
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe lattice reconstruction of Cs-introduced FAPbI1.80Br1.20 enables improved stability for perovskite solar cells†
Shuang Chen,
Lu Pan,
Tao Ye,
Nuo Lei,
Yijun Yang and
Xi Wang*
and
Xi Wang*
Department of Physics, School of Science, Key Laboratory of Luminescence and Optical Information, Ministry of Education, Beijing Jiaotong University, Beijing, 100044, China. E-mail: xiwang@bjtu.edu.cn
First published on 20th January 2021
Abstract
Inorganic–organic hybrid perovskite solar cells (PSCs) have stirred up a new research spree in the field of photovoltaics due to its high photoelectric conversion efficiency and simple preparation process. In recent years, the research of inorganic–organic hybrid PSCs has been widely reported, among which FA+/Cs+ PSCs are especially outstanding. However, there are few reports explaining the lattice structural change mechanism of CsxFA1−xPbI1.80Br1.20 PSCs from the view of chemical bonds. In this work, a facile method of 15% Cs+ cations partially substituting FA+ cations has been presented to enhance the structural stability and photovoltaic performances of FAPbI1.80Br1.20 PSCs. The partial incorporation of Cs+ in FAPbI1.80Br1.20 resulted in a more beneficial tolerance factor and inhibited the deep defect state of elemental Pb. More importantly, it inhibited the phase transition from the cubic black α-phase to the hexagonal yellow δ-phase of FAPbI1.80Br1.20. Moreover, the power conversion efficiency (PCE) of Cs0.15FA0.85PbI1.80Br1.20 PSCs achieved a substantial improvement. The stability also achieved a remarkable promotion, which was demonstrated by X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD) and Nuclear Magnetic Resonance (NMR). These analyses indicate that 15% Cs+ can induce the lattice shrinkage, reduce the specific traps and inhibit the phase transition, thus improving the structural stabilities of Cs0.15FA0.85PbI1.80Br1.20 PSCs under atmosphere and calefaction. These results provide an effective way for fabricating stable and efficient inorganic–organic perovskite solar cells with promising properties.
Introduction
Since the path-breaking work reported in 2009,1 perovskite solar cells (PSCs) have attracted wide attention because of their fast-increasing power conversion efficiency (PCE). In addition, the highest PCE quickly reached a certified record value of 25.2%.2 Generally, an organic–inorganic hybrid perovskite can be expressed with an ABX3 formula composed of organic or inorganic cation A, divalent metal ion B and halogen ion X, where A = (CH3NH3+ methylammonium (MA); CH3(NH2)2+ formamidinium (FA); Cs+),3–13 B = (Pb2+; Sn2+; Bi3+),14,15 and X = (Cl−; Br−; I−).5,16 There are two key factors in the development of perovskite solar cells: stability and PCE. However, the PSCs of MAPbI3 are degraded when there is contact with moisture, and trapped states are formed when light is induced. In addition, the release of FA+ or Cs+ is not as significant as MA+.17 Based on the situation above, their potential for commercial applications are diminishing.18–24 Compared with the MAPbI3 perovskite, FAPbI3 has better performance due to its optimal redshifted bandgap and higher absorption coefficient.25,26 However, the structural stability of pure FAPbI3 is poor at room temperature since it will transform into the non-photoactive perovskite hexagonal δ-phase (“yellow phase”),27–29 influenced by the solvent and environment humidity conditions.5,30,31 Consequently, thermal and air instabilities are the largest bottlenecks for the commercialization of FAPbI3-based PSCs.Materials in the configuration of ABX3 can form different crystal structures according to the size of the A cation, and the interaction with the corner-sharing BX6 octahedron. In 1926, Goldschmidt introduced the concept of the tolerance factor to describe the mismatch between equilibrium bond lengths of A–X and B–X bonds in perovskite structures.32 The tolerance factor of the inorganic–organic hybrid halide perovskite tends to present an orthogonal structure when t < 0.8, and a hexagonal structure when t > 1, which are both photoinactive.31,32 Materials with tolerance factors in the range of 0.8 to 1.0 belong to regular cubic perovskite structures (photoactive). It is well known that FAPbI3 has a large tolerance factor (t > 1), and is stabilized as a hexagonal structure. Simultaneously, CsPbI3 has a small tolerance factor (t < 0.8) and is more stable as an orthorhombic structure at room temperature.32 Therefore, mixing A site cations has become an effective strategy within the organic–inorganic perovskite system.31 In the cation-mixed perovskite, Cs with the atomic diameter of 1.81 Å is much smaller than that of FA (2.79 Å) and MA (2.70 Å).33 Therefore, we reduced the tolerance factor from t > 1 to 0.8 < t < 1.0 by introducing a smaller Cs cation into the lattice of FAPbI3. We also determined the appropriate amount of addition, aiming to obtain the black perovskite phase. To date, several studies have investigated the mixing of cations with Cs.34 Choi et al. proposed a Cs+/MA+ mixture and theoretically proved that embedding a small amount of Cs+ in the MAPbI3 structure can improve the film morphology and light absorption, and increase the energy difference between the valence band of the perovskite and the LUMO level of PCBM, yielding a PCE value of 7.7%.35 Park et al. reported that the Cs+/FA+ mixture showed good temperature and humidity stability.25 Snaith et al. used a mixture of FA+ and Cs+-replaced MA+ to depress the light-induced phase segregation.7 Yi et al. explained the improvement of the structural stability. They pointed out that due to the stability of the entropy, Cs+ can effectively promote the crystallization of the black phase in the FA-based perovskite.36 From the conclusions of these investigations, the introduction of Cs+ is beneficial for the material to form a cubic black perovskite phase. However, few studies focused on explaining why the addition of cesium ions into FAPbI3 improves the lattice structural stability, and there are few explanations on the underlying mechanisms.
In this work, CsxFA1−xPbI1.80Br1.20 perovskite compounds were synthesized to reveal the influences on the structural stability of perovskite with different addition amounts of Cs+. Perovskite crystal configurations with different proportions of Cs+/FA+ are discussed, and XPS, XRD and NMR are combined to further prove our conjecture. From XPS spectra, with the increase of the Cs ion addition, the depth defects of Pb gradually disappear, and the crystal structure tends to change to the cubic phase. Subsequent XRD measurements show that the Cs+ doping shrinks the lattice, and inhibits the transition to the non-photoactive δ-phase. As a result, we determine a perfect ratio of Cs+/FA+ and successfully prepared Cs0.15FA0.85PbI1.80 Br1.20, which exhibited a relatively high PCE of 16.2% when used in PSCs. Moreover, a cubic black perovskite phase was formed, and its stability is greatly improved.
Experimental section
Materials and fabrication
Formamidinium iodide (FAI, >99.5%) was obtained from Greatcell Solar Australia Pty., Ltd. Dimethylsulfoxide (DMSO, 99.7%) and N,N-dimethylformamide (DMF, 99.8%) were purchased from Acros Organics. Chlorobenzene (99.8%), titanium(IV) isopropoxide (TTIP, 99.999%) and cesium iodide (CsI, 99.999%) were obtained from Sigma-Aldrich. Lead(II) iodide (PbI2, 99.99%) and lead(II) bromide (PbBr2, 99%) were purchased from Tokyo Chemical Industry (TCI). All chemicals were used as received without further purification.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 mass ratio of mesoporous titanium dioxide and anhydrous ethanol. For the deposition of the mixed formamidinium–Cs lead iodide (CsxFA1−xPbI1.80Br1.20) films, CsxFA1−xPbI1.80Br1.20 solution was prepared by precursor solutions containing FAI (1.3 M), CsI (1.3 M), PbI2 (1.3 M) and PbBr2 (1.3 M) in a DMF:DMSO ratio of 4:1 (v/v). The solution was then stirred for 5 minutes and heated at 50 °C until it was completely dissolved, and formed a transparent and uniform yellow solution. The spiro-OMeTAD solution was prepared by dissolving 74 mg spiro-OMeTAD, 28.5 μL 4-tert-butylpyridine (Sigma-Aldrich), 17.5 μL of a stock solution of 520 mg mL−1 lithium bis-(trifluoromethylsulphonyl) imide (Sigma-Aldrich) in acetonitrile (Sigma-Aldrich), and 29 μL of a stock solution of 200 mg mL−1 tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)-cobalt(III) tris(bis(trifluoromethylsulfonyl) imide) (Sigma-Aldrich) in acetonitrile in 1 mL anhydrous chlorobenzene.
:6 mass ratio of mesoporous titanium dioxide and anhydrous ethanol. For the deposition of the mixed formamidinium–Cs lead iodide (CsxFA1−xPbI1.80Br1.20) films, CsxFA1−xPbI1.80Br1.20 solution was prepared by precursor solutions containing FAI (1.3 M), CsI (1.3 M), PbI2 (1.3 M) and PbBr2 (1.3 M) in a DMF:DMSO ratio of 4:1 (v/v). The solution was then stirred for 5 minutes and heated at 50 °C until it was completely dissolved, and formed a transparent and uniform yellow solution. The spiro-OMeTAD solution was prepared by dissolving 74 mg spiro-OMeTAD, 28.5 μL 4-tert-butylpyridine (Sigma-Aldrich), 17.5 μL of a stock solution of 520 mg mL−1 lithium bis-(trifluoromethylsulphonyl) imide (Sigma-Aldrich) in acetonitrile (Sigma-Aldrich), and 29 μL of a stock solution of 200 mg mL−1 tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)-cobalt(III) tris(bis(trifluoromethylsulfonyl) imide) (Sigma-Aldrich) in acetonitrile in 1 mL anhydrous chlorobenzene.Characterization
The surface morphology and section morphology of the samples were obtained by scanning electron microscope (SEM; SU 8010) at 10 kV. Powder X-ray diffraction (XRD, PANalytical Empyrean diffractometer) with a Cu-Kα source (λ = 1.5406 Å) measurements were used to analyze the crystal features of the samples. X-ray photoelectron spectroscopy (XPS) analysis was performed on ESCALAB250XI (Thermo Fisher Scientific). The ultraviolet visible (UV-Vis) absorption spectra were collected on a UV-Vis spectrophotometer (UV-2600). Photoluminescence (PL) spectra and time-resolved PL (TRPL) were measured with an Edinburgh Instrument FLS 980 spectrophotometer. The liquid-state 1H-NMR spectroscopy measurements and solid-state 13C-NMR spectroscopy measurements were collected on a Bruker Avance III 400 HD. J–V characteristics were measured by using a solar simulator (SAN-EI Electric XES-301S) and a Keithley 2400 sourcemeter under standard simulated AM 1.5 illumination. The solar cells were masked with metal apertures to define the active areas, which were typically 0.09 or 0.08 cm2.Results and discussion
Morphology and optical characterization of CsxFA1−xPbI1.80Br1.20 (x = 0.05, 0.15, 0.25) perovskites
