
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-assembly of plasmonic nanoparticles: methods and applications
Jan Krajczewski
 *,
Robert Ambroziak
and
Andrzej Kudelski
*
*,
Robert Ambroziak
and
Andrzej Kudelski
*
University of Warsaw, Faculty of Chemistry, 1 Pasteur St., 02-093 Warsaw, Poland. E-mail: jkrajczewski@chem.uw.edu.pl; akudel@chem.uw.edu.pl
First published on 12th January 2021
Abstract
In this review article, various methods for the light-induced manipulation of plasmonic nanoobjects are described, and some sample applications of this process are presented. The methods of the photo-induced nanomanipulation analyzed include methods based on: the light-induced isomerization of some compounds attached to the surface of the manipulated object causing formation of electrostatic, host–guest or covalent bonds or other structural changes, the photo-response of a thermo-responsive material attached to the surface of the manipulated nanoparticles, and the photo-catalytic process enhanced by the coupled plasmons in manipulated nanoobjects. Sample applications of the process of the photo-aggregation of plasmonic nanosystems are also presented, including applications in surface-enhanced vibrational spectroscopies, catalysis, chemical analysis, biomedicine, and more. A detailed comparative analysis of the methods that have been applied so far for the light-induced manipulation of nanostructures may be useful for researchers planning to enter this fascinating field.
 Jan Krajczewski | Jan Krajczewski is an associate professor in Molecular Interactions and Spectroscopy group at the Faculty of Chemistry, University of Warsaw. His main research interest is the synthesis and characterization of metallic and plasmonic nanostructures and their application in optical spectroscopies. |
 Robert Ambroziak | Robert Ambroziak is pursuing his PhD at the University of Warsaw, Poland. His main research interest is SERS and the synthesis of nanostructured semiconductor substrates coated with silver and gold nanomaterials. |
 Andrzej Kudelski | Andrzej Kudelski is a professor at the University of Warsaw, Poland. His area of expertise is synthesis of electromagnetic nanoresonators for surface-enhanced Raman scattering (SERS) measurements and applications of SERS in biochemical and medical analysis. |
1. Introduction
Manipulating ever smaller objects makes progress in miniaturization possible, and so developing new methods by which we can force small objects to arrange as we want on the nanoscale, or even on the atomic scale, is the subject of research of many groups around the world. The first method of nanomanipulation developed involved mechanical manipulation.1 Later, it was found that nanostructures can be manipulated with the use of an electromagnetic wave. In 2018, the Nobel Prize in physics was awarded for the creation of nanooptical tweezers,2 and further solutions have followed, including methods of manipulating nano- and micro-objects with the use of a magnetic field3 and manipulating objects smaller than 100 nm optically.4Technologies for manipulating atomic objects and nanoclusters allow for the creation of many interesting individual nanostructures. With the help of AFM, for example, one can create circuits from gold nanowires that can be used to characterize electrical transport properties at the nanoscale, and to create other nanostructures.5 Techniques for creating single nanocrystals of a given shape,6 attaching semiconductor electrodes to single nanocrystals,7 and producing atomic semiconductor quantum emitters8 have also been developed. With AFM, one can isolate mRNA from a single cell.9 Similarly, laser nanotweezers can trap single nanoparticles10,11 or 2D nanostructures so that they can be manipulated.12 This was also realized for single proteins13 or whole cells.14 Unfortunately, when using AFM or optical nanotweezers, only a limited number of nano- and micro-objects can be manipulated simultaneously (in many cases only a single object). In this work, we describe various light-induced methods of rearranging plasmonic nanoobjects. Some of the methods described, which utilize photochemical reactions, allow to transform a very large number of nanoobjects at the same moment; practically, a bulk chemical reaction can be carried out.
Directed manipulation (including bulk self-assembly) of nanoobjects has been studied by many groups, so, large number of related research articles and excellent review articles are available, for example ref. 15–18. Because the issue of directed manipulation of nanoobjects is very broad, in this review article we focus only on light-induced manipulations of plasmonic nanoparticles. Plasmonic properties of manipulated objects make some techniques of manipulation significantly more effective, or even possible. Moreover, the transformation of plasmonic nanostructures is used significantly more often than the transformation of other systems. The aggregation and de-aggregation of plasmonic nanoobjects significantly changes their optical properties, and this makes many important applications of light-induced transformations of such objects possible.19–21 When a plasmonic nanostructure (meaning a nanoobject made from a plasmonic metal – in general, a metal with a negative real and a small positive imaginary dielectric constant at the frequency of the illuminating radiation) is irradiated by an electromagnetic wave of the proper frequency, strong oscillations of conduction electrons are induced. This induced oscillation (a quantum of such plasma oscillation is called a plasmon) can be treated as an oscillating electric dipole that generates an additional electric field; a situation in which such strong oscillations of surface plasmons are obtained is called surface plasmon resonance (SPR). For example, for a spherical metal nanoparticle, the magnitude of the induced dipole (p) is proportional to:22
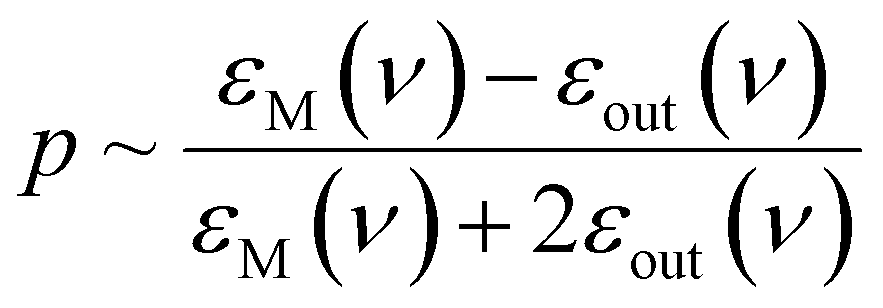
2. Photo-assembly of plasmonic nanoparticles
The photo-induced nanomanipulation of plasmonic nanoparticles can be realized using many different methods, for example, methods based on: the light-induced isomerization of some compounds that cause formation of electrostatic or host–guest bonds, the photo-induced formation of covalent bonds, the photo-response of a thermo-responsive material, and the photo-catalytic processes enhanced by the coupled plasmons excited in manipulated nanoobjects. The photo-induced nanomanipulations induced by the cis–trans isomerization of the azobenzene moiety seem to be currently the most often used methods of nanomanipulations based on photo-isomerization. Therefore, the photo-isomerization of azobenzene is described in more detail and the selected applications of photo-assembly of nanoobjects based on the change in the molecular size and the dipole moment induced by the cis–trans isomerization of the azobenzene moiety are presented in a separate chapter. Selected nanomanipulations based on the change of ionic interactions induced by light-induced isomerization of another compounds are presented in the second chapter. The other chapters present nanomanipulations based on: the photo-induced formation of covalent bonds, the photo-response of a thermo-responsive material, and the photo-catalytic processes enhanced by the coupled plasmons excited in manipulated nanoobjects.2.1. Photo-assembly based on change in the molecular size and the dipole moment realized by the cis–trans isomerization of the azobenzene moiety
A large group of organic molecules that can be used to carry out the process of photo-switching nanoparticles is the azobenzenes. It is well known that azobenzene-based compounds undergo a trans–cis isomerization around the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. The trans isomer of azobenzene is more stable by approximately 50 kJ mol−1 (ref. 23) than the cis isomer (which has a distorted configuration and is less delocalized than the trans configuration), and the barrier to isomerization in the ground electronic state is approximately 100 kJ mol−1.24 Therefore, in darkness and at room temperature, cis-azobenzene relaxes very slowly back to the stable trans form. However, the two isomers of azobenzene can be switched each other with relatively good efficiency by irradiation using light having particular wavelengths: ultraviolet light, which corresponds to the energy gap of the π–π* transition, for trans-to-cis conversion; and blue light, whose energy fits the n-π* transition, for cis-to-trans isomerization. Trans-azobenzene is planar and cis-azobenzene is non-planar, with a C–NN–C dihedral angle of 173.5°.25 The isomerization process itself runs through a linear transition state. This state is obtained not only by the inversion of the α angle but also by a rotation around the ω dihedral angle (see Fig. 1a).26,27 A more complex isomerization route has been also suggested,28 where the phenyl rings maintain their relative positions with a simultaneous rotation around the NN bond. This requires a simultaneous twisting around the axis of the C–N bonds. This type of transition is known as the “hula twist”.28
N bond. The trans isomer of azobenzene is more stable by approximately 50 kJ mol−1 (ref. 23) than the cis isomer (which has a distorted configuration and is less delocalized than the trans configuration), and the barrier to isomerization in the ground electronic state is approximately 100 kJ mol−1.24 Therefore, in darkness and at room temperature, cis-azobenzene relaxes very slowly back to the stable trans form. However, the two isomers of azobenzene can be switched each other with relatively good efficiency by irradiation using light having particular wavelengths: ultraviolet light, which corresponds to the energy gap of the π–π* transition, for trans-to-cis conversion; and blue light, whose energy fits the n-π* transition, for cis-to-trans isomerization. Trans-azobenzene is planar and cis-azobenzene is non-planar, with a C–NN–C dihedral angle of 173.5°.25 The isomerization process itself runs through a linear transition state. This state is obtained not only by the inversion of the α angle but also by a rotation around the ω dihedral angle (see Fig. 1a).26,27 A more complex isomerization route has been also suggested,28 where the phenyl rings maintain their relative positions with a simultaneous rotation around the NN bond. This requires a simultaneous twisting around the axis of the C–N bonds. This type of transition is known as the “hula twist”.28
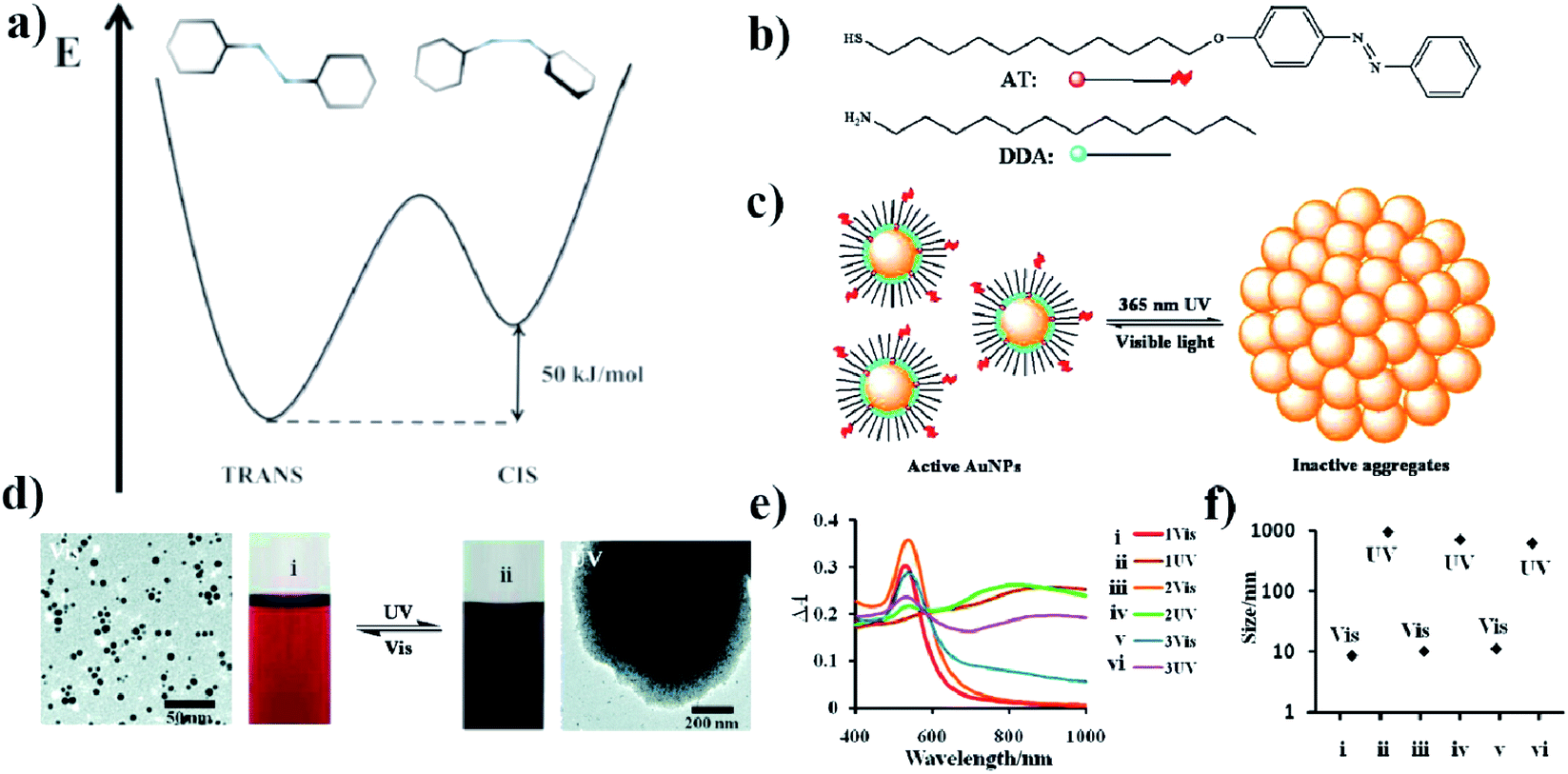 | ||
| Fig. 1 (a) Energy diagram showing the relative energy of both the cis and trans forms of azobenzene. (b) Molecular structure of DDA surfactant and the structure of photo-responsive azobenzene–thiol ligand (AT), (c) schematic representation of a photo-switchable system utilizing gold nanoparticles, (d) TEM micrographs showing dispersed and aggregated gold nanoparticles with corresponding typical color change of the reaction mixture after UV and VIS irradiation, (e) change in UV-VIS extinction spectra induced by VIS and UV irradiation, (f) sizes of free and aggregated gold nanoparticles measured by DLS (reprinted with permission from ref. 32. Copyright 2010 the Owner American Chemical Society). | ||
Photo-switchers are usually formed using substituted azobenzenes containing the mercapto group, since such compounds can be relative easily attached to a gold and silver surface (thiols react chemically with the surface of gold and silver, forming very strong Au–S or Ag–S bonds, respectively). In some cases, instead of using azobenzenes with an attached mercapto group, molecules are used that are initially equipped with another stable moiety, such as the thioester group, which, after releasing the protective group, is transferred into the thiol moiety. For example, an azo-biphenyl linker is often used as a passive acetyl ester form that can be transformed into a thiol compound in alkaline conditions.29 It has been observed for many slightly different systems that irradiating a solution containing Au nanoparticles modified with an azo compound with 450 nm light leads to the isomerization of the azo compound and the precipitation of the Au nanoparticles. On the other hand, irradiating such precipitated Au nanoparticles with 366 nm light leads to a phase transition from the solid to the colloidal phase. Such structural changes arise from the trans–cis isomerization of the azo compounds under light irradiation. A more detailed discussion of the mechanism of this photo-rearrangement is given below, with a detailed description of examples of such rearrangements.
One of the first photo-switchable system based on trans–cis isomerization of azobenzene has been constructed by Sidhaye et al.30 This group used a benzyldimethylstearylammoniumchloride (BDSAC) azo compound to construct photo-switched gold nanostructures.30 The trans form had a length of 3 nm whereas the cis form had a length of 2 nm. Gold nanoparticles capped by BDSAC formed a close knit network. Each BDSAC molecule was attached to two gold nanoparticles by the cysteine groups (this compound is also strongly bonded to the gold surface due to the formation of Au–S bond) that are present on both ends of this molecule. After UV irradiation, the distance between the nanoparticles decreased. This process can be easily monitored by UV-VIS spectroscopy. Gold nanoparticles coated by BDSAC exhibited two bands: at 560 nm, related to the SPR of gold nanoparticles, and at 365 nm, attributed to the azo compound in the trans conformation. UV irradiation led to a red shift of the position of the SPR band and the formation of a new absorption band at 320 nm related to the cis conformation of the azo compound. After irradiation with visible light, the spectrum of the system came back to its initial form. The presence of the additional capping agent was able to block the possibility of the trans–cis isomerization due to spatial restriction.
