
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdditive-free photo-mediated oxidative cyclization of pyridinium acylhydrazones to 1,3,4-oxadiazoles: solid-state conversion in a microporous organic polymer and supramolecular energy-level engineering†
Kyung-su Kima,
You Kyoung Chunga,
Hyunwoo Kima,
Chae Yeon Haa,
Joonsuk Huh abc and
Changsik Song*a
abc and
Changsik Song*a
aDepartment of Chemistry, Sungkyunkwan University, 2066 Seobu-ro, Janan-gu, Suwon-si, Gyeonggi-do 16419, Republic of Korea. E-mail: songcs@skku.edu
bSchool of Advanced Institute of Nanotechnology, Sungkyunkwan University, Suwon 16419, Republic of Korea
cInstitute of Quantum Biophysics, Sungkyunkwan University, Suwon 16419, Republic of Korea
First published on 7th January 2021
Abstract
We discovered the efficient catalyst-free, photo-mediated oxidative cyclization reaction of bis-p-pyridinium benzoyl hydrazone (BH1) to 2-pyridinium-5-phenyl-1,3,4-oxadiazoles. This photoreaction is remarkable because it does not require additives (e.g., bases, strong oxidants, or photocatalysts), which are essential in previous reports, and proceeds very effectively even with solid-state microporous organic polymers. Interestingly, we found that the inclusion complexation of BH1 with cucurbit[7]uril (CB7) interferes with the photo-induced electron transfer from BH1 to molecular oxygen through modification of the LUMO energy level, thus inhibiting the photo-medicated oxidative cyclization.
Introduction
Acylhydrazones are well known to be converted from the E-isomer to Z-isomer under visible or UV light irradiation, and reversibly return to the E-isomer by thermal treatment1,2 (Fig. 1a). Moreover, acylhydrazones can be transformed to oxadiazole derivatives by the oxidative cyclization reaction under UV-irradiation including a photocatalyst or under the condition of using a strong oxidizing agent. 1,3,4-Oxadiazoles are an essential class of five-membered heterocyclic compounds that have been intensively studied for biological and medicinal applications.3,4 There are two general methods for the construction of 1,3,4-oxadiazoles from acylhydrazones via oxidative cyclization. The first method for oxidative cyclization of acylhydrazones to 1,3,4-oxadiazole is to use strong oxidants such as Dess–Martin reagent, ceric ammonium nitrate, bis(trifluoroacetoxy)iodobenzene, (diacetoxyiodo)benzene, or (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO).5–11 The second method is photo-driven oxidative cyclization using a photocatalyst.12,13 However, these synthetic methods for 1,3,4-oxadiazoles suffer from by-product generation caused by harsh reaction conditions or the necessity for separation and purification steps. Thus, the development of facile, mild, and additive-free photo-mediated protocols for 1,3,4-oxadiazole seems highly demanding.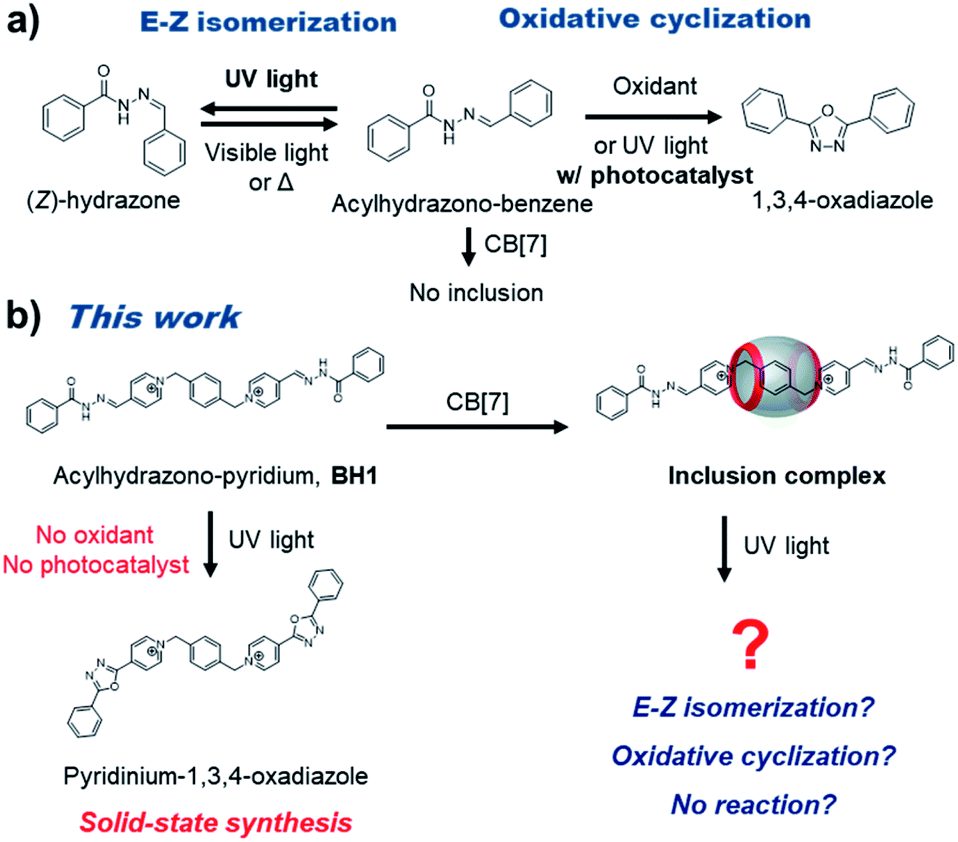 | ||
| Fig. 1 (a) E–Z isomerization and oxidative cyclization of acylhydrazone. (b) Catalyst-free photo-mediated oxidative cyclization reaction of pyridinium acylhydrazone, and the effect of inclusion complexation on photoreaction. | ||
Herein, we discovered the photo-mediated oxidative cyclization of pyridinium acylhydrazone conjugates, which forms 1,3,4-oxadiazole without additional additives (base, strong oxidant, or photocatalyst) (Fig. 1b). In our previous report,14 bis-p-pyridinium benzoyl hydrazone (BH1, a cationic guest molecule) showed pH-dependent supramolecular assembly and dynamic covalent exchange reactions (transamination). Initially, we tested the effect of inclusion complexation of BH1 with cucurbit[7]uril (CB7) to stimuli-responsiveness and self-assembly. Cucurbit[n]urils (CBn), which consists of glycoluril units linked by methylene bridges, are considered to be suitable candidates for the complexation with cationic molecules because of their electronegative carbonyl functional groups and hydrophobic cavity with portals. Interestingly, the pyridinium acylhydrazone conjugate appeared to form well the inclusion complex with CB7, which affected its supramolecular polymerization. What is more interesting, however, is that the photo-mediated oxidative cyclization seems to be affected by the inclusion complexation, presumably via energy level modification. The CBn-catalyzed or -templated photochemical reactions such as photohydrolysis15 and photodimerizations16–19 have been intensively investigated, but in most cases CBn promotes photoreaction by providing a cavity as a molecular container for effective spatial arrangement of reactants. On the other hand, the control of photoreaction through energy level tuning by encapsulation of reactants with CB7, not by spatial arrangement, is relatively rare and our work seems to provide with a noteworthy strategy to control photo-mediated chemical transformations.
