
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroxyethyl starch based smart nanomedicine
Huimin Wang†
a,
Hang Hu†b,
Hai Yanga and
Zifu Li
 *acd
*acd
aNational Engineering Research Center for Nanomedicine, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China. E-mail: zifuli@hust.edu.cn
bNational and Local Joint Engineering Research Center for High-efficiency Refining and High-quality Utilization of Biomass, School of Pharmaceutical Engineering and Life Sciences, Changzhou University, Changzhou 213164, People's Republic of China
cKey Laboratory of Molecular Biophysics of Ministry of Education, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China
dHubei Key Laboratory of Bioinorganic Chemistry and Materia Medical, Huazhong University of Science and Technology, Wuhan, 430074, China
First published on 14th January 2021
Abstract
In the past decades, the vigorous development of nanomedicine has opened up a new world for drug delivery. Hydroxyethyl starch (HES), a clinical plasma volume expander which has been widely used for years, is playing an attracting role as drug carriers. Compared with all other polysaccharides, HES has proven its unique characteristics for drug delivery platforms, including good manufacture practice, biodegradability, biocompatibility, abundant groups for chemical modification, excellent water solubility, and tailorability. In this review, an overview of various types of HES based drug delivery systems is provided, including HES–drug conjugates, HES-based nano-assemblies, HES-based nanocapsules, and HES-based hydrogels. In addition, the current challenges and future opportunities for design and application of HES based drug delivery systems are also discussed. The available studies show that HES based drug delivery systems has significant potential for clinical translation.
 Huimin Wang | Huimin Wang is currently a master candidate under the supervision of Prof. Zifu Li at Huazhong University of Science and Technology. She received her bachelor degree in pharmaceutical engineering at China Three Gorges University in 2019. Her current research interests focus on the hydroxyethyl starch based smart nanomedicine. |
 Zifu Li | Professor Zifu Li received B.S. degree at Huazhong University of Science and Technology in 2008 and PhD degree at the Chinese University of Hong Kong in 2012. From 2013 to 2015, he worked as a postdoctoral fellow at University of Alberta. He then joined in Georgia Institute of Technology as a research scientist. Since 2016, he has been a full professor at Huazhong University of Science and Technology. His group studies mechano-nanooncology and smart nanomedicine. |
1. Introduction
Since 1980s, nanotechnology has been attracting much attention and has been applied to many fields like electronics, mechanics, biomedical engineering, and so on. Especially in the biomedical field, nanomedicine is booming.1 Compared with traditional chemotherapeutics, loading free drugs within nanoparticles will improve their pharmacokinetics and biodistribution, so as to realize targeted drug delivery and reduced side effects.2 Over the past decades, great efforts have been devoted to design smart nanomedicine. So far, a series of nanoparticle-based therapeutics have already been approved such as Doxil®, Abraxane®, and Lipusu®.3 Nonetheless, these emerging nano-formulations exhibited limited benefits compared with traditional free drugs, which may partially be ascribed to the non-sufficient drug release at the targeted sites.4,5 Among the nanomedicines on the market, most are modified with polyethylene glycol (PEG), which is welcomed for being used as an in vivo “invisible” material since PEG can effectively avoid the adsorption of plasma proteins.6 Nanomedicine surface modified by PEG has become the gold standard in the field of nanomedicine, and there are tremendous works in PEGylation.7 The PEG modification does prolong the circulation time of the drug in vivo, but the drawbacks cannot be ignored. Firstly, PEG is non-degradable in vivo, resulting in cumulative toxicity.8 Besides, PEG may trigger an immune response. The repeated administration would cause the nanomedicine to be quickly cleared.9 PEG also has limited functional groups for chemical modification.10 This is one of the reasons why polysaccharide has been considered as an alternative to PEG.Polysaccharide is a kind of biopolymer constructed from sugar monomers and linked by O-glycosidic bonds. Polysaccharides contain a large amount of hydroxyl groups, which provides opportunities for adequate drug conjugation. Polysaccharides can be divided into two main categories: natural and semisynthetic. Natural polysaccharides can be obtained from abundant sources such as animals, plants, algae and microorganisms. Semisynthetic polysaccharides are usually produced by chemical or enzymatic modification of the parent macromolecules. Thus, they are available at large scale and are relatively cheap.11,12 In addition, polysaccharides also have good biocompatibility and biodegradability.13,14 Polysaccharides are a large family including chitosan, pullulan, dextran, hyaluronic acid, heparin, and to name a few. Not only do they have the above desirable properties, some of them also have unique biological activities. For example, chitosan has bioactivity as an antioxidant, an antimicrobial and an antifungal, while hyaluronic acid is able to bind to CD44 receptors.15,16 However, most of polysaccharides have unavoidable shortcomings, such as immunogenicity, insufficient water solubility and/or toxic side effects.17,18 As a kind of polysaccharide, hydroxyethyl starch (HES) has attracted great attention in the field of drug delivery due to its excellent physiochemical and biochemical properties. Used as clinical plasma substitute for more than fifty years, HES has good manufacture practice, good biocompatibility and biodegradability, showing promising prospects in the field of drug delivery.