Grzybowski et al. investigated the photo-switching properties of a dithiol biazo compound: 4,4′-bis(11-mercaptoundecanoxy)azobenzene (ADT).31 This compound can combine two gold nanoparticles with a diameter of ca. 5.6 nm through the thiol linking groups. Aggregation occurs in a certain range of conditions when the sample is irradiated with UV light (365 nm, 0.7 mW cm−2). According to Grzybowski et al., the joining of nanoparticles starts from the appearance of a dipole moment and dipole–dipole interactions in a solvent with limited polarity. In such a situation, the two particles are brought closer together, and a bond can form between the sulfur and the gold. One of the factors affecting the stability of aggregates is the degree of coverage of the nanoparticles with ADT. This value can be regulated by using a different ratio of dodecylamine (DDA) and ADT. It has been shown that, when the surface coverage of ADTs on the gold nanoparticles is high (above 75%), the nanoparticles do not aggregate, even after prolonged UV irradiation. According to Grzybowski et al., this is due to a steric hindrance. In turn, even when only 16 ADT molecules are attached to a gold nanoparticle, the aggregation of the nanoparticles can be carried out under UV irradiation in a toluene: methanol (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1/v:v) solution. In such conditions, small crystals of aggregates with a size of about 1 μm are formed. Crystals with a very small ADT content are unstable, and break down as soon as the UV is turned off. The decomposition of the crystals results from the low strength of the dipole interactions between the small number of molecules. The amount of sulfur–gold bonds formed is too small to resist the energy of the thermal movements, and this causes the crystals to decompose into single nanoparticles. In turn, re-irradiation leads to the formation of the same crystals again. The authors tested 15 such cycles and did not observe any changes in the structure of the crystals formed. The transition from “free” nanoparticles to crystals and again to “free” nanoparticles took about 10 minutes. Aggregation can be reversed only for systems with a low ADT content. Crystals formed during UV exposure from nanoparticles covered by a large number of ADT molecules are stable – the cessation of the light exposure does not cause the aggregates to break down. In fact, heating the solution to 100 °C, prolonged sonication and transferring the crystals to a number of other polar and non-polar solvents did not cause them to decompose, either. Even the use of other alkyl monothiols did not affect the crystals. This work shows how precisely the defined conditions should be selected to allow some systems to be reversibly switched between two stages.
:1/v:v) solution. In such conditions, small crystals of aggregates with a size of about 1 μm are formed. Crystals with a very small ADT content are unstable, and break down as soon as the UV is turned off. The decomposition of the crystals results from the low strength of the dipole interactions between the small number of molecules. The amount of sulfur–gold bonds formed is too small to resist the energy of the thermal movements, and this causes the crystals to decompose into single nanoparticles. In turn, re-irradiation leads to the formation of the same crystals again. The authors tested 15 such cycles and did not observe any changes in the structure of the crystals formed. The transition from “free” nanoparticles to crystals and again to “free” nanoparticles took about 10 minutes. Aggregation can be reversed only for systems with a low ADT content. Crystals formed during UV exposure from nanoparticles covered by a large number of ADT molecules are stable – the cessation of the light exposure does not cause the aggregates to break down. In fact, heating the solution to 100 °C, prolonged sonication and transferring the crystals to a number of other polar and non-polar solvents did not cause them to decompose, either. Even the use of other alkyl monothiols did not affect the crystals. This work shows how precisely the defined conditions should be selected to allow some systems to be reversibly switched between two stages.
Using ADT, DDA, 11-(4-(phenylazo)phenoxy)-1-undecane-thiol (AT) and gold nanoparticles, Grzybowski et al. also constructed a somewhat different photo-switched system.32 They modified 5.5 nm gold nanoparticles with ADT, DDA and AT (see Fig. 1b). The surface coverage of nanoparticles with AT was relatively low, so most of the surface of the nanoparticles was covered with DDA (see Fig. 1c). After irradiating the sample with UV light at λ = 365 nm and the intensity of 50 mW cm−2, the isomerization of the azobenzene group of ADT from trans to cis occurred, which increased the dipole moment of ADT to about 5 D. In a non-polar solvent, this leads to the aggregation of the nanoparticles (see Fig. 1d and e). However, after turning off the UV light, a re-transition of the azobenzene from cis to trans isomer was observed, which led to a breakdown of the agglomerates. The rate of disintegration of the agglomerates can be significantly increased by irradiating the sample using white light with an intensity of 100 mW cm−2; in such conditions, within about 1–2 minutes “free” nanoparticles are obtained again (see Fig. 1e and f). Recently, photoswitchable systems using azobenzene-functionalized polydopamine/gold nanoparticle composite materials, and based on large change in molecular size and dipole moment due to trans-to-cis azobenzene isomerization has been constructed by Kunfi et al.33
Another photo-switchable system based on cis–trans azobenzene isomerization has been constructed by Housni and co-workers in 2010.34 These authors covered gold nanoparticles with a water-soluble copolymer formed from N,N-dimethylacrylamide (DMA) and N-4-phenylazophenylacrylamide (Azo). Poly(DMA-co-Azo) exhibits cis–trans photo-isomerization after UV/VIS light irradiation. Without UV irradiation, at temperatures below the low critical solution temperature (LCST), the polymer adopts a stable trans structure, and the polymer-covered nanoparticles suspension is also stable under such conditions. Upon UV radiation, the azobenzene moiety isomerizes to the cis form, the LCST temperature of the system goes down and, as a result, the polymer chains are no longer soluble in water, which lead to an aggregation of the nanoparticles. The addition of free polymer chains to the solution allows for a reversible photo-controlled aggregation of gold nanoparticles. Photo-isomerization can be monitored, for example, using UV-VIS spectroscopy. The trans isomer of azobenzene exhibits an absorption band centered at 350 nm, while the cis form is at 440 nm. Therefore, in the case of a high polymer concentration, the suspension of gold nanoparticles exhibits, in addition to the SPR band at 520 nm, and absorption band of the trans form of the polymer at 350 nm. After UV irradiation, the band at 350 nm disappears, while a new band at 440 nm is formed. Moreover, the position of the SPR band shifts to the longer wavelength. The reverse spectral changes were observed after VIS irradiation. This system exhibits good reversibility even after five full cycles.
An interesting application of azobenzene in the construction of photo-switched systems was presented in the publication of Ginger et al.35 This group synthesized a sol of gold nanoparticles and divided the obtained sol into two batches. Both batches were modified with DNA oligomers complementary to each other – to chemically attach DNA chains to the gold nanoparticles, the DNA chains were modified on one side with a thiol group. In one batch of nanoparticles, the DNA oligomers were additionally modified with azobenzene. The modification consisted of introducing azobenzene groups between some nucleotides. These two batches of nanoparticles prepared in such a way were mixed, and this was followed by a crosslinking and aggregation of nanoparticles due to the combination of complementary DNA strands. The time needed to complete the aggregation was about 4 hours. The sample was then stirred and illuminated with UV light with a wavelength of 330 nm and a flux of 0.83 mW cm−2 (the temperature of the solution was 45 °C). After 1 hour of irradiation, the solution again took on the color characteristic of the presence of non-aggregated gold nanoparticles. Exposing the non-aggregated sample to blue light with a wavelength of 470 nm and a flux of 11 mW cm−2 led after about 2 h to the re-aggregation of the sample. These processes were repeated 3 times. Measurements of the UV-VIS spectra after subsequent cycles showed the reversibility of the re-aggregation process, since the intensity of the plasmonic band at 526 nm decreased only slightly in subsequent measurements. Azobenzene was responsible for this aggregation – re-aggregation process; under the influence of UV light, azobenzene passes into the cis form and prevents the combination of complementary DNA pairs. This was confirmed by an experiment in which nanoparticles with azobenzene-modified DNA were irradiated before mixing them with nanoparticles connected to a complementary DNA strand. In this case, there was no aggregation. The nanoparticles would only aggregate if complementary DNA strands were present. Therefore, a possible application of the system presented in the work of Ginger et al. is to determine whether two samples of DNA contain complementary or non-complementary DNA strands.
Lysyakova et al. modifies gold nanoparticles prepared by the method of laser ablation with an azobenzene-containing surfactant (4-butyl-4′-(trimethylammoniumhexyloxy)-azobenzene) bromide.36 The functionalization of the surface of gold nanoparticles by means of an azo-surfactant leads to a positive zeta potential, and hence to the stability of the suspension of nanoparticles due to ionic repulsion. Formed by UV irradiation the cis form of the surfactant is more bulky, and does not interact strongly with the gold surface, therefore such capping molecules are removed from the surface of the nanoparticles. In this case, the zeta potential decreases to 0, which leads to an aggregation of the nanoparticles. TEM analysis confirmed the formation of 100 nm cluster-like aggregates with a simultaneous blue shift of the SPR band.
Köhntopp et al. formed on gold nanoparticles mixed azobenzenethiolate–alkylthiolate monolayers.37 Their calculations showed that, typically, 100 thiol molecules were attached to the gold nanoparticles with an average diameter of 4 nm. For photo-switching, a sol of functionalized nanoparticles was irradiated using radiation with a wavelength of 365 or 455 nm. It was found that, when azoligands having a longer carbon chain were used, the efficiency of the trans to cis isomerization increased, while the efficiency of cis to trans isomerization decreased. The observed aggregation of functionalized gold nanoparticles is attributed to the attractive dipole to dipole interactions between the cis forms of the azobenzenes. The high dipole moment of this system (3.2 D) led to a weaker solvent stabilization of the gold nanoparticles in the non-polar solvent (toluene), and hence to their aggregation. It was found that using alkylthiols with longer chains allows for better solvation, and thereby prevents aggregation. Alkylthiols with shorter chains permit closer contact between the cis forms of the azobenzenes, and thereby stronger dipole interactions. Additionally, the tendency to aggregation increases with higher azobenzene surface concentrations, due to the higher dipole density. The samples could be photo-switched back and forth between two photo-stationary states without any visible photo-degradation, even after several cycles. Subsequently, it was presented that such photo-isomerization could be monitored using femtosecond time-resolved absorption spectroscopy.38 The results obtained showed an almost unchanged behavior of the azo compounds attached to the gold nanoparticles in comparison with the free molecules in solution.
In another approach, small gold nanoclusters (Au25) were coated with azobenzene C3 alkyl monothiol.39 It was found that for such a system, a transformation from trans to cis form is realized after exposure to 345 nm light, and the reverse process is observed after exposure to 435 nm light. Attractive dipole–dipole interactions, which occur only for the cis form, lead to a self-assembly of nano-clusters covered with ligands containing the azobenzene moiety in the cis form. In the case of small clusters, they form a disk-like superstructure. The AFM analysis showed the formation of a superstructure with an average thickness of 50 nm and a diameter of 800 nm after 240 minutes of illumination. The formation of aggregates was also confirmed by the DLS measurements. The average size of the nanostructures formed after 120 minutes of illumination was 250 nm, and increased to 550 nm after 180 minutes, finally reaching a maximum size of 1.5 μm after 240 minutes of illumination. TEM tilt micrographs and their corresponding three-dimensional reconstruction suggest that the superstructures formed are disc-like colloidal particles. A decrease in the size of the agglomerates was observed after irradiation with 435 nm light.
In toluene the aggregation of a sol of gold nanoparticles coated simultaneously with a thiol derivative of azobenzene and a thiol derivative of ethylenediamine can be influenced not only by UV irradiation, but also by introducing carbon dioxide.40 Aggregation under the influence of CO2 is caused by the formation of a more polar complex with a derivative of ethylenediamine. Cessation of the irradiation of the sample and gas passage do not cause the dissolution of the aggregates for at least 30 min. Interestingly, these factors do not have to occur simultaneously. To aggregate, a sample can be first exposed to UV and then to the flow of CO2; the reverse order is also effective. Sonication causes the aggregates to break down when the CO2 flow is turned off and the sample is exposed to UV light. Exposing the sample to white light also leads to a breakdown of the agglomerates. In each of these cases, the sample can be re-aggregated by treating it with UV light and carbon dioxide. This behavior is observed only when the coverage of nanoparticles with a diamine derivative is about 25%. Otherwise, the aggregation of the nanoparticles is not reversible.
Biswas et al. synthesised some alkoxy azobenzene mesogenic thiols with different lengths of alkane chain and applied these compounds as ligands to modify the surface of gold nanoparticles.41 In the next step, the photo-switching properties of the systems obtained were analysed. It has been shown that the irradiation of samples of such modified nanoparticles in chloroform with a 5 mW cm−2 flux of UV radiation having a wavelength of 365 nm induces a trans to cis isomerization after about 44 s, regardless of the length of the alkane chain; however, the return to the trans isomer in conditions without light is different for different lengths of the alkyl chains. The thermal transition from the cis to trans isomer takes 91, 100 and 125 min for alkyl chains with 4, 5 and 6 carbon atoms, respectively.41
Two other azobenzene compounds that have been used to construct photo-switchable gold nanostructures are 4-(3-mercaptopropyl-1-oxy)azobenzene and 4-(3-mercaptopropyl-1-oxy)-4'-(dimethylamino)azobenzene.42 For 4-(3-mercaptopropyl-1-oxy)-4'-(dimethylamino)azobenzene, irradiation with UV light (λ = 365 nm, flux 0.7 mW cm−2) causes a transition from the cis to trans isomer, whereas irradiation with blue light (λ = 420 nm, flux 1 mW cm−2) causes an isomerization from trans to cis (such behavior is opposite to the changes observed for the majority of other azo compounds). Although the isomerization is not complete, gold nanoparticles with a diameter of 2.5 nm functionalized with 4-(3-mercaptopropyl-1-oxy)-4'-(dimethylamino)azobenzene aggregate in a non-polar solvent under influence of blue light. The agglomeration is caused by the isomerization of the azobenzene moiety from the trans to cis form, which increases the dipole moment of the molecule. Exposing the sample to UV radiation causes a transition from the cis to trans form and the dispersion of aggregated nanoparticles. Manna et al. carried out an interesting experiment in which gold nanoparticles with a diameter of 2.5 nm were functionalized with 4-(3-mercaptopropyl-1-oxy)-4'-(dimethylamino)azobenzene, and nanoparticles with a diameter of 5.5 nm were functionalized with 4-(3-mercaptopropyl-1-oxy)azobenzene which, after UV irradiation, isomerized from the trans to the cis form, whereas irradiation with blue light caused an inverse isomerization (typical behavior for azo compounds). After irradiation of such a mixture of differently functionalized nanoparticles with blue light, a partial aggregation of 2.5 nm particles was observed, while UV irradiation led to a breakdown of those aggregates and to the aggregation of 5.5 nm nanoparticles. It has been observed that the aggregation of 5.5 nm nanoparticles begins on existing 2.5 nm aggregates. The reverse process causes the formation of 2.5 nm aggregates on existing 5.5 nm aggregates. An interesting result was observed when nanoparticles were covered with both types of ligands. Exposing such a system to UV radiation led to aggregation, while the exposure to blue light did not change this state; the reverse order of exposure led to the same results. This was caused by the transition from trans to cis of one of the ligands, regardless of the selected wavelength of the radiation used. Such a system can be restored to a dispersed state by heating to 45 °C, which leads to the transition of both ligands from the cis to the trans isomer.