Experimental
Materials
Pyridinium hydrazone conjugates, H1 and BH1 were prepared following the published procedures.14 Cucurbit[7]uril (CB7) was purchased from CBTECH (Pohang, Korea). 1-Adamantanamine, benzohydrazide, 4-pyridinecarboxaldehyde, p-xylene dibromide, and iodomethane were commercially available and used as received.Instruments
Ultraviolet-visible (UV-Vis) absorption data were acquired on a UV-1800 (Shimadzu) spectrophotometer in spectroscopic grade solvents without further purification. NMR measurements were performed in standard 5 mm NMR tubes at 300 K. All the data were acquired on a Bruker AVANCE III HD 500 and 700 MHz NMR spectrometer. Solid-state 13C CP/MAS NMR spectra of PH-MOP and Oxz-MOP were recorded on a 400 MHz solid state NMR spectrometer (AVANCE III HD, Bruker, Germany) at KBSI Western Seoul center. Two-dimensional ROE spectra were measured with a spectral width of 8 kHz in 2 K data points using 16 scans for each of the 512 t1 increments with 300 ms as a mixing time. Cyclic voltammetry (CV) was performed using an Epsilon electrochemical analyzer (EC Epsilon, BASi), and carried out in a three electrodes system, with Ag/Ag+ as the reference electrode, Pt foil as the counter electrode, and the platinum working electrode, with a scan rate of 100 mV s−1. For the photo-mediated oxidative cyclization reaction, pyridinium acylhydrazones were UV-irradiated with 342 nm using the Lumatec Superlite 410. Scanning electron microscopy (SEM) images were captured on a JEOL 7800 field emission SEM (FE-SEM) operated at an accelerating voltage of 15 kV. FT-IR spectra were recorded using a spectrometer Vertex 70 (Bruker Optic, Billerica, MA, USA), equipped with a diamond ATR unit.Synthesis of microporous organic polymer (MOP)
Pyridinium acylhydrazone based MOP was synthesized by ball milling method. Bis-(pyridinium aldehyde) 1 (1.0 g, 2.1 mmol) and benzene-1,3,5-tricarbohydrazide 2 (0.35 g, 1.4 mmol) were added to a 25 mL stainless steel vessel with two stainless steel balls (diameter 15 mm). The vessel containing the substrates was vibrated at 20 Hz for 60 min at room temperature. The reacted powder was collected and washed by DMSO, DMF, acetone and MeOH. Yield: 1.2 g (88%).General procedure for the photosynthesis of 1,3,4-oxadiazole derivatives from H1, BH1, and PH-MOP
1,3,4-Oxadiazole derivatives were synthesized by photo-mediated oxidative cyclization reaction of pyridinium acylhydrazones (H1, BH1 and PH-MOP). A pyridinium acylhydrazone was dissolved in DMSO (0.1 M) and then UV-irradiated with 342 nm for 4 h using the Lumatec Superlite 410. After completion of the reaction monitored by 1H NMR analysis except PH-MOP, the solvent was reduced, and then the residue was recrystallized from DMSO. Yield: 18–33 mg (83–92%).Computational calculations
To obtain the local geometries and UV-Vis spectra of the interested molecules, we performed density functional theory (DFT) calculations by using a hybrid functional B3LYP and 6-311+g* basis set implemented in Gaussian 16 program. We also used the escf module integrated into Turbomole 7.3 program utilizing the resolution of identity (RI) method under the Conductor-like Screening Model (COSMO) condition in order to consider a DMSO-solvent-mediated effect for the excited state calculations.Results and discussion
Catalyst-free photo-mediated oxidative cyclization of pyridinium acylhydrazones to 1,3,4-oxadiazoles
In order to investigate the photo-reactivity of pyridinium acylhydrazones such as E–Z isomerization, we performed the time-dependent UV-irradiation (342 nm) experiment for methylpyridinium benzoyl hydrazone H1 (0.66 mM in DMSO) (Fig. 2a). H1 can be considered as a model for BH1 since it is a dimer of H1 with a 1,4-phenylene linker. Upon the UV-irradiation of H1 solution, we observed that the UV-Vis absorption band of H1 became blue-shifted (∼19 nm), and the intensity of the absorption band at 348 nm considerably decreased (Fig. S1†). Of note, similar changes of optical properties can be observed during the E–Z isomerization process of acylhydrazones.1,2 However, after performing thermal treatment and visible light irradiation experiments, we can confirm that a certain irreversible photoreaction occurred that was not E–Z isomerization. Fig. 2b shows the 1H NMR spectra of H1 (0.66 mM in DMSO) under UV-irradiation. After UV-irradiation for 1 h, the initial proton signals for H1 decreased, and at the same time several new proton peaks were observed in the spectra. After UV-irradiation for 2 h, the proton peaks of H1 completely disappeared, and only the newly observed peaks remained. We confirmed that H1 changed to 2-methylpyridinium-5-phenyl-1,3,4-oxadiazole (Oxa-H1) by UV-irradiation using 1H (Fig. S2†) and 13C NMR (Fig. S3†), and high-resolution mass spectrometry (Fig. S4†) analyses. Similar with the case of H1, time-dependent UV-Vis absorption measurements and NMR experiments of BH1 were also performed after UV-irradiation (342 nm). The UV-Vis absorption spectra of BH1 (0.33 mM in DMSO) under UV-irradiation (Fig. S5†) showed blue-shifted absorption (∼65 nm), which was similar to the patterns of H1, and they showed a more apparent isosbestic point at 295 nm. After the UV-irradiation of BH1 for 2 h, the 1H NMR spectra of BH1 showed that the initial proton peaks of BH1 disappeared and those of newly generated bis-p-(pyridinium 1,3,4-oxadiazole) (Oxa-BH1) remained (Fig. S6†).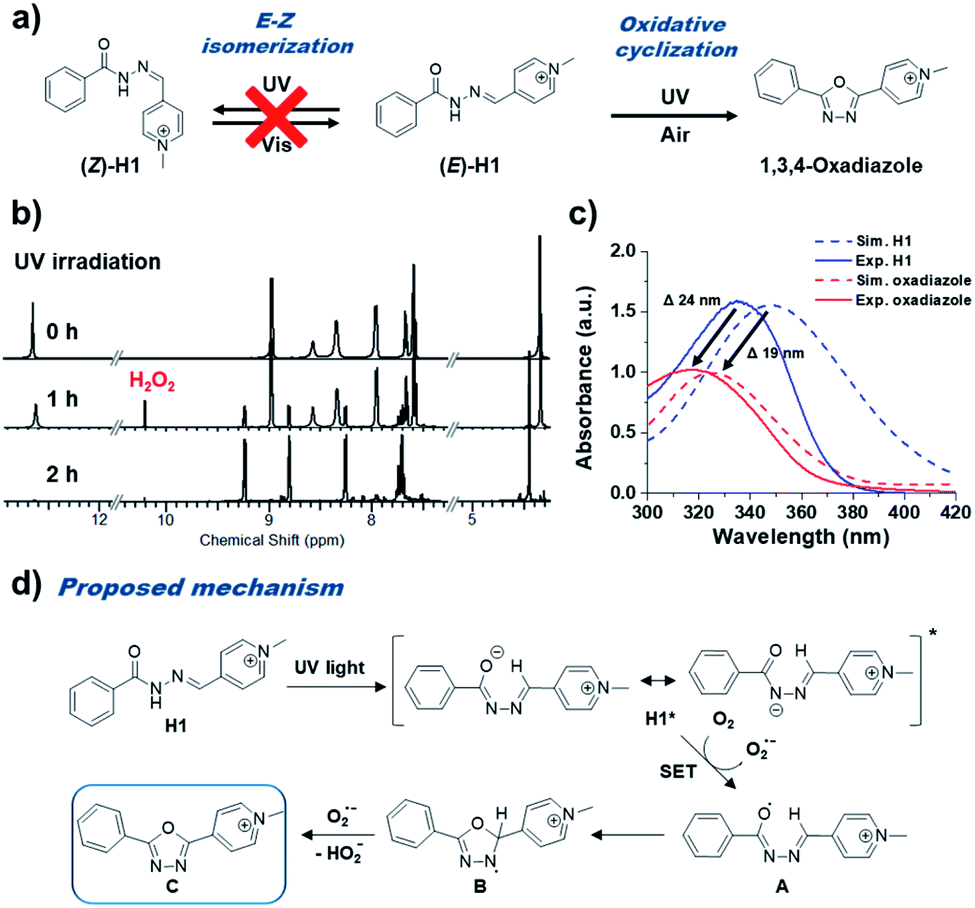 | ||
| Fig. 2 (a) Catalyst-free photo-mediated oxidative cyclization from pyridinium acylhydrazones to 1,3,4-oxadiazoles. (b) Irradiation time-dependent 1H NMR spectra of H1 (500 MHz, 0.66 mM in DMSO-d6). (c) Simulated and experimental UV-Vis absorption spectra of H1 and 2-methylpyridinium-5-phenyl-1,3,4-oxadiazole. (d) Proposed mechanism of photo-mediated oxidative cyclization of pyridinium acylhydrazone, H1, to 1,3,4-oxadiazole. | ||
Of note, this reaction is important because it has simple, economic, and environmental advantages compared to the previously reported methods for obtaining 1,3,4-oxadiazole from hydrazone. There is no need to worry about side reactions because the proposed approach does not use strong oxidizing agent; in addition, because no additional additives are used, less effort is required for purification. We expected that electronic modulation by the introduction of pyridinium into acylhydrazone would enable this additive-free photoreaction.
To support our experimental results observed by UV-Vis spectra and photoreaction, the DFT calculations of electronic states of H1 and 1,3,4-oxadiazole in a DMSO solution were performed using B3LYP and 6-311+G* basis set (Fig. S7†). When H1 was converted to 1,3,4-oxadiazole, a blue-shift of the absorption band (from 348 nm to 324 nm) similar to the experimental result was confirmed in simulated UV-Vis spectra (from 335 nm to 316 nm) (Fig. 2c). A possible mechanism of photo-mediated 1,3,4-oxadiazole synthesis from pyridinium acylhydrazone is represented in Fig. 2d. Upon the absorption of UV light, pyridinium acylhydrazone, H1, is excited to H1*, which can be easily deprotonated owing to the electron-accepting ability of conjugated pyridinium to form a zwitterion. A single electron transfer (SET) from H1* to oxygen generates acyl radical A, which forms B by intramolecular cyclization. The generated superoxide radical anion (O2−) by SET from H1* attacks the proton of tertiary carbon of B to produce 1,3,4-oxadiazole, C. In fact, we confirmed the existence of superoxide radical anion (O2−) during the photo-mediated oxidative cyclization reaction by the observation of hydrogen peroxide in NMR spectra (10.2 ppm). It was observed that hydrogen peroxide was initially formed at the same level as the product 1,3,4-oxadiazole derivative in the photo-induced oxidative cyclization of H1 (Fig. S2†). However, as shown in Fig. 2b, it appears that hydrogen peroxide decomposed over time under UV irradiation conditions, presumably due to photolysis of hydrogen peroxide. Of note, this oxidative cyclization reaction is carried out without additional strong oxidants or photocatalysts, which were necessary in previous studies.5–13
We attempted to apply the additive-free, photo-mediated oxidative cyclization method to the synthesis of oxadiazole-based microporous organic polymer (Oxz-MOP) under air. Our strategy is to obtain Oxz-MOP through a solid-state conversion of a pyridinium-acylhydrazone-based microporous organic polymer (PH-MOP), which can be prepared by facile condensation reaction between bis-(pyridinium aldehyde) 1 and benzene-1,3,5-tricarbohydrazide 2. Furthermore, we were able to synthesize PH-MOP successfully under ball milling condition at room temperature with a reactor frequency of 20 Hz for 1 h (Fig. 3a). Ball milling is a mechano-chemical method that has been considered an attractive tool for synthesizing various organic materials,20,21 due to advantages such as environment-friendliness, fast speed and simplicity. The 13C solid-state CP/MAS NMR spectra of PH-MOP revealed the similar patterns with that of BH1 (Fig. S8†), which means that PH-MOP were well synthesized resulting from hydrazone formation reactions between 1 and 2. Furthermore, to investigate the solid-state conversion of PH-MOP to oxadiazole-based microporous organic polymer (Oxz-MOP) by additive-free photo-mediated oxidative cyclization under air, we performed Fourier transform infrared (FT-IR) analysis of PH-MOP before and after UV-irradiation (Fig. 3b). In the FT-IR spectra of PH-MOP after UV-irradiation for 4 h, we could confirm that the disappearance of the N–H stretching peak at 3445 cm−1, and decrease of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1681 cm−1, which is consistent with the results with BH1 (Fig. S9†). The 13C solid-state CP/MAS NMR spectra of PH-MOP after UV-irradiation for 4 h supported also that photo-mediated conversion from PH-MOP to Oxz-MOP was occurred even in the solid state (Fig. 3c and d). Carbon signal of amide carbonyl carbon from PH-MOP at 167.7 ppm was changed to two broad peaks, which were speculated to originate from two carbons of 1,3,4-oxadiazole ring in Oxz-MOP. This photoreaction is noteworthy in that it provides a new bypass pathway to solve the difficulties in the synthesis and processing of aromatic poly(oxadiazole) due to the problem of poor solubility and high glass transition temperature (Tg).22–26
O stretching at 1681 cm−1, which is consistent with the results with BH1 (Fig. S9†). The 13C solid-state CP/MAS NMR spectra of PH-MOP after UV-irradiation for 4 h supported also that photo-mediated conversion from PH-MOP to Oxz-MOP was occurred even in the solid state (Fig. 3c and d). Carbon signal of amide carbonyl carbon from PH-MOP at 167.7 ppm was changed to two broad peaks, which were speculated to originate from two carbons of 1,3,4-oxadiazole ring in Oxz-MOP. This photoreaction is noteworthy in that it provides a new bypass pathway to solve the difficulties in the synthesis and processing of aromatic poly(oxadiazole) due to the problem of poor solubility and high glass transition temperature (Tg).22–26
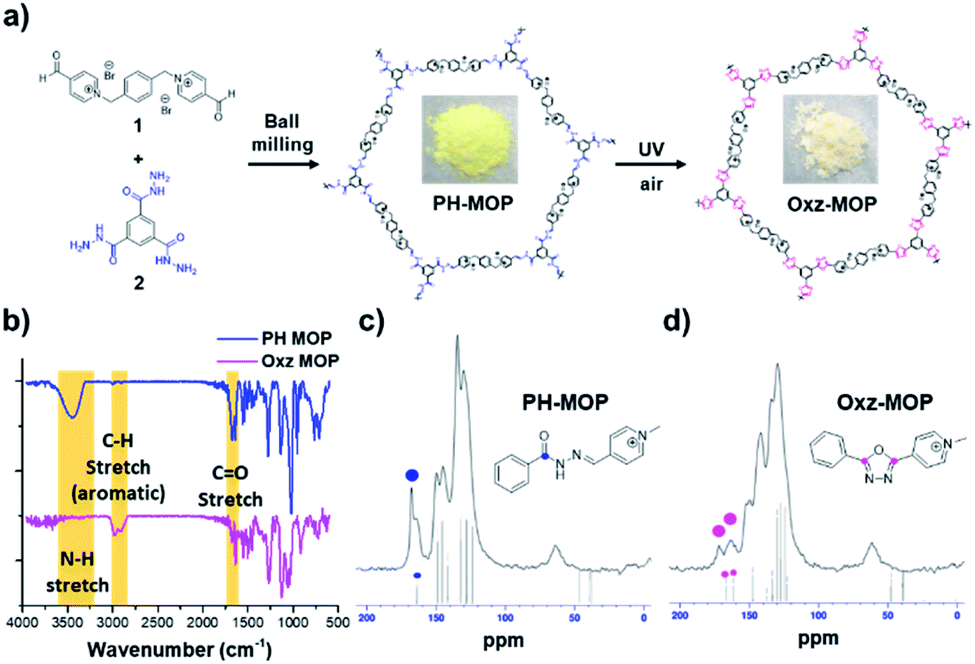 | ||
| Fig. 3 (a) Synthesis of pyridinium acylhydrazone based microporous organic polymer (PH-MOP) by ball milling method, and 1,3,4-oxadiazole based MOP (Oxz-MOP) by photo-mediated oxidative cyclization of PH-MOP in the solid-state. (b) FT-IR spectra of PH-MOP (blue-line) and Oxz-MOP (pink-line). Solid-state 13C CP/MAS NMR of (c) PH-MOP and (d) Oxz-MOP. | ||
Inclusion complexation of BH1 with CB7 and inhibited photo-mediated oxidative cyclization reaction: mechanistic insight
Previously, our group reported bis-p-pyridinium benzoyl hydrazone BH1 (Fig. 4), which exhibited a stimuli-responsive helical wire-type self-assembly by direct intermolecular interactions such as dipole–dipole, π–π, CH–π, and van der Waals interactions.14 We tested whether BH1 made an inclusion complex with CB7 (Fig. 4a) and its effect on the photo-mediated oxidative cyclization. In the UV-Vis absorption spectra of BH1 in DMSO (0.33 mM), the maximum absorption band initially at 340 nm showed a hypsochromic shift (∼10 nm) and slightly increased absorptivity upon the addition of an increasing amount of CB7 (Fig. 4b). This blue-shift in UV-Vis absorption spectra possibly originated from the changed microenvironment of BH1 from bulk to macrocyclic upon encapsulation with CB7.27,28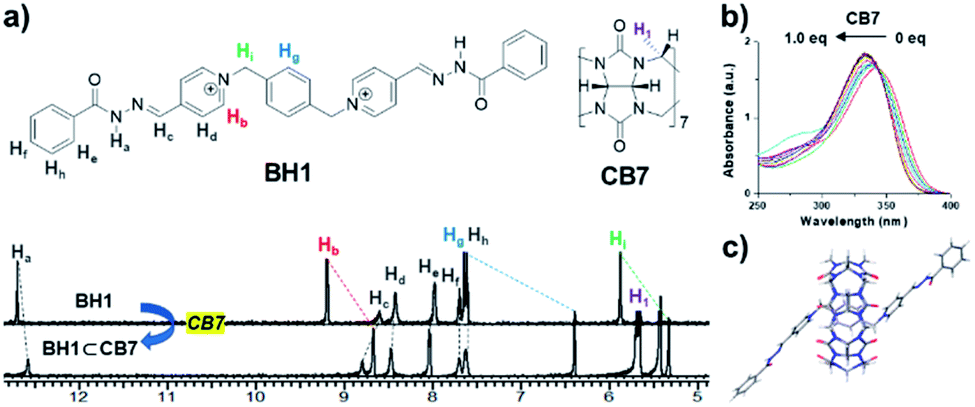 | ||
| Fig. 4 (a) 1H NMR spectra of BH1 (500 MHz, 1.0 mM in DMSO-d6) in the absence and presence of 1.0 equiv. of CB7. (b) UV-Vis absorption spectra of BH1 in DMSO (0.33 mM) upon the addition of CB7. (c) Energy-minimized structure for BH1⊂CB7 obtained by DFT calculation. | ||
1H NMR titration experiments clearly showed the complexation event between BH1 and CB7. BH1 (1.0 mM in DMSO-d6) was titrated against CB7, and 1H NMR measurements were performed, which allowed to determine the equilibrium and binding constant (K) (Fig. 4a and S10†). Upon the addition of CB7, BH1 formed only a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex (Fig. S11†) with a considerable upfield shift in the protons of 1,4-xylylene and pyridinium, which indicated that CB7 with shielding cavity make an internal complex with BH1.29,30 Owing to the slow exchange in complexation between CB7 and BH1 on the NMR time scale, we monitored the amount of formed inclusion complexes, BH1⊂CB7, by integrating each NMR signal of a free and bound state of BH1. The binding constant (K) of BH1 for CB7 was calculated by fitting using eqn (1),31 which allowed to determine the concentration of BH1⊂CB7, which was obtained by the multiplication of BH1⊂CB7 fraction and total concentration of BH1 ([BH1]0).
:1 complex (Fig. S11†) with a considerable upfield shift in the protons of 1,4-xylylene and pyridinium, which indicated that CB7 with shielding cavity make an internal complex with BH1.29,30 Owing to the slow exchange in complexation between CB7 and BH1 on the NMR time scale, we monitored the amount of formed inclusion complexes, BH1⊂CB7, by integrating each NMR signal of a free and bound state of BH1. The binding constant (K) of BH1 for CB7 was calculated by fitting using eqn (1),31 which allowed to determine the concentration of BH1⊂CB7, which was obtained by the multiplication of BH1⊂CB7 fraction and total concentration of BH1 ([BH1]0).
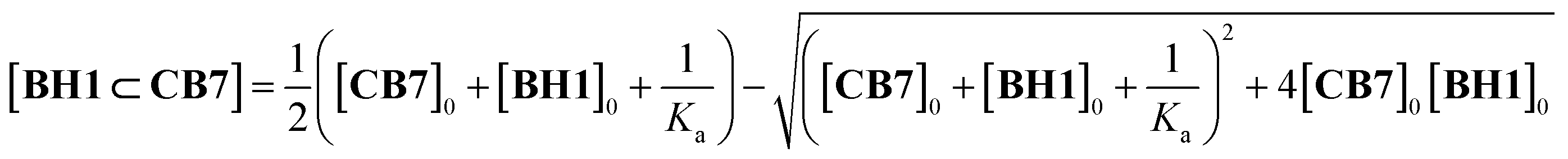 | (1) |
Compared to a previously study on the binding constant of the bis(pyridinium)-xylylene moiety with CB7,32 we obtained a relatively lower K value (1.53 ± 0.82 × 104 M−1), which was presumably due to solvent DMSO; this solvent fits better into the cavity of a hydrophobic binding site than water.33 To re-evaluate the binding affinity of CB7 to BH1, the data obtained from UV-Vis titration experiments of BH1 versus CB7, monitored at 330, 340, 360, 366, and 378 nm, were fitted according to the 1:1 binding isotherm on the website http://supramolecular.org (Fig. S12†),34 which showed a similar binding constant (4.05 × 104 M−1). The 2-D rotating-frame Overhauser effect spectroscopy (ROESY) NMR experiment was performed for the detailed structural characterization of inclusion complexes (BH1⊂CB7). As shown in Fig. S13,† ROE cross peaks between the proton of CB7 (H1) and protons of bis(pyridinium)-1,4-xylylene on BH1 (Hb and Hg) were clearly observed, which supported the formation of the 1:1 complex between BH1 and CB7, in which CB7 was symmetrically centrally positioned on BH1. Assuming 1:1 complexation, we were able to calculate the energy-minimized structure of the inclusion complex (BH1⊂CB7) in the ground state using the density functional theory (DFT) calculation at B3LYP/6-311+G*. As shown in Fig. 4c, centrally positioned CB7 in the xylylene unit and Z-like structures with two anti-parallel planes of pyridium-acylhydrazone conjugates were observed. These structural characteristics have features that are similar to those in previous studies on inclusion complexation between the bis(pyridinium)-1,4-xylylene unit and CB7.35,36
UV-irradiation (342 nm) time-dependent UV-Vis absorption measurements and NMR experiments of BH1⊂CB7 were performed. Inclusion complexes, BH1⊂CB7 (0.33 mM in DMSO-d6), which were prepared by the simple mixing of BH1 (0.66 mM in DMSO-d6) and CB7 (0.66 mM in DMSO-d6) solutions with 1:1 stoichiometry, were excited at 342 nm by UV-irradiation. Unlike the behavior in UV-Vis absorption spectra of BH1 under UV light irradiation, the UV-Vis spectra of BH1⊂CB7 did not show any shift in absorption band, isosbestic point, or increase in absorption at 275 nm (Fig. 5a). The 1H NMR spectra of BH1⊂CB7 (0.66 mM in DMSO-d6), which were irradiated by UV light for 4 h, did not show a significant difference compared to the initial NMR spectra of BH1⊂CB7 (Fig. S14†). Additional experiments on the photo-mediated oxidative cyclization reaction by varying the content of BH1⊂CB7 in the solution of BH1 were carried out (Fig. 5b and S15†). The UV-irradiation time-dependent 1H NMR spectra of 20% content of BH1⊂CB7 in the solution of BH1 showed that only unbound BH1 was changed to photo-reacted oxadiazole, while the total amount of bounded BH1 (BH1⊂CB7) remained. However, for 40% content of BH1⊂CB7 and above, we could not observe photo-reacted oxadiazole from BH1 in 1H NMR spectra, while the ratio of the amount of BH1 and BH1⊂CB7 was maintained. The isosbestic point, i.e., the existence of an equilibrium between BH1 and photo-reacted oxadiazole, was also not observed in UV-Vis spectra. Therefore, we concluded that BH1 encapsulated into CB7 was inhibited by the photo-mediated oxidative cyclization reaction, which also reduced the photo-reactivity of unbounded BH1 (Fig. 5b).
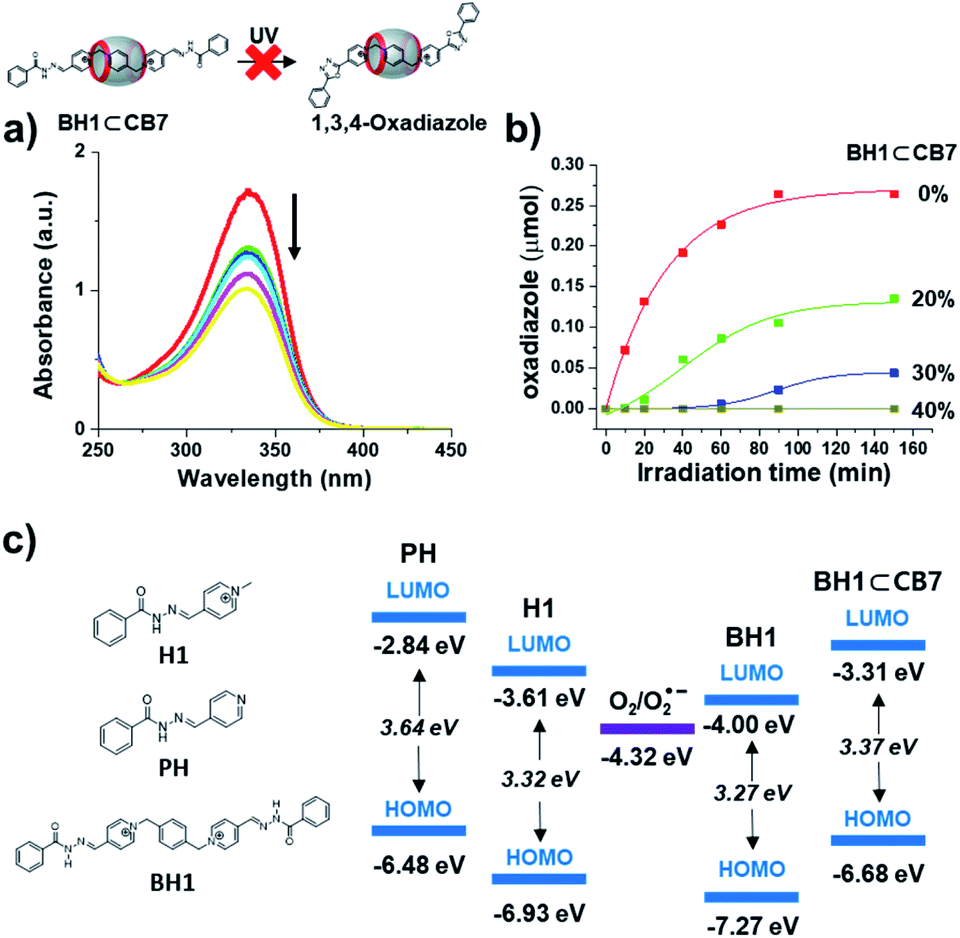 | ||
| Fig. 5 (a) UV-Vis spectra of BH1⊂CB7 upon the 342 nm UV light irradiation (black arrows indicate the direction of absorptivity with an increase in irradiation time). (b) Amount of formed oxadiazole after the photo-mediated oxidative cyclization reaction. (c) The LUMO and HOMO energy diagram of PH, H1, BH1, and BH1⊂CB7, and the reduction potential of triplet oxygen. | ||
We have not yet understood this phenomenon, but one thing to note is that in the presence of BH1⊂CB7 there seems to be an induction period or retardation effect, which intensifies with increasing CB7 content. Recently, Kaifer and coworkers reported the strong effect of the terminal carboxylate groups on the dissociation kinetics of bis(pyridinium)-xylylene32 Through competitive binding analysis, the kinetic constants for dissociation were assessed; anionic carboxylates appear to act as a kinetic barrier to exchange reactions, but dissociation was much faster in the acid form of carboxyl groups. Similarly, an exchange experiment of BH1⊂CB7 was performed with a competing guest molecule, adamantanamine (binding constant for CB7, 4.23 ± 1.00 × 1012 M−1), using NMR titration (Fig. S16†). The NMR spectrum of BH1⊂CB7 changed to that of BH1 within 30 minutes after the addition of adamantanamine, indicating that BH1 was completely exchanged to adamantanamine in the cavity of CB7 and the exchange kinetics appeared very quickly. Thus, although the complex BH1⊂CB7 is thermodynamically stable, it is kinetically labile so that the guest exchange can be relatively fast. These hypotheses require further investigation, but their relatively fast kinetics may influence the photo-mediated oxidative cyclization reaction of BH1 in the presence of less than stoichiometric CB7. In other words, guest exchanges in the cavity of CB7 between adjacent BH1 molecules can occur at a relatively faster rate than oxidative cyclization reactions, thereby suppressing photo-mediated reactions below a stoichiometric amount.
To investigate the reason why photoreaction only occurred in pyridinium acylhydrazone conjugates (H1 and BH1) and not in pyridine acylhydrazone conjugate (PH) (Fig. S17†) and inclusion complexes (BH1⊂CB7), and whether photoreaction was affected by their HOMO and LUMO energy levels, we measured the redox potentials of H1, BH1, PH, and BH1⊂CB7 by cyclic voltammetry (Fig. S18†). The LUMO energy levels of H1, BH1, PH, and BH1⊂CB7 were obtained from reduction potentials at 1.19, 1.19, 1.96, and 1.43 V vs. Fc/Fc+, respectively [ELUMO = −(Ered + 4.8 V)]. To calculate the HOMO energy level, their UV-Vis absorption spectra were used to estimate the HOMO–LUMO energy gap (ΔE). The calculated HOMO and LUMO energies of H1, BH1, PH, and BH1⊂CB7 are shown in Fig. 5c. The LUMO energies of pyridinium acylhydrazones H1 and BH1 were −3.61 eV and −4.00 eV, respectively, which were significantly lower than those of pyridine acylhydrazone PH (−2.84 eV) and the inclusion complex BH1⊂CB7 (−3.37 eV). All LUMO energies are slightly above the reduction potential for triplet oxygen (O2) (−4.32 eV).
Based on the obtained redox potentials and the proposed mechanism for photo-induced electron transfer from acylhydrazone to O2 (Fig. 2d), we could conclude that the electron transfer from photo-excited state to O2 occurs much favourably in H1 and BH1 due to the small energy difference between their LUMO and the reduction potential of O2, compared to the case of PH and BH1⊂CB7. According to Marcus theory, which explains the rate of electron transfer reactions between donor and acceptor, the electron transfer processes can be classified into three types of regions according to the rate constant versus reaction free energy: normal, activation-less, and inverted regions.37–40 Among them, the ‘inverted region’ is where the electron transfer becomes slower down as the reaction becomes more exothermic. Apparently, it is consistent with the results of our system since the rate of photo-induced electron transfer seems slower as the energy gap becomes larger. It is likely that the photo-induced electron transfer occurs via ‘inner sphere’ mechanism or bonded electron transfer41–44 through pre-organization of oxygen molecules and the excited H1* or BH1*, although this hypothesis requires further investigation. It was noteworthy that in our additive-free photo-mediated oxidative cyclization reaction, the rate of electron transfer could be controlled by tuning the energy levels through structural variation and supramolecular approach (inclusion complexation).
Conclusions
We described the photo-mediated oxidative cyclization reaction of pyridinium acylhydrazones to 1,3,4-oxadiazole without a photocatalyst or strong oxidant. This photoreaction proceeded by single electron transfer from excited pyridinium acylhydrazones to molecular oxygen, generating superoxide radical anion (O2−). The photo-mediated oxadiazole formation reaction of pyridinium acylhydrazones proved effective even in solid-state microporous organic polymers. Moreover, the reaction could be completely inhibited by inclusion complexation with CB7 via supramolecular engineering of LUMO energy levels.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Nano Material Development Program (2012M3A7B4049677) and Basic Science Research Programs (2019R1A2C1004256 and 2020R1A6A3A01100092) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology, Republic of Korea. We also thank Prof. Ki Hyun Kim (School of Pharmacy, Sungkyunkwan University) for HRMS measurements.Notes and references
- B. Bai, M. Zhang, N. Ji, J. Wei, H. Wang and M. Li, Chem. Commun., 2017, 53, 2693–2696 RSC.
- D. J. Van Dijken, P. Kovaricek, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982–14991 CrossRef CAS.
- F. A. Omar, N. M. Mahfouz and M. A. Rahman, Eur. J. Med. Chem., 1996, 31, 819–825 CrossRef CAS.
- A. S. Aboraia, H. M. Abdel-Rahman, N. M. Mahfouz and M. A. El-Gendy, Bioorg. Med. Chem., 2006, 14, 1236–1246 CrossRef CAS.
- C. Dobrota, C. C. Paraschivescu, I. Dumitru, M. Matache, I. Baciu and L. L. Ruta, Tetrahedron Lett., 2009, 50, 1886–1888 CrossRef CAS.
- P. Gao and Y. Y. Wei, J. Chem. Res., 2013, 506–510 CrossRef CAS.
- S. Guin, T. Ghosh, S. K. Rout, S. A. Banerjee and B. K. Patel, Org. Lett., 2011, 13, 5976–5979 CrossRef CAS.
- C. C. Paraschivescu, A. G. Coman, C. C. Anghel and M. Matache, Rev. Roum. Chim., 2015, 60, 339–343 Search PubMed.
- M. Dabiri, P. Salehi and M. Baghbanzadeh, Tetrahedron Lett., 2006, 47, 6983–6986 CrossRef CAS.
- C. C. Paraschivescu, M. Matache, C. Dobrota, A. Nicolescu, C. Maxim, C. Deleanu, I. C. Farcasanu and N. D. Hadade, J. Org. Chem., 2013, 78, 2670–2679 CrossRef CAS.
- R. Y. Yang and L. X. Dai, J. Org. Chem., 1993, 58, 3381–3383 CrossRef CAS.
- B. Kurpil, K. Otte, M. Antonietti and A. Savateev, Appl. Catal., B, 2018, 228, 97–102 CrossRef CAS.
- A. K. Yadav and L. D. S. Yadav, Tetrahedron Lett., 2014, 55, 2065–2069 CrossRef CAS.
- K.-s. Kim, H. J. Cho, J. Lee, S. Ha, S. G. Song, S. Kim, W. S. Yun, S. K. Kim, J. Huh and C. Song, Macromolecules, 2018, 51, 8278–8285 CrossRef CAS.
- J. Smitka, A. Lemos, M. Porel, S. Jockusch, T. R. Belderrain, E. Tesarova and J. P. Da Silva, Photochem. Photobiol. Sci., 2014, 13, 310 RSC.
- S. Y. Jon, Y. H. Ko, S. H. Park, H.-J. Kim and K. Kim, Chem. Commun., 2001, 1938 RSC.
- M. Pattabiraman, L. S. Kaanumalle, A. Natarajan and V. Ramamurthy, Langmuir, 2006, 22, 7605 CrossRef CAS.
- N. Barooah, B. C. Pemberton and J. Sivaguru, Org. Lett., 2008, 10, 3339 CrossRef CAS.
- F. Biedermann, I. Ross and O. A. Scherman, Polym. Chem., 2014, 5, 5375 RSC.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- S. Hsiao and G. Liou, Polym. J., 2002, 34, 917–924 CrossRef CAS.
- B. Schulz and E. Leibnitz, Acta Polym., 1992, 43, 343–347 CrossRef CAS.
- M. D. Losip, M. Bruma, I. Ronova, M. Szesztay and P. Muller, Eur. Polym. J., 2003, 39, 2011–2021 CrossRef.
- S. Janietz and S. Anlauf, Macromol. Chem. Phys., 2002, 203, 427–432 CrossRef CAS.
- S. Janietz, S. Analauf and A. Wedel, Synth. Met., 2001, 122, 11–14 CrossRef CAS.
- G. Zhang, D. Gao, J. Chao, S. Shuang and C. Dong, Dyes Pigm., 2009, 82, 40–46 CrossRef CAS.
- D. Banik, J. Kuchlyan, A. Roy, N. Kundu and N. Sarkar, J. Phys. Chem. B, 2015, 119, 2310–2322 CrossRef CAS.
- P. H. Dixit, R. V. Pinjari and S. P. Gejji, J. Phys. Chem. A, 2010, 114, 10906–10916 CrossRef CAS.
- S. Li, H. Yin, G. Martinz, I. W. Wyman, D. Bardelang, D. H. Macartney and R. Wang, New J. Chem., 2016, 40, 3484–3490 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- I. Neira, M. D. Garcia, C. Peinador and A. E. Kaifer, J. Org. Chem., 2019, 84, 2325–2329 CrossRef CAS.
- C. Senac, S. Desgranges, C. Contino-Pepin, W. Urbach, P. F. J. Fuchs and N. Taulier, ACS Omega, 2018, 3, 1014–1021 CrossRef CAS.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- L. Mei, Z. N. Xie, K. Q. Hu, L. Y. Yuan, Z. Q. Gao, Z. F. Chai and W. Q. Shi, Chem.–Eur. J., 2017, 23, 13995–14003 CrossRef CAS.
- V. Sindelar, K. Moon and A. E. Kaifer, Org. Lett., 2004, 6, 2665–2668 CrossRef CAS.
- E. H. Yonemoto, G. B. Saupe, R. H. Schmehl, S. M. Hubig, R. L. Riley, B. L. Iverson and T. E. Mallouk, J. Am. Chem. Soc., 1994, 116, 4786–4795 CrossRef CAS.
- C. Turro, J. M. Zaleski, Y. M. Karabatsos and D. G. Nocera, J. Am. Chem. Soc., 1996, 118, 6060–6067 CrossRef CAS.
- H. Imahori, K. Tamaki, D. M. Guldi, C. P. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607–2617 CrossRef CAS.
- M. Tachiya, J. Phys. Chem., 1993, 97, 5911–5916 CrossRef CAS.
- X. Amashukeli, N. E. Gruhn, D. L. Lichtenberger, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2004, 126, 15566–15571 CrossRef CAS.
- T. M. Bockman, Z. J. Karpinski, S. Sankararaman and J. K. Kochi, J. Am. Chem. Soc., 1992, 114, 1970–1985 CrossRef CAS.
- J. K. Kochi, Acc. Chem. Res., 1992, 25, 39–47 CrossRef CAS.
- S. J. Skoog and W. L. Gladfelter, J. Am. Chem. Soc., 1997, 119, 11049–11060 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09581h |
| This journal is © The Royal Society of Chemistry 2021 |