of plasmid PET-28a–AK. M: DNA marker; 1: A380C; 2: T379N/A380C. (B) Agarose gel electrophoresis of mutation PCR. M: DNA marker. 1: A380C; 2: T379N/A380C. (C) Agarose gel electrophoresis of bacterial PCR. M: DNA marker. 1: A380C; 2: T379N/A380C. (D) Purification of AK by ÄKTA. f1, f2: crude enzyme sample after flowing the His Trap; f3: purified enzyme sample. (E) SDS-PAGE and western blot. M: protein marker; 1: crude enzyme sample; 2–3: f1; 4–5: f2; 6: f3; 7: western blot. | ||
2.2. Kinetics and enzymatic properties
The kinetic analysis and enzymatic properties of WT (wild-type CpAK), A380C, and T379N/A380C mutant strains were shown in Fig. 3. As shown in Fig. 3A, the reaction rate of WT, single mutant A380C, and double mutant T379N/A380C was 3.28, 40.51, and 80.7 U mg−1 min−1, respectively. Compared to WT, the enzyme activities of the mutants were increased by 12.35-fold and 22.79-fold, respectively. The Km values of WT, A380C, and T379N/A380C were 4.17, 3.57, and 2.89 mM, respectively. The Km value decreased gradually, indicating that the enzyme–substrate affinity was enhanced. The n value of WT was 1.54, which was a typical allosteric enzyme, manifesting positively synergistic. The n values of A380C and T379N/A380C were 1.21 and 1.46, respectively. The smaller is the n value, the stronger is the resistance to feedback inhibition.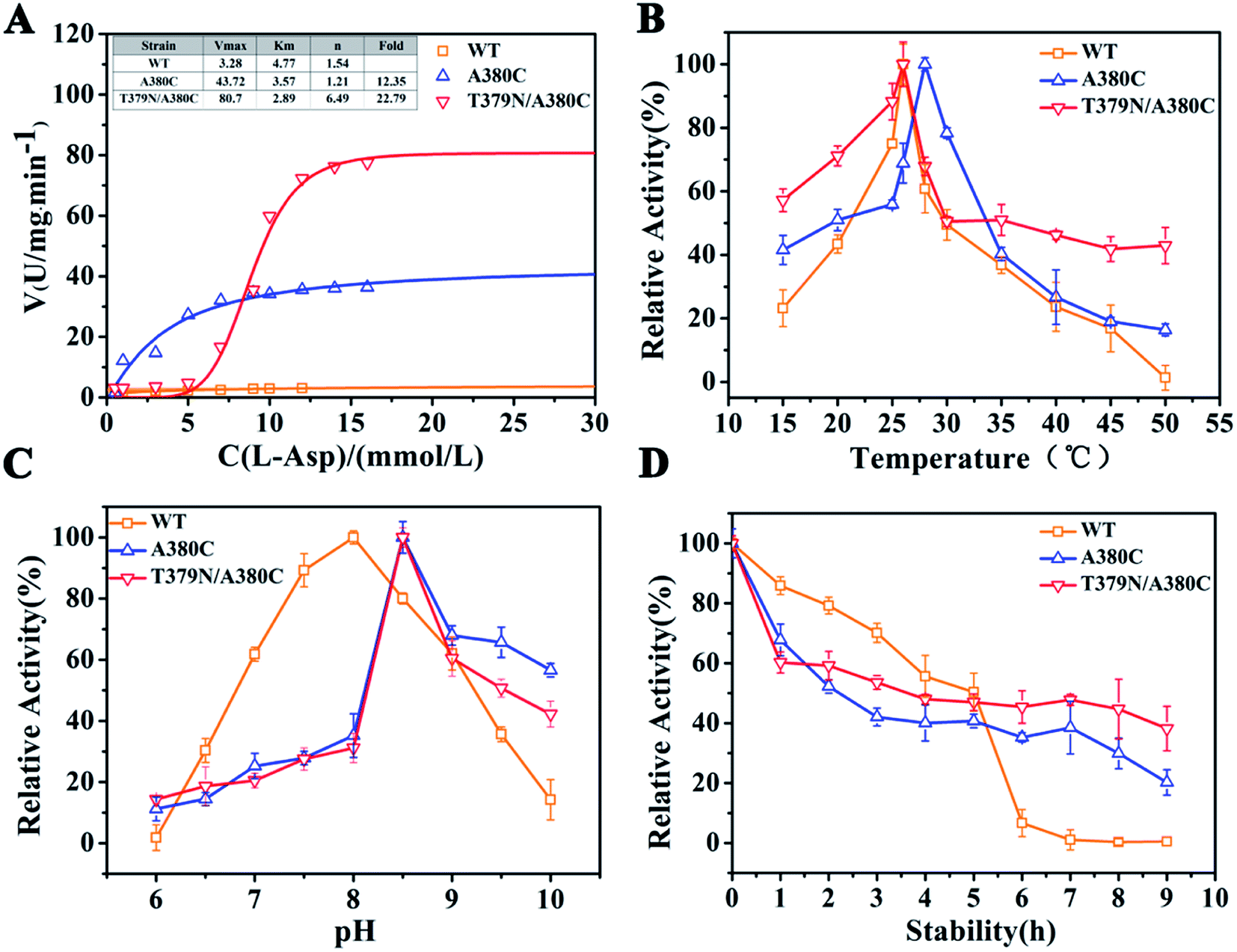 | ||
| Fig. 3 Dynamic analysis and enzymatic properties. (A) Dynamic analysis. (B) The optimum temperature. (C) The optimum pH. (D) The thermal stability. | ||