An interesting photo-switchable system based on gold nanorods was developed by Wang and co-workers.43 This system does not require any additional functionalization of the surface of the gold nanostructures other than stabilization by the cetyl trimethylammonium bromide (CTAB) surfactant, which is commonly used in the synthesis of gold nanorods. The mechanism making this system photo-switchable is based on the application of an anionic azobenzene derivative, (4-phenylazo-phenoxy)-acetate (AzoNa), as a co-adsorbent. The suspension of gold nanorods stabilized by CTAB exhibits a stable positive zeta potential of 32.8 mV. The addition of the trans form of AzoNa led to a fast and significant decrease in the value of the zeta potential, to 4.0 mV, which led to the rapid aggregation of gold nanorods. The reverse process (decomposition of the aggregates) can be induced by UV irradiation and subsequent aggregation of the nanorods by irradiation using visible light. The aggregation/de-aggregation processes are connected with the trans–cis isomerization of AzoNa. The trans form of AzoNa is highly hydrophobic, and could therefore penetrate into the alkane chains of the CTAB, while the cis form of the AzoNa is squeezed out of the CTAB due to high hydrophilicity and steric limitations. The same process was successfully conducted using spherical gold nanoparticles, and so it can be reasonably expected that this strategy could be applicable to nanoparticles of various shapes.
In another study, Huebner et al. covered gold nanoparticles with a thin layer of polymer produced from a methacrylate type monomer with an azobenzene sidechain.44 It is well known that gold nanoparticles catalyze the thermal cis–trans isomerization by an electron transfer45 (see Fig. 2a). It was found that, in the case of a polymer chain attached to the surface of gold nanoparticles, the isomerization occurred 1.7 times faster than in the case of free polymer chains. Such an isomerization could be achieved by irradiation with 365 nm light (see Fig. 2b). The cis form of the polymer is more polar, and therefore the cis polymer itself and nanoparticles covered by the polymer containing azobenzene moieties in the cis form partially precipitate from the toluene solution. Aggregation was confirmed by TEM (see Fig. 2c and d) and DLS (see Fig. 2e and f) measurements. It was found that the distance between particles increased along with the length of the polymer chain. In the case of a polymer having a lower average molecular mass (13 kD), the TEM micrographs showed densely packed gold nanoparticles. In the case of the polymer with a higher molar mass (35 kD), the density of nanoparticle packages was lower. This could be explained by the increasing shell thickness with longer polymer chains.
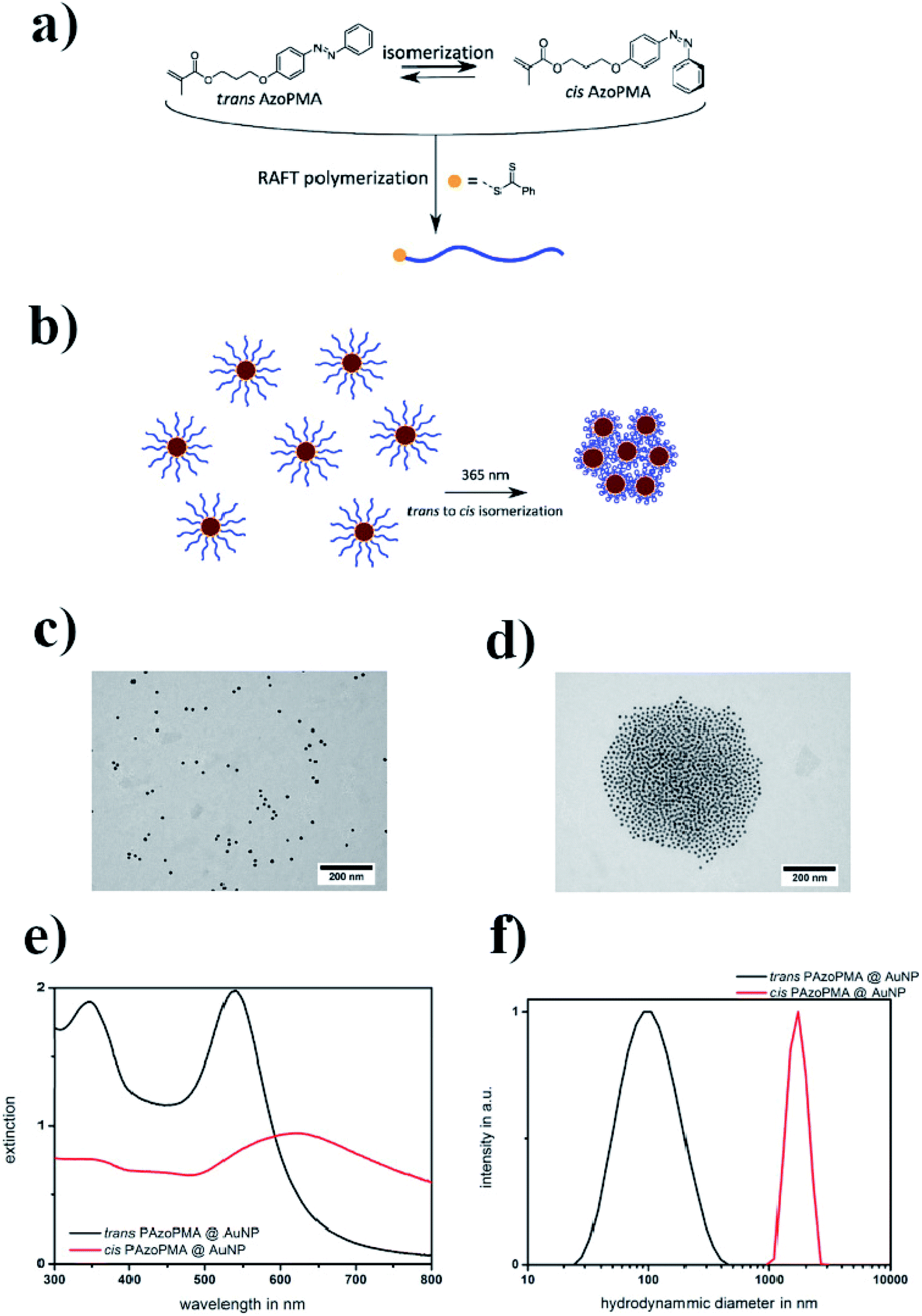 | ||
| Fig. 2 (a) Structure of the AzoPMA monomer in its trans and cis states, (b) schematic illustration of the photo-aggregation of gold nanoparticles coated with poly-AzoPMA, (c and d) TEM micrographs showing gold nanoparticles coated with poly-AzoPMA, before (c) and after (d) exposure to light, (e) UV-VIS extinction spectra of trans-type (black) and cis-type (red) poly-AzoPMA grafted on gold nanoparticles in toluene, (f) particle size distributions obtained by DLS from trans-poly-AzoPMA and cis-poly-AzoPMA grafted onto gold nanoparticles (reprinted with permission from ref. 44. Copyright 2016 the Owner Elsevier). | ||
As mentioned above, the photo-isomerization of azobenzene is induced by UV-VIS irradiation. However, in experiments with biological samples, some components, such as tissue or blood, were also able to absorb light from this region, which makes azobenzene isomerization significantly more challenging where certain organic molecules are present in the system. A promising method of carrying out the cis to trans photo-isomerization of azobenzene, which has been introduced to some tissues, is the application of upconversion NaYF4 nanoparticles doped with Yb3+, Tm3+ or Er3+.46 Such upconversion nanoparticles can be excited using near-infrared radiation (within the transparency window of many tissues), and they emit visible light. For example, nanoparticles doped with a Tm3+ activator emit in a range from 450 to 480 nm, while nanoparticles doped with an Er3+ activator emit in a range from 525 to 545 nm. The spectral changes related to the cis and trans conformers confirm the successful isomerization after exposure to near-infrared radiation. In the case of nanoparticles doped with Er3+, the rate of isomerization was actually very fast due to the fact that the absorption band of the azo compound overlaps with the emission band of the upconverting nanoparticles. Therefore, such a system could also be used for the photo-assembly of plasmonic nanoparticles based on a cis to trans isomerization.
2.2. Photo-assembly based on change of ionic interactions – realizations based on photo-transformation of another compounds
Ikkala et al. coated 11 nm gold nanoparticles with (11-mercaptoundecyl)-N,N,N-trimethyl-ammonium bromide (MUTAB), which forms positively charged ions.47 To this sol, they added photoacid spiropyran which, without exposure to light, is in the form of zwitterion ions. When the sample is exposed to light having a wavelength of 420 nm, the zwitterions transform into anions. These anions are adsorbed on the surface of the positively-charged modified gold nanoparticles, which creates a hydrophobic coating around the nanoparticles, and, as a consequence, the hydrophobisation and aggregation of the nanoparticles. The time it takes to switch to a hydrophobic form under the influence of light is 5 min. To disperse the nanoparticles again, it is necessary to store them in the dark for an hour. A UV-VIS test was carried out to monitor this process. Nanoparticles stable in an aqueous solution have a plasmon band at 520 nm. After exposure to light, this band shifts to 557 nm, which suggests that the aggregation of nanoparticles has occurred. Such a mechanism has been confirmed by SEM and zeta potential measurements. The aggregation rate of nanoparticles during exposure to light having a wavelength of 420 nm was examined as a function of time and temperature. It was shown that, at 50 °C, the aggregation time for a given radiation flux was 10 s, and at 15 °C the aggregation time was 20 min. In addition, the use of this method makes it possible to transfer nanoparticles between the inorganic and the organic phase only by means of exposure to 420 nm light. The aqueous solution of nanoparticles was orange in color, pure toluene over water was colorless, but after irradiation the aqueous phase turned yellow and the toluene phase changed pink. These changes were attributed to a transition of nanoparticles to the toluene phase, while the yellow color of the aqueous phase corresponded to the color of the modified spiropyran dissolved in the water. An interesting application of this system is the possibility of changing the wetting property of a modified gold surface. A gold plate covered with a MUTAB monolayer reveals the contact angle for water equal to 0°. When irradiation begins, the contact angle also begins to change, and becomes 60°. At the same time, pure gold shows a constant contact angle of 20°. Light-induced switching between the hydrophilic and hydrophobic state of a modified gold surface have a wide range of applications. All described details are presented in Fig. 3.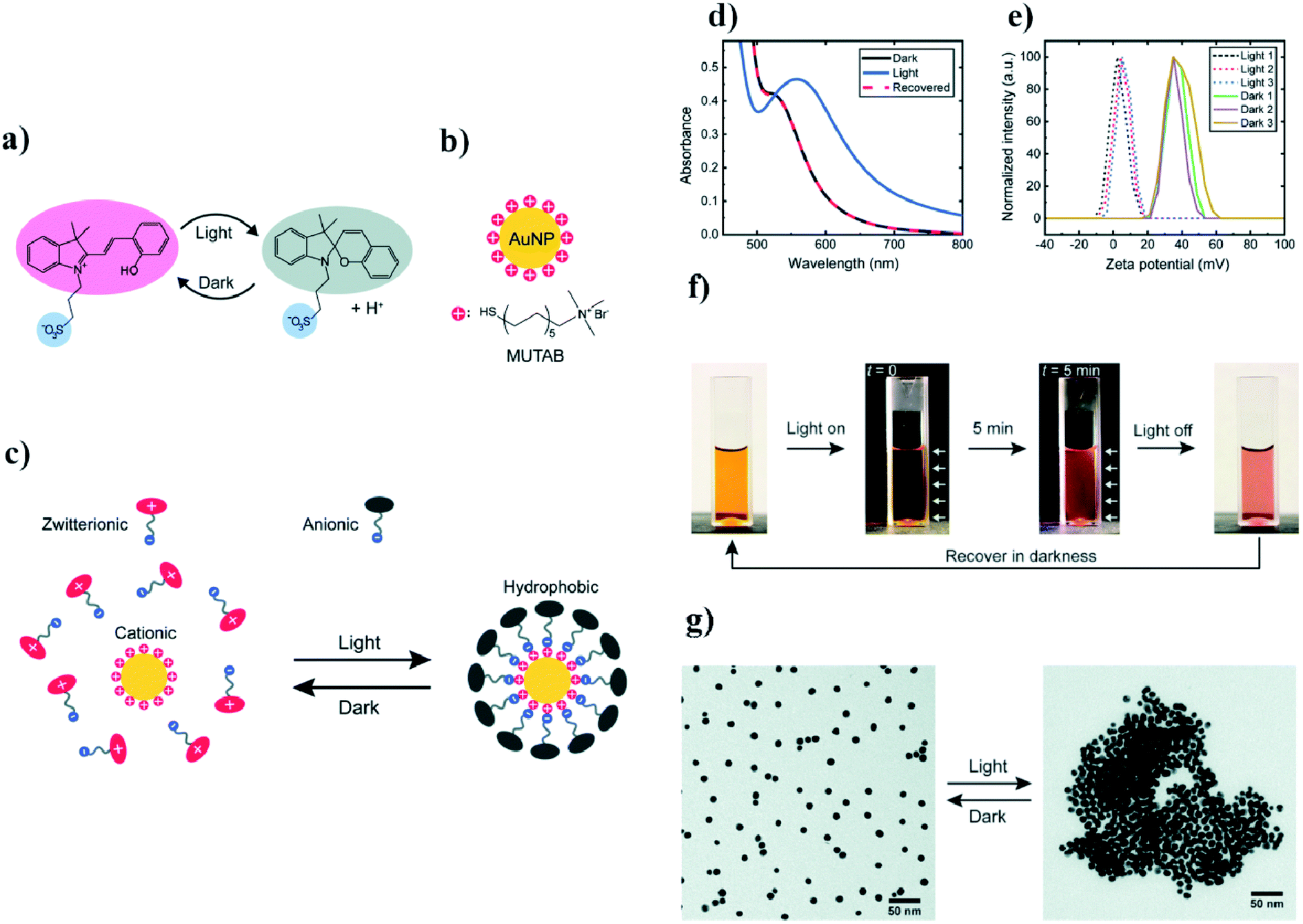 | ||
| Fig. 3 (a) Diagram of light-induced reversible switching between the zwitterionic and anionic forms of a photoacid, (b) chemical structure of the cationic surfactant MUTAB, (c) diagram of light-induced adsorption of the anionic form of the photoacid on a modified surface of gold nanoparticles, (d) UV-VIS extinction spectra of photoacid-modified gold nanoparticles before and after light irradiation, (e) diagram illustrating the zeta potential change in gold nanoparticles after three full cycles of irradiation, (f) photographs of photoacid-modified gold nanoparticles (the white arrows indicate the direction of irradiation), (g) TEM micrographs showing dispersed and agglomerated gold nanoparticles before and after irradiation (reprinted with permission from ref. 47. Copyright 2019 the Owner The Royal Society of Chemistry). | ||
A similar approach based on spiropyran was described by Ikbal and co-workers in 2017.48 The formation of the zwitterionic form of spiropyran during irradiation led to a rapid aggregation of modified nanoparticles based on electrostatic interactions. The aggregated nanoparticles could be re-dispersed by irradiation with visible light, or by thermal treatment in darkness.
Typically, a photo-assembly process is very slow, and sometimes it takes even many hours to transform a system from one state to another. A very fast photo-assembly process, where the transformation occurs practically immediately after excitation, was invented by Yucknovsy and co-workers in 2019.49 Yucknovsy et al. functionalized semi-spherical gold nanoparticles having an average diameter of 3.8 nm with 6-mercaptohexanoic acid (MHA).49 It was found that the aggregation of such nanoparticles could take place after the addition of a solution of HCl – in this case, the aggregation was due to the hydrogen bonding between the –COOH groups of chemisorbed MHA. Re-aggregation (formation of individual gold nanoparticles) could be also induced by the light. For this purpose, to a sol of modified gold nanoparticles, pyranine (HPTS) and 6-methoxyquinoline (6MQ) were added. HPTS is a water-soluble photoacid which, in the excited state, is able to release a proton. 6MQ is a water-soluble photobase which, in the excited state, can accept a proton. The absorption maximum of HPTS is located at 405 nm, while for 6MQ it is at 326 nm. Another important parameter is the values of pKa for the photoacid and photobase. The pKa should be significantly different for the ground and excited states. The pKa of a ground state of HPTS is equal to 7.4, while for the excited state it is equal to 0.4, whereas the pKa values for the photobase are 5.18 and 11.8, respectively. The pH of the solution of gold nanoparticles was tuned to 4.5, which means that the MHA monolayer was partially deprotonated at these conditions, and therefore the charge of the surface of the nanoparticles stabilizing the sol is negative. At pH = 4.5 HPTS is protonated, while 6MQ is deprotonated. After the proper irradiation, molecules of HPTS are transferred into the excited state and release protons. Those protons can be captured by the negatively-charged carboxyl groups of chemisorbed MHA, which leads to their neutralization and the aggregation of nanoparticles, mediated by hydrogen bonding. The excitation of the 6MQ leads to a capturing of protons by this compound, to the dissociation of the carboxyl groups from chemisorbed MHA, and to the re-dispersion of the nanoparticles. In darkness, photoacid and photobase undergo instant neutralization, which makes the process reversible. Because the charge recombination is very fast, the aggregation of the nanoparticles in this process is very rapid. As in other cases, the process of aggregation can be studied by UV-VIS spectroscopy – aggregation causes a red shift in the SPR band. After functionalization, the SPR band of the sol of nanoparticles was located at 513 nm. The formation of aggregates increased extinction at ca. 800 nm. Also dynamic light scattering (DLS) can be used to study this process. The hydrodynamic diameter of the dispersed gold nanoparticles was 35 nm, while light activation led to the formation of aggregates with an average hydrodynamic diameter of 200 nm.
In another approach used to carry out photo-aggregation, gold nanoparticles synthesized in an organic solvent were stabilized by an adsorbed mixed layer composed of inert non-ionic hexaethylene glycol monododecyl ether and a photolyzable anionic surfactant hexylphenylazosulfonate (C6PAS).50 UV irradiation led to the breakdown of the photoactive C6PAS, which in turn led to a reduction in colloid stability, and hence to a flocculation of gold nanoparticles. In the UV-VIS spectrum measured for this colloid before irradiation, one can observe two strong bands: at 420 nm, related to the absorption of C6PAS molecules, and an SPR band of gold nanoparticles at 520 nm; these bands are not observed in the UV-VIS spectrum measured for the irradiated system. This is direct evidence for the complete photolysis of the C6PAS and for the flocculation of plasmonic nanoparticles. The light-induced aggregation of nanoparticles was also confirmed by TEM measurements. Where there is an optimal ratio of the photodestructive surfactant to the inert surfactant, the aggregation can be reverse.
Cheng et al. stabilized gold nanoparticles by a derivative of boronic acid called SPB.51 In terms of molecular structure, SPB is a typical pH-responsive surfactant with a phenylboronic moiety in the hydrophilic part. In neutral and alkaline conditions, SPB exists in an ionic form, while in acidic conditions it adopts a non-ionic form. Therefore, SPB can be employed to stabilize nanoparticles in neutral and basics environments. In order to make this system photo-responsible, Cheng et al. added diphenyliodonium nitrate to a sol of nanoparticles stabilized with SPB.51 Under UV irradiation, the diphenyliodonium nitrate was transformed to other compounds, including nitric acid. The formation of nitric acid led to a decrease in the pH of the sol, and to the transformation of the ionic form of SPB to the non-ionic one, which did not stabilize the nanoparticles effectively, and hence led to the aggregation of the nanoparticles. As in many other experiments with plasmonic nanoparticles, their aggregation can be easily proved by the observed change in the recorded UV-VIS extinction spectrum: the initial sol (pH = 6.7) exhibits an SPR band at 523 nm, whereas after the exposure to UV radiation, the position of the SPR band shifted to 563 nm and the pH of the solution decreased to 4.7. TEM measurements also confirmed that photo-aggregation took place. Before irradiation, mainly single semi-spherical nanoparticles with an average size of 20 nm were observed, whereas after irradiation, large agglomerates composed of hundreds of nanoparticles predominated. Increasing the pH of the sol to above 6.7 reverses this aggregation.
2.3. Photo-assembly based on covalent interactions
Assembling of nanoparticles can also be performed by creating new covalent interactions. This method is very attractive for making aggregates because of the high stability and durability of the chemical bonds, even after many important parameters of the reaction conditions are changed, e.g. by the addition of various amounts of different salts or a change in pH or temperature. One method for carrying out photo-assembly, which is based on the creation of chemical bonds, utilizes the reaction between o-nitrobenzyl alcohol and benzylamine in aqueous solution.52 Lai et al. carried out such a reaction in the presence of 23 nm semi-spherical gold nanoparticles.52 Some of the gold nanoparticles were modified with o-nitrobenzyl thiol derivative (TNB), while the other nanoparticles were functionalized by the thiol derivative of benzyaldehyde (TBA). UV radiation led to the formation of indazolone linkages between the nanoparticles modified with TNB and TBA, and therefore to their agglomeration (see Fig. 4a and b). The SPR band of the non-aggregated gold nanoparticles is located at 520 nm. Irradiation for 5 minutes of the sol of such nanoparticles using radiation with a wavelength of 310 nm led to a slight increase in its extinction at 550 nm, while 10 minutes of irradiation led to a noticeable reduction in the intensity of the SPR band at 520 nm and the formation of a new band at 650 nm, due to the plasmon excitation in the nanochains of nanoparticles formed (for this mode, the excitation was along the long axis of the formed structure) (see Fig. 4c). Further irradiation led to an increase in the intensity of this plasmon band and to shifts thereof to longer wavelengths (finally, to 675 nm), which indicates a lengthening of the nanochains. Aggregation was also confirmed by the TEM analysis, from which one can estimate that the distance between successive nanoparticles is about 1.5 ± 0.4 nm. This value is smaller than the size of the indazolone linker formed (3.6 nm), which suggests that organic linkers are in a molecular folding state. The main disadvantage of this method is that it is not reversible.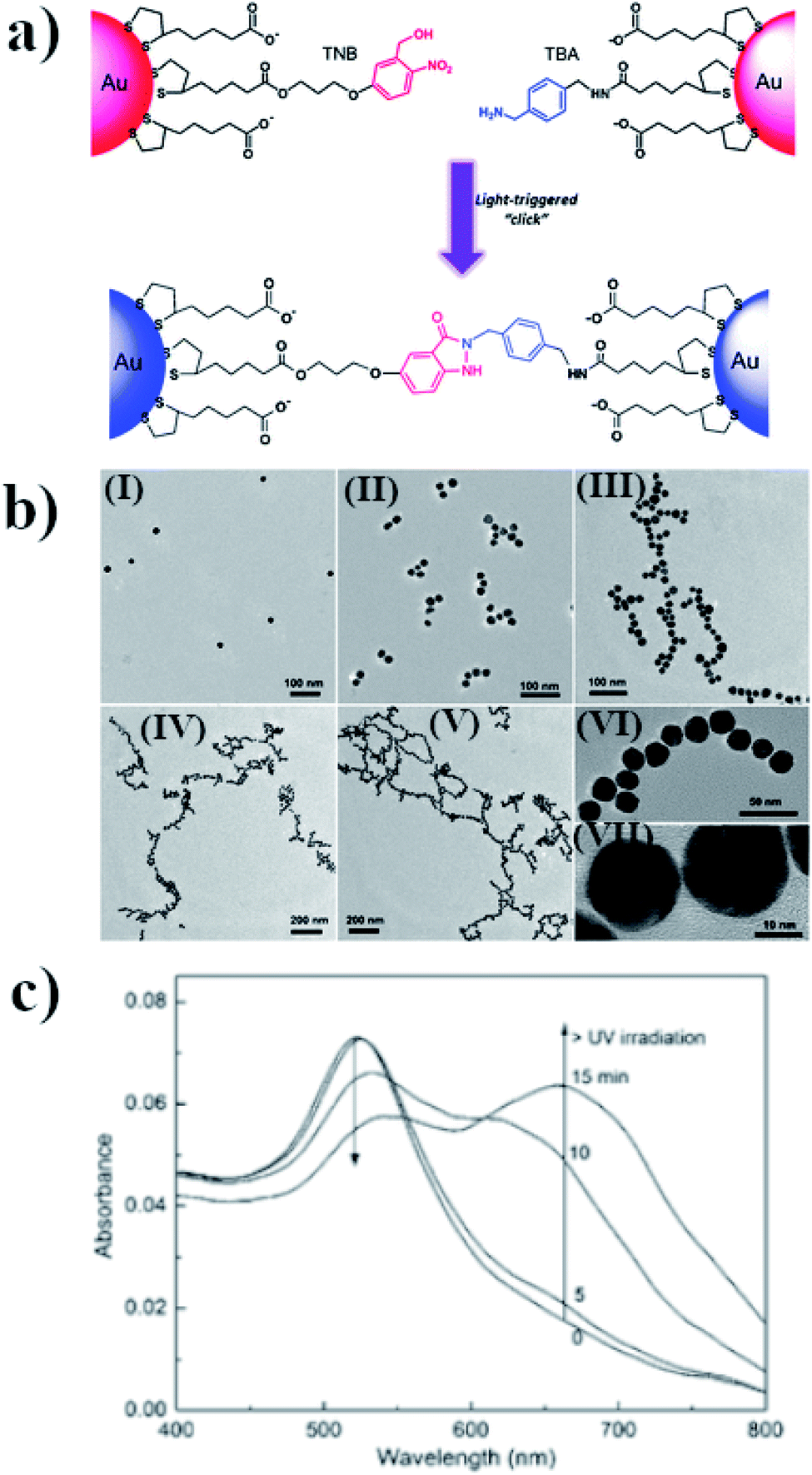 | ||
| Fig. 4 (a) Schematic illustration of light-triggered covalent assembling of gold nanoparticles, (b) TEM micrographs of gold nanoparticles after UV irradiation with increasing time: (I) 0, (II) 5, (III) 10, (IV) 15 and (V) 30 min. Images (VI) and (VII) are HRTEM characterizations of the nano-chain of gold nanoparticles obtained, (c) UV-VIS extinction spectra of modified gold nanoparticles after irradiation with increasing time (reprinted with permission from ref. 52. Copyright 2016 the Owner Elsevier). | ||
In another approach, a light-induced Diels–Alder cycloaddition of photocaged dimers like o-quinodimethanes or photoenols and maleimides was used to link plasmonic nanoparticles.53 Enols, or more formally alkenols, are a type of reactive structure that is represented as an alkene with a hydroxyl group attached to one end of the alkene double bond. The generation of enols often involves the removal of a hydrogen adjacent to the carbonyl group; in the case of photoenols, this hydrogen adjacent is caused by light.54 Irradiation of the o-quinodimethanes precursor leads to intramolecular hydrogen abstraction followed by a bond reorganization, to form highly reactive dienes which permit the Diels–Alder reaction with electron deficient alkines.55 This method can be applied for the aggregation of gold nanoparticles.56 For this purpose, gold nanoparticles were functionalized by mercaptoundecanol (MUD). Subsequently, via esterification, the photoenol (PE) moiety was attached to the surface of the modified Au nanoparticles. TEM, FT-IR and 1H-NMR analyses revealed that semi-spherical gold nanoparticles used, having a diameter of 3 nm, were coated on average by 209 MUD ligands, while the amount of photoenol was estimated as 10 photoenol molecules per gold nanoparticle. The MUD-PE were then mixed with maleimide. The reaction was carried out in dimethylformamide. A photo-induced Diels–Alder reaction occurs after illumination by radiation having a wavelength in a range of from 315 to 400 nm. This method can be applied for the photochemical formation of aggregates of gold nanoparticles on some modified (prepared) substrates.
The other class of photochromic ligands that can be useful for photo-switching of plasmonic nanoparticles based on formation of new chemical bonds are diarylethenes. These molecules undergo a reversible cyclization reaction upon interaction with UV and visible light in less than 10 ps.57 The diarylethene compounds could be modified by a thiol group to enhance their affinity to the metallic surface.58 The open ring form is colorless in toluene. After illumination by a 313 nm radiation it undergo a photochromic reaction and form a closed-ring isomer. The colorless solution became a blue-purple and a new absorption band at 578 nm appears. The conversion rate after UV irradiation is high. Also the diarylethene polymer could be used to photo-switching of nanoparticles.59 The photo-cyclization reactivity of the diarylethenes ligand attached to the gold nanoparticles decrease when shortening the distance between the surface of the gold nanoparticles and the diarylethene chromophore.
Another example of light triggered covalent assembly of gold nanoparticles is based on tetrazole – alkene photo click chemistry.60 The gold nanoparticles was decorated by 2,5-diphenyltetrazole and methacrylic acid. This allows for formation of covalently cross-linked aggregates after light (λ = 405 nm) irradiation. The aggregates formation was confirmed by the TEM and DLS measurements.
Photo-assembly utilizing formation of covalent bonds can be also based on the photo-dimerization of linking compounds attached to the surface of plasmonic nanoparticles. Itoh et al. realized such type of a photo-assembly utilizing the photo-dimerization of thymine units immobilized on the surface of gold nanoparticles (it is well known that molecules of thymine photo-dimerize after UV irradiation).61 The aggregation of gold nanoparticles can be deduced from a shift and decrease in the intensity in the maximum of the SPR band of the irradiated, modified gold nanostructures. Precipitation of the gold nanostructures was observed after 72 hours of irradiation. This means that, after this time of irradiation, the aggregates formed were too large to remain in solution. A TEM analysis showed that aggregates with an average size of 0.25 μm are formed after 22 hours of irradiation, while after 72 hours the average size of the aggregates exceeded 1 μm.61
The first successful performance of a reversible photo-induced aggregation of gold nanoparticles based on the photo-dimerization of a coumarin derivative was carried out by He and co-workers in 2016.62 For this purpose, a thiolated coumarin derivative (7-(11-mer-captoundecanoxy)coumarin) was immobilised on the surface of gold nanoparticles (with an average size of 7.6 nm and an SPR band located at 523 nm) and was used as a photo-responsive linker. The processes of photo-induced aggregation and de-aggregation were carried out in tetrahydrofuran. Tetrahydrofuran was used as an electron donor to yield the charge-separated coumarin singlet excimer, ensuring effective photo-cleavage for the rapid disassembly of the aggregates of the nanoparticles. The assembly/disassembly of the system could be repeated with full recovery, four times. It was found that irradiating the solution containing modified gold nanoparticles for 72 hours using radiation with a wavelength of 365 nm led to a red shift in the SPR band, from 523 nm to 556 nm. Irradiating the unmodified gold nanoparticles under the same conditions did not lead to any observable change in their optical properties. A TEM analysis showed that the modified gold nanoparticles before irradiation did not form aggregates, whereas the irradiated (using radiation having a wavelength of 365 nm) modified nanoparticles formed aggregates. The size of the clusters formed increased with increasing exposure time. The aggregates could be disassembled by illumination using UV radiation with a wavelength of 254 nm. It was found that illuminating the solution with such radiation for 20, 40 and 60 minutes led to a blue shift in the position of the SPR band to 550, 536 and 523 nm, respectively. A more detailed analysis showed that aggregation process described above is only observed when the surface concentration of the coumarin moiety is above a certain threshold. The main disadvantageous of the coumarin linker is that it is irreversibly damaged by strong UV radiation.
One can also construct systems in which more than one kind of linking bonds have to be created to form a connection between nanoparticles. An example63 of such a system is a sol of gold nanoparticles functionalized by pillar[5]arens terminated with a thiol moiety (and therefore attached to the surface of the gold nanoparticles by Au–S bonds) to which anthracene modified with a quaternary amine terminated carbon chain has been added. Due to the host–guest interactions, the quaternary amine can penetrate into the cavity formed by the pillar[5]arens, while the anthracene groups can dimerize under the influence of UV radiation, which leads to the aggregation of the nanoparticles. Raising the temperature increases the number of free nanoparticles by decreasing the number of host–guest interactions. Heating the system to 60 °C for 20 h, or exposing it to radiation having a wavelength of 300 nm, leads to a dissociation of the anthracene dimers and the decomposition of the aggregates of nanoparticles. The anthracene dimers are re-formed if the system is re-exposed to UV radiation with a wavelength longer than 360 nm. Zhou et al. carried out 4 cycles of aggregation–de-aggregation, in which they illuminated this system with radiation having a wavelength of 360 nm for aggregation, and with radiation having a wavelength of 300 nm for de-aggregation. The surface plasmon band on the UV-VIS spectrum appeared at the same wavelengths in subsequent cycles. Zhou et al. suggested that this system could be applied in catalysis, to recover metal catalytic nanoparticles by aggregation after carrying out the reaction. Moreover, the aggregation of this material can be controlled not only by the application of appropriate irradiation, but also by changing the temperature.