As can be seen in the scanning electron microscope (SEM) top view images shown in Fig. 1a–c, the average grain size was larger for the Cs0.15FA0.85PbI1.80Br1.20 (hereinafter referred to “Cs0.15”) films versus the Cs0.05FA0.95PbI1.80Br1.20 (hereinafter referred to “Cs0.05”) films. However, compared to the perovskite films with 25% Cs ions (see Fig. 1c), the Cs0.25FA0.75PbI1.80Br1.20 (hereinafter referred to “Cs0.25”) perovskite grain gradually breaks into a smaller particle size. It demonstrates that the appropriate amount of Cs+-doped perovskite film has good compactability and crystallinity. In the XRD spectra, it can be seen that the full-width at half-maximum of the Cs0.15 sample is less than that of the Cs0.05 sample, which corresponds to the grain enlargement in SEM. However, under high doping concentration (Cs0.25 sample), Cs+ could not be completely included in the crystal lattice of the FAPbI3 perovskite,37,38 and the Cs0.25 curve in XRD is shifted to a high angle, which leads to the decrease of the grain size.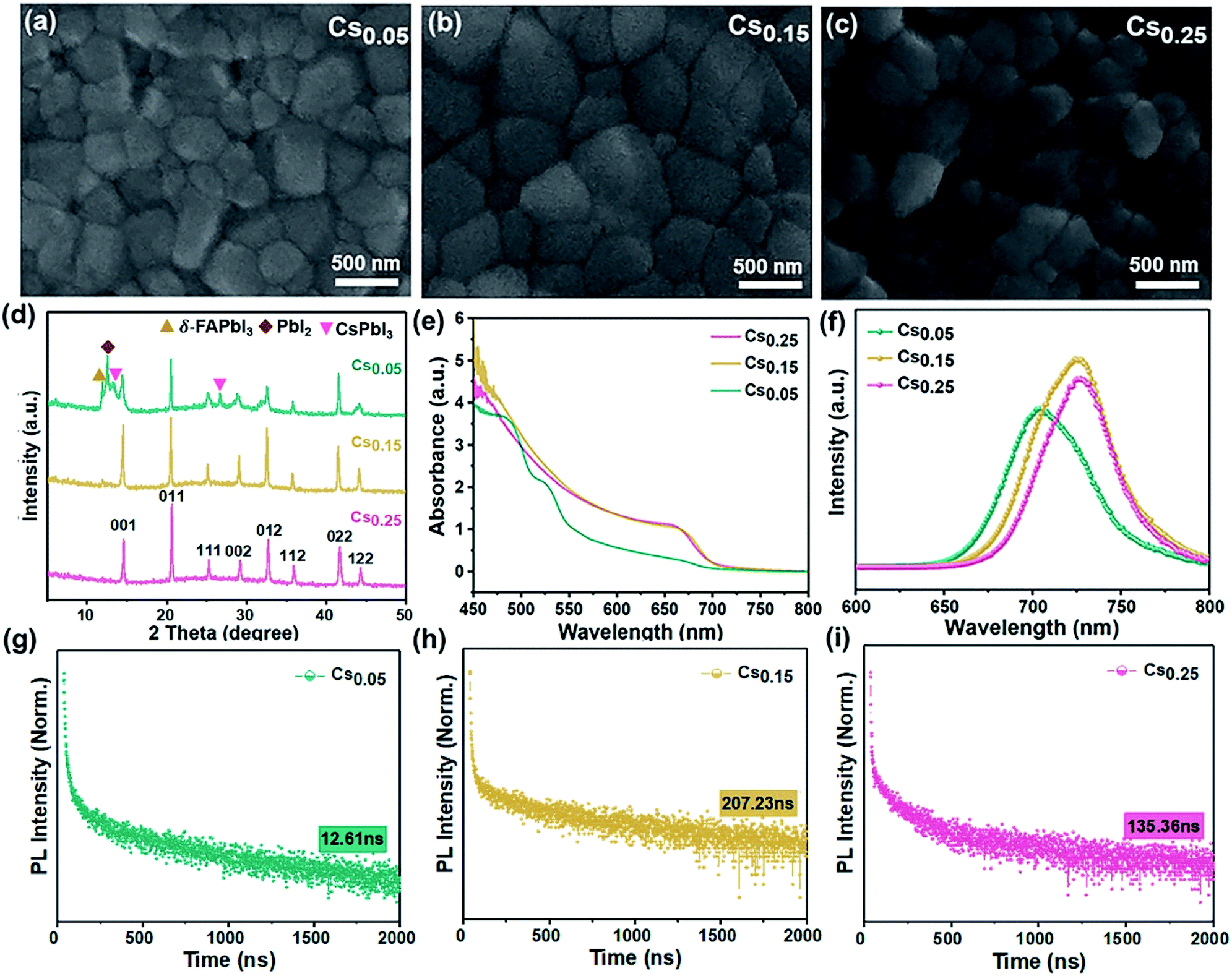 | ||
| Fig. 1 SEM images, XRD and optical characterization of the CsxFA1−xPbI1.80Br1.20 compounds. (a–c). SEM images of the perovskite films prepared by perovskite seed growth with different concentration ratios. (a) Cs0.05FA0.95PbI1.80Br1.20 (Cs0.05). (b) Cs0.15FA0.85PbI1.80Br1.20 (Cs0.15). (c) Cs0.25FA0.75PbI1.80Br1.20 (Cs0.25). (d) XRD spectra of the perovskite upon addition of Cs investigating the series CsxFA1−xPbI1.80Br1.20. The figure represents the perovskite crystal face of the α-phase. (e) The corresponding UV-Vis absorbance spectra and (f) PL spectra. (g–i) TRPL spectra with a log scale for the y axis. (g) Cs0.05FA0.95PbI1.80Br1.20 (Cs0.05). (h) Cs0.15FA0.85PbI1.80Br1.20 (Cs0.15). (i) Cs0.25FA0.75PbI1.80Br1.20 (Cs0.25). | ||
To understand the modification of the FAPbI1.80Br1.20 perovskite solar cells after Cs ion doping, the evolution of the crystal structure is revealed. Fig. 1d is the XRD curves of Cs0.05, Cs0.15, Cs0.25 films after annealing at 100 °C. Consistent with the SEM results, the Cs0.05 films contained the peaks of the yellow δ-phase of FAPbI3, impurity PbI2 and CsPbI3. However, for the Cs0.15 films, the δ-phase was effectively suppressed and the full-width at half-maximum (FWHM) of the XRD peaks were decreased accordingly because of the increased peak intensity, indicating an increase in the film crystallinity. As the Cs+ content is 25%, the diffraction peak shifts to the direction of a larger diffraction angle (Fig. S1†), which indicates a further reduction of the unit cell volume (Fig. 1c),39 and it produces the lattice distortion. The XRD spectra of other samples with different Cs+/FA+ ratios (Fig. S2 and S3†) show the appearance of the impurity phase with the hexagonal non-photoactive δ-phase and PbI2 after annealing. However, there is no obvious formation of the photoactive pure FAPbI3 phase in the XRD spectra (Fig. S4†). Comparing Fig. 1d and S4†, it is noted that the peaks of the δ-FAPbI3 non-photoactive phase appear in the original FAPbI1.80Br1.20 and Cs0.05, but do not appear in the diffraction patterns of the Cs0.15 and Cs0.25-doped samples, showing that the formation of the non-photoactive phase is inhibited after doping.40 However, the relative intensity of the Cs0.25 perovskite peak decreases compared with the Cs0.15 sample, proving that excess Cs+ could destroy the perovskite crystal lattice. In conclusion, the introduction of 15% Cs+ content can be proved to better forming the cubic black perovskite phase. This demonstrates that the introduction of an appropriate proportion of Cs+ can thermodynamically inhibit the phase transition and impurities generation, which is conducive to the formation of the α perovskite phase, thus reaching a new equilibrium state of the system, which is beneficial to preparing pure and defect-free perovskite films.41
The effect of adding different proportions of Cs+ on the absorption performance of the FAPbI1.80Br1.20 perovskite was studied by optical absorption method (ultraviolet-visible (UV-Vis) absorption). As can be seen from Fig. 1e, influenced by the structural instability, the light absorption performance of the Cs0.05 sample is poorer and the photoactive phase is not formed. As for the Cs0.15 and Cs0.25 samples, an identical absorption at 450–800 nm can be seen. However, the Cs0.15 film showed a stronger absorption over all wavelengths compared with the Cs0.25 sample.
On this basis, the steady state photoluminescence (PL) spectra of the Cs+-doped perovskite films with different proportions were studied (see Fig. 1f). The samples of Cs0.15 and Cs0.25 were accompanied by slight blue shifts, corresponding to the local micro-zoom of XRD spectra (Fig. S1†), which show that the characteristic peak of perovskite at 14° shifts with the increase of the doping amount of Cs ions. Moreover, the PL intensity of the Cs0.15 sample is the highest, proving that there are fewer internal traps and the charge recombination rate is slow. We further performed time-resolved PL (TRPL) decay measurements on various perovskite films (Fig. 1g–i). The TRPL carrier lifetime of the Cs0.05, Cs0.15, and Cs0.25 samples are 12.61 ns, 207.23 ns and 135.36 ns (see the measurements in Table 1), respectively. The life expectancy of the Cs0.15 perovskite is the longest, indicating that the replacement of FA+ with 15% Cs+ could decrease the defects and restrain the nonradiative recombination,42,43 leading to enhanced charge carrier transport. Upon further increasing the Cs+ doping concentration to 25%, the carrier lifetime is sharply decreased, which may be due to the introduction of defects by excess Cs ions.
| Perovskite | A1 (%) | τ1 (ns) | A2 (%) | τ2 (ns) | Lifetime (ns) |
|---|---|---|---|---|---|
| Cs0.25FA0.75PbI1.80Br1.20 | 19.80 | 2.30 | 1.29 | 164.00 | 135.36 |
| Cs0.15FA0.85PbI1.80Br1.20 | 44.30 | 2.47 | 10.84 | 216.77 | 207.23 |
| Cs0.05FA0.95PbI1.80Br1.20 | 32.84 | 6.35 | 0.02 | 261.87 | 12.61 |
In addition, this work studied the optical properties of the samples with other Cs+ proportions. From the UV-Vis absorption, PL and TRPL measurements, comparing the CsxFA1−xPbI1.80Br1.20 perovskite (x ≠ 0) and pure FA-based perovskite (x = 0), the pure FA-based perovskite has a low light absorption intensity with no photoactive phase formation after annealing (Fig. S5a†). However, the CsxFA1−xPbI1.80Br1.20 (x = 0.10, 0.20) perovskites showed the remarkably steep absorption onset (Fig. S6a and S7a†). In addition, the PL spectrum was widened with a relatively shorter lifetime (Fig. S5b and c; S6b and c; S7b and c†). The detailed lifetime data values are shown in Table S1†, which indicate that the electron and hole transmission speed are slow and the carrier recombination is fast, reflecting the formation of defects.
Thermal stability tests of CsxFA1−xPbI1.80Br1.20 (x = 0.05, 0.15, 0.25) perovskites
The XRD results and photographs of the perovskite films with different Cs+ ratios heated at 200 °C from 0 to 2 hours in the glove box are shown in Fig. 2. The Cs0.05 and Cs0.25 perovskite films began to turn yellow after such intense thermal stress tests. From the detailed XRD spectra of the Cs0.05 and Cs0.25 samples, the intensity of the peaks on behalf of the δ-FAPbI3 phase increases with the increase of heating time (Fig. 2c, S8 and S9†). At the same time, the impurity phase of hexagon PbI2 (P63mc) appears in all three samples due to the incremental release of organic compounds and halides. In the process of heating at 200 °C, the perovskite phase gradually disappears, and impurities such as CsPbI3 and PbI2-FAI phases appear. On the other hand, the Cs0.15 perovskite film remained in the dark state and did not significantly fade. The hexagonal non-photoactive δ-phase cannot be seen in Fig. 2b, indicating that the degradation rate of the thin films is slower. Therefore, it can be concluded that the Cs0.15FA0.85PbI1.80Br1.20 perovskite film formed by 15% Cs+ substitution has higher thermal stability. | ||
| Fig. 2 Mixed-halide perovskite films heated on a 200 °C hotplate inside a N2-filled glovebox for 2 hours. XRD spectra of (a) Cs0.25FA0.75PbI1.80Br1.20 (Cs0.25), (b) Cs0.15FA0.85PbI1.80Br1.20 (Cs0.15), and (c) Cs0.05FA0.25PbI1.80Br1.20 (Cs0.05). The figure represents the perovskite crystal face of the α-phase. (d) Photo images. | ||
Air stability tests of the CsxFA1−xPbI1.80Br1.20 (x = 0.05, 0.15, 0.25) perovskites
All of these films are aged under natural environmental conditions (25 °C, air humidity 55–60%, and ambient light) without encapsulation. The morphology and structure changes of the films after aging can be observed in Fig. 3. The XRD patterns of Cs0.25 are shown in Fig. 3a. With the exposure time increased under an atmosphere of oxygen and moisture, the peaks assigned to δ-FAPbI3, CsPbI3 and PbI2-FAI emerged with enhanced intensity (more details are shown in Fig. S10†). This phenomenon occurs because the size mismatch between Cs+ and FA+ is large, resulting in phase separation. The results demonstrate that the release of FA+ from Cs0.25 is accompanied by a phase transition, which can be proved by the changes of the film state in Fig. 3d. In addition, the variation tendency in the sample of Cs0.15 (Fig. 3b) is almost the same with Fig. 3a. The main difference is that the intensity of the emerged PbI2 shows a downward trend with the increases in exposure time, and the peak intensity of δ-FAPbI3, CsPbI3, and PbI2-FAI is relatively low. The results show the lower release of FA+ and the lower number of phase transitions (Fig. 3b). In Fig. 3c (Cs0.05), there are peaks of δ-FAPbI3, CsPbI3, and PbI2 that appear at the beginning. Soon afterwards, the peak intensity of PbI2 declines and the peak intensities of δ-FAPbI3 and CsPbI3 increase, while the peak intensity of the α perovskite phase decreases. The phenomenon demonstrates that with the increase in the exposure time, the CsxFA1−xPbI1.80Br1.20 perovskite is gradually decomposed with FA+ released, leading to the appearance and enhancement of the δ-FAPbI3 and CsPbI3 peaks accompanied by a phase transition. It can be seen more intuitively from Fig. 3d that when 5% Cs+ and 25% Cs+ are introduced, the colour of the perovskite films change more obviously with longer exposure time in the air, and gradually changes from the mirror-like black surface state to a coarsened state, indicating an obvious degradation process from the cubic black perovskite to hexagonal yellow photoinactive phase. From Fig. 3, it can be concluded that the photoactive black phase of the Cs0.15 perovskite is entropy-stable at room temperature, thus inhibiting the formation of the hexagonal δ-phase of the FA-based perovskite. This explanation is consistent with recent reports of using Cs+/FA+ mixtures as effective cations.25,32,36 In addition, we evaluated the UV absorption characteristics of Cs0.05, Cs0.15 and Cs0.25 (Fig. S11–S13†). Fig. 3d shows the perovskite films of Cs0.05, Cs0.15 and Cs0.25 exposed to air from 0 to 72 hours. Combined with the optical absorption spectra of different Cs+ proportions, it can be clearly seen that the perovskite films without Cs+ (Fig. S5a†) and Cs0.05 (Fig. S11†) do not show the characteristics of the photoactive phase absorption, and the absorption intensity of Cs0.05 and Cs0.25 (Fig. S11 and S13†) shows a significant decline. On the other hand, Cs0.15 remained dark with no significant fading. Although some degradation is visible, it is far less pronounced than that for Cs0.05 and Cs0.25 (Fig. S12†). | ||
| Fig. 3 Stability test (environmental conditions: 25 °C, 55–60% humidity) XRD spectra of (a) Cs0.25FA0.75PbI1.80Br1.20 (Cs0.25), (b) Cs0.15FA0.85PbI1.80Br1.20 (Cs0.15), (c) Cs0.05FA0.95PbI1.80Br1.20 (Cs0.05) thin films stored for 120 hours. The figure represents the perovskite crystal face of the α phase. (d) Photo images. | ||
Therefore, this study proves that adding Cs+ is beneficial to inhibiting the yellow phase formation, thus enhancing the thermal stability and air stability of the FAPbI1.80Br1.20 perovskite.
X-ray photoelectron spectroscopy (XPS) spectra and crystal structure transformation mechanism of perovskites
Fig. 4a–c are schematic diagrams of the crystal structure transformation of the samples after introducing 5%, 15% and 25% Cs+ into the lattice of FAPbI3, respectively. It can be seen from Fig. 4a that when a small amount of Cs+ is introduced (Cs0.05), the perovskite structure is formed with a lattice distortion and a depth defect state of Pb, no cubic black phase perovskite structure is formed. The existence of a Pb–Pb bond in XPS also proves this point of view (Fig. 4g). When an excess amount of Cs+ is introduced (Cs0.25), the cubic structure is formed with the disappearance of the Pb elemental defects, but the arrangement of the crystal cells is not continuous (Fig. 4c). Consistent with the results in Fig. 4c, as confirmed in Fig. S1†, when the proportion of Cs+ increases from 15% to 25%, it can be seen that the intensity of the perovskite peak decreases except for the (011) peak, and especially (001) and (002) peaks. A large number of theoretical studies have found that the low exponent non-polar (001) surface of the perovskite structure has the most stable surface orientation. Comparing the relative intensities of the (001) and (011) crystal facets, it can be concluded that the Cs0.25 perovskite crystal facet is poorly ordered, forming a discontinuous crystal structure. A stable cubic perovskite crystal structure can only be formed when the appropriate proportion of 15% Cs+ is introduced and the tolerance factor is adjusted to the arrangement between 0.8 to 1.0 (Fig. 4b). At the same time, incorporating Cs+ into FAPbI3 can enhance the stability of α-FAPbI3. As can be seen from the hydrogen spectrum in Fig. 4d–f, with the increase of Cs+, Cs+ only acts on the functional group R = NH2+ in FA+, and the peak position is shifted to the left, proving that the electron cloud is distributed around the proton, causing a low density and a low shielding effect. In addition, it can be seen that the ability to attract electrons of the material increases, resulting in a weaker FA–I bond. The NMR samples are tested immediately after preparation, and the solvent is deuterium instead of DMSO with no other protons influence. Since there is only one carbon element in FA+, no significant change is observed in the carbon spectrum (Fig. S14–S16†).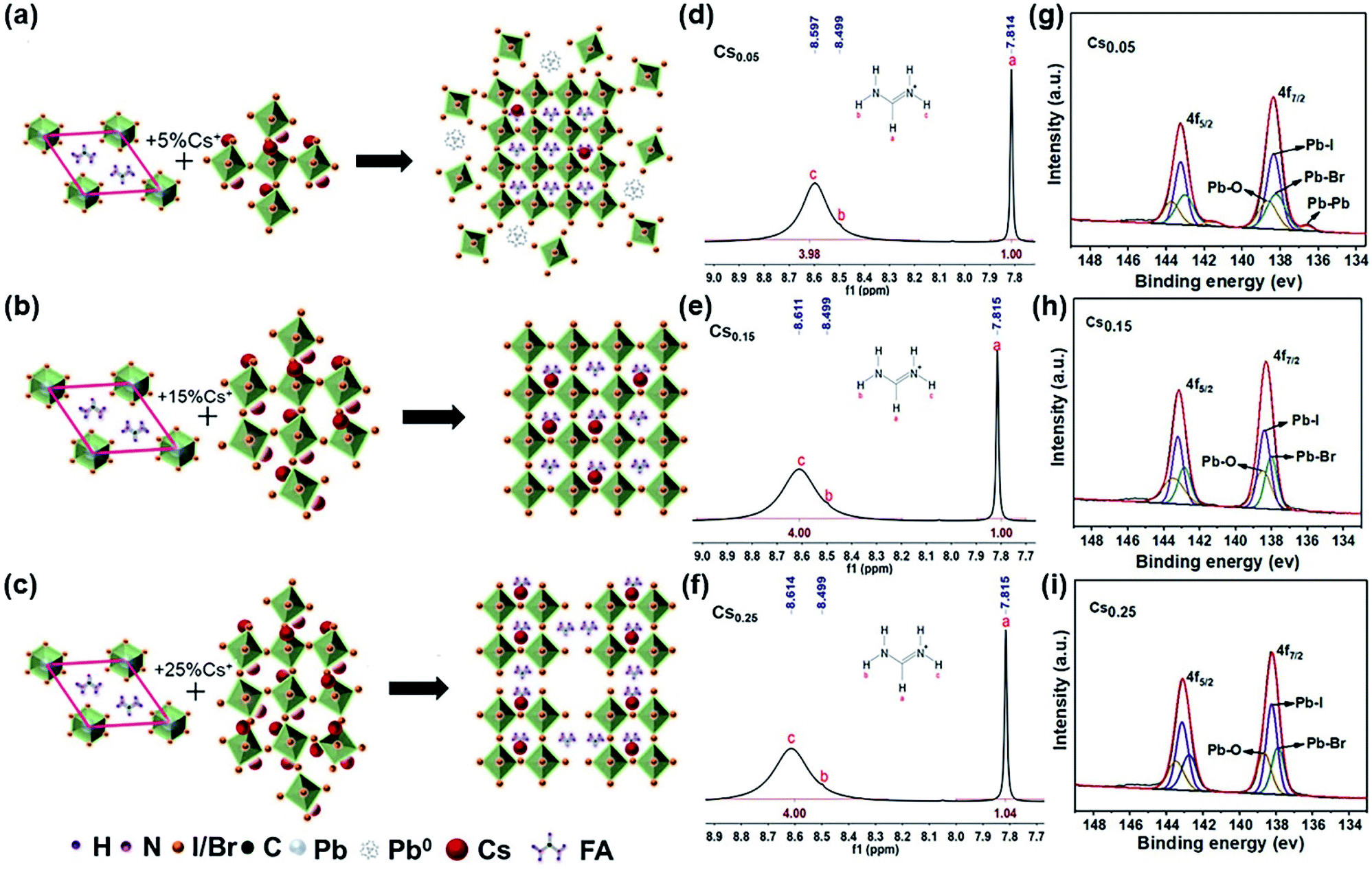 | ||
| Fig. 4 Schematic diagram of the influence of Cs+ on the lattice size of the FAPbI1.80Br1.20 perovskite phase. (a) 5% Cs+. (b) 15% Cs+. (c) 25% Cs+. Liquid-state 1H-NMR spectra of the CsxFA1−xPbI1.80Br1.20 precipitate powder in the deuterium generation of DMSO. (d) x = 0.05 (Cs0.05). (e) x = 0.15 (Cs0.15). (f) x = 0.25 (Cs0.25). The XPS spectrum of Pb 4f5/2 and Pb 4f7/2 of CsxFA1−xPbI1.80Br1.20 thin films with different Cs concentrations. (g) x = 0.05 (Cs0.05). (h) x = 0.15 (Cs0.15). (i) x = 0.25 (Cs0.25). | ||
The XPS spectrum of the CsxFA1−xPbI1.80Br1.20 samples were measured, and the results are shown in Fig. 4g–i. When the Cs+ content is only 5%, the binding energy (BE) at 143.25 eV and 138.38 eV are assigned to 4f5/2, 4f7/2 of Pb2+, respectively. In addition, the lower binding energy at 141.5 eV and 136.5 eV around the shoulder peaks are affiliated with metallic Pb0. The appearance of the Pb–Pb bond leads to the reduction of the Pb–I bond binding energy. The Pb–Pb bond (Pb elemental) is a major deep defect state that can influence the stability of perovskite, which is not conducive to the promotion of the efficiency and stability of solar cells.44 With the increase of the Cs+ cation, the Pb–Pb bond decreases gradually and the binding energy of the Pb–I bond increases. This is because the increase of the Cs+ content will lead to the lattice and volume contraction. It indicates that the addition of Cs+ weakens the effect of the R = NH2 functional group on the Pb–I bond in FA+, and increases the binding energy of the Pb–I bond in the system. Meanwhile, XPS tests were also conducted under 0–48 hours under air exposure. During the exposure, moisture and oxygen will react on the perovskite film, which causes the I− and Pb2+ to become chemically active and initiate the perovskite decomposition, finally resulting in PCE deterioration. The influence of Pb0 and I0 defects on perovskite was studied by XPS analysis. In the annealing process of the perovskite preparation, the I0 peak cannot be directly observed in the XPS spectrum because of its volatility. Hence, we determined the presence of I0 and I− by looking at the shift in the BE of I3d. As can be seen from Fig. S17b,† the binding energy of I3d (48 h) shifts towards a lower value compared with I3d (0 h) in the Cs0.15 sample, proving that I− is well preserved in this sample. However, the peaks of I3d (48 h) in both Fig. S17a and c† tend to shift towards high BE, proving the presence of I0 in perovskite.44 Similarly, Fig. S18† is an XPS spectrum of Pb4f. When the exposure time reaches 48 h, the only form is Pb2+ in Fig. S18b†, while the Pb0 substance appears in Fig. S18a and c†. It further shows that Cs0.15 forms a more stable cubic perovskite crystal structure. In addition, according to the XPS spectra of Cs 3d3/2 and Cs 3d5/2 of the CsxFA1−xPbI1.80Br1.20 films with different Cs+ concentrations (Fig. S19–S21†), the binding energy of the Cs–I bond and Cs–Br bond decreases with the increase of the Cs+ concentration, showing that the increase of the Cs+ content is beneficial for the formation of the black perovskite phase. Finally, the total XPS of the CsxFA1−xPbI1.80Br1.20 films with different Cs+ concentrations are shown in Fig. S22.† As can be seen from the figure, the peak intensity of Cs 3d increases significantly with the increase of the Cs+ content.
Photovoltaic performance based on the different Cs+ ratios of perovskite solar cells
Fig. 5 shows the photovoltaic performance based on the different Cs+ ratios of PSCs. The device structure is shown in Fig. S23†. With the electron transport layer of compact TiO2, the electrons generated by light absorption in the perovskite layer can be transferred to the FTO electrode. On top of the mp-TiO2 layer is a highly uniform perovskite active layer (∼300 nm thick). Spiro-OMeTAD and metallic Au were deposited on the active layer as hole transport materials and photocathode, respectively. | ||
| Fig. 5 Device stability and efficiency of PSCs. (a) The J–V characteristics of the champion cells based on different perovskite films under simulated AM 1.5 radiation. (b) Original performance based on the CsxFA1−xPbI1.80Br1.20 perovskite with different ratios (x = 0.05, 0.10, 0.15, 0.20, 0.25). (c) The stability performance of the PSCs based on the CsxFA1−xPbI1.80Br1.20 perovskite with different ratios (x = 0.05, 0.15, 0.25), under the environmental condition (25 °C, 55–60% humidity). (d) The stability performance of the PSCs based on the CsxFA1−xPbI1.80Br1.20 perovskite with different ratios (x = 0.05, 0.15, 0.25), stored in a N2-filled glovebox. The PCE evolution of the CsxFA1−xPbI1.80Br1.20 perovskite films heated on a 200 °C hotplate inside for 2 hours. | ||
The J–V characteristics of the champion cells based on different perovskite films are shown in Fig. 5a. The corresponding photovoltaic parameters are summarized in Table 2, in which the best cell of the Cs0.15 perovskite exhibits a short-circuit current density (Jsc) of 18.5 mA cm−2, fill factor (FF) of 74%, open-circuit voltage (Voc) of 1.18 V and PCE of 16.2%. Specifically, the Jsc increased from 15.4 mA cm−2 (Cs0.05 device) to 18.5 mA cm−2 (Cs0.15 device), and the Voc increased from 1.04 V to 1.18 V. The increase of Voc and Jsc may be due to the lower occurrence of electron–hole recombination and the more efficient charge carrier transport. However, the device performance does not increase continually with more Cs+ ion doping into the mixed perovskite system. When the content of Cs+ increases to 25%, the efficiency of the device degraded from 16.2% to 14.5%. This may be caused by the redundant Cs+ in the system, which restrained the charge carrier transport.45 Finally, it is obvious that the Cs0.15 perovskite has the best device performance. The statistics of the CsxFA1−xPbI1.80Br1.20 devices with different Cs+ ratios are shown in Fig. 5b, showing that the average PCEs for the Cs0.05, Cs0.10, Cs0.15, Cs0.20 and Cs0.25 devices are 9.9%, 13.7%, 16.2%, 15.0% and 14.5%, respectively. The steady power output for this champion device (Cs0.15) was sustained at 15.7%, measuring at the fixed voltage of 0.86 V close to the maximum power point (Fig. S24†), and the J–V curves of the champion device (Cs0.15) in reverse and forward scan directions (Fig. S25†) with negligible hysteresis. Fig. S26† shows the corresponding incident photon-to-electron conversion efficiency (IPCE). Fig. 5c is the stability performance of the Cs0.05, Cs0.15 and Cs0.25 perovskites under environmental condition (25 °C, 55–60% humidity). The air environment stability of the Cs0.15 perovskite is superior to that of the Cs0.05 and Cs0.25 cells, maintaining 90% of their initial PCE after 4 days under an environmental condition of 25 °C and 55–60% humidity. With the 15% Cs+ addition in the FAPbI1.80Br1.20 perovskite, the devices are relatively stable, proven by the improved Voc, Jsc and FF stability (Table 2). As shown in Fig. 5d, the thermal stability of the Cs0.15 devices retain 50% of their original PCE after 2 hours, while the Cs0.05 devices retain only 10% of their original PCE.
| Perovskite | Jsc (mA cm−2) | Voc (V) | FF | PCE (%) |
|---|---|---|---|---|
| Cs0.25FA0.75PbI1.80Br1.20 | 17.7 | 1.15 | 0.71 | 14.5 |
| Cs0.20FA0.80PbI1.80Br1.20 | 17.9 | 1.15 | 0.73 | 15.0 |
| Cs0.15FA0.85PbI1.80Br1.20 | 18.5 | 1.18 | 0.74 | 16.2 |
| Cs0.10FA0.90PbI1.80Br1.20 | 18.4 | 1.11 | 0.67 | 13.7 |
| Cs0.05FA0.75PbI1.80Br1.20 | 15.4 | 1.04 | 0.62 | 9.9 |
Conclusions
In summary, this work used the substitution of 15% Cs+ for FA+ to form Cs0.15FA0.85PbI1.80Br1.20 perovskite solar cells, achieving high efficiency PSCs with a stable PCE of 16.2%. The crystal structure transformation of the Cs0.05, Cs0.15 and Cs0.25 perovskites was monitored for the first time through XPS, XRD and NMR characterizations. From the XPS spectra, the depth defects of Pb gradually disappear as 15% Cs is introduced. After 48 h placement in the air environment, the peak of elemental Pb does not appear, indicating that the crystal structure of the cubic phase was better formed with no phase transition. XRD and TRPL measurements also demonstrate that the Cs+ doping shrinks the lattice and inhibits the production of the non-photoactive δ-phase, thus enhancing the charge carrier lifetime. Moreover, the Cs0.15-based PSCs devices maintained 90% of the initial PCE in air (environmental condition 25 °C, 55–60% humidity) for 4 days, and 50% of the initial PCE when heated in a 200 °C hotplate for 2 hours, showing the enhanced thermal and air stabilities compared with other devices. This work provides a novel way for understanding the lattice structural change mechanism of inorganic–organic perovskites, showing great potential for fabricating a new type of PSCs devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the Fundamental Research Funds for the Central Universities (Grant No. 2018JBZ107 and 2019RC021). This work was also supported financially by the National Natural Science Foundation of China (Grant No. 91961125 and 51802013), the “Key Program for International S&T Cooperation Projects of China” from the Ministry of Science and Technology of China (Grant No. 2018YFE0124600). The authors also appreciate the support from the “Excellent One Hundred” project of Beijing Jiaotong University.References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- National Renewable Energy Laboratory (NREL), Best Research Cell Efficiency Chart, https://www.nrel.gov/pv/cell-efficiency.html, accessed, October, 2020 Search PubMed.
- K. H. Lee, S. J. Kim, H. S. Park, B. W. Lim, B. Lee, Y. J. Park, W. Nam and N. H. Hur, RSC Adv., 2020, 10, 38055–38062 RSC.
- D. Liu, W. Zha, R. Yuan, B. Lou and R. Sa, RSC Adv., 2020, 10, 36734–36740 RSC.
- T. Ye, S.-L. Lim, X. Li, M. Petrović, X. Wang, C. Jiang, W.-P. Goh, C. Vijila and S. Ramakrishna, Sol. Energy Mater. Sol. Cells, 2018, 175, 111–117 CrossRef CAS.
- C.-H. Kuan, P.-T. Kuo, C.-H. Hou, J.-J. Shyue and C.-F. Lin, RSC Adv., 2020, 10, 35898–35905 RSC.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner and M. B. Johnston, Science, 2016, 351, 151–155 CrossRef CAS.
- F. Lu, D. Yi, S. Liu, F. Zhan, B. Zhou, L. Gu, D. Golberg, X. Wang and J. Yao, Angew. Chem., Int. Ed., 2020, 59, 17712–17718 CrossRef CAS.
- H. Zhou, Q. Chen, G. Li, S. Luo, T. b. Song, H. S. Duan, Z. Hong, J. B. You, Y. S. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS.
- T. Ye, S. Ma, X. Jiang, L. Wei, C. Vijila and S. Ramakrishna, Adv. Funct. Mater., 2017, 27, 1606545 CrossRef.
- S. H. Chang, P.-C. Tseng, S.-E. Chiang, J.-R. Wu, Y.-T. Chen, C.-J. Chen, C.-T. Yuan and S.-H. Chen, Sol. Energy Mater. Sol. Cells, 2020, 210, 110478 CrossRef CAS.
- T. Ye, L. Pan, Y. Yang, Q. Liang, Y. Lu, M. Sui, D. Golberg and X. Wang, Chem. Mater., 2020, 32, 3235–3244 CrossRef CAS.
- K. Yan, M. Long, T. Zhang, Z. Wei, H. Chen, S. Yang and J. Xu, J. Am. Chem. Soc., 2015, 137, 4460–4468 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS.
- F. Lu, W. Xie, D. Yi, Y. Wang, F. Zhang, Y. Xu, B. Zhou, S. Liu, X. Wang and J. Yao, CCS Chem, 2020 DOI:10.31635/ccschem.020.202000659.
- A. F. Akbulatov, L. A. Frolova, N. N. Dremova, I. Zhidkov, V. M. Martynenko, S. A. Tsarev, S. Y. Luchkin, E. Z. Kurmaev, S. M. Aldoshin, K. J. Stevenson and P. A. Troshin, J. Phys. Chem. Lett., 2020, 11, 333–339 CrossRef CAS.
- M. Kot, M. Vorokhta, Z. Wang, H. J. Snaith, D. Schmeißer and J. I. Flege, Appl. Surf. Sci., 2020, 513, 145596 CrossRef CAS.
- M. Saliba, T. Matsui, J. Y. Seo, K. Domanski, J. P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
- W. L. Leong, Z. E. Ooi, D. Sabba, C. Yi, S. M. Zakeeruddin, M. Graetzel, J. M. Gordon, E. A. Katz and N. Mathews, Adv. Mater., 2016, 28, 2439–2445 CrossRef CAS.
- Y. Hu, T. Qiu, F. Bai, W. Ruan and S. Zhang, Adv. Energy Mater., 2018, 8, 1703620 CrossRef.
- T. Ye, X. Wang, X. Li, A. Q. Yan, S. Ramakrishna and J. Xu, J. Mater. Chem. C, 2017, 5, 1255–1260 RSC.
- S. Seo, S. Jeong, C. Bae, N. G. Park and H. Shin, Adv. Mater., 2018, e1801010 CrossRef.
- H. Zhang, J. Xiao, J. Shi, H. Su, Y. Luo, D. Li, H. Wu, Y.-B. Cheng and Q. Meng, Adv. Funct. Mater., 2018, 28, 1802985 CrossRef.
- J.-W. Lee, D.-H. Kim, H.-S. Kim, S.-W. Seo, S. M. Cho and N.-G. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D'Haen, L. D'Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. D. Angelis and H.-G. Boyen, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS.
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 RSC.
- J. W. Lee, D. J. Seol, A. N. Cho and N. G. Park, Adv. Mater., 2014, 26, 4991–4998 CrossRef CAS.
- Y. Deng, Q. Dong, C. Bi, Y. Yuan and J. Huang, Adv. Energy Mater., 2016, 6, 1600372 CrossRef.
- M. Saliba, T. Matsui, J. Y. Seo, K. Domanski, J. P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- Z. Li, M. Yang, J.-S. Park, S.-H. Wei, J. J. Berry and K. Zhu, Chem. Mater., 2015, 28, 284–292 CrossRef.
- A. Amat, E. Mosconi, E. Ronca, C. Quarti, P. Umari, M. K. Nazeeruddin, M. Grätzel and F. De Angelis, Nano Lett., 2014, 14, 3608–3616 CrossRef CAS.
- Y. Xue, J. Tian, H. Wang, H. Xie, S. Zhu, B. Zheng, C. Gao and X. Liu, RSC Adv., 2018, 8, 25645–25652 RSC.
- H. Choi, J. Jeong, H.-B. Kim, S. Kim, B. Walker, G.-H. Kim and J. Y. Kim, Nano Energy, 2014, 7, 80–85 CrossRef CAS.
- C. Yi, J. Luo, S. Meloni, A. Boziki, N. Ashari-Astani, C. Grätzel, S. M. Zakeeruddin, U. Röthlisberger and M. Grätzel, Energy Environ. Sci., 2016, 9, 656–662 RSC.
- H. Wang, H. Wu, Y. Xian, G. Niu, W. Yuan, H. Li, H. Yin, P. Liu, Y. Long, W. Li and J. Fan, ACS Appl. Mater. Interfaces, 2019, 11, 13812–13821 CrossRef CAS.
- L. Chen, Y. Y. Tan, Z. X. Chen, T. Wang, S. Hu, Z. A. Nan, L. Q. Xie, Y. Hui, J. X. Huang, C. Zhan, S. H. Wang, J. Z. Zhou, J. W. Yan, B. W. Mao and Z. Q. Tian, J. Am. Chem. Soc., 2019, 141, 1665–1671 CrossRef CAS.
- A. K. K. Kyaw, D. H. Wang, C. Luo, Y. Cao, T.-Q. Nguyen, G. C. Bazan and A. J. Heeger, Adv. Energy Mater., 2014, 4, 1301469 CrossRef.
- S. Chen, T. Zhang, X. Liu, J. Qiao, L. Peng, J. Wang, Y. Liu, T. Yang and J. Lin, J. Mater. Chem. C, 2020, 8, 3351–3358 RSC.
- X. Xia, W. Wu, H. Li, B. Zheng, Y. Xue, J. Xu, D. Zhang, C. Gao and X. Liu, RSC Adv., 2016, 6, 14792–14798 RSC.
- Y. Tu, X. Yang, R. Su, D. Luo, Y. Cao, L. Zhao, T. Liu, W. Yang, Y. Zhang, Z. Xu, Q. Liu, J. Wu, Q. Gong, F. Mo and R. Zhu, Adv. Mater., 2018, 30, e1805085 CrossRef.
- M. Petrovic, T. Ye, V. Chellappan and S. Ramakrishna, ACS Appl. Mater. Interfaces, 2017, 9, 42769–42778 CrossRef CAS.
- L. Wang, H. Zhou, J. Hu, B. Huang, M. Sun, B. Dong, G. Zheng, Y. Huang, Y. Chen, L. Li, Z. Xu, N. Li, Z. Liu, Q. Chen, L. D. Sun and C. H. Yan, Science, 2019, 363, 265–270 CrossRef CAS.
- T. Ye, A. Bruno, G. Han, T. M. Koh, J. Li, N. F. Jamaludin, C. Soci, S. G. Mhaisalkar and W. L. Leong, Adv. Funct. Mater., 2018, 28, 1801654 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09294k |
| This journal is © The Royal Society of Chemistry 2021 |