In this review, we would like to introduce the characteristics of HES and their clinical applications briefly and summarize the progress of HES based smart nanomedicine for disease control, in order to provide a better understanding of HES based smart nanomedicine and its clinical translation potential.
2. Characteristics and clinical applications of HES
HES is a semisynthetic polysaccharide, which has been prepared by reacting amylopectin in waxy corn or potato starch with ethylene oxide. HES has hyper-branched structure. There are mainly three steps in the synthesis of HES: hydrolysis, hydroxyethylation and ultrafiltration, where hydroxyethylation helps increase water solubility of starch, reduce viscosity, and slow down α-amylase-mediated hydrolysis.19 As shown in Fig. 1A, HES consists of two kinds of glycosidic bonds, α 1–4 and α 1–6. According to the average molecular weight, degree of substitution and substitution position of hydroxyethyl, HES can be divided into many types.20–23 In the blood, HES is hydrolyzed mainly by the α-amylase, the speed of which is related to the degree of substitution of the hydroxyethyl group and the ratio of C2/C6. In consideration of steric hindrance effect, higher hydroxyethyl substitution and higher C2/C6 ratio lead to the slower hydrolysis rate.24–26 HES is mainly excreted through the kidney. After being hydrolyzed by α-amylase in the blood to a molecular weight less than the threshold of the kidney (≈70 kDa), it can be filtered through the glomerulus and excreted in the urine.27 Therefore, HES exhibits good biocompatibility as well as safety. At the same time, it is revealed that HES themselves are nanoparticles with a spherical shape, as shown in Fig. 1B. To be specific, the hydrodynamic diameter (Dh) of HES 70/0.5, HES 130/0.4 and HES 200/0.5 are 15.4 ± 0.6 nm, 15.5 ± 0.7 nm and 20.6 ± 0.6 nm, respectively. To sum up, from Fig. 1C, the HES hydration diameter increases with the increase of the HES molecular weight, and decreases with the increase of the HES concentration, indicating that HES molecules are soft particles and can be compressed.28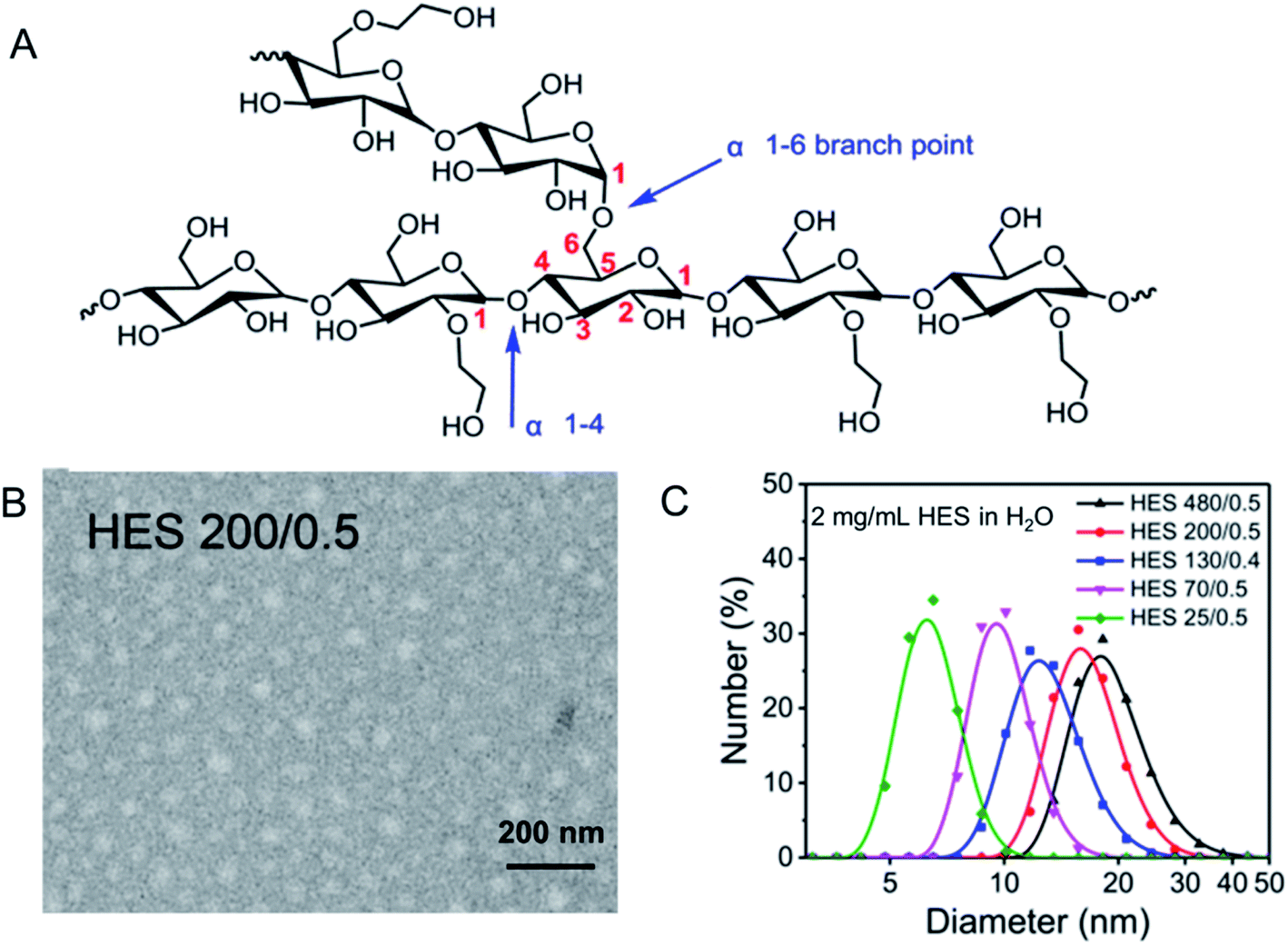 | ||
| Fig. 1 Structure and characterization of HES. (A) Structure of HES; (B) TEM image of HES 200/0.5; (C) size distribution of HES in H2O. Reprinted with permission from ref. 28. Copyright 2019. Royal Society of Chemistry. | ||
HES has several unique pharmacological effects. Firstly, it can improve plasma osmotic pressure and hemodynamics. Several animal experiments have shown that HES can quickly, continuously, and reliably correct hypovolemia, ensure systemic and microcirculation perfusion, thus maintaining organ oxygen supply and normal function.29,30 Due to a strong volume expansion effect, HES can significantly improve hemodynamic parameters, and even higher doses of HES 130/0.4 used for expansion treatment is safe and effective. Generally, low molecular weight HES has low expansion strength.31,32 Secondly, it works on improving hemorheology. At low blood volume, due to the increase of blood viscosity, the blood flow slows down and the perfusion of organs is affected. HES has the beneficial effects of stabilizing osmotic pressure, optimizing tissue perfusion, improving oxygen supply, and preventing injury to endothelial cells after hypoxia, thereby improving tissue perfusion and oxygenation by diluting blood, reducing blood viscosity, and improving hemodynamics.33 Additionally, the improvement of hemorheological indexes such as erythrocyte aggregation and plasma viscosity by HES solution with low molecular weight and low degree of substitution was better than that of HES solution with high molecular weight. For example, 130/0.4 can significantly reduce blood viscosity in patients with an acute ischemic stroke.34 Thirdly, HES also has anti-inflammatory effects. It can reduce the inflammatory response, thereby reducing the damage to tissues. Polymorphonuclear neutrophils (PMN) are important non-specific immune cells in vivo, and are also the main effector cells of inflammatory injury. During the inflammatory response, PMN respiratory burst intensifies, producing a large amount of toxic substances such as oxygen free radicals, causing tissue damage, which is significantly reduced by HES with low molecular weight.35–37 Then, the adhesion of PMN to endothelial cells is the basis and key step of inflammatory response. It was found that HES with different physical and chemical properties could inhibit the adhesion of PMN-endothelial cell (EC).38 Furthermore, HES can significantly down-regulate pro-inflammatory mediators and up-regulate the release of anti-inflammatory mediators.39,40 Fourthly, it plays a role in affecting coagulation function. HES is able to reduce red blood cell aggregation and blood viscosity, thereby reducing vascular resistance and improving blood coagulation status. However, the effect of HES on coagulation function is mainly related to its substitution level and molecular mass and the mechanism is unclear.41 Hwang et al. revealed that HES with high molecular weight has significant impact on coagulation function, while HES 130/0.4 has no significant effect on platelet adhesion, aggregation and coagulation.42,43 Last but not least, it also has an inhibitory effect on tumor metastasis to some extent. By down-regulating the expression of matrix metalloproteinase 2 (MMP2) and MMP9, which are associated with tumor cell adhesion, invasion and migration, HES realizes the suppression of tumor metastasis.44,45 In addition to this, the circulating tumor cells (CTCs) are another major cause of tumor metastasis and the platelets activation may promote CTCs survival and increase the metastasis potential.46 It was found that HES 200/0.5 infusion would result in an inhibition effect on platelets activation, thereby decreasing CTCs of patients undergoing colorectal cancer radical surgery.47
The unique biological properties of HES make it quickly attract the attention of researchers in all aspects. HES has realized wide application in clinic. Table 1 summarizes the clinical use of HES as plasma expenders, cryoprotectant agents and organ preservation solutions. Clinical used under the trade name VitaHES, Infukoll and Voluven in German, and Hespan in US, HES solutions have been used as plasma expanders for 20 years with fast and powerful volume expansion effect.48 By increasing plasma volume, improving hemodynamics, HES thereby improves cardiac output and oxygen delivery value, and improving internal microcirculation.33 HES is degraded by α-amylase after injected in vivo, and when the molecular weight of some particles is less than 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, it is quickly discharged through the glomerular filtration membrane. The expansion capacity of HES is mainly determined by the molecular weight, and the residence time in the body depends on the degree of hydroxyethylation of starch. To achieve unity of effectiveness and safety, studies have shown that the molecular weight of HES 130/0.4 is close to the ideal renal threshold level, which has lower plasma and tissue accumulation, and has little effect on platelet aggregation and coagulation. Accordingly, HES is clinically used for operation, massive blood loss and septic shock, etc.31
000, it is quickly discharged through the glomerular filtration membrane. The expansion capacity of HES is mainly determined by the molecular weight, and the residence time in the body depends on the degree of hydroxyethylation of starch. To achieve unity of effectiveness and safety, studies have shown that the molecular weight of HES 130/0.4 is close to the ideal renal threshold level, which has lower plasma and tissue accumulation, and has little effect on platelet aggregation and coagulation. Accordingly, HES is clinically used for operation, massive blood loss and septic shock, etc.31
| Clinical use | Mechanisms | Advantages | Limitations | Ref. |
|---|---|---|---|---|
| Plasma expanders | Increase plasma volume | Fast and powerful | Influence coagulation function | 33 and 48 |
| Cell cryopreservation and culture | Absorb water molecules without phase transition | Nontoxicity | Not enough effective alone | 49 and 50–52 |
| Organ preservation | Reduce tissue edema | Great safety | Just as a supplementary component | 53 and 54 |
To improve cell, tissue and organ storage, the safety of material for cell cryopreservation is crucial. A major disadvantage of traditional cryoprotectant agents (CPAs) is the toxicity. Low-molecular-permeability CPAs such as DMSO, glycerol, etc. can remove water from intracellular molecules, leading to unnecessary interactions between proteins and resulting in protein denaturation. In contrast, macromolecular HES is used as a non-penetrating cryoprotectant. It participates in cell dehydration in the extracellular space and minimizes the formation of intracellular ice crystals. The cryoprotective effect of HES depends on its ability to absorb water molecules and keep these thermally inert in glassy state without experiencing any phase transition during cooling.49 Research results showed that cryopreservation solution containing 11.5% HES can improve the survival rate of human red blood cells. Another study reported cell recovery of 97% and more than 80% saline stability of human red blood cells after cryopreservation with 14% HES. At the same time, the parameters of hemolysis, saline stability, osmotic fragility and morphological changes of canine red blood cells frozen in 12.5% HES were significantly better than the traditional 20% glycerol solution.50–52 Therefore, HES has a good application prospect in cell cryopreservation.
In addition to cell preservation, HES also shows certain protective ability in tissue cryopreservation. The presence of HES during the storage helps limit hepatic proteolysis and the ensuing metabolic impairment whereas cryopreservation without HES stimulates oxygen consumption, protein degradation, decreases glucose production and promotes intracellular volume reduction. HES not only acts on the hydration state of the liver, but also affects the metabolism, thereby maintaining the structure of the liver.53 As for the preservation of the trachea, in the presence of 20% glycerol plus HES group, the trachea retained all histological and mechanical characteristics, and the original donor epithelial cells were less depleted and the submucosal glands were less inflammatory. These all provide guarantee for the next organ transplantation.54
3. HES as drug carrier
In view of the successful applications in clinical settings, there has been considerable interests in developing HES-based drug delivery systems.55 In addition to the characteristics of good hydrophilicity, biocompatibility, and biodegradablity, HES as an amylopectin is structurally similar to glycogen (a branched glucose storage polymer in humans) and this is one of the reasons why HES lacks immunogenicity.56 For the HES polymer, no unfavourable accumulation has been observed in the liver or spleen. Besides, it can improve hemodynamic and enhance the oxygen supply for tissue, thus to change the microcirculation, as well as reduce concurrent inflammatory reactions and inhibit tumor metastasis. Among the various kinds of HES, HES 130/0.4 is mostly studied and widely used, for the reason that it has excellent systemic tolerance, high renal excretion rate, and little influence on coagulation.As shown in Fig. 2, HES can conjugate with small molecule chemotherapeutics under simple and convenient reactions, thus increasing the water solubility and stability of some hydrophobic drugs such as doxorubicin (DOX), paclitaxel (PTX), camptothecin (CPT) et al. The application of some responsive chemical bonds is able to realize the selective release of drug to specific sites, which greatly reduces the toxic side effects. Nano drug delivery system based on HES like HES–polycaprolactone (HES–PCL) and HES–polylactide (HES–PLA) can be used for the delivery of both hydrophobic and hydrophilic drugs, achieving long circulation and targeted delivery of drugs in vivo. Especially for protein, the decoration of HES successfully extends the half-time. As a preeminent nanocarrier, there are series of brilliant works about HES boosted already in recent years.
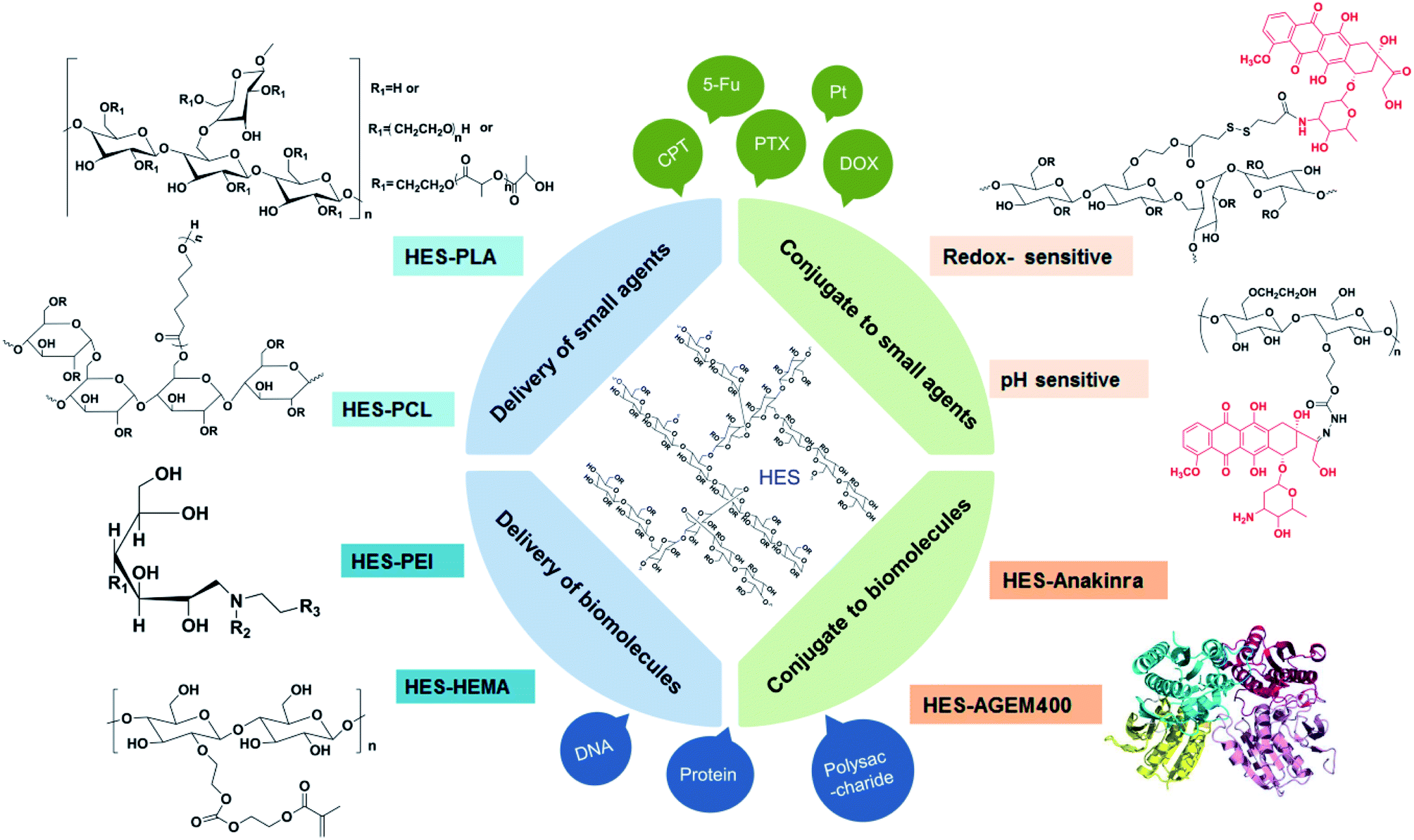 | ||
| Fig. 2 HES-based drug delivery systems. | ||
3.1 HES conjugate
Then, the conjugation of methotrexate (MTX) to HES realizes the first application of antifolate covalently conjugated to HES in experimental anticancer treatment.58 Activation of the carboxyl groups of MTX made it possible to react directly with the hydroxyl groups of HES glycosyl units. It was proposed that HES–MTX conjugates, due to their hydrodynamic size range (about 15.2 nm) were able to avoid renal clearance. The surfaces of HES–MTX conjugates had a negative charge, resulting in a longer half-life in plasma and consequently increased tumor accumulation of HES–MTX conjugate via the enhanced permeability and retention (EPR) effect. The experiment in vivo performed on the MV-4-11 leukaemia model revealed the high antitumor efficacy of HES–MTX conjugate compared to nonconjugated ones. Specifically, HES–MTX revealed significant activity in inhibiting the growth of MV-4-11 tumors from day 11 to the end of the experiment. The well-established therapeutic efficacy of HES–MTX suggests that the conjugate has clinical translation potential. However, the pharmacokinetics and biodistribution of HES–MTX need to be further studied to clarify the enhanced antitumor effect.
The above study created new opportunities in the design of HES-based innovative drug–polymer conjugates. DOX, which can interact with DNA by intercalation to inhibit the transcription and replication of DNA, thereby inhibiting the growth and proliferation of tumor cells, is widely used in chemotherapy for the treatment of various tumors including hematological malignancies as well as for many types of carcinoma and soft tissue sarcomas. However, the clinical use of DOX is hindered by the limitations such as the short half-life in blood and severe toxicity to heart and renal.59,60 To improve the above drawbacks, Li et al. firstly synthesized three intracellular acid-sensitive HES![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) DOX conjugates through an efficient Schiff base reaction between the aldehyde group (–CHO) in oxidized HES and the amino group (–NH2) of DOX.61 Based on the fact that both the intratumoral (pH 6.8) and intracellular microenvironments (endosome: pH 5.0–6.5, lysosome: pH 4.0–5.0) are acidic for that cancer cells produce lactic acid from glucose