2.4. Photo-assembly based on host–guest interactions
The mechanism of the change in the aggregation stage of nanoparticles upon irradiation can also be based on host–guest interactions. For example, using β-cyclodextrin as a host, and a compound containing two azobenzene units on ends as a guest, Wu et al. constructed a system which, upon irradiation, can be effectively transferred from the aggregated stage into separate nanoparticles. Wu et al.64 added solutions of (E)-1-(4-(4-bromobutoxy)phenyl)-2-phenyldiazene (BiAzo) and thiol-modified β-cyclodextrin to a sol of gold nanorods. Thiol-modified water-soluble cyclodextrins attached to the gold nanorods via the Au–S bonds. BiAzo has two azobenzene units on its ends, which can enter the hydrophobic cavity of β-cyclodextrin (see Fig. 5a). In this way, it is possible to link two cyclodextrins via a BiAzo molecule. The formation of aggregates of gold nanorods (see Fig. 5c) leads to a red shift (of 51 nm) in the position of the SPR band in comparison with the position of the SPR band for the non-aggregated nanoparticles. Irradiating the sample of aggregated nanorods with UV radiation and simultaneous sonication caused a transition of the BiAzo from the trans to cis conformation, which in turn caused the azobenzene units to exit the cavity in the cyclodextrin molecules and break down the aggregate. As expected, de-aggregation caused a blue shift in the SPR band (by 46 nm) (see Fig. 5b). De-aggregation was also observed after the addition of adamantanamine hydrochloride, which has a higher affinity for cyclodextrins than does BiAzo, and therefore removes the linking moieties, making the gold nanoparticles bond together. The main disadvantage of this system is the non-reversibility of the de-aggregation/aggregation processes. The host–guest complex is formed before the cyclodextrins bond with the nanoparticles, and after the nanorods combine with the cyclodextrins, because it is very unlikely that the BiAzo will connect two cyclodextrins connected to various gold nanoparticles.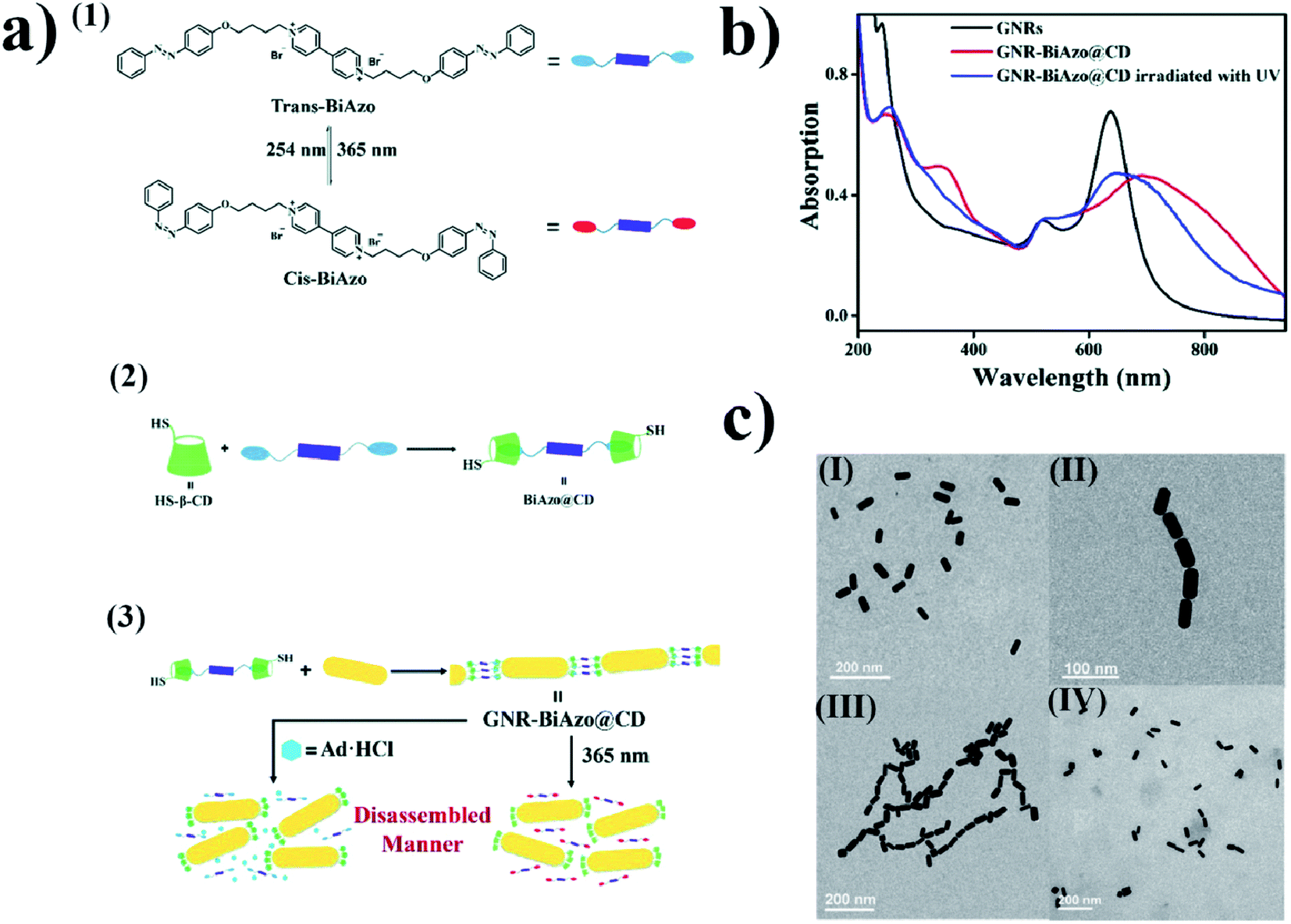 | ||
| Fig. 5 (a) (1) Diagram showing cis–trans isomeric transition of BiAzo compound under the influence of light, (2) formation of a complex from trans BiAzo and cyclodextrins with attached thiol groups, (3) diagram showing formation of aggregates of gold nanorods. (b) UV-VIS extinction spectra of: gold nanorods before the addition of a BiAzo compound with cyclodextrins (GNRs black), gold nanorods with the addition of a complex of the trans BiAzo compound with cyclodextrins (GNR-BiAzo@CD red), and gold nanorods with the addition of a complex of the trans BiAzo compound with cyclodextrins after UV irradiation (GNR-BiAzo@CD irradiated with UV blue). (c) TEM images of gold nanorods: (I) without BiAzo@CD complex, (II) with BiAzo@CD complex, and (III) with BiAzo@CD complex after UV irradiation (reprinted with permission from ref. 64. Copyright 2017 the Owner The Royal Society of Chemistry). | ||
A similar approach has been applied by Stricker et al.65 This group added water-soluble arylazopyrazoles (AAPs) to a sol of cyclodextrin-functionalized gold nanoparticles.65 It was found that the cis isomer of the AAPs did not fit the cavity formed by the cyclodextrin and hence could not effectively interact with the cyclodextrin molecules, whereas the trans isomer fit very well into the cavity and formed host–guest complexes with molecules of cyclodextrins. Hence, if this isomer of AAPs is present in a sol of cyclodextrin-functionalized gold nanoparticles, aggregation of the nanoparticles occurs. The trans–cis isomerization occurs upon UV radiation (λ = 365 nm), and this process is reversible when the sample is illuminated with light having a wavelength of 520 nm. This system is reversible for at least a few cycles of trans–cis isomerization. Based on this idea, Niehues and co-workers developed a method for the end-to-end gold nanorods photoassembly.66 The ends of nanorods were modified by molecules of β-cyclodextrin. In this case, the azobenzene molecules are replaced by the AAP linker. AAP having an E structure forms an inclusion complex with β-cyclodextrin. Interaction with light leads to change of the conformation of AAP to Z structure and prevents this interaction what leads to redispersion of nanorods.
Gold nanoparticles could be also reversible attached to the surface of multiwalled carbon nanotubes.67 For this purpose, gold nanoparticles were functionalized with α-cyclodextrin, while multiwalled carbon nanotubes were modified with an azobenzene derivative. In dark, the –NN– bond of azobenzene is in trans conformation and, in this conformation, azobenzene is able to form an inclusion complex with α-cyclodextrin. After formation of the inclusion complex the gold nanoparticles were attached to the surface of nanotubes. The aqueous solution of formed composite is stable even for few months. However, as proved by the TEM measurements, the UV irradiation (λ = 365 nm) leads to detachment of gold nanoparticles from the surface of carbon nanotubes, because the azobenzene change the conformation to the cis one which does not form an inclusion complex with α-cyclodextrin. In dark, the azobenzene again transforms into the trans form, and as a result, gold nanoparticles are again attached to the surface of multiwalled carbon nanotubes.
2.5. Photo-assembly based on photo-thermo-response
Photo-assembly is often realized using thermo-responsive materials. For example, Fava et al. functionalized the ends of gold nanorods (produced in a CTAB solution) with thiol terminated molecules of poly(N-isopropylacrylamide) (p-NIPAM).68 The p-NIPAM is selectively attach to the curved ends of the nanorods because the interaction of the molecules of CTAB with the ends of the gold nanorods was significantly weaker, and therefore the thiol terminated p-NIPAM was preferentially adsorbed at such places.69 In the first experiments, Fava et al. showed that heating a sol of gold nanorods modified with p-NIPAM and stabilized with CTAB to 45 °C led to the formation of chains of nanorods assembled end-to-end. This was due to the fact that heating above the lower critical solution temperature (LCST) of the NIPAM polymer led to their coil globule transitions. This aggregation is fully reversible: cooling the system down to room temperate causes the re-aggregation of the nanoparticles. The aggregation could also be obtained by illumination using 803 nm laser radiation. This process is also reversible. An analogous procedure has been applied to spherical gold nanoparticles.70 It was shown that irradiating them for 5 minutes with a 532 nm laser beam with an average power of 5 W led to an increase in the temperature of the sample above 35 °C. This temperature is high enough to collapse the p-NIPAM chain, and hence induce the aggregation of the nanoparticles. For this system, a very large shift in the position of the SPR band was observed: from 539 nm to 670 nm after just 30 s of irradiation, and up to 750 nm after a longer irradiation time. This means that in this system the plasmon coupling is strong, which suggests that the gold nanoparticles are very closely packed. After cooling, the position of the SPR band changed back to the initial value.The above approach was also used to change the distances between the gold nanorods deposited on the surface of the p-NIPAM microspheres.71,72 It was found that, at temperatures above 50 °C, the p-NIPAM microspheres collapsed, leading to a mutual approach of the nanorods deposited on their surface. The process is also fully reversible. So far, this effect has only been achieved by changing the temperature, although one can expect that a similar effect should also occur after irradiation. In such a case, the interaction between the light and the plasmonic gold nanorods should lead to the generation of heat, and subsequently to the collapse of the p-NIPAM microspheres.
To construct photo-switchable devices, gold nanoparticles are usually used. In some cases, however, other plasmonic structures are employed, silver ones especially. The rationale behind using silver nanoparticles is that, under certain conditions, they can generate even ten times more heat than gold nanoparticles.73 An interesting example of the application of non-gold plasmonic structures has been described by Han et al.74 This group illuminated a sample of Ag–Fe3O4 dimers coated with p-NIPAM with solar radiation that passed through a filter transferring only that part of the spectrum between 400 and 700 nm. The UV and IR part of the spectrum were eliminated to avoid heating caused by the absorption of water molecules, so practically the only source of the heat in this system was the heat generated on the plasmonic nano-structures during the absorption of light. Han et al. found that the absorption of radiation by the plasmonic nanoparticles (and hence localized heating) caused a collapse in the thermo-responsive polymer chain and a clustering of the dimers into spherical aggregates, which facilitated their magnetic separation.74
In another approach, core–shell Ag–Au nanoparticles have been used to construct photo-switchable devices.75 Surface plasmon resonance for core–shell Ag@Au nanoparticles is obtained in a significantly wider frequency range than for analogous gold nanoparticles, and the local field enhancement generated in the proximity of such nanostructures is also significantly larger than in the case of analogous gold structures, whereas surface gold makes the system biocompatible, as in the case of pure gold nanoparticles. In an experiment carried out by Patra et al., a solution containing such nanoparticles was deposited on a 40 nm thick layer of silver evaporated onto the surface of a prism.75 The silver film was illuminated in the Kretschmann configuration by a laser beam, which allowed plasmon polarons (SPP) to be generated on the surface of the silver film. The aggregation of Ag@Au nanoparticles occurred by a local heating of the environment, which caused the nanoparticles to move towards the region having a higher temperature. The formation time for a 2 μm aggregate of nanoparticles was 30 min, although the vast majority of nanoparticles were already in the area after 16 min. The aggregates thus produced were tested for stability. Turning off the laser caused the aggregates to break down, while turning it on again led to re-aggregation. These aggregation/re-aggregation processes were carried out five times. A very interesting property of this system is the possibility of shifting the aggregates simply by shifting the position of the laser beam. The aggregates were stable during this process.
An interesting modification of the system described above was proposed by Zheng et al.76 This group illuminated a gold substrate that was in contact with a solution containing triangular gold nanoparticles and cetyltrimethylammonium chloride (CTAC) using a laser beam having a wavelength of 532 nm. The laser illumination caused a local heating of the environment and the thermophoresis and migration of CTA+ ions towards regions having a lower temperature. Because the nanoparticles also had a positive charge, they migrated in the opposite direction, towards regions having higher temperatures, which meant that the nanoparticles accumulated in the center of the laser dot. This method permits the use of a laser beam of much lower power than in the method described above, and there is no requirement to adjust the wavelength of the radiation to the SPR of the nanoparticles. Aggregations were observed after significantly shorter times – even after just a few seconds. The limitation of this method is the need to carefully choose the power of the laser. Too little power will not result in an appropriate temperature gradient, and too much power will induce significant convection movements, which can cause the aggregates to break down. This photo-aggregation is reversible; the aggregates decompose after the laser is turned off. Manipulation of the position of the aggregates is realized by moving the laser spot. After an aggregate is formed, it becomes a source of heat due to the excitation of the plasmons by the laser beam. This additional thermoelectric effect increases the stability of the aggregate.
In further articles, Zheng and co-workers demonstrated more possibilities of optical assembly of colloidal nanoparticles. This group formed some colloidal matter of diverse colloidal sizes (from subwavelength scale to micrometre scale) with versatile configurations and tuneable bonding strengths and lengths.77 Then, this group developed opto-thermoelectric nanotweezers that allowed to capture and manipulate metal nanoparticles of a wide range of sizes and shapes at single-particle resolution.78 Spectroscopic response of trapped nanoparticles can be selectively characterised in situ using dark-field optical imaging.78 This group also developed optical nanomanipulation on solid substrates via optothermally-gated photon nudging for the versatile manipulation and dynamic patterning of a variety of colloidal particles on a solid substrate at nanoscale accuracy.79 The scheme presenting the mechanism of the photomanipulation and the real-time manipulation of a 300 nm Au nanoparticle is presented in Fig. 6.80 In all mentioned above experiments CTAC was used as anionic surfactant having strong photothermal response.
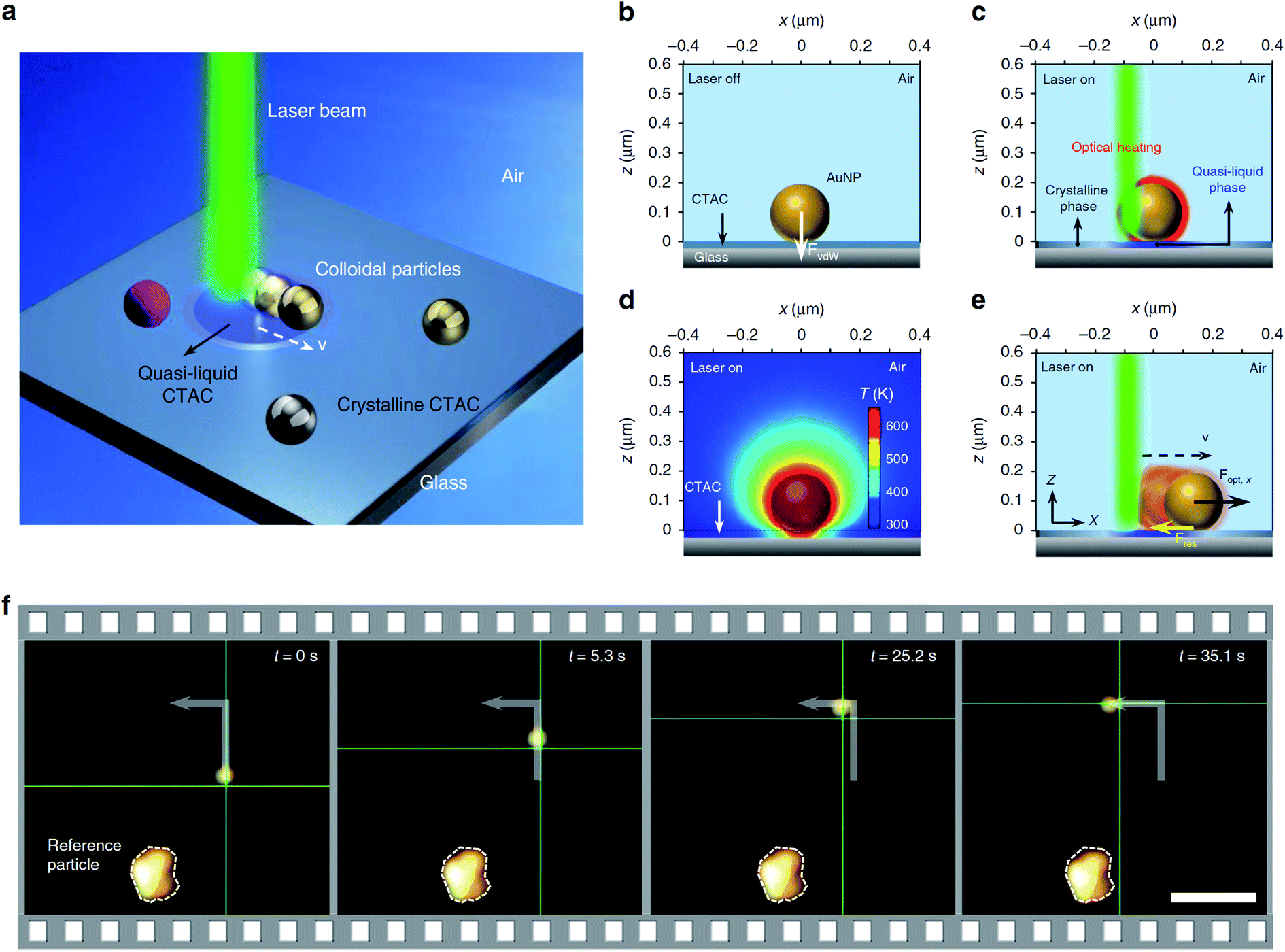 | ||
| Fig. 6 Illustration of the general concept of optothermally-gated photon nudging. (a) Schematic illustration of the system. (b) A 200 nm Au nanoparticle placed (and bonded by van der Waals interactions) on glass substrate with the CTAC layer. (c) The optical heating under laser illumination induces a localised phase transition in the surrounding CTAC layer. CTAC turns into a quasi-liquid phase and releases the bond with Au nanoparticle. (d) The simulated temperature distribution around illuminated 200 nm Au nanoparticle (incident power: 1 mW; laser beam size: 0.8 μm). (e) Au nanoparticle moves against the laser beam with an in-plane optical force Fopt and a resistant force Fres. (f) Sequential dark-field optical images showing real-time manipulation of a 300 nm Au nanoparticle. The green crosshair indicates the position of the laser beam. Scale bar: 5 μm. Reproduced from ref. 80 (Li et al.), licensed under a Creative Commons Attribution 4.0 International License. | ||
2.6. Photo-aggregation based on photocatalytic reaction
The simplest method of photo-aggregation, however nonreversible and giving relatively unreproducible aggregates of plasmonic nanoparticles is realized by the irradiation of the sol of plasmonic nanoparticles that also contains the metal precursor and the reducing agent. This method of aggregation is especially useful for silver nanoparticles. Although photo-aggregation of silver nanoparticles based on photocatalytic selective deposition of silver is nonreversible and the shape and size control of the formed aggregates is very poor, this method of aggregation seems to be very promising for producing highly active substrates for surface-enhanced Raman scattering (SERS) measurements – for details see Chapter 3.2.It was shown that when a sol of silver nanoparticles containing some amount of silver cations (which are, for example, created by the partial dissolution of Ag nanoparticles when the Ag sol contains dissolved oxygen) and a proper reducing agent (for example citrates) is irradiated, a photocatalytic reduction of Ag+ occurs, which causes preferential deposition of silver at some places of the silver nanostructure. The efficient photocatalytic reduction of Ag+ occurs only at such places of silver nanoclusters, at which strong surface plasmons are excited (usually sharp edges of plasmonic nanostructures and narrow slits between plasmonic objects81). When two illuminated silver nanoparticles touch (or almost touch) each other, there is a large increase in the intensity of the electromagnetic field in the narrow slit between them. This causes that silver cations are effectively photo-chemically reduced in the formed slit and the deposited silver “glues” silver nanograins, which leads to their agglomeration. The influence of illumination on the morphology of illuminated silver sol is presented in Fig. 7.82
 | ||
| Fig. 7 The influence of illumination on the morphology of silver sol synthesized by the reduction of AgNO3 with NaBH4 in the presence of sodium citrate. TEM micrographs show silver nanoparticles present in the sol: (a) before illumination, (b) in the sol illuminated for 1 day. This figure was prepared on the basis of TEM images from ref. 82. | ||
3. Sample applications of photo-assembled plasmonic nanoparticles
3.1 Catalysis
A promising application of reversible aggregation controlled by light is to use this process to facilitate the recovery of a catalyst from a post-reaction mixture. After the process has been performed, the nanoparticles of the catalyst dispersed in the reaction mixture (the solution in which the reaction was carried out) are agglomerated under the influence of light, which significantly facilitates the separation of the nanoparticles of the catalyst so that they can be reused. The recovery of nanoparticles reduces the costs of the process and the consumption of sometimes scarce materials. Another application of photo-switchable catalysts is the ability to terminate a chemical reaction at the point of your choice. This facilitates the control of the relative amounts of substrates and products in the reaction mixture.Wei et al. presented an interesting example of the application of reversible aggregation in the synthesis of 4-methoxybenzyloxy-diphenylsilane.32 The reaction between 4-methoxybenzaldehyde and diphenylsilane was carried out in the presence of gold nanoparticles functionalized by dodecylamine and photoswitchable 11-(4-(phenylazo) phenoxy)-1-undecane-thiol. The rate constant for this reaction was determined as equal to 9 × 10−4 mM−1 min−1. After exposure to UV radiation, the reaction rate constant dropped to 1 × 10−5 mM−1 min−1. A quick (approx. 2 minutes) switch-on is realized by exposing the sample to white light. This system allows for 3 switch on–off cycles. Subsequent cycles do not allow for such effective deactivation of the reaction by UV radiation, as the nanoparticles stop aggregating so effectively. This application of photoswitchable nanoparticles is an example of how a reaction can be interrupted at the point of your choice, and then continued on demand.
In some cases, agglomerates of nanoparticles are significantly more efficient as a catalyst than isolated nanoparticles. Zhao et al.83 showed that the light-induced formation of aggregates of nanoparticles (called by this group nanoflasks) accelerate certain chemical reactions. The first reaction in nanoflasks described by this group was the acid-catalyzed hydrolysis of acetal to aldehyde in the presence of 6 nm gold nanoparticles functionalized by a compound containing azobenzene moiety. The reaction was carried out in water-saturated toluene. It was shown that the use of aggregates of nanoparticles led to a multiple increase in the reaction rate. When no nanoparticles were present in the sample, or no UV radiation was used to form aggregates, the reaction was not catalyzed. The aggregation of the nanoparticles leads to an increase in the reaction rate by a few orders of magnitude. It was also proven that heating from the excitation of plasmons did not play a major role in increasing the reaction rate. Zhao et al. explained the increase in the reaction rate by the accumulation of polar substrates inside the agglomerates (causing a significant increase in their local concentrations) which occurred due to the creation of dipole moments by the light-induced isomerization of azobenzene.83 In another experiment (dimerization of hydroxyanthracene), they demonstrated that carrying out the agglomeration of nanoparticles (the formation of nanoflasks) allowed to obtain products having a specific structure. During the reaction without nanoflasks, an anti-isomer was mainly formed, but in the presence of aggregates of nanoparticles, a sym-isomer was obtained. In all the above-mentioned cases, the isolated nanoparticles can be recovered by irradiating the samples with white light, which causes the agglomerates to break down.
3.2 Surface-enhanced optical spectroscopies
The interaction of light of an appropriate frequency with plasmonic nanoparticles leads to the excitation of collective oscillations of the electron plasma in the conductive band of the nanoparticles. Such electron oscillations lead to a very large local increase in the intensity of the electromagnetic field. Therefore, plasmonic nanoparticles are often used to increase the efficiency of certain optical processes whose efficiency depends on the intensity of the electromagnetic field. For example, Raman scattering (Raman scattering enhanced by plasmonic nanoparticles is called surface-enhanced Raman scattering – SERS), infra-red absorption (the enhanced process is called surface-enhanced infra-red absorption – SEIRA) and fluorescence (the enhanced process is called metal-enhanced fluorescence – MEF). Theoretical calculations confirmed by experiments have shown that the highest field enhancement is induced in the gap(s) between plasmonic nanoparticles.84 These especially active places in the field enhancement are called “hot-spots”85,86 (see Fig. 8a). As mentioned above, an enhancement of the electromagnetic field causes an increase in the efficiency of many optical processes (see Fig. 8b). For example, the increase in the intensity of the SERS signal is proportional to the square of the enhancement of the excitation wave and to the square of the enhancement of the scattered wave – which means that the SERS enhancement is roughly proportional to the fourth power of the field enhancement.87 In the case of SEIRA, the SEIRA enhancement factor is roughly proportional to the second power of the field enhancement.88 Since agglomeration significantly increase the local field enhancement (due to the creation of a large number of slits between the plasmonic nanoparticles), photoswitchable nanoparticles can be successfully used to perform switchable (on and off) SERS and SEIRA experiments.89 For example, Wang et al. analyzed the SERS spectra of rhodamine 6G and 4-aminothiophenol adsorbed on gold nanorods that had been made photoswitchable by the ionic interactions between CTAB and AzoNA (for more details, see Chapter 2.1).43 SERS measurements revealed that the Raman spectra of rhodamine 6G and 4-aminothiophenol recorded when gold nanorods were in the aggregated state were 11 and 17 times more intense, respectively, than when the nanorods were in a dispersed state.43 Using this system, Wang et al. completed 3 full photo-cycles that led to “switching on” and “switching off” the SERS spectra. | ||
| Fig. 8 (a) An electric field distribution around two nanoparticles close to each other (reprinted with permission from ref. 73. Copyright 2004 the Owner The Journal of Chemical Physics). (b) The SERS spectrum of rhodamine 6G with a concentration of 10 μM obtained during progressive aggregation over time. A 660 nm laser beam was used as an excitation source and spectra were recorded every 15 seconds (reprinted with permission from ref. 76. Copyright 2016 the Owner The Royal Society of Chemistry). | ||
Patra et al. showed that using a reversible formation of aggregates of Ag@Au nanoparticles near a silver substrate (the mechanism of this photo-aggregation was based on photo-thermo-response), one can record Raman spectra even of individual molecules of rhodamine 6G and Nile blue75 (for details concerning this photo-aggregation, see Chapter 2.5). Moreover, in their experiment, the same laser beam was used to create aggregates and to excite Raman scattering. Also, a system utilizing photo-thermo-response developed by Zheng et al. has been used for the light-induced “switching on” and “switching off” of SERS measurements.76 As described in detail in Chapter 2.5, this system was constructed from a gold substrate in contact with a solution containing triangular gold nanoparticles and CTAC. When the gold substrate was illuminated by a laser beam having an appropriate wavelength, a very fast and efficient aggregation was observed – the gold nanoparticles accumulated in the center of the laser dot. This led to a significant increase in the intensity of the SERS spectra of rhodamine 6G, which was used as a Raman probe.76
As mentioned in Chapter 2.6, especially dedicated for producing of SERS substrates is the simplest method of photo-aggregation of silver nanoparticles, it means irradiation of silver sol in the presence of air. Although this method gives relatively unreproducible aggregates of silver nanoparticles (see Fig. 7), this transformation is very simple to carry out and gives highly SERS-active aggregates of silver nanoparticles (for details concerning the mechanism of this photo-aggregation, see Chapter 2.6). Therefore, this method seems to be very useful from the practical point of view. The change of the optical properties of the silver sol during illumination with the white light is illustrated by Fig. 9.81 The same figure illustrates the increase in the intensity of the SERS spectrum measured using such photo-transferred Ag colloid.
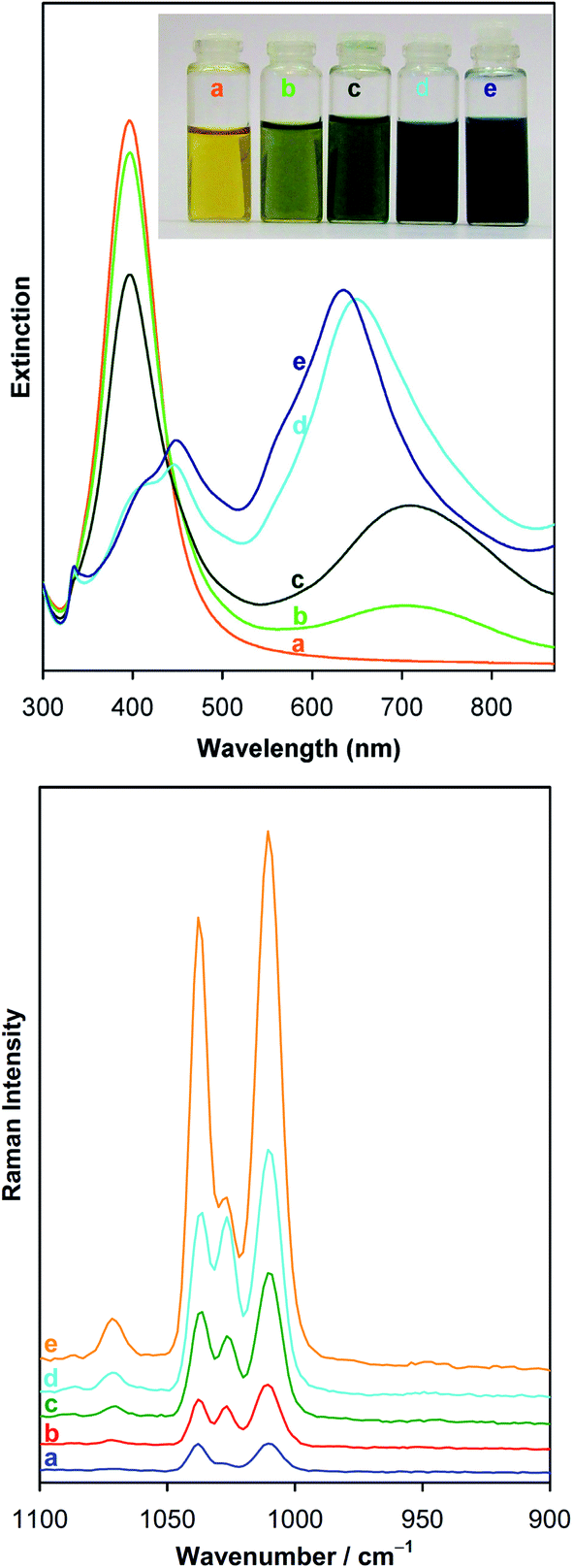 | ||
| Fig. 9 Upper panel: temporal evolution during illumination with the white light of the extinction spectra of silver sol synthesized by the reduction of AgNO3 with NaBH4 in the presence of sodium citrate. (a) Freshly synthesized sol, (b) sol irradiated for 1 day, (c) sol irradiated for 2 days, (d) sol irradiated for 5 days, and (e) sol irradiated for 7 days. The inset shows the visual appearance of the sols, for which extinction spectra were measured. Bottom panel: SERS spectra of pyridine adsorbed on photo-transferred sol. (a) Freshly synthesized sol, (b) sol irradiated for 1 day, (c) sol irradiated for 2 days, (d) sol irradiated for 3 days, and (e) sol irradiated for 6 days. Measurements were carried out using excitation radiation having wavelength of 532 nm. Spectra are presented at the same scale, vertically shifted for the sake of clarity (reprinted with permission from ref. 81. Copyright 2014 the Owner Elsevier). | ||
3.3 Photothermal therapy (PTT)
A very promising application of the light-induced aggregation of gold nanoparticles is the in vivo formation of photosensitizers for photothermal therapy (PTT). In PTT, the photosensitizer generates heat upon irradiation. Locally increasing the temperature above certain threshold leads to the death of cells in a vicinity of the photosensitizer. The optimal wavelength for PTT is the NIR region, where the human body exhibits the lowest absorption. This region is known as a ‘biological window’. An interesting example of the application of light-induced aggregation for PTT is presented in the work of Cheng and co-workers.90 This group covered gold nanoparticles having a diameter of 20 nm with polyethylene glycol. It was found that the polyethylene glycol transformed into carbene upon irradiation using a 405 nm laser beam. Reactive carbene moieties form covalent bonds, leading to the formation of covalently cross-linked particle aggregates. The hydrodynamic size of the nano-structures quickly increased with irradiation time, and reaches 346 nm after 25 minutes of irradiation. Also, the position of the SPR band shifted from an initial value of 520 nm to 700–900 nm (into the desired NIR region, where the human body exhibits the lowest absorption). Subsequently, the aggregates formed were irradiated with 808 nm laser radiation. Cheng et al. found that, after 5 minutes of irradiation of the aqueous solution containing aggregates of gold nanoparticles (with a radiation flux of 1 W cm−2), the temperature increased to 57.1 °C, while in the case of irradiation of a solution containing non-aggregated nanoparticles, the temperature increased only to 30.5 °C. These results clearly show that, after the photochemical aggregation of gold nanoparticles, effective thermal treatment can be achieved using the safe flux density of the 808 nm laser beam, whereas in the case of the non-aggregated nanoparticles no evident cytotoxicity was observed. This method was also successfully used in vitro with a 4T1 cell line. It was shown that tumors holding aggregated nanoparticles could be completely eliminated by irradiation using a 808 nm laser beam. In the control samples containing non-aggregated nanoparticles, the size of the tumor increased with time. Similar approach was based on photo-click chemistry of tetrazole–alkene system.60 The dispersed gold nanoparticles exhibit SPR band at 528 nm, however, after illumination by 405 nm radiation, gold nanoparticles aggregate and the SPR band is shifted into longer wavelength, at around 700 nm. It was found that when the sample is irradiated using 808 nm laser beam with the average power of 1 W cm−2, after 10 minutes of illumination the temperature of the aqueous solution of cross-linked gold nanoparticles was raised to 52.4 °C, while the temperature of the dispersed nanoparticles reached only 28.6 °C. The optical-thermal conversion efficiencies for cross-linked gold nanoparticles is much higher (78.3%) than that for dispersed nanoparticles (10.6%). Formed nanoaggregates can be effectively used for the photothermal therapy of cancer cells.3.4 Photo-controlled release of selected compounds
Another application of photo-switchable systems composed of gold nanoparticles is the photo-controlled release of amino acid derivatives.91 Ipe et al. constructed such a system using gold nanoparticles coated with spiropyran. As mentioned above, in darkness, the majority of molecules of spiropyran exist in a “closed” spiro form, which is colorless and non-polar. UV irradiation leads to the photoisomerization of spiropyran to the merocyanine form (which is connected with the opening of one aromatic ring), which is zwitterionic and hence polar. A reversible reaction is realized after irradiation using light with a wavelength of 520 nm. Due to the zwitterionic character of the merocyanine form, spiropyran in this form could bind to the charged molecules. Therefore, Ipe et al. decided to conduct the photo-switching of spiropyran on gold in the presence of various amino acids and their derivatives, such as L-tryptophan, L-tyrosine and L-DOPA.91 These compounds form complexes with spiropyran in the merocyanine form. Ipe et al. found that after irradiation of these complexes with light having a wavelength of 520 nm, the spiropyran was transferred to the closed spiro form, the complexes dissociated, and molecules of the amino acids were released. The local concentrations of the amino acids could be regulated by changing the number of spiropyran molecules attached to the gold surface. Such a method of the photo-controlled release of selected compounds has potential for medical applications, for example, to photo-control the release of drugs.In another experiments gold nanorods with poly(N-isopropylacrylamide) hydrogel was used to light triggered drug release.92 As targeted molecules doxorubicix (DOX) and curcumin (CUR) were tested. The DOX was encapsulated in the nanogels, while CUR and gold nanorods were embedded into the hydrogel. The heat induced shrinkage of hydrogel and then drug release. It was found that the release drug quantity decrease with an increasing amount of poly(N-isopropylacrylamide). The heating of the system could be also caused by NIR laser beam. It was found that the temperature increased from 25 °C to 65 °C after 7 minutes of the irradiation using 808 nm laser beam with the power density of 1150 mW cm−2. The efficiency of the drug released can be controlled by tuning laser power. This allows for drug releasing on demand. CUR molecules embedded into hydrogel was released first, while DOX encapsulated in nanogels was related subsequently.
3.5 Examples of other applications
Photo-switchable systems can be also used to construct self-erasing (after some time) notes.93 A self-erasing sheet was constructed by Grzybowski et al. from gold nanoparticles (5.6 nm in diameter) functionalized with dodecylamine and 1-(4-(phenylazo)phenoxy)-1-undecane-thiol (AT). Modified gold nanoparticles were placed in a 150 micron thick layer of organogel and then laminated into flexible poly(ethylene terephthalate) sheets coated with poly(vinyl chloride). The color of the resulting material corresponded to the nanoparticles in a “standard” solution. UV-VIS measurements confirmed the absence of aggregates of nanoparticles in the material. Exposing the obtained sheets to UV radiation led to a local change in color, from red to purple or blue. This was due to the transition of AT from the trans to cis form, and induced by this transition formation of agglomerates of nanoparticles. Cessation of the exposure to UV radiation led to a complete removal of a drawing within 9 hours. Faster removal (after 60 seconds) of a drawing is realized by exposing the sample to white light. A similar effect is obtained by heating the material. All these activities cause the azobenzene moiety to go back from the cis to the trans form, and the aggregates of nanoparticles to break down. The color of the resulting material after UV exposure depends on the UV radiation dose. For example, after a short exposure (0.8 s) the irradiated area was purple, after 4 s exposure violet, and after 10 s exposure blue. These times can be modified by increasing or decreasing the intensity of the UV flux. The obtained material is very durable, the authors postulate the possibility of repeating the process hundreds of times on one sample.Gold nanoparticles modified in such a way that they have been rendered photo-responsive can be also used to construct sensors that “remember” whether the system has ever exceeded the melting temperature of the matrix in which modified nanoparticles have been dispersed. Klajn et al. constructed such a sensor by suspending gold nanoparticles functionalized by dithiol trans azobenzene in solutions of various solvents with different melting points.94 The obtained sols were then frozen and irradiated using UV radiation. Irradiation induced a transition of the azobenzene moiety from the trans to cis form, which would cause an aggregation of the gold nanoparticles if they were dispersed in a liquid. Aggregation did not occur in the case of the irradiation of nanoparticles in a solid matrix. However, if the irradiated matrix with the trapped gold nanoparticles is melted, an irreversible aggregation of the nanoparticles occurs and the color of the sample changes. This new color persists even after re-freezing the matrix containing the nanoparticles. This behavior allows to determine whether the melting temperature of the matrix has been achieved or not. Using a system containing many sensors prepared from solvents with different freezing temperatures permits a more precise determination of the highest temperature the system reached, without the use of electronic monitoring.
The process of the photo-agglomeration of plasmonic nanoparticles is often also affected by other compounds present in the surrounding solution, and so this process can be used to construct chemically resettable logic systems that are capable of performing AND, OR, and INHIBIT logic operations and of detecting the species influencing the process of agglomeration. For example, Liu et al. constructed such a system using gold nanoparticles covered with spiropyran.95 Under the influence of UV radiation spiropyran isomerizes into an open-chain form that effectively complexes some metal ions by a phenolate group. Thus, under certain conditions, the addition of Cu2+ ions to a solution containing gold nanoparticles functionalized by spiropyran and following UV irradiation (Cu2+/UV 1/1) leads to agglomeration and a change in the color of the solution from red to purple, whereas only UV irradiation (Cu2+/UV 0/1), or only the addition of Cu2+ ions (Cu2+/UV 1/0), does not lead to agglomeration. Therefore, such a system can be defined as an AND gate, where the purple color is treated as a response (1), and no color change as a response (0). Many ions have been tested for this: Al3+, Ba2+, Ca2+, Cd2+, Co2+, Cr3+, Cu2+, Fe2+, Fe3+, Hg2+, Mg2+, Mn2+, Ni2+, Pb2+, and Zn2+. It has been shown that, at low concentrations, only Cu2+ gives such effects, whereas at higher concentrations all the metal cations listed above give a similar response. In all the cases studied, the aggregation of nanoparticles was reversible when exposed to white light. This system can be also used to detect the presence of Cu2+ ions in aqueous media.95
4. Conclusion
In this review work, we described various methods of the light-induced photo-manipulation (especially aggregation) of plasmonic nanoparticles (see Fig. 10), as well as some typical applications of this process. The methods of the photo-induced nanomanipulation analyzed include methods based on: the light-induced isomerization of some compounds attached to the surface of the manipulated objects causing formation of electrostatic, host–guest or covalent bonds or other structural changes, the photo-response of a thermo-responsive material attached to the surface of the manipulated nanoparticles, and the photo-catalytic process enhanced by the coupled plasmons in manipulated nanoobjects. Sample applications of the process of the photo-aggregation of plasmonic nanosystems are also presented, including applications in surface-enhanced vibrational spectroscopies, catalysis, chemical analysis, biomedicine, and more. Despite the fact that work in this field has been going on for many years, many problems still need to be solved. In the case of precise manipulating of plasmonic nanoobjects there is still a problem with increasing the accuracy of manipulation and increasing the size of 2D and 3D objects obtaining by photo-manipulation. A significant problem in applications of photo-manipulation in the construction of substrates for surface-enhanced spectroscopic measurements is usually the high spectroscopic background generated by the compounds used to build such systems. Also for other applications (medical or in catalysis), there is a need to look for new photosensitive compounds to perform photo-manipulation to increase stability of the system and to reduce toxicity to the body (where important). Therefore, despite the relatively long activity in this field, there are still many problems to be solved. This means that developing of new, more accurate, more stable, less toxic or generating lower spectral background systems for photo-manipulation are still needed and developing such new materials will open up new possibilities. We hope that presented in this review a detailed comparative analysis of the methods that have been applied so far for the light-induced manipulation of nanostructures and some examples of applications of such systems may be useful for researchers planning to enter this fascinating field.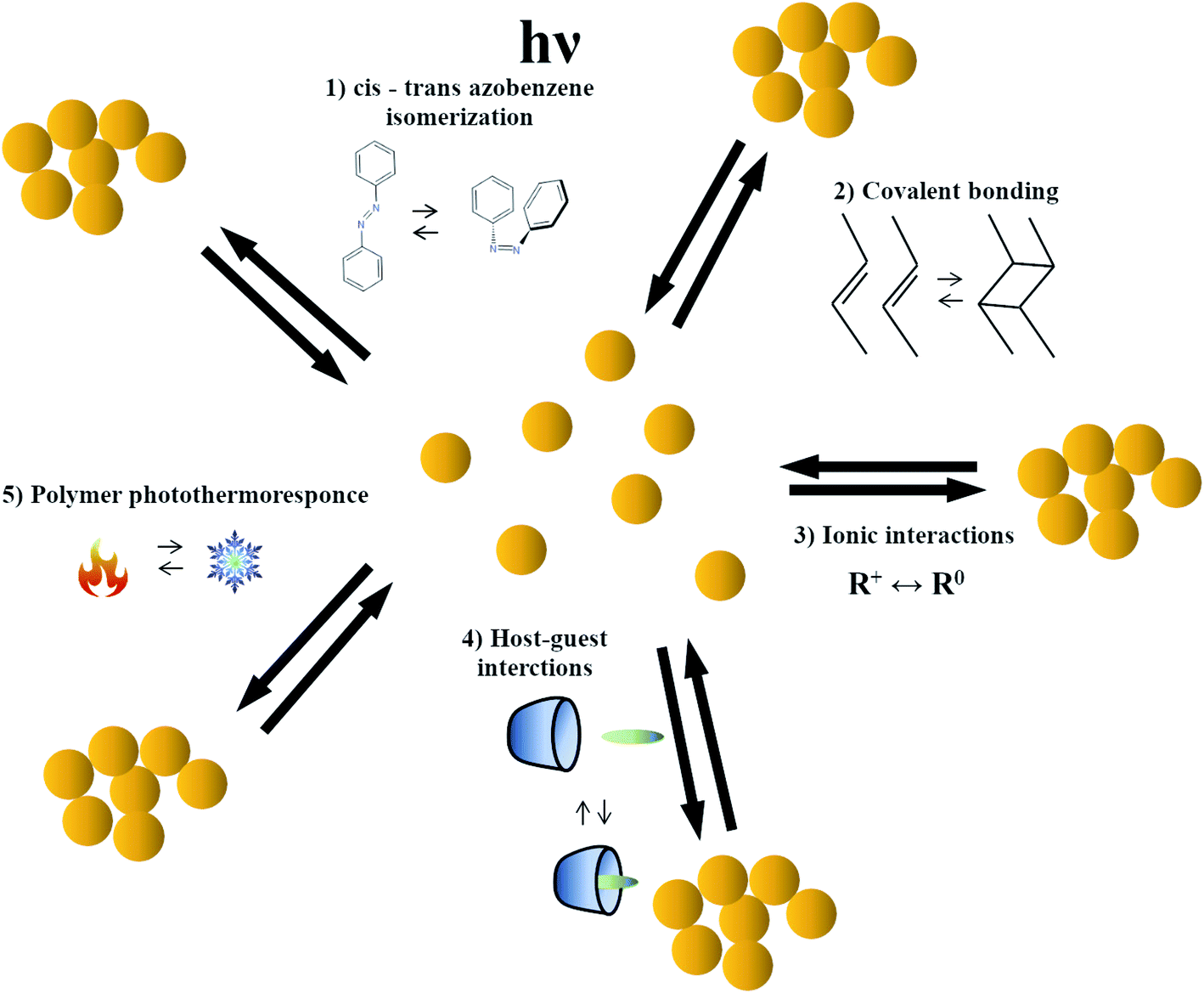 | ||
| Fig. 10 Schematic illustration of some mechanisms of the photo-aggregation of nanoparticles. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Warsaw, the Faculty of Chemistry.References
- M. Makulavicius, O. Balitskyi, R. Urbonas, A. Dzedzickis, V. Bučinskas, A. Petronis, M. Kovalenko and I. Morkvenaite-Vilkonciene, Recent Advances in Mechanical Micro- and Nanomanipulation, Automation 2020: Towards Industry of the Future, ed. R. Szewczyk, C. Zieliński and M. Kaliczyńska, Springer, Cham, 2020, pp. 248–256 Search PubMed.