The optimal temperature of WT, A380C, and T379N/A380C was 26, 28, and 26 °C, respectively (Fig. 3B). The optimal temperature of the A380C variant has risen. Although it has not changed for the double mutant T379N/A380C, its thermal stability increased and retained about 50% of the enzyme activity at 50 °C, which was beneficial to the late fermentation production.9 The optimal pH of WT, A380C, and T379N/A380C was 8.0, 8.5, 8.5, respectively, demonstrating that the optimal pH was not improved after mutation (Fig. 3C).
The half-life values of A380C and T379N/A380C mutant strains were 2.22 and 3.27 h, respectively, which were both lower than that of WT (4.5 h), suggesting the relative enzymatic activity values were reduced in Fig. 3D. However, the relative enzyme activity of the mutant strains was higher than that of WT after 6 h, and especially T379N/A380C still maintained above 40% activity after 8 h, implying that the mutations could enhance the thermostability on the whole.
As reported, WT was inhibited by the synergistic feedback of threonine and lysine in a dose-dependent manner.16–19 As can be seen from Table 1, the enzyme activity of mutant A380C was improved under different concentration of inhibitors, and even activated at high concentration of Lys, Lys + Thr, Lys + Met, Thr + Met, and Lys + Thr + Met, which was consistent with the decrease of the n value in the kinetic analysis. Compared with WT,16 mutants had a more obvious disinhibition effect, and the activation effect of mutant is mainly contributed by Lys inhibitor. The activation effect was very clear with the concentration of Lys at 10 mM, indicating that the modified site 380 can effectively remove the inhibition effect. This result lays the foundation for our subsequent research on the allosteric regulation of Lys.
| Enzyme | A380C | T379N/A380C | ||||||
|---|---|---|---|---|---|---|---|---|
| Inhibitors | Relative activity (%) | Concentration (mM) | ||||||
| 0.2 | 1 | 5 | 10 | 0.2 | 1 | 5 | 10 | |
| a The bold indicates activation. | ||||||||
| Control | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Lys | 94.80 ± 0.97 | 95.74 ± 1.43 | 99.70 ± 0.54 | 103.54 ± 1.32 | 96.99 ± 0.56 | 98.78 ± 4.95 | 106.87 ± 0.56 | 115.84 ± 1.81 |
| Thr | 78.26 ± 1.19 | 80.94 ± 3.06 | 82.14 ± 3.56 | 87.80 ± 3.53 | 94.59 ± 3.26 | 97.06 ± 0.68 | 97.84 ± 1.95 | 98.12 ± 0.44 |
| Met | 95.06 ± 1.69 | 97.71 ± 0.85 | 98.30 ± 2.6 | 98.13 ± 1.89 | 90.47 ± 0.12 | 96.33 ± 0.56 | 98.12 ± 0.44 | 99.47 ± 0.53 |
| Lys + Thr | 98.58 ± 2.96 | 99.09 ± 3.13 | 105.48 ± 3.17 | 109.82 ± 3.35 | 92.02 ± 0.44 | 97.44 ± 0.96 | 103.57 ± 1.12 | 105.48 ± 0.97 |
| Lys + Met | 97.88 ± 1.47 | 98.56 ± 2.34 | 105.36 ± 2.45 | 99.12 ± 4.34 | 89.61 ± 1.35 | 93.29 ± 2.65 | 97.74 ± 3.45 | 114.35 ± 1.96 |
| Thr + Met | 97.31 ± 0.86 | 98.00 ± 1.78 | 113.70 ± 3.35 | 98.19 ± 3.98 | 81.87 ± 2.02 | 90.79 ± 0.75 | 104.07 ± 2.53 | 92.16 ± 0.65 |
| Lys + Thr + Met | 74.04 ± 4.66 | 80.12 ± 1.03 | 86.26 ± 1.07 | 102.77 ± 3.81 | 86.97 ± 1.85 | 99.21 ± 1.91 | 110.01 ± 1.24 | 111.92 ± 2.57 |
As observed from Table 1, the resistant feedback-inhibition of T379N/A380C was significantly improved with respect to A380C, among which the activation effect was obvious under the action of 5–10 mM Lys, Lys + Thr, and Lys + Met, especially under the action of 10 mM Lys, indicating that the activation effect was regulated by Lys and weakened after mutation. The double mutation may further affect the allosteric regulation of Lys.
The affinity between CpAK and the ligand was further analyzed by MST. As shown in Table 2, the Kd values of T379N/A380C, A380C, and WT with inhibitor Lys were 9.24, 5.32, and 1.32 mM, respectively. After mutation, the increased Kd values between inhibitor Lys and variant revealed the lower affinity with inhibitor Lys. The double mutant T379N/A380C had the lowest affinity with the inhibitor Lys, so the disinhibition was relatively obvious. The Kd values between WT, A380C, and T379N/A380C with substrate Asp (named WT + Asp, A380C + Asp, T379N/A380C + Asp) were 655.08, 313.32, and 309.7 mM, respectively. The decreased Kd values indicated the binding of AK to the substrate Asp was increased. The Kd values between WT, A380C, and T379N/A380C and Lys + Asp (named WT + Lys + Asp, A380C + Lys + Asp, T379N/A380C + Lys + Asp) were 701.5, 447.69, and 99.64 mM, respectively. The decreased Kd values demonstrate strong affinity and high maximum reaction rate. The inhibition of AK was improved after mutation, which was consistent with the inhibitor data (shown in Table 1). The Kd values of WT + Asp was less than that of WT + Lys + Asp, manifesting its greater affinity, which further identified that Lys played an important role in the allosteric regulation. However, the Kd value of the mutant strain was significantly lower than that of WT, indicating that the modification around the inhibitor Lys site could eliminate the feedback-inhibition and well regulate substrate binding through allosteric regulation. In the following discussions, molecular dynamics simulation was used for in-depth illustration.
| Enzymes | Kd (mM) | ||
|---|---|---|---|
| Lys | Asp | Lys + Asp | |
| WT | 1.32 ± 0.00 | 655.08 ± 0.90 | 701.25 ± 1.24 |
| A380C | 5.32 ± 0.01 | 313.32 ± 0.79 | 447.69 ± 0.55 |
| T379N/A380C | 9.24 ± 0.00 | 309.70 ± 0.23 | 99.64 ± 0.07 |
2.3. Computational analysis
 | ||
| Fig. 4 Detailed interaction between the substrate Asp and AK residues. (a) WT system. (b) WT + Lys system. | ||
 | ||
| Fig. 5 The interaction energy between the substrate Asp and AK protein. | ||
3. Materials and methods
3.1. Experimental materials
Recombinant plasmid pET-28a–AK from the Escherichia coli was preserved by our laboratory. The plasmid extraction kit, protein electrophoresis marker, nucleic acid electrophoresis marker, and DpnI digestive enzyme were purchased from the TaKaRa Company (Japan). His Trap HP 5 mL was bought from General Electric Company (USA), and His-Tag (27 × 108) Mouse mAb (HRP conjugate) (art. no. 9991S) from Cell Signaling Technology (USA). Monolith™ Protein Labeling Kit RED-NHS and Capillaries for Monolith NT115 Standard Capillary were bought from NanoTemper Technology Co., Ltd. (Beijing, China).3.2. Construction of mutant strains
To construct mutants of CpAK, the sites T379N and A380C were subjected to site-directed mutagenesis. According to the design principle of site-directed mutagenesis, the mutant primers were as follows: A380 – 5′GCTTCCATGAACTCNNNGGTAACACCTGG3′, 5′CCCAGGTGTTACCNNNGAGTTCATGGAAG3′; T379N/A380C – 5′CAGAGCTTCCATGAACTCACANNNAACACCTGGGTGAG3′, 5′CAGTCTCACCCAGGTGTTNNNTGTGAGTTCATGGAAGC3′. PCRs were performed to amplify the target fragment sequences employing the recombinant plasmid pET-28a–AK as the template. The resulting plasmid were then digested by DpnI enzyme under the conditions: DpnI enzyme 0.3 μL, buffer 2 μL, PCR product 2 μL, and at 37 °C for 2 h. The PCR products were transformed into E. coli BL21 cells by the method of heat shock, which contained an ice bath for 5 min, hot compress at 42 °C for 90 s, and an ice bath for 2 min again, adding 900 μL Luria–Bertani (LB) solid medium without resistance. After the cells were incubated at 37 °C, 160–170 rpm for 90 min, centrifuged at 8000 rpm for 2 min, and finally 800 μL supernatant were discarded.Monoclonal strains expressing mutant AK with high-enzyme activity were achieved by high-throughput technique.16 The high-throughput method was as follows: the mutant strains were collected as much as possible, cultivated in the 96-well plate (200 μL LB culture medium) at 37 °C, 180 rpm for 12 h, and then 20 μL strains were transferred to a new 96-well plate for inducing 4.5 h at 37 °C, 200 rpm. After 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) was added into the strains which were induced at 30 °C, 130 rpm for 8 to 9 h. The cells were gathered by centrifugation at 3500 rpm for 45 min, and then frozen at −80 °C for 0.5–1 h and thawed at 30 °C for 0.5–1 h. The crude enzymes could be obtained and their enzyme activity were determined in the absorbance at 600 nm by high throughput screening.
The strains with high-enzyme activity were amplified with the cloned primers (5′GGAATTCCATATGGCCCTGGTCGTACAGAA3′, 5′GGAATTCTTAGCGTCCGGTGCCTGCAT3′) and PCR amplification of bacterial liquid was carried out. After the products were verified by 1% agarose gel electrophoresis. The CpAK gene replacement mutant strains were further verified by DNA sequencing and for subsequent use.
3.3. Purification and validation of AK
The bacteria solution of successful sequencing was induced by 1 mM isopropyl-β-D-thiogalactoside (IPTG) overnight at 23 °C. The cells were subsequently collected after centrifugation at 4 °C, 8000 rpm for 10 min, and suspended in 10 mL PBS (pH 7.4). The suspension was centrifuged again for 10 min after ultrasonic crushing, and purified with ÄKTA. The conditions were as follows: (1) equilibration: PBS of 10 CV (column volume); flow rate, 5 mL min−1; pressure, 0.4 MPa; (2) sample application: sample volume, 6 CV; flow rate, 2 mL min−1, pressure, 0.4 MPa; (3) equilibration: PBS of 25 CV; (4) elution: elution was performed by a linear gradient of buffer B (500 mM imidazole) and buffer A (20 mM imidazole). The concentration of buffer B was from 0 to 35% during 20 CV and then eluted by 100% with 25 CV. The solution was 4 mL per tube, and the purified liquid was verified by SDS-PAGE and western blot.3.4. Kinetic assays
The reaction system (1 mL) contained at the final concentration: 1.6 mM MgSO4, 800 mM KCl, 0.5–16 mM L-Asp, 10.4 mM ATP (dissoluted by 25 mM Tris–HCl), 100 mM Tris–HCl buffer solution (pH 8.0), 10 mM β-mercaptoethanol, and 800 mM NH4OH. The water was instead of L-Asp as blank control. The reaction was generated by adding 50 μL purified enzyme at 28 °C for 30 min, and stopped by adding FeCl3 solution (three parallel, below the same) and the absorbance were determined at 540 nm. The nonlinear curve was fitted by the Hill equation (V = Vmax (Sn)/(Kn + Sn)) using Origin 8.5 software. In the equation, the n value represents the Hill coefficient; the K value is equal to the substrate concentration at half the maximum rate of enzymatic reaction; Vmax and V indicate the maximum velocity and reaction rate, respectively; S means the substrate concentration.3.5. Determination of enzymology properties
The reaction was performed through adding 50 μL purified enzyme at different temperature gradients (15, 20, 25, 26, 28, 30, 35, 40, 45, and 50 °C) and different pH gradients (6.0, 6.5, 7.0, 7.5, 8.0, 8.5, 9.0, 9.5, and 10.0) for 30 min, severally. It was terminated by adding FeCl3 and measured absorbance at 540 nm. L-Asp was instead of water as negative control. The highest enzyme activity was defined as 100%.The reaction was performed through adding 50 μL purified enzyme at the optimum temperature and pH. The initial enzyme activity was defined as 100% and then measured every one hour in total 10 times. The half-life period was defined by the half of the original enzyme activity.
The reaction was performed through adding 20 μL purified enzyme and 20 μL substrate inhibitors (Lys, Thr, Met, Lys + Thr, Lys + Met, Thr + Met, and Lys + Thr + Met) at 0.2, 1, 5, and 10 mM. The reaction was finished by adding volume FeCl3 after 30 min and the absorbance was measured at 540 nm. Water was instead of the substrate inhibitor as blank control. The enzyme activity of blank control was defined as 100%.
3.6. Microscale thermophoresis (MST) analysis
The purified AK concentration was adjusted to 2–20 μM with labeled buffer. The solid fluorescent dye (NT-647-NHS) was appended with 100% DMSO (30 μL) and dissolved thoroughly after vortex vibration. Then the dyes were adjusted to 2–3 folds of the protein concentration with the marker buffer. Protein and the solid fluorescent dye were mingled in equal proportion for dark reaction 30 min, and then purified according to the purification kit operation. The unlabeled substrate Asp and inhibitor Lys were diluted in the enzymatic activity measurement system, and the final concentration ranged from 0.5 M to 15 M and from 0.1 M to 3 M, respectively, which they were incubated with the labeled protein (40 nM) at room temperature for 10 min. Then the samples were loaded into the Standard Capillaries from Nano Temper Technologies. Thermophoresis was measured 30 min after incubation at 28 °C on Monolith NT.115.22 The dissociation constant (Kd) values were fitted by means of the MO. Affinity analysis software.3.7. Molecular dynamics simulations
In our previous studies,17,18 the homology model for CpAK was obtained. Here we directly used the homology model, including the ligands (Asp and ATP) and inhibitor (Lys). The mutants of AK were generated by Discovery Studio (DS).23 All of the missing atoms in the complex are automatically added by the VMD psfgen plugin.24 Then more TIP3P molecules25 extending up to 10 Å from the solute in each direction were added by the solvate plugin in VMD, forming a rectangular solvated system. The salt concentration of the system was set to 154 mM by the addition of Na+ and Cl− counterions.All the molecular dynamics (MD) were performed by the package NAMD 2.12.26 Topology and force field parameters were assigned from the CHARMM36 parameter set.27 The van der Waals and short-range electrostatic interactions were gradually turned off at a distance range between 9 and 10 Å. The long-range electrostatic interactions were described by the particle mesh Ewald (PME).28 Every simulation system was first energy minimized with 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps of conjugate gradients, by restraining the positions of the protein backbone and ligand atoms with a force constant of 1 kcal mol−1 Å−2. After that, at the same restraint, the temperature of the system was, using Langevin thermostat,29 gradually increased from 100 to 300 K at an increment rate of 10 ps K−1 within the simulation time of 2 ns. Next, 1 ns MD simulation was carried out to ensure the stability of the system's temperature in the canonical (NVT) ensemble, and then 2.5 ns NPT MD simulations were performed in which the pressure of the system was coupled to a reference pressure of 1 bar with the modified Nosé–Hoover–Langevin piston method.30 In the process, the restraint potential function for the system was gradually turn off by progressively decreasing the scaling factor to 0. Finally, at least 110 ns free NPT MD simulations were conducted. The MD trajectories were analyzed using VMD. Root-mean-square deviation (rmsd) of the Cα atoms was calculated.
000 steps of conjugate gradients, by restraining the positions of the protein backbone and ligand atoms with a force constant of 1 kcal mol−1 Å−2. After that, at the same restraint, the temperature of the system was, using Langevin thermostat,29 gradually increased from 100 to 300 K at an increment rate of 10 ps K−1 within the simulation time of 2 ns. Next, 1 ns MD simulation was carried out to ensure the stability of the system's temperature in the canonical (NVT) ensemble, and then 2.5 ns NPT MD simulations were performed in which the pressure of the system was coupled to a reference pressure of 1 bar with the modified Nosé–Hoover–Langevin piston method.30 In the process, the restraint potential function for the system was gradually turn off by progressively decreasing the scaling factor to 0. Finally, at least 110 ns free NPT MD simulations were conducted. The MD trajectories were analyzed using VMD. Root-mean-square deviation (rmsd) of the Cα atoms was calculated.
4. Conclusions
Corynebacterium pekinense AK was successfully modified and two mutants A380C and T379N/A380C with high enzyme activity (12.35-fold and 22.79-fold as compared to CpAK, respectively) were constructed. The structural and biochemical analyses such as enzyme activity are used by both experimental and theoretical methods. The microscale thermophoresis results showed that the mutations in sites T379 and 380 can resist the feedback inhibition of Lys. Moreover, the molecular dynamics simulation has given a reasonable explanation of the inhibitor mechanism of inhibitor Lys as well as the feedback inhibition-resistant of mutations. The binding of Lys with WT resulted in conformational changes of CpAK and a larger distance between the phosphorus atom of ATP and the oxygen atom of Asp (dPi–O = 4–5 Å), and thus, was detrimental for the catalytic reaction. However, mutations can present the conformational transformation and some key residues such as G168, R203, and D193 play an important role in maintaining the substrates binding with CpAK and further enhance the enzyme activity. The Kd value of Asp and AK experimented by MST for WT, A380C + Lys, and T379N/A380C + Lys is decreased and the affinity of substrate Asp and AK is enhanced, which is in good agreement with the increased electrostatic interaction energy. The present work is useful for the further study of phosphorylation mechanism of aspartate kinase, and it provides the rational engineering of the biosynthesis pathway of amino acid in the production of Asp-derived amino acids.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is supported by the Natural Science Foundation of China (Grant No. 31771957) and Excellent Doctoral Research Fund of Jilin Agricultural University (Grant No. 202023317). Dr X. Liu has acknowledged the computing resources from high performance computing center in Institute of Theoretical Chemistry, Jilin University.References
- G. N. Cohen, The Common Pathway to Lysine, Methionine, and Threonine, in Amino Acids: Biosynthesis and Genetic Regulation, ed. K. M. Herrmann and R. L. Somerville, Addison-Wesley, Reading, MA, 1983, pp. 147–171 Search PubMed.
- A. Yoshida, T. Tomita, T. Kurihara, S. Fushinobu, T. Kuzuyama and M. Nishiyama, Structural Insight into Concerted Inhibition of α2β2-Type Aspartate Kinase from Corynebacterium glutamicum, J. Mol. Biol., 2007, 368, 521–536 CrossRef CAS.
- M. Kotaka, J. Ren, M. Lockyer, A. R. Hawkins and D. K. Stammers, Structures of R- and T-state Escherichia coli aspartokinase III. Mechanisms of the allosteric transition and inhibition by lysine, J. Biol. Chem., 2006, 281, 31544–31552 CrossRef CAS.
- B. A. Manjasetty, M. R. Chance, S. K. Burley, S. Panjikar and S. C. Almo, Crystal structure of Clostridium acetobutylicum aspartate kinase (CaAK): an importantallosteric enzyme for amino acids production, Biotechnol. Rep., 2014, 3, 73–85 CrossRef.
- X. Liu, A. G. Pavlovsky and R. E. Viola, The structural basis for allosteric inhibition of a threonine sensitive aspartokinase, J. Biol. Chem., 2008, 283, 16216–16225 CrossRef CAS.
- M. Nishiyama, M. Kukimoto, T. Beppu and S. Horinouchi, An operon encoding aspartokinase and purine phosphoribosyltransferase in Thermus flavus, Microbiology, 1995, 141, 1211–1219 CrossRef CAS.
- S. E. Rognes, P. J. Lea and B. J. Miflin, S-Adenosylmethionine—a novel regulator of aspartate kinase, Nature, 1980, 287, 357–359 CrossRef CAS.
- A. Y. Robin, D. Cobessi, G. Curien, M. Robert-Genthon, J. L. Ferrer and R. Dumas, A new mode of dimerization of allosteric enzymes with ACT domains revealed by the crystal structure of the aspartate kinase from Cyanobacteria, J. Mol. Biol., 2010, 399, 283–293 CrossRef CAS.
- A. Yoshida, T. Tomita, T. Kuzuyama and M. Nishiyama, Mechanism of Concerted Inhibition of α2β2-type Hetero-oligomeric Aspartate Kinase from Corynebacterium glutamicum, J. Mol. Biol., 2010, 285, 27477–27486 CAS.
- L. Aravind and E. V. Koonin, Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches, J. Mol. Biol., 1999, 287, 1023–1040 CrossRef CAS.
- C. C. Li, M. J. Yang, L. Liu, T. Li, C. T. Peng, L. H. He, Y. J. Song, Y. B. Zhu, Y. L. Shen, J. Yang, N. L. Zhao, C. Zhao, Q. X. Zhou, H. Li, M. Kang, A. P. Tong, H. Tang and R. Bao, Mechanistic insights into the allosteric regulation of Pseudomonas aeruginosa aspartate kinase, Biochem. J., 2018, 475, 1107–1119 CrossRef CAS.
- E. J. Hasenoehrl, D. R. Sajorda, L. Berney-Meyer, S. Johnson, J. M. Tufariello, T. Fuhrer, G. M. Cook, W. R. Jacobs Jr and M. Berney, Derailing the aspartate pathway of Mycobacterium tuberculosis to eradicate persistent infection, Nat. Commun., 2019, 10, 4215–4226 CrossRef.
- S. Hwang, C. Y. Kim, S.-G. Ji, J. Go, H. Kim, S. Yang, H. J. Kim, A. Cho, S. S. Yoon and I. Lee, Network-assisted investigation of virulence and antibiotic-resistance systems in Pseudomonas aeruginosa, Sci. Rep., 2016, 6, 26223 CrossRef CAS.
- Z. Xu, X. Fang, T. K. Wood and Z. J. Huang, A systems-level approach for investigating Pseudomonas aeruginosa biofilm formation, PLoS One, 2013, 8, e57050 CrossRef CAS.
- Q. Yang, K. Yu, L. Yan, Y. Li, C. Chen and X. Li, Structural view of the regulatory subunit of aspartate kinase from Mycobacterium tuberculosis, Protein Cell, 2011, 2, 745–754 CrossRef CAS.
- C. J. Han, S. M. Liu, C. L. Liu, X. Y. Xie, L. Fang and W. H. Min, The mutant T379L of novel aspartokinase from Corynebacterium pekinense: a combined experimental and molecular dynamics simulation study, Process Biochem., 2019, 83, 77–85 CrossRef CAS.
- Y. Gao, C. Han, C. Liu, J. Wang, L. Zhao, L. Fang and W. Min, Enzymatic characterization and molecular mechanism of a novel aspartokinase mutant M372I/T379W from Corynebacterium pekinense, RSC Adv., 2019, 9, 21344–21354 RSC.
- C. J. Han, L. Fang, C. L. Liu, Y. N. Gao and W. H. Min, Construction of novel Aspartokinase mutant A380I and its characterization by molecular dynamics simulation, Molecules, 2018, 23, 3379 CrossRef.
- M. Huang, X. Li, J.-W. Zou and D. J. Timson, Role of Arg228 in the Phosphorylation of Galactokinase: The mechanism of GHMP Kinases by Quantum Mechanics/Molecular Mechanics Studies, Biochemistry, 2013, 52, 4858–4868 CrossRef CAS.
- P. Ojeda-May, Y. Li, V. Ovchinnikov and K. Nam, Role of Protein Dynamics in Allosteric Control of the Catalytic Phosphoryl Transfer of Insulin Receptor Kinase, J. Am. Chem. Soc., 2015, 137, 12454–12457 CrossRef CAS.
- J. McClory, G.-X. Hu, J.-W. Zou, D. J. Timson and M. Huang, Phosphorylation Mechanism of N-Acetyl-L-glutamate Kinase, a QM/MM Study, J. Phys. Chem. B, 2019, 123, 2844–2852 CrossRef CAS.
- H. G. Kazemier, K. Paeschke and P. M. Lansdorp, Nucleic Acids Res., 2017, 45, 5913–5919 CrossRef CAS.
- Discovery Studio 4.1, Accelrys, San Diego, CA, 2014 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, Vmd: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, Scalable Molecular Dynamics with Namd, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS.
- R. B. Best, X. Zhu, J. Shim, P. E. M. Lopes, J. Mittal, M. Feig and A. D. MacKerell Jr, Optimization of the Additive Charmm All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, Ψ and Side-Chain X1 and X2 Dihedral Angles, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle Mesh Ewald: An N·Log(N) Method for Ewald Sums in Large Systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- S. E. Feller, Y. Zhang, R. W. Pastor and B. R. Brooks, Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method, J. Chem. Phys., 1995, 103, 4613–4621 CrossRef CAS.
- W. G. Hoover, Canonical Dynamics: Equilibrium Phase-Space Distributions, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09153g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |