
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn active and stable multifunctional catalyst with defective UiO-66 as a support for Pd over the continuous catalytic conversion of acetone and hydrogen†
Yingjie Hua,
Yuxin Meia,
Baining Lin*a,
Xuhong Dua,
Fan Xua,
Huasheng Xieb,
Kang Wangb and
Yonghua Zhou *a
*a
aCollege of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China. E-mail: baininglin@csu.edu.cn; zhouyonghua@csu.edu.cn
bCangzhou Dahua Group Company, Ltd, Cangzhou 061000, China
First published on 21st December 2020
Abstract
The one-pot synthesis of methyl isobutyl ketone (MIBK) and methyl isobutyl methanol (MIBC) from acetone and hydrogen is a typical cascade reaction comprised of aldol condensation-dehydration-hydrogenation. Pd loss and aggregation during long term operation are typical problems in industrial application. In this paper, an active and stable catalyst was achieved with defective UiO-66 as a support for Pd, which was synthesized with the ratio 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of ZrOCl2·8H2O to ZrCl4 as Zr-precursors. The resultant Pd catalyst remained active for at least 1000 h with a MIBK + MIBC selectivity of 84.87–93.09% and acetone conversion of 45.26–53.22% in a continuous trickle-bed reactor. Besides the increased Brønsted acid amount generated by the defect sites was favorable for the activity, the cavity confinement in the UiO-66 (R = 15:1) structure also efficiently prevented Pd loss and aggregation during the long term run. The contrast of the characterization of the fresh and used Pd/UiO-66 (R = 15:1) indicated that the deactivation of the catalyst was attributed to carbonaceous accumulation on the catalyst surface, which could be easily regenerated by calcination. This work supplied a new alternative for the design and utilization of industrial catalysts for MIBK and MIBC synthesis.
:1 of ZrOCl2·8H2O to ZrCl4 as Zr-precursors. The resultant Pd catalyst remained active for at least 1000 h with a MIBK + MIBC selectivity of 84.87–93.09% and acetone conversion of 45.26–53.22% in a continuous trickle-bed reactor. Besides the increased Brønsted acid amount generated by the defect sites was favorable for the activity, the cavity confinement in the UiO-66 (R = 15:1) structure also efficiently prevented Pd loss and aggregation during the long term run. The contrast of the characterization of the fresh and used Pd/UiO-66 (R = 15:1) indicated that the deactivation of the catalyst was attributed to carbonaceous accumulation on the catalyst surface, which could be easily regenerated by calcination. This work supplied a new alternative for the design and utilization of industrial catalysts for MIBK and MIBC synthesis.
1. Introduction
Methyl isobutyl ketone (MIBK) is one of the most important chemical products widely used in the coatings, pharmaceutical, and petrochemical industries.1 Methyl isobutyl methanol (MIBC), synthesized via MIBK hydrogenation, is an excellent medium-boiling solvent and used as a raw material for organic synthesis and mineral floaters. With the development of the global economy, the demand for MIBK and MIBC in Asia, especially China and South Korea, will increase further, due to the pull of rubber antioxidant 4020 and high-solid coatings.At present, MIBK is mainly manufactured by a one-pot process, composed of three steps, say, acetone condensation catalyzed by acid or base to diacetone alcohol (DAA), DAA dehydration catalyzed by acid to mesityl oxide (MO), and MO hydrogenation catalyzed by Pd to MIBK.2,3 Normally, MIBC could also be generated in this one-pot process. To date, the only commercialized multifunctional catalyst is Pd/resin, which possess both the acid and Pd sites, which delivered an acetone conversion of 25–50% with a MIBK selectivity of 70–90% at 1–10 MPa and 120–150 °C.4–7 Nevertheless, due to the poor thermal stability of the resin, the Pd loss and aggregation during long term operation is inevitable, which leads to the deactivation of catalyst. Aiming at solving this problem, although lots of bifunctional catalysts, including Pd/metal oxide,8,9 Pd/molecular sieve and Pd/resin,10,11 Ni-based,12,13 Cu-based,14 and Pt-based,15 have been reported during the recent decades, the high active and stable catalyst is still desirable.
Metal–organic frameworks (MOF), in particular Zr-based MOF,16–18 have been extensively investigated for acid catalyzed reactions such as citronellal cyclization,19 esterification,20 transfer hydrogenation,21 and cross-aldol condensation.22 Besides the acid property, the cage structure of MOF also allows it to act as the excellent support for metal nanoparticles.23–25 UiO-66, as a kind of traditional Zr-based MOF, contains metal nodes composed of a zirconium oxide complex bridged by terepthalic acid ligands and possesses the high thermal stability.26 In addition, it is well recognized that the defect sites of UiO-66, mainly the missing linkers, correlates closely with the catalytic performance, since the acid–base property arises mainly from the defect sites. Therefore, to achieve the high performance of UiO-66 in catalysis, more defects are desirable to be created. In common, the defects in UiO-66 can be tailored via adjusting the synthesis conditions, including incorporation of modulators,27–29 thermal activation/dehydration,30 metal cation substitution31 and linker modification.32
In this paper, the utilizing of the mixture Zr-precursor of ZrOCl2·8H2O and ZrCl4 was firstly developed to create defect-rich UiO-66 and then Pd was loaded thereon to fabricate the multifunctional catalyst for acetone hydrogenation reaction. As expected, the catalytic performance results exhibited that a high active and stable catalyst was achieved, with the at least 1000 h stability with MIBK + MIBC selectivity of 84.87–93.07% and acetone conversion of 45.26–53.22%.
2. Experimental
2.1 Materials
All chemicals and reagents were obtained from commercial sources and used as received, unless otherwise stated. Specifically, zirconium chloride (ZrCl4), zirconyl chloride octahydrate (ZrOCl2·8H2O), glacial acetic acid (HAc), palladium chloride (PdCl2) and 1,4-benzenedicarboxylic acid (BDC) were purchased from Aladdin Reagent (Shanghai) Company of China, and anhydrous N,N-dimethylformamide (DMF) sodium chloride (NaCl2) and methanol was purchased from Sinopharm Chemical Reagent Co., Ltd.2.2 Catalyst preparation
UiO-66 was prepared following a procedure previously reported33 with slight modifications. In a typical synthetic process, 3.28 g of ZrCl4 and 2.32 g of BDC were first added in 160 mL of DMF. Then, 1 mL of deionized water and 24 mL of HAc were successively dropped into the mixture and stirring was initiated until all precursors were completely dissolved. The obtained mixture was transferred to a 500 mL of hydrothermal reaction vessel with Teflon liner, and kept at 120 °C for 24 h under static conditions. After cooling to room temperature, the solid MOF were collected via centrifugation and thoroughly washed with a mixture of methanol and DMF (v/v = 1:4) for three times, and methanol for two times. Finally, the obtained sample was calcined in N2 atmosphere under 180 °C for 3 h and denoted by UiO-66.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :y). The synthesis technique of UiO-66 (R = x:y) was similar to that of UiO-66, except replacing ZrCl4 with the mixture Zr-precursor. Typically, mix ZrOCl2·8H2O and ZrCl4 in a mass ratio of 0:1, 1:1, 3:1, 7:1, 9:1, 15:1, 19:1 and 1:0 respectively. The obtained samples were denoted by UiO-66 (R = x:y), and x:y represented the mass ratio of ZrOCl2·8H2O to ZrCl4.:y) catalysts. The Pd catalysts were prepared by a wet impregnation technique. At room temperature, UiO-66 or UiO-66 (R = x:y) was added into the Na2PdCl4 aqueous solution and the mixture was subsequently stirred for 4 h, followed by reduction by a KBH4 solution for 30 min and washing with ethanol aqueous solution (volume ratio 1:1) to neutral. Eventually, vacuum drying at 50 °C was carried out. The obtained samples were denoted by Pd/UiO-66 and Pd/UiO-66 (R = x:y) catalysts, respectively.
:y). The synthesis technique of UiO-66 (R = x:y) was similar to that of UiO-66, except replacing ZrCl4 with the mixture Zr-precursor. Typically, mix ZrOCl2·8H2O and ZrCl4 in a mass ratio of 0:1, 1:1, 3:1, 7:1, 9:1, 15:1, 19:1 and 1:0 respectively. The obtained samples were denoted by UiO-66 (R = x:y), and x:y represented the mass ratio of ZrOCl2·8H2O to ZrCl4.:y) catalysts. The Pd catalysts were prepared by a wet impregnation technique. At room temperature, UiO-66 or UiO-66 (R = x:y) was added into the Na2PdCl4 aqueous solution and the mixture was subsequently stirred for 4 h, followed by reduction by a KBH4 solution for 30 min and washing with ethanol aqueous solution (volume ratio 1:1) to neutral. Eventually, vacuum drying at 50 °C was carried out. The obtained samples were denoted by Pd/UiO-66 and Pd/UiO-66 (R = x:y) catalysts, respectively.2.3 Catalyst characterizations
Fourier transform infrared (FT-IR) spectra was measured on a Nicolet 6700 Fourier transform spectrometer (Nicolet Magana Co.). Scanning electron microscopy (SEM) images was recorded on JSM-7610FPlus. A structural characterization was obtained by X-ray diffraction (XRD). XRD patterns of catalysts were recorded between 5° and 50° (2θ) on a Bruker D8 Advance A25 diffractometer with monochromatic Cu Kα radiation (λ = 0.154 nm). Thermogravimetric (TG) analysis was carried out in a TA Instruments TGA Q500 at a heating rate of 10 °C min−1 to 800 °C in air. The measurements of the acidity and basicity of the catalysts were accomplished by Autosorb-iQ-C equipped with a thermal conductivity detector. The sample was pretreated in helium gas at 400 °C for 2 h and then cooled down to 50 °C. The surface of the materials was saturated with NH3 (or CO2) for 60 min. TPD profile of NH3 (or CO2) was recorded at the rate of 10 °C min−1 from 50 °C to 800 °C by a He flow. Pd mass content was measured with a PerkinElmer Inductively Coupled Optical Emission Spectrometer (ICP), model Optima 5300DV, equipped with a peristaltic pump and across-flow nebulizer, coupled to a Ryton double pass spray chamber of the Scott type. Before analysis, Pd/UiO-66 (R = x:y) was immersed in aqua regia solution (v(HCl + HNO3)/vH2O = 20:30) to dissolve Pd into solution. The CO chemisorption measurements were performed in MICROMERITICS AutoChem II 2920 equipment. Before analysis, the catalysts were reduced under 10% H2/Ar flow at 200–250 °C for 3 h, and subsequently under He (50 mL min−1) flow at 150 °C for 1 h to blow away H2 adsorbed on the surface of catalysts. Then, CO chemisorption analysis using double isotherm methodology was performed at 35 °C and metal dispersion (or the particle size) was calculated. Potentiometric acid–base titrations were completed with a Metrohm Titrando 905 autotitrator equipped with Dosino 800 20 and 10 mL dosing units. X-ray photoelectron spectroscopy (XPS) was performed using an ESCALAB250Xi instrument with an Al Kα X-ray source (1486.6 eV), under about 2 × 10−9 mbar at room temperature and a pass energy of 20 eV.
2.4 Catalytic testing
Acetone hydrogenation reaction was performed in a batch reactor with an inner volume of 40 mL. The reactor was equipped with a mechanically driven agitator, thermocouples, and inlet and outlet gas purge lines. Typically, 5 mL of acetone and experimentally desired amount of catalyst were added to the reactor chamber and mixed. The reactor was tightly sealed and purged three times with H2 to remove air. Then the reactor was pressurized with H2 at 0.75 MPa, and the reaction proceeded at various temperatures and time. After the reaction finished, cold-water quenching was applied to the reactor to stop the reaction. The liquid product was separated from the solid residue (if formed) and the catalyst by filtration.The performance of acetone hydrogenation was also tested in a continuous trickle-bed reactor using 6 mL catalyst. The reaction conditions, reaction temperature, hydrogen pressure, liquid hour space velocity (LHSV), and feed ratio of acetone to H2, were optimized. The long-term run of the catalyst in the trickle-bed reactor was performed under the following conditions: 0.18 mL min−1 of acetone flow, 12 mL min−1 of hydrogen flow, 120 °C, 2.0 MPa and LHSV of 1.8 h−1. A Shimadzu GC-2010 gas chromatograph (GC) equipped with a flame ionization detector (FID) and a 30 m × 0.25 mm × 0.25 μm Insert Cap WAX capillary column was used to determine the amounts of acetone, MIBK, MIBC, MO, DAA, and others in the product. Total analysis time was 20 min. The definition of acetone conversion (xA), selectivity to MIBK + MIBK (SBK+BC) and MIBK + MIBC yield (YBK+BC) were calculated according to the following formulas.
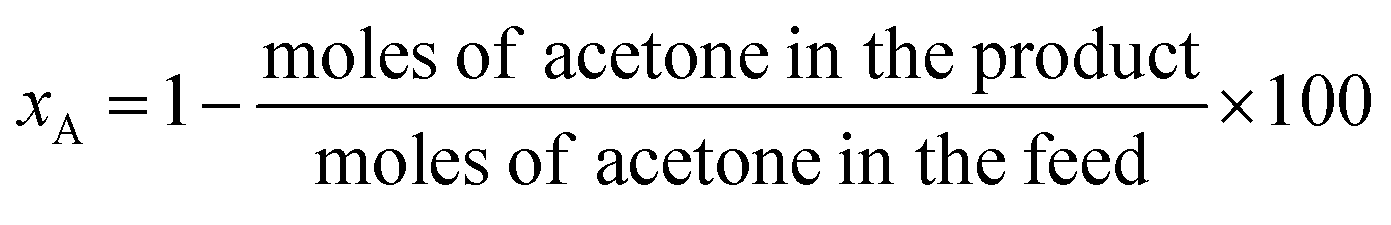
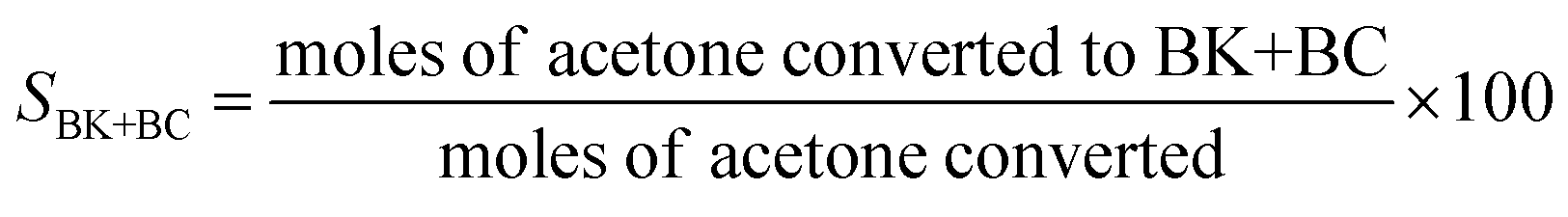
| YBK+BC = xA × SBK+BC/100 |
3. Results and discussion
3.1 Catalytic performance
The catalytic performance of the Pd catalysts loaded on various UiO-66 (R = x:y) for acetone hydrogenation in the batch reactor are shown in Table 1. It is observed that Pd/UiO-66 delivers an acetone conversion of 25.31%, and a MIBK + MIBC selectivity of 94.00%. Comparatively, Pd/UiO-66 (R = 1:0) exhibits an acetone conversion of 34.00%, and a MIBK + MIBC selectivity of 92.72%. This indicates that both UiO-66 and UiO-66 (R = 1:0) possess the acid–base property for acetone condensation. By increasing the ratio of ZrOCl2·8H2O to ZrCl4 in the raw material from 1:1 to 19:1, the acetone conversion of Pd/UiO-66 (R = x:y) catalyst increases firstly and then drops gradually. In addition, the selectivity increases at the beginning and then varies little. Among them, Pd/UiO-66 (R = 15:1) exhibits the highest MIBK + MIBC yield of 51.95% with an acetone conversion of 54.44% and a MIBK + MIBC selectivity of 95.38%.
| Catalyst | Acetone conversion (%) | Selectivity (%) | MIBK + MIBC yield (%) | |||||
|---|---|---|---|---|---|---|---|---|
| MIBK | MIBC | MO | DAA | IPA | Others | |||
| a T = 120 °C, reaction time = 2 h. IPA is the abbreviation of isopropanol. Others is mainly C9+. 0.5 wt% Pd loading. | ||||||||
| Pd/UiO-66 | 25.31 | 92.67 | 1.33 | 0.31 | 2.77 | 1.52 | 1.40 | 23.79 |
| Pd/UiO-66 (R = 1:1) |
11.18 | 85.38 | 0.46 | 0.10 | 7.54 | 5.35 | 1.17 | 9.59 |
| Pd/UiO-66 (R = 3:1) |
20.46 | 87.54 | 0.89 | 0.81 | 3.08 | 4.80 | 2.88 | 18.09 |
| Pd/UiO-66 (R = 7:1) |
37.12 | 91.32 | 2.93 | 0.93 | 1.24 | 1.45 | 2.13 | 34.98 |
| Pd/UiO-66 (R = 9:1) |
39.02 | 91.96 | 2.56 | 1.04 | 1.35 | 1.26 | 1.83 | 36.88 |
| Pd/UiO-66 (R = 15:1) |
54.44 | 90.90 | 4.48 | 0.64 | 0.56 | 0.31 | 3.11 | 51.95 |
| Pd/UiO-66 (R = 19:1) |
34.64 | 90.88 | 1.96 | 0.93 | 1.88 | 1.50 | 2.85 | 32.16 |
| Pd/UiO-66 (R = 1:0) |
34.00 | 90.82 | 1.90 | 0.65 | 1.80 | 3.00 | 1.83 | 31.52 |
The catalytic performances of Pd/UiO-66 (R = 15:1) catalysts with different palladium loading are shown in Table 2. UiO-66 (R = 15:1) support itself shows the acetone conversion of 29.51%, the MIBK + MIBC selectivity of 9.72%, and MO selectivity of 68.13%. This indicates that UiO-66 (R = 15:1) support itself indeed only has acid–base function, without hydrogenation function. When Pd loading is higher than 0.3 wt%, MO is almost completely converted. With the increase of the amount of Pd, there is a climax of the MIBK + MIBC yield. It is noted that too much Pd is not beneficial to the MIBK + MIBC yield due to the decreased acetone conversion, probably due to the coverage of the acid–base sites by Pd. Therefore, 0.5 wt% Pd/UiO-66 (R = 15:1) catalyst is chosen here for further investigation in the trickle-bed reactor. We compare the performance of Pd/UiO-66 (R = 15:1) with those reported in a batch liquid phase process as shown in Table S1.† It is seen that Pd/UiO-66 (R = 15:1) delivers the best performance among them under the mild reaction conditions (Table S1†).34–37
:1) in the batch reactora
| Catalyst | Acetone conversion (%) | Selectivity (%) | MIBK + MIBC yield (%) | |||||
|---|---|---|---|---|---|---|---|---|
| MIBK | MIBC | MO | DAA | IPA | Others | |||
| a T = 120 °C, reaction time = 2 h. IPA is the abbreviation of isopropanol. Others is mainly C9+. | ||||||||
| UiO-66 (R = 15:1) |
29.50 | 9.51 | 0.21 | 68.13 | 2.41 | 0.32 | 19.42 | 2.87 |
| 0.3wt% Pd/UiO-66 (R = 15:1) |
47.07 | 80.95 | 3.37 | 6.94 | 0.80 | 0.24 | 7.70 | 39.69 |
| 0.4wt% Pd/UiO-66 (R = 15:1) |
53.64 | 87.60 | 4.63 | 2.59 | 0.77 | 0.07 | 4.34 | 46.99 |
| 0.5wt% Pd/UiO-66 (R = 15:1) |
54.44 | 90.90 | 4.48 | 0.64 | 0.56 | 0.31 | 3.11 | 51.95 |
| 0.6wt% Pd/UiO-66 (R = 15:1) |
52.67 | 88.60 | 4.86 | 1.07 | 0.67 | 0.28 | 4.52 | 49.22 |
| 0.7wt% Pd/UiO-66 (R = 15:1) |
52.22 | 90.71 | 2.52 | 1.91 | 0.70 | 0.34 | 3.82 | 48.68 |
| 0.8wt% Pd/UiO-66 (R = 15:1) |
38.62 | 90.72 | 2.54 | 2.11 | 1.34 | 0.23 | 3.06 | 36.02 |
| 0.9wt% Pd/UiO-66 (R = 15:1) |
31.71 | 90.60 | 2.03 | 2.21 | 1.34 | 1.47 | 2.35 | 29.37 |
The reaction conditions, including the reaction temperature, hydrogen pressure, LHSV and mole ratio of C3H6O/H2, over 0.5 wt% Pd/UiO-66 (R = 15:1) were optimized via the orthogonal experiments with four factors and four levels. The effects of these factors on the catalytic performance are shown in Tables S2 and S3.† It is seen that as for 0.5 wt% Pd/UiO-66 (R = 15:1) operated in a trickle-bed reactor, the reaction temperature is the most significant one among the four factors. More experiment results in Table 3 indicate that the increase of reaction temperature from 80 °C up to 160 °C leads to an increase of the acetone conversion from 32.95% to 56.89%, accompanied by a volcanic-changed MIBK selectivity. The decline of MIBK selectivity at higher temperature is due to the formation of heavier condensation products (C9+).3 In addition, it is noted that more MIBK is hydrogenated further to MIBC with the increase of temperature. Except the reaction temperature, the mole ratio of C3H6O/H2 is the second significant factor on the catalyst performance. Considering the yield of MIBK + MIBC, the mole ratio of C3H6O/H2 should be maintained at 1:2, consistent with the theoretic value. Therefore, the optimal reaction conditions of Pd/UiO-66 (R = 15:1) catalyst is reaction temperature of 120 °C, hydrogen pressure of 2 MPa, LHSV of 1.8 h−1 and mole ratio of C3H6O/H2 of 1:2, which deliver the highest MIBK + MIBC yield of 51.95%.
:1) at different temperature in the trickle-bed reactora
| Temperature (°C) | Acetone conversion (%) | Selectivity (%) | MIBK + MIBC yield (%) | |||||
|---|---|---|---|---|---|---|---|---|
| MIBK | MIBC | MO | DAA | IPA | Others | |||
| a The catalyst volume is 6 mL, P = 2.0 MPa, H2 flow rate = 12 mL min−1, and time on stream = 2 h. IPA is the abbreviation of isopropanol. Others is mainly C9+. | ||||||||
| 80 | 32.95 | 87.03 | 1.47 | 5.70 | 3.38 | 0.12 | 2.30 | 29.16 |
| 90 | 35.13 | 86.11 | 1.60 | 6.95 | 2.48 | 0.11 | 2.75 | 30.81 |
| 100 | 40.43 | 87.08 | 2.89 | 4.80 | 1.18 | 0.52 | 3.53 | 36.37 |
| 110 | 49.13 | 91.63 | 3.72 | 0.63 | 1.11 | 0.61 | 2.30 | 46.84 |
| 120 | 54.44 | 90.90 | 4.48 | 0.64 | 0.56 | 0.31 | 3.11 | 51.95 |
| 130 | 54.59 | 89.11 | 4.53 | 0.62 | 0.52 | 1.68 | 3.54 | 50.11 |
| 140 | 54.53 | 87.69 | 4.55 | 0.59 | 0.49 | 2.87 | 3.81 | 50.30 |
| 150 | 55.15 | 85.43 | 4.91 | 0.57 | 0.40 | 3.40 | 5.29 | 48.82 |
| 160 | 56.89 | 82.69 | 4.97 | 0.56 | 0.41 | 3.87 | 7.50 | 49.87 |
The long-term stability of 0.5 wt% Pd/UiO-66 (R = 15:1) catalyst in the trickle-bed reactor under the optimal conditions was investigated (Fig. 1). After 1000 h run, the acetone conversion drops by 7.96% from 53.22% to 45.26%, and the MIBK + MIBC selectivity drops by 8.22% from 93.09% to 84.87%. Zhu4 reported that Pd/S (PS-GO) catalyst was tested for 1000 h and the acetone conversion dropped from 50.85% to 39.71% by 10.14% and the MIBK selectivity dropped from 77.86% to 73.47% by 4.39%. Duan3 showed that the TiO2–SiO2/SiO2 (F)-1&Pd/Cor catalyst was active for 300 h with a MIBK selectivity of 83–93% at 26–36% acetone conversion. Comparatively, the catalyst in this paper displays the better stability than those reported in literature, presenting the prospect for industrial application.
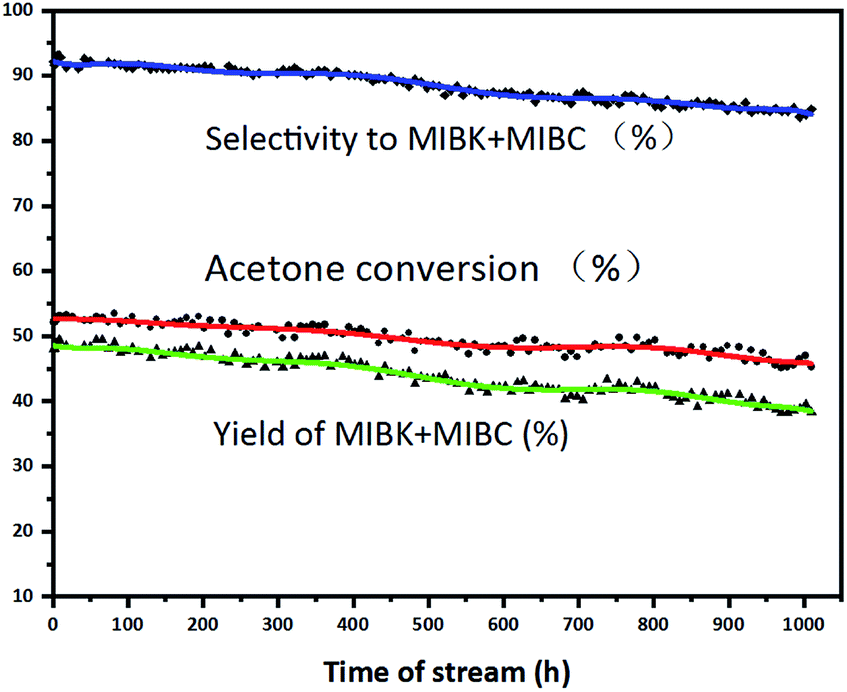 | ||
| Fig. 1 Stability of 0.5 wt% Pd/UiO-66 (R = 15:1) catalyst. Reaction conditions: volume of catalyst = 6 mL, T = 120 °C, P = 2.0 MPa, H2 flow rate = 12 mL min−1 and LHSV = 1.8 h−1. | ||
3.2 Characterizations of catalysts
The XRD patterns of UiO-66, UiO-66 (R = 15:1) and Pd/UiO-66 (R = 15:1) in Fig. 2 exhibit the characteristic peaks of the typical UiO-66 structure.16 No additional peaks can be observed, indicating that no impure crystalline phases are formed. These suggest that the variation of the Zr precursors and the Pd loading process have no influence on the bulk structure of UiO-66. In addition, it is noted that no characteristic peak of Pd at 2θ = 40.1° and 46.7° can be detected due to the quite low loading amount of Pd.
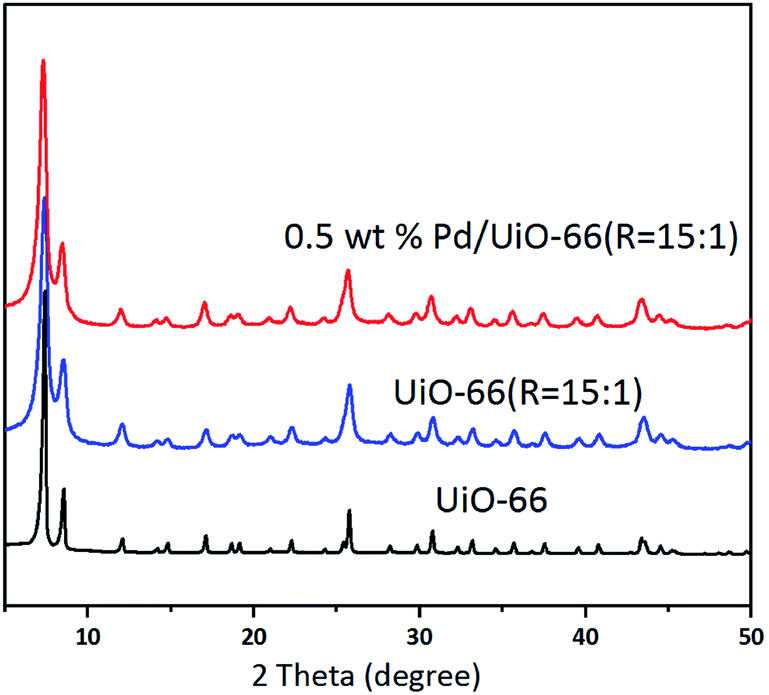 | ||
| Fig. 2 XRD patterns of UiO-66, UiO-66 (R = 15:1) and Pd/UiO-66 (R = 15:1). | ||
The SEM images of UiO-66 and UiO-66 (R = 15:1) are shown in Fig. 3. Obviously, two samples display different morphology. The UiO-66 granules display the cubic octahedron with the edge length size of around 150 nm (Fig. 3a and b). Comparatively, UiO-66 (R = 15:1) sample exhibits the amorphous morphology composed of flaky granules (Fig. 3c and d). It is noteworthy that although the macroscopic morphology of UiO-66 and UiO-66 (R = 15:1) seems quite different, both of them possess the same crystal structure revealed by XRD. It is known that the crystalline process could be accelerated and small UiO-66 particles with several nanometers were formed when using ZrOCl2·8H2O as Zr-precursor.37 Therefore, in this paper, the particle size of UiO-66 (R = 15:1) is much smaller than UiO-66. This is beneficial for the acetone hydrogenation reaction due to the readily accessibility of active sites on UiO-66 (R = 15:1).
 | ||
| Fig. 3 SEM images of UiO-66 (a and b) and UiO-66 (R = 15:1) (c and d). | ||
The BET specific surface area and the porous structure of UiO-66 and UiO-66 (R = 15:1) are evaluated by N2 adsorption–desorption experiment (Fig. 4 and Table S4†). UiO-66 exhibits the type I isotherm in the region of low relative pressure, indicating the main existence of micropores. The BET specific surface area and the total pore volume are estimated to be 1677 m2 g−1 and 0.88 cm3 g−1, respectively. The narrow distribution of the pore size justifies that the micropores come from the intrinsic structure of UiO-66. According to literature, the theoretical pore volume of UiO-66 is 0.77 cm3 g−1 and the surface area is 1160 m2 g−1.17,33,38 Comparatively, UiO-66 (R = 15:1) displays the type II isotherm with the hysteresis loop, suggesting the main existence of mesopores, which probably are the secondary particle piled pores of formed by flaky granules. This was further confirmed by the pore size distribution calculated from the N2 isotherms. Accordingly, the BET specific surface area of UiO-66 (R = 15:1) is 1068 m2 g−1, the total pore volume is 1.03 cm3 g−1 and the proportion of mesopores volume increases.
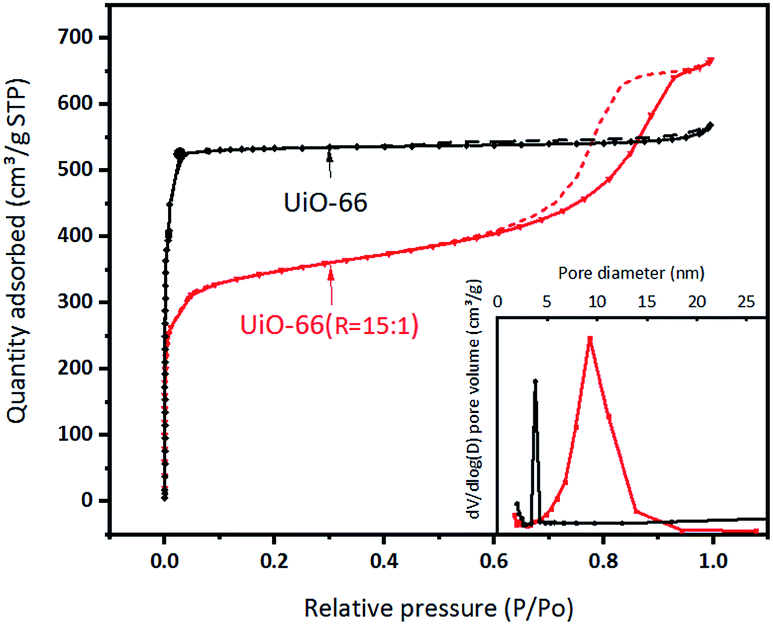 | ||
| Fig. 4 N2 adsorption–desorption isotherms and pore distribution curves of UiO-66 and UiO-66 (R = 15:1). | ||
The thermal stability of UiO-66 and UiO-66 (R = 15:1) determined by TG measurement is shown in Fig. 5. The TG curve of UiO-66 reveal three broad areas representing weight loss. The first area is linked to the removal of water between 30 °C and 100 °C. And the second one is caused by the removal of the residual solvent N,N-dimethylformamide and/or of the unreacted terephthalic acid between 100 °C and 480 °C. The third one is assigned to the decomposition of the framework between 400 °C and 600 °C. The final solid product after thermal treatment in air is 6ZrO2.41 It is observed from Fig. 5 that UiO-66 and UiO-66 (R = 15:1) have almost the same TG profiles, suggesting their similar thermal stability.
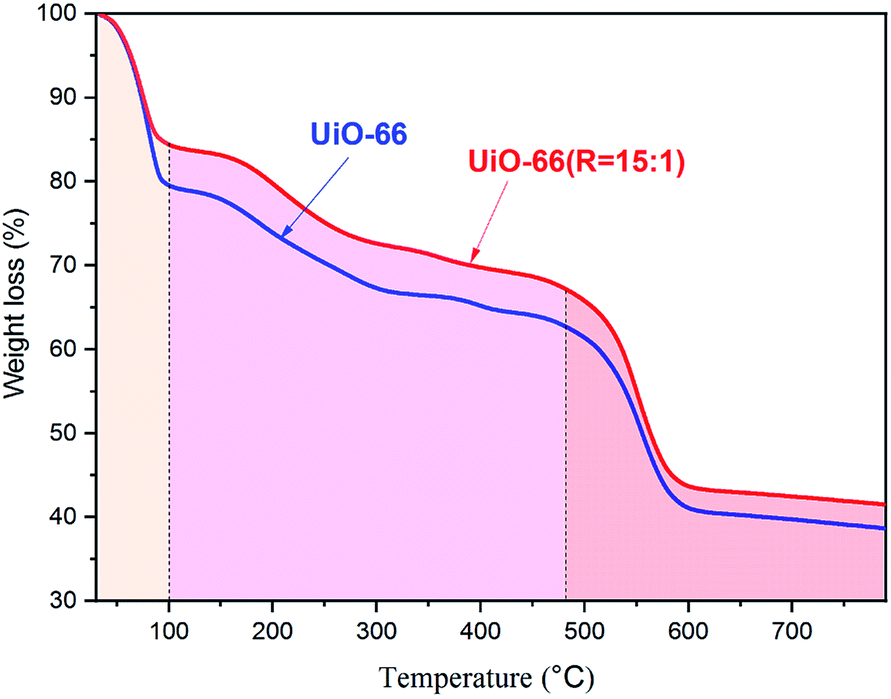 | ||
| Fig. 5 TG curves of UiO-66 and UiO-66 (R = 15:1) in air. | ||
The FT-IR spectra of UiO-66 and UiO-66 (R = 15:1) in Fig. 6 show that both samples have the characteristic peaks of UiO-66. The sharp peak at around 1400 cm−1 represents the symmetric stretching of the O–C–O carboxylate group of BDC ligands, and the asymmetric stretching vibration is located at about 1587 cm−1. The peak at 1670 cm−1 is assigned to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the BDC ligands. The peak at 1503 cm−1 represents the CC vibrations of benzene rings.16 At the lower range of wavenumbers, the strong peaks at 848 cm−1, 745 cm−1, and 656 cm−1 are attributed to the vibrations of O–H and C–H bonds in the BDC ligands. These peaks ranged in 848–656 cm−1 overlap with the Zr–O modes. In addition, the gap between the two peaks of around 1400 cm−1and 1500–1700 cm−1 can give us the information about the coordination modes of BDC and Zr atoms.39,40 When the gap is larger than 200 cm−1, BDC acts as a monodentate ligand to Zr atoms. And the gap in the range of 130–200 cm−1 indicates that BDC behaves as a bridging ligand. Alternatively, when the gap is in the range of 50–150 cm−1, BDC acts as a bidentate ligand to Zr atoms.42,43 Fig. 6 shows that there is a strong splitting of 187 cm−1 with a weak one of 103 cm−1 for UiO-66 and UiO-66 (R = 15:1). This indicates that the coordination modes of BDC in UiO-66 and UiO-66 (R = 15:1) are mainly consisted of bridging ligands with a small amount of bidentate ligands.
O of the BDC ligands. The peak at 1503 cm−1 represents the CC vibrations of benzene rings.16 At the lower range of wavenumbers, the strong peaks at 848 cm−1, 745 cm−1, and 656 cm−1 are attributed to the vibrations of O–H and C–H bonds in the BDC ligands. These peaks ranged in 848–656 cm−1 overlap with the Zr–O modes. In addition, the gap between the two peaks of around 1400 cm−1and 1500–1700 cm−1 can give us the information about the coordination modes of BDC and Zr atoms.39,40 When the gap is larger than 200 cm−1, BDC acts as a monodentate ligand to Zr atoms. And the gap in the range of 130–200 cm−1 indicates that BDC behaves as a bridging ligand. Alternatively, when the gap is in the range of 50–150 cm−1, BDC acts as a bidentate ligand to Zr atoms.42,43 Fig. 6 shows that there is a strong splitting of 187 cm−1 with a weak one of 103 cm−1 for UiO-66 and UiO-66 (R = 15:1). This indicates that the coordination modes of BDC in UiO-66 and UiO-66 (R = 15:1) are mainly consisted of bridging ligands with a small amount of bidentate ligands.
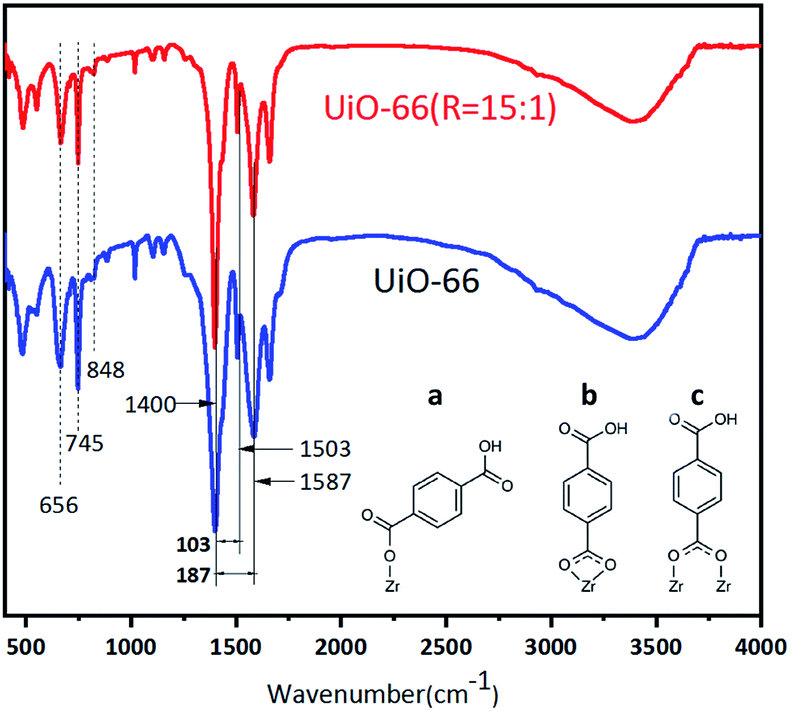 | ||
| Fig. 6 FT-IR spectra of UiO-66 and UiO-66 (R = 15:1). (a) Monodentate, (b) bidentate, (c) bridging. | ||
The defects sites in the MOF materials could be identified by acid–base titration method.44,45 The acid–base titration curves of UiO-66 and UiO-66 (R = 15:1) are measured as shown in Fig. 7. To more accurately estimate the equivalence points in the titration curves, the first derivative curves are fit to Lorentzian-type peaks. It is observed that both samples display the titration curves with three apparent inflection points, which correspond to three types of protons (μ3-OH, Zr–OH2, and Zr–OH).46,47 Among them, Zr–OH2 and Zr–OH sites are generated as the defects sites by missing linker in the synthesis process. Therefore, the missing linker number can be further calculated from Fig. 7. The calculation process is shown in Tables S5 and S6.† And the missing linker numbers of UiO-66 and UiO-66 (R = 15:1) are estimated to be 1.58 and 1.89, respectively. This suggests that more defect sites are introduced into UiO-66 (R = 15:1) than UiO-66 via utilizing the precursor mixture of ZrCl4 and ZrOCl2·8H2O. In addition, according to the calculation results, the percentage of Zr–OH in defects increases from 65.9% to 83.4% in UiO-66 (R = 15:1) compared with UiO-66.
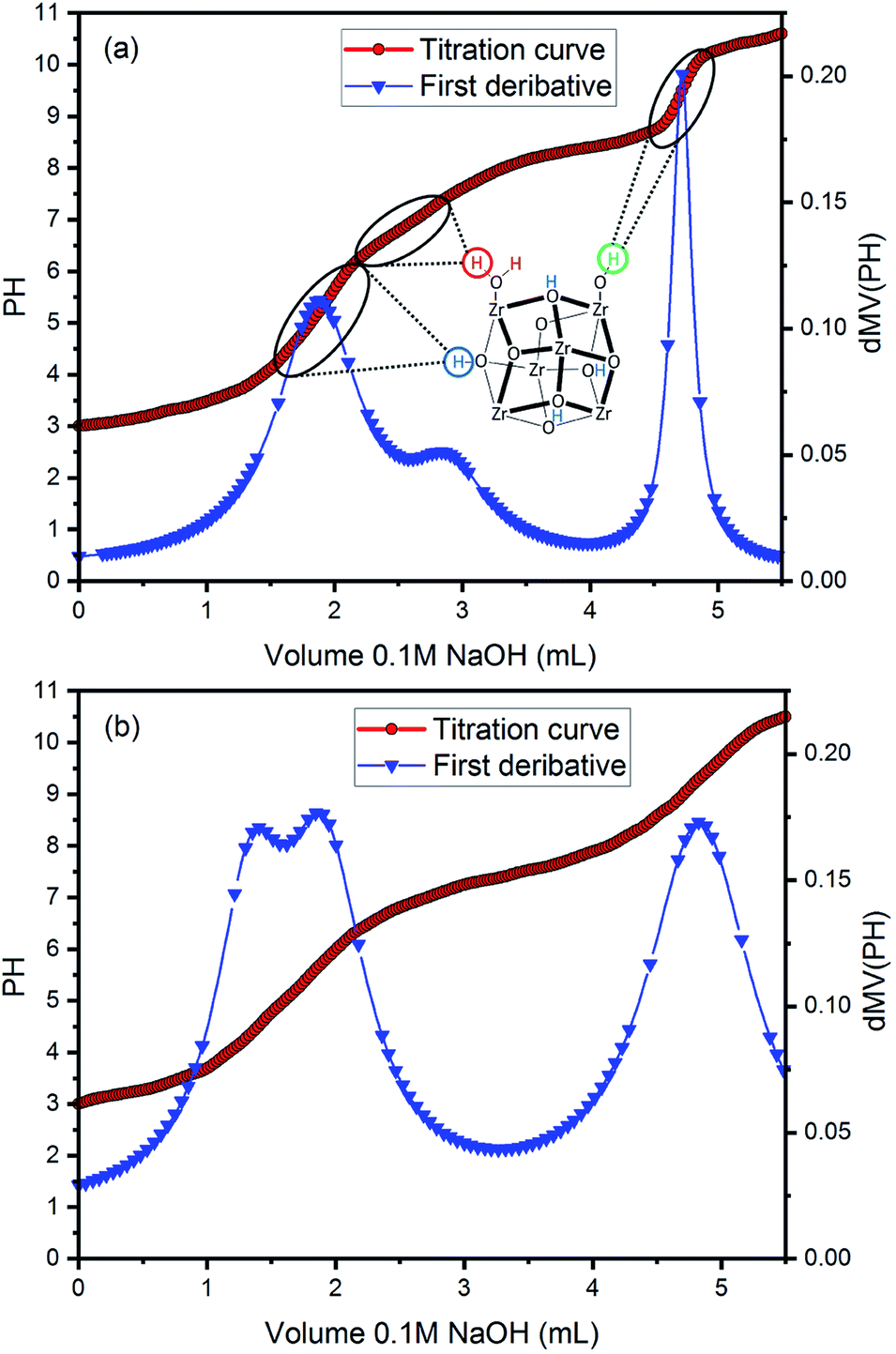 | ||
| Fig. 7 Acid–base titration curve and first derivative curve for (a) UiO-66 and (b) UiO-66 (R = 15:1). | ||
To correlate the nature of defect sites of UiO-66 and UiO-66 (R = 15:1) and the acid–base property required for acetone condensation, the NH3-TPD and CO2-TPD were carried out as shown in Fig. 8. It is well known that both Zr–OH and Zr–OH2 sites belong to typical Brønsted acid.48 The 6-fold-coordinated Zr sites on the Zr6 nodes of UiO-66 are Lewis acid. In Fig. 8a, the broad peak can be deconvoluted into three peaks. The peak centered at about 290 °C can be attributed to the Lewis acid of Zr sites, and the other two peaks can be assigned to the Brønsted acid sites of Zr–OH and Zr–OH2. It is noted that the amount of Brønsted acid sites in UiO-66 (R = 15:1) is obviously increased, while the strength of Lewis acid sites is almost constant compared with UiO-66. It was previously reported that the change of specific coordination number of Zr has a minor effect on the Lewis acid strength of the active site.49–52
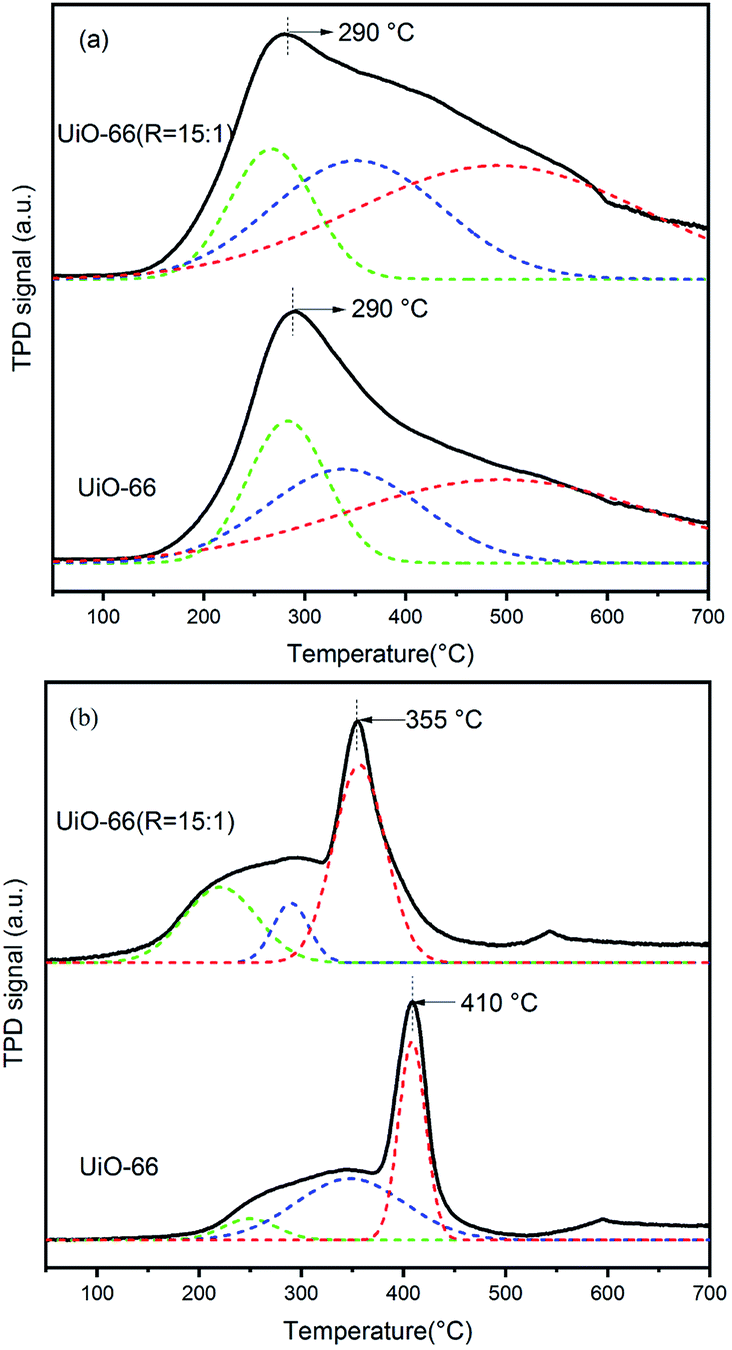 | ||
| Fig. 8 NH3-TPD (a) and CO2-TPD (b) profiles of UiO-66 and UiO-66 (R = 15:1). | ||
As for base sites, the μ3-O on the Zr6 nodes was normally regarded as the Lewis base sites. When the missing linker number is higher, the unsaturated coordinative number of Zr atoms becomes higher and leads to the shortened length of Zr–O bond, which further results in the weakening of the Lewis basicity. This can be reflected by the peak shift to lower temperature from 410 °C of UiO-66 to 355 °C of UiO-66 (R = 15:1) in the CO2-TPD profiles in Fig. 8b. In addition, the other broad peak shifting to lower temperature could be attributed to other kinds of basic sites related to oxygen in the framework.
In a word, compared with UiO-66, UiO-66 (R = 15:1) exhibits the obviously increased amount of Brønsted acid sites, mainly Zr–OH and Zr–OH2 sites, caused by the increasing of missing linker number. For acetone condensation and dehydration reactions, except for base site, it is intensively reported that Brønsted acid is the typical catalytic site for the reaction, for instance sulfonate ion.5 In this paper, although the basicity of UiO-66 (R = 15:1) is weakened, the enhanced Brønsted acid amount leads to the enhancement of the cascade reaction.
The comprehensive contrast of the characterizations of UiO-66 and UiO-66 (R = 15:1) has been presented in the above text, which help us to understand the reason for the better performance of Pd/UiO-66 (R = 15:1). To further reveal the nature of the activity drop of Pd/UiO-66 (R = 15:1) after 1000 h run, the TG curves, FT-IR spectra, NH3-TPD and CO2-TPD profiles of the fresh and used catalysts are presented as follows. From the contrast of the TG curves of the fresh and used Pd/UiO-66 (R = 15:1) in Fig. 9, it is seen that an increased weight loss 26% ranged in 100–500 °C is observed, suggesting the existence of the carbonaceous accumulation on the used catalyst. Except this, the crystal structure of UiO-66 (R = 15:1) has not changed even after 1000 h run, since the decomposition temperature of the framework at 500 °C is the same as the fresh one. In addition, the structure stability of UiO-66 (R = 15:1) can also been identified by the contrast of FT-IR spectra in Fig. S1.† The change of the acid–base property of fresh and used Pd/UiO-66 (R = 15:1) is revealed by NH3-TPD and CO2-TPD profiles in Fig. S2.† After 1000 h run, the Brønsted acid amount of the used Pd/UiO-66 (R = 15:1) decreases, and the acidity and basicity is strengthened as shown in Fig. S2b.† About the reason for the change of acidity and basicity, we assume that the long chain alcohol produced during the reaction may be co-adsorbed by the adjacent zirconium atoms, leading to the reduction of the acid sites. Then, the alcohol further deprotonate to form alkoxide, and the proton is transferred to the μ3 oxygen to strengthen its basicity. Hence, compared with the fresh catalyst, the CO2 desorption peak on the used Pd/UiO-66 (R = 15:1) shifts to a higher temperature.
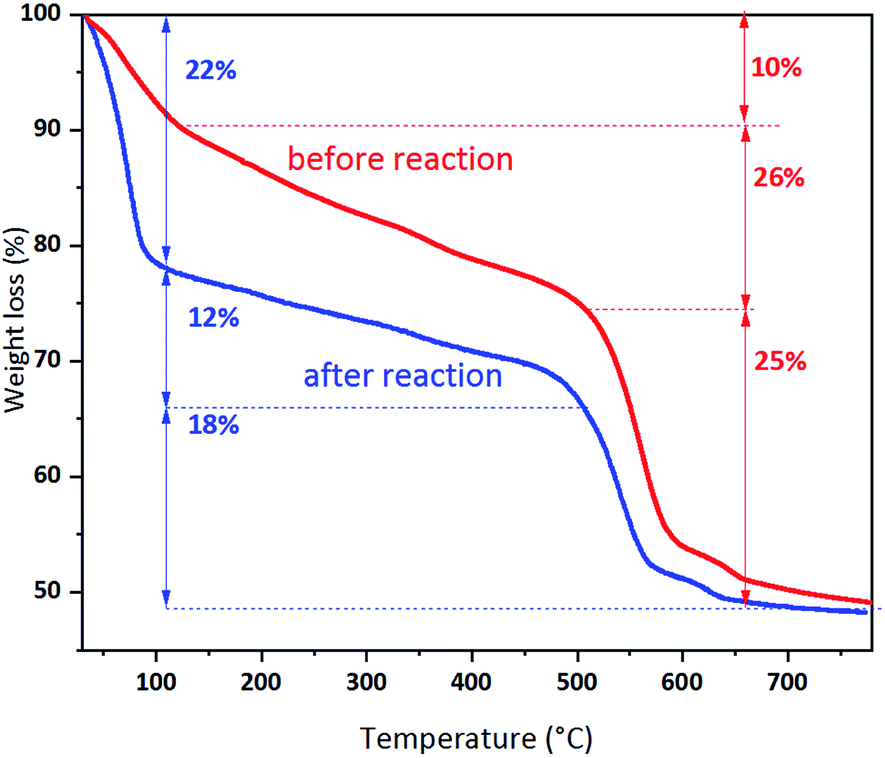 | ||
| Fig. 9 TG curves of Pd/UiO-66 (R = 15:1) before and after 1000 h reaction in air. | ||
In addition, the surface chemical state of Pd, Zr and O on Pd/UiO-66 and Pd/UiO-66 (R = 15:1) was characterized by XPS as shown in Fig. S3.† The existence of C, O, Pd and Zr elements on Pd/UiO-66 and Pd/UiO-66 (R = 15:1) is identified in Fig. S3(a).† The Pd 3d and Zr 3p spectra are present in Fig. S3(b).† The two strong peaks in Fig. S3(b)† are assigned to Zr 3p3/2 and Zr 3p1/2, respectively.53 And the two weak peaks at 335.6 and 341.1 eV can be attributed to Pd 3d5/2 and Pd 3d3/2, respectively, which are partially overlapped by the two peaks of Zr 3p. The similar XPS results were previously reported by Guan53 and Yang.54 It is observed in Fig. S3(b)† that the binding energy of Pd species on both samples is slightly higher than the standard value of 335.3 eV of Pd metal, probably attributed to the interaction between Pd and the UiO-66 framework.54 As for Zr 3d and O1s spectra in Fig. S3(c and d),† no difference is observed between Pd/UiO-66 and Pd/UiO-66 (R = 15:1).
Furthermore, the Pd mass percent and Pd dispersion of fresh Pd/UiO-66, fresh and used Pd/UiO-66 (R = 15:1) were characterized by ICP and CO chemisorption as shown in Table S7.† The two samples present the close Pd mass percent. And the average particle size of Pd on Pd/UiO-66 is estimated to be 3.05 nm, slightly larger than that of 2.20 nm on Pd/UiO-66 (R = 15:1). Considering the contrast of the Pd average particle size and the surface chemical state between Pd/UiO-66 and Pd/UiO-66 (R = 15:1), we can regard that there is no obvious difference in view of the Pd particles loaded thereon. In addition, the condensation of acetone is normally regarded as the rate-determining step, instead of the hydrogenation step, for the overall cascade reactions. Therefore, the gap of catalytic performance between Pd/UiO-66 and Pd/UiO-66 (R = 15:1) can be mainly ascribed to the difference of acid and base sites.
As for the fresh Pd/UiO-66 (R = 15:1), the Pd mass percent is 0.486% with an average Pd particle size of 2.20 nm. Comparatively, as for the used Pd/UiO-66 (R = 15:1), the Pd mass percent drops to 0.442% (by 9.05%) accompanied by the slight aggregation of Pd particle to 2.73 nm. The change of the Pd mass and particle size is quite smaller than those reported in literature,3,4 This suggests that the phenomena of metal loss and aggregation of Pd particles can be efficiently inhibited by loading Pd in the small cavities of UiO-66 (R = 15:1), resulting in the excellent stability in long term run. Therefore, the reason for the deactivation of Pd/UiO-66 (R = 15:1) can be mainly attributed to the carbonaceous accumulation, instead of the Pd loss and aggregation. The used Pd/UiO-66 (R = 15:1) catalyst after 1000 h run was regenerated in our experiments. It was calcined in air flow with 10 mL min−1 of at 150 °C for 1 h followed by reduced in H2 flow with 10 mL min−1 at 150 °C for 1 h. Then, the catalytic performance of the regenerated catalyst was tested again under the same reaction conditions. The regenerated catalyst exhibited an acetone conversion of 53.23% and a MIBK + MIBC selectivity of 94.19%, close to the initial performance. This indicates the deactivation of Pd/UiO-66 (R = 15:1) is reversible and can be readily regenerated by calcination. This is valuable for the prolongation of the service life of Pd catalyst.
4. Conclusions
The cage structure, high thermal stability and the adjustability of acid–base property of UiO-66 allowed it to act as an excellent candidate to build multifunctional catalysts. In this paper, the defective UiO-66 was synthesized with the optimal ratio 15:1 of ZrOCl2·8H2O to ZrCl4 as Zr-precursors firstly. Then, Pd was loaded thereon and tested in a typical cascade reaction, the one-pot synthesis of methyl isobutyl ketone and methyl isobutyl methanol from acetone and hydrogen. The Pd/UiO-66 (R = 15:1) catalyst exhibited a stable run for at least 1000 h with a MIBK + MIBC selectivity of 84.87–93.09% and acetone conversion of 45.26–53.22% in a continuous trickle-bed reactor. Compared with pure UiO-66, the improvement of the activity can be attributed to the increased Brønsted acid amount caused by the introduced defect sites. And the confinement of UiO-66 (R = 15:1) structure efficiently prevented the Pd loss and aggregation during the long term run. The contrast of the characterizations of the fresh and used Pd/UiO-66 (R = 15:1) indicated that the deactivation of the catalyst was attributed to carbonaceous accumulation on the catalyst surface. This belongs to the reversible deactivation and the catalyst could be easily regenerated by calcination. This work supplied a new alternative for the design and utilization of industrial catalysts for MIBK and MIBC synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant 21676303) and the Fundamental Research Funds for the Central Universities of Central South University (Grant 2020zzts415).References
- N. Takarroumt, M. Kacimi, F. O. Bozon-Verduraz and L. F. Liotta, J. Mol. Catal. A: Chem., 2013, 377, 42–50 CrossRef CAS.
- M. Mediavilla, L. Melo, Y. Díaz, A. Albornoz and A. Llanos, Microporous Mesoporous Mater., 2008, 116, 627–632 CrossRef CAS.
- D. Hui, W. Zhihui, C. Lifeng, L. Baining and Z. Yonghua, Ind. Eng. Chem. Res., 2018, 57, 12358–12366 CrossRef.
- Y. Zhu, B. Lin, Y. Hu, Z. Cai and Z. Yonghua, Mol. Catal., 2019, 478, 110609 CrossRef.
- W. Nicol and E. L. D. Toit, Chem. Eng. Process., 2004, 43, 1539–1545 CrossRef CAS.
- S. Talwalkar and S. Mahajani, Appl. Catal. A., 2006, 302, 140–148 CrossRef CAS.
- E. D. Toit, R. Schwarzer and W. Nicol, Chem. Eng. Sci., 2004, 59, 5545–5550 CrossRef.
- L. M. Gandía, R. Malm, R. Marchand, R. Conanec, Y. Laurent and M. Montes, Appl. Catal. A., 1994, 114, L1–L7 CrossRef.
- J. J. Gamman, S. D. Jackson and F. A. Wigzell, Ind. Eng. Chem. Res., 2010, 49, 8439–8443 CrossRef CAS.
- S. M. Yang and Y. M. Wu, Appl. Catal. A., 2000, 192, 211–220 CrossRef CAS.
- A. Chakrabarti and M. M. Sharma, React. Polym., 1993, 20, 1–45 CrossRef CAS.
- A. C. C. Rodrigues, J. L. F. Monteiro and C. A. Henriques, C. R. Chim., 2009, 12, 1296–1304 CrossRef CAS.
- R. Unnikrishnan and S. Narayanan, Mol. Catal., 1999, 144, 173–179 CrossRef CAS.
- V. Chikán, á. Molnár and K. Balázsik, J. Catal., 1999, 184, 134–143 CrossRef.
- G. Waters, O. Richter and B. Kraushaar-Czarnetzki, Ind. Eng. Chem. Res., 2006, 45, 6111–6117 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS.
- Y. Bai, Y. Dou, L. H. Xie, W. Rutledge, J. R. Li and H. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- F. Vermoortele, M. Vandichel, B. V. d. Voorde, R. Ameloot, M. Waroquier, P. V. V. Speybroeck and P. D. Edition, Angew. Chem. Int. Edit., 2012, 124, 4887–4890 CrossRef.
- F. G. Cirujano, A. Corma and F. X. L. Xamena, Catal. Today, 2014, 257, 213–220 CrossRef.
- A. H. Valekar, K. H. Cho, S. K. Chitale, D. Y. Hong, G. Y. Cha, U. H. Lee, D. Hwang, C. Serre, J. S. Chang and Y. Hwang, Green Chem., 2016, 18, 4542–4552 RSC.
- J. Hajek, M. Vandichel and B. V. Voorde, J. Catal., 2015, 331, 1–12 CrossRef CAS.
- H. Fei and S. Cohen, Chem. Commun., 2014, 50(37), 4810–4812 RSC.
- L. Chen, R. Luque and Y. Li, Chem. Soc. Rev., 2017, 46, 4614–4620 RSC.
- Q. Yang, Q. Xu and H. L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gándara and H. B. Bürgi, Angew. Chem., 2015, 128, 11314–11319 CrossRef.
- O. V. Gutov, M. G. Hevia, E. C. Escudero-Adán and A. Shafir, Inorg. Chem., 2015, 54, 8396–8400 CrossRef CAS.
- Z. Hu, I. Castano, S. Wang, Y. Wang, Y. Peng, Y. Qian, C. Chi, X. Wang and D. Zhao, Cryst. Growth Des., 2016, 16, 2295–2301 CrossRef CAS.
- L. Yuan, M. Tian, J. Lan, X. Cao and X. Wang, Chem. Commun., 2018, 54, 370–373 RSC.
- G. C. Shearer, S. Forselv, S. Chavan and S. Bordiga, Top. Catal., 2013, 56, 770–782 CrossRef CAS.
- Z. Niu, Q. Guan, Y. Shi, Y. Chen, Q. Chen, Z. Kong, P. Ning, S. Tian and R. Miao, New J. Chem., 2018, 42, 19764–19770 RSC.
- Y. Wu, Q. Li, D. J. Rutstrom, M. Zhuravleva and C. Melcher, Phys. Status Solidi Rapid Res. Lett., 2017, 1700403, 1–4 Search PubMed.
- D. Jiang, G. Fang, Y. Tong, X. Wu, Y. Wang, D. Hong, W. Leng, Z. Liang, P. Tu, L. Liu, K. Xu, J. Ni and X. Li, ACS Catal., 2018, 8, 11973–11978 CrossRef CAS.
- R. Ma, Y. Li, G. Wu, Y. He, J. Feng, Y. Zhao and D. Li, Chinese. J. Catal., 2018, 39, 1384–1394 CrossRef CAS.
- F. Al-Wadaani, E. F. Kozhevnikova and I. V. Kozhevnikov, J. Catal., 2008, 257, 199–205 CrossRef CAS.
- D. Wang, Z. Wang, B. Zahng, H. Sun, P. Wu, Z. Gao and W. Yang, Chem. React. Eng. Technol., 2017, 33(2), 89–297 Search PubMed.
- P. Wang, S. Bai, J. Zhao, P. Su, Q. Yang and C. Li, ChemSusChem, 2012 Dec, 5, 2390–2396 Search PubMed.
- L. Hao, X. Li, M. J. Hurlock, X. Tu and Q. Zhang, Chem. Commun., 2018, 54, 11817–11820 RSC.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. R. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- P. Ghosh, Y. J. Colon and R. Q. Snurr, Chem. Commun., 2014, 50, 11329–11331 RSC.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- F. Verpoort, T. Haemers, P. Roose and J. P. Maes, Appl. Spectrosc., 1999, 53, 1528–1534 CrossRef CAS.
- K. B. Lausund and O. Nilsen, Nat. Commun., 2016, 7, 13578 CrossRef CAS.
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A., 2016, 4, 1479–1485 RSC.
- T. J. Bandosz, M. Laskoski, J. Mahle, G. Mogilevsky and G. W. Wagner, J. Phys. Chem. C, 2012, 116, 11606–11614 CrossRef CAS.
- T. G. Glover, G. W. Peterson, J. B. DeCoste and M. A. Browe, Langmuir, 2012, 28, 10478–10487 CrossRef CAS.
- G. Ye, D. Zhang, X. Li, K. Leng, W. Zhang, J. Ma, Y. Sun, W. Xu and S. Ma, ACS. Appl. Mater. Inter., 2017, 9, 34937–34943 CrossRef CAS.
- Y. Liu, R. C. Klet, J. T. Hupp and O. Farha, Chem. Commun., 2016, 52, 7806–7809 RSC.
- L. Hao, X. Li, M. Hurlock and X. Tu, Chem. Commun., 2018, 54, 11817–11820 RSC.
- B. Bueken, N. V. Velthoven and T. Willhammar, Chem. Sci., 2017, 8, 3939–3948 RSC.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gándara, H.-B. Bürgi and O. M. Yaghi, Angew. Chem., 2015, 127, 11314–11319 CrossRef.
- J. Hajek, B. Bueken and M. Waroquier, ChemCatChem, 2017, 9, 2203–2210 CrossRef CAS.
- Q. Guan, B. Wang, X. Chai, J. Liu, J. Gu and P. Ning, Fuel, 2017, 205, 130–141 CrossRef CAS.
- Y. Yang, D. Deng, D. Sui, Y. Xie, D. Li and Y. Duan, Nanomaterials, 2019, 9, 1698–1704 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09217g |
| This journal is © The Royal Society of Chemistry 2021 |