- D. G. Grier, Nature, 2003, 424, 810–816 CrossRef CAS.
- Q. Cao, Q. Fan, Q. Chen, C. Liu, X. Han and L. Li, Mater. Horiz., 2020, 7, 638–666 RSC.
- Y. C. Li, H. B. Xin, H. X. Lei, L. L. Liu, Y. Z. Li, Y. Zhang and B. J. Li, Light: Sci. Appl., 2016, 5, 1–9 Search PubMed.
- M. Moreno-Moreno, P. Ares, C. Moreno, F. Zamora, C. Gómez-Navarro and J. Gómez-Herrero, Nano Lett., 2019, 19, 5459–5468 CrossRef CAS.
- Z. Zeng and J.-C. Tan, ACS Appl. Mater. Interfaces, 2017, 9, 39839–39854 CrossRef CAS.
- X. Zheng, A. Calò, E. Albisetti, X. Liu, A. S. M. Alharbi, G. Arefe, X. Liu, M. Spieser, W. J. Yoo, T. Taniguchi, K. Watanabe, C. Aruta, A. Ciarrocchi, A. Kis, B. S. Lee, M. Lipson, J. Hone, D. Shahrjerdi and E. Riedo, Nat. Electron., 2019, 2, 17–25 CrossRef CAS.
- C. Palacios-Berraquero, D. M. Kara, A. R.-P. Montblanch, M. Barbone, P. Latawiec, D. Yoon, A. K. Ott, M. Loncar, A. C. Ferrari and M. Atatüre, Nat. Commun., 2017, 8, 15093 CrossRef CAS.
- X. Li, Y. Tao, D.-H. Lee, H. K. Wickramasinghe and A. P. Lee, Lab Chip, 2017, 17, 1635–1644 RSC.
- K. Svoboda and S. M. Block, Opt. Lett., 1994, 19, 930 CrossRef CAS.
- K. Ajito and K. Torimitsu, Appl. Spectrosc., 2002, 56, 541–544 CrossRef CAS.
- J. Zhang, H. I. Kim, C. H. Oh, X. Sun and H. Lee, Appl. Phys. Lett., 2006, 88, 053123 CrossRef.
- D. Verschueren, X. Shi and C. Dekker, Small Methods, 2019, 3, 1800465 CrossRef.
- H. Tan, H. Hu, L. Huang and K. Qian, Analyst, 2020, 145, 5699–5712 RSC.
- L. Lin, E. H. Hill, X. Peng and Y. Zheng, Acc. Chem. Res., 2018, 51, 1465–1474 CrossRef CAS.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS.
- R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203 RSC.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS.
- K. Kołątaj, J. Krajczewski and A. Kudelski, Environ. Chem. Lett., 2020, 18, 529–542 CrossRef.
- J. Krajczewski and A. Kudelski, Front. Chem., 2019, 7, 410 CrossRef CAS.
- J. Krajczewski, K. Kołątaj and A. Kudelski, RSC Adv., 2017, 7, 17559–17576 RSC.
- P. G. Etchegoin and E. C. Le Ru, in Surface Enhanced Raman Spectroscopy, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, pp. 1–37 Search PubMed.
- F.-W. Schulze, H.-J. Petrick, H. K. Cammenga and H. Klinge, Z. Phys. Chem., 1977, 107, 1–19 CrossRef CAS.
- P. Cattaneo and M. Persico, Phys. Chem. Chem. Phys., 1999, 1, 4739–4743 RSC.
- Y. Ye, J. Pang, X. Zhou and J. Huang, Comput. Theor. Chem., 2016, 1076, 17–22 CrossRef CAS.
- J. Dokić, M. Gothe, J. Wirth, M. V. Peters, J. Schwarz, S. Hecht and P. Saalfrank, J. Phys. Chem. A, 2009, 113, 6763–6773 CrossRef.
- A. Muždalo, P. Saalfrank, J. Vreede and M. Santer, J. Chem. Theory Comput., 2018, 14, 2042–2051 CrossRef.
- M. Quick, A. L. Dobryakov, M. Gerecke, C. Richter, F. Berndt, I. N. Ioffe, A. A. Granovsky, R. Mahrwald, N. P. Ernsting and S. A. Kovalenko, J. Phys. Chem. B, 2014, 118, 8756–8771 CrossRef CAS.
- C. Raimondo, F. Reinders, U. Soydaner, M. Mayor and P. Samorì, Chem. Commun., 2010, 46, 1147–1149 RSC.
- D. S. Sidhaye, S. Kashyap, M. Sastry, S. Hotha and B. L. V. V. Prasad, Langmuir, 2005, 21, 7979–7984 CrossRef CAS.
- R. Klajn, K. J. M. Bishop and B. A. Grzybowski, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10305–10309 CrossRef CAS.
- Y. Wei, S. Han, J. Kim, S. Soh and B. A. Grzybowski, J. Am. Chem. Soc., 2010, 132, 11018–11020 CrossRef CAS.
- A. Kunfi, R. Bernadett Vlocskó, Z. Keresztes, M. Mohai, I. Bertóti, Á. Ábrahám, É. Kiss and G. London, Chempluschem, 2020, 85, 797–805 CrossRef CAS.
- A. Housni, Y. Zhao and Y. Zhao, Langmuir, 2010, 26, 12366–12370 CrossRef CAS.
- Y. Yan, J. I. L. Chen and D. S. Ginger, Nano Lett., 2012, 12, 2530–2536 CrossRef CAS.
- L. Lysyakova, N. Lomadze, D. Neher, K. Maximova, A. V. Kabashin and S. Santer, J. Phys. Chem. C, 2015, 119, 3762–3770 CrossRef CAS.
- A. Köhntopp, A. Dabrowski, M. Malicki and F. Temps, Chem. Commun., 2014, 50, 10105 RSC.
- A. Köhntopp, M. Dittner and F. Temps, J. Phys. Chem. Lett., 2016, 7, 1088–1095 CrossRef.
- J. V. Rival, Nonappa and E. S. Shibu, ACS Appl. Mater. Interfaces, 2020, 12, 14569–14577 CrossRef CAS.
- J.-W. Lee and R. Klajn, Chem. Commun., 2015, 51, 2036–2039 RSC.
- T. K. Biswas, S. M. Sarkar, M. M. Yusoff and M. L. Rahman, J. Mol. Liq., 2016, 214, 231–237 CrossRef CAS.
- D. Manna, T. Udayabhaskararao, H. Zhao and R. Klajn, Angew. Chem., Int. Ed., 2015, 54, 12394–12397 CrossRef CAS.
- Q. Wang, D. Li, J. Xiao, F. Guo and L. Qi, Nano Res., 2019, 12, 1563–1569 CrossRef CAS.
- D. Huebner, C. Rossner and P. Vana, Polymer, 2016, 107, 503–508 CrossRef CAS.
- J. H. Yoon and S. Yoon, Phys. Chem. Chem. Phys., 2011, 13, 12900 RSC.
- S. Schimka, D. T. Klier, A. L. de Guereñu, P. Bastian, N. Lomadze, M. U. Kumke and S. Santer, J. Phys.: Condens. Matter, 2019, 31, 125201 CrossRef.
- H. Zhang, J. Muhammad, K. Liu, R. H. A. Ras and O. Ikkala, Nanoscale, 2019, 11, 14118–14122 RSC.
- M. Ikbal, D. Balogh, E. Mervinetsky, R. Sfez and S. Yitzchaik, J. Phys. Chem. C, 2017, 121, 27176–27181 CrossRef CAS.
- A. Yucknovsky, S. Mondal, A. Burnstine-Townley, M. Foqara and N. Amdursky, Nano Lett., 2019, 19, 3804–3810 CrossRef CAS.
- A. Vesperinas, J. Eastoe, S. Jackson and P. Wyatt, Chem. Commun., 2007, 3912–3914 RSC.
- Y. Cheng, J. Dong and X. Li, Langmuir, 2018, 34, 6117–6124 CrossRef CAS.
- J. Lai, Y. Xu, X. Mu, X. Wu, C. Li, J. Zheng, C. Wu, J. Chen and Y. Zhao, Chem. Commun., 2011, 47, 3822–3824 RSC.
- T. Pauloehrl, G. Delaittre, V. Winkler, A. Welle, M. Bruns, H. G. Börner, A. M. Greiner, M. Bastmeyer and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2012, 51, 1071–1074 CrossRef CAS.
- T. E. Lehmann, G. Müller and A. Berkessel, J. Org. Chem., 2000, 65, 2508–2516 CrossRef CAS.
- T. Gruendling, K. K. Oehlenschlaeger, E. Frick, M. Glassner, C. Schmid and C. Barner-Kowollik, Macromol. Rapid Commun., 2011, 32, 807–812 CrossRef CAS.
- L. Stolzer, A. S. Quick, D. Abt, A. Welle, D. Naumenko, M. Lazzarino, M. Wegener, C. Barner-Kowollik and L. Fruk, Chem. Commun., 2015, 51, 3363–3366 RSC.
- H. Miyasaka, T. Nobuto, A. Itaya, N. Tamai and M. Irie, Chem. Phys. Lett., 1997, 269, 281–285 CrossRef CAS.
- K. Matsuda, M. Ikeda and M. Irie, Chem. Lett., 2004, 33, 456–457 CrossRef CAS.
- H. Nishi and S. Kobatake, Macromolecules, 2008, 41, 3995–4002 CrossRef CAS.
- H. Xia, Y. Gao, L. Yin, X. Cheng, A. Wang, M. Zhao, J. Ding and H. Shi, ChemBioChem, 2019, 20, 667–671 CrossRef CAS.
- H. Itoh, A. Tahara, K. Naka and Y. Chujo, Langmuir, 2004, 20, 1972–1976 CrossRef CAS.
- H. He, M. Feng, Q. Chen, X. Zhang and H. Zhan, Angew. Chem., Int. Ed., 2016, 55, 936–940 CrossRef CAS.
- Q. Zhou, B. Zhang, D. Han, R. Chen, F. Qiu, J. Wu and H. Jiang, Chem. Commun., 2015, 51, 3124–3126 RSC.
- J. Wu, Y. Xu, D. Li, X. Ma and H. Tian, Chem. Commun., 2017, 53, 4577–4580 RSC.
- L. Stricker, E. C. Fritz, M. Peterlechner, N. L. Doltsinis and B. J. Ravoo, J. Am. Chem. Soc., 2016, 138, 4547–4554 CrossRef CAS.
- M. Niehues, P. Tegeder and B. J. Ravoo, Beilstein J. Org. Chem., 2019, 15, 1407–1415 CrossRef CAS.
- Z. Wang, Z. Li and Z. Liu, J. Phys. Chem. C, 2009, 113, 3899–3902 CrossRef CAS.
- D. Fava, M. A. Winnik and E. Kumacheva, Chem. Commun., 2009, 2571–2573 RSC.
- P. K. Jain, S. Eustis and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 18243–18253 CrossRef CAS.
- T. Ding, V. K. Valev, A. R. Salmon, C. J. Forman, S. K. Smoukov, O. A. Scherman, D. Frenkel and J. J. Baumberg, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5503–5507 CrossRef CAS.
- M. Das, N. Sanson, D. Fava and E. Kumacheva, Langmuir, 2007, 23, 196–201 CrossRef CAS.
- M. Karg, I. Pastoriza-Santos, J. Pérez-Juste, T. Hellweg and L. M. Liz-Marzán, Small, 2007, 3, 1222–1229 CrossRef CAS.
- J. Qiu, Y.-C. Wu, Y.-C. Wang, M. H. Engelhard, L. McElwee-White and W. D. Wei, J. Am. Chem. Soc., 2013, 135, 38–41 CrossRef CAS.
- H. Han, J. Y. Lee and X. Lu, Chem. Commun., 2013, 49, 6122–6124 RSC.
- P. P. Patra, R. Chikkaraddy, R. P. N. Tripathi, A. Dasgupta and G. V. P. Kumar, Nat. Commun., 2014, 5, 4357 CrossRef CAS.
- L. Lin, X. Peng, M. Wang, L. Scarabelli, Z. Mao, L. M. Liz-Marzán, M. F. Becker and Y. Zheng, ACS Nano, 2016, 10, 9659–9668 CrossRef CAS.
- L. Lin, J. Zhang, X. Peng, Z. Wu, A. C. H. Coughlan, Z. Mao, M. A. Bevan and Y. Zheng, Sci. Adv., 2017, 3, e1700458 CrossRef.
- L. Lin, M. Wang, X. Peng, E. N. Lissek, Z. Mao, L. Scarabelli, E. Adkins, S. Coskun, H. E. Unalan, B. A. Korgel, L. M. Liz-Marzán, E.-L. Florin and Y. Zheng, Nat. Photonics, 2018, 12, 195–201 CrossRef CAS.
- J. Li, Y. Liu, L. Lin, M. Wang, T. Jiang, J. Guo, H. Ding, P. S. Kollipara, Y. Inoue, D. Fan, B. A. Korgel and Y. Zheng, Nat. Commun., 2019, 10, 5672 CrossRef CAS.
- J. Li, Y. Liu, L. Lin, M. Wang, T. Jiang, J. Guo, H. Ding, P. S. Kollipara, Y. Inoue, D. Fan, B. A. Korgel and Y. Zheng, Nat. Commun., 2019, 10, 1–9 CrossRef.
- J. Krajczewski, V. Joubert and A. Kudelski, Colloids Surf., A, 2014, 456, 41–48 CrossRef CAS.
- J. Krajczewski, K. Kołątaj and A. Kudelski, Chem. Phys. Lett., 2015, 625, 84–90 CrossRef CAS.
- H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kucanda, D. Manna, P. K. Kundu, J. W. Lee, P. Král and R. Klajn, Nat. Nanotechnol., 2016, 11, 82–88 CrossRef CAS.
- E. Hao and G. C. Schatz, J. Chem. Phys., 2004, 120, 357–366 CrossRef CAS.
- H. Wei and H. Xu, Nanoscale, 2013, 5, 10794 RSC.
- Y. Zheng, T. Thai, P. Reineck, L. Qiu, Y. Guo and U. Bach, Adv. Funct. Mater., 2013, 23, 1519–1526 CrossRef CAS.
- E. C. Le Ru and P. G. Etchegoin, Chem. Phys. Lett., 2006, 423, 63–66 CrossRef CAS.
- R. F. Aroca, D. J. Ross and C. Domingo, Appl. Spectrosc., 2004, 58, 324A–338A CrossRef CAS.
- R. Ambroziak, J. Krajczewski, M. Pisarek and A. Kudelski, ACS Omega, 2020, 5, 13963–13972 CrossRef CAS.
- X. Cheng, R. Sun, L. Yin, Z. Chai, H. Shi and M. Gao, Adv. Mater., 2017, 29, 1604894 CrossRef.
- B. I. Ipe, S. Mahima and K. G. Thomas, J. Am. Chem. Soc., 2003, 125, 7174–7175 CrossRef CAS.
- S. Jiang, K. Wang, Y. Dai, X. Zhang and F. Xia, Macromol. Mater. Eng., 2019, 304, 1900087 CrossRef.
- R. Klajn, P. J. Wesson, K. J. M. Bishop and B. A. Grzybowski, Angew. Chem., Int. Ed., 2009, 48, 7035–7039 CrossRef CAS.
- R. Klajn, K. P. Browne, S. Soh and B. A. Grzybowski, Small, 2010, 6, 1385–1387 CrossRef CAS.
- D. Liu, W. Chen, K. Sun, K. Deng, W. Zhang, Z. Wang and X. Jiang, Angew. Chem., Int. Ed., 2011, 50, 4103–4107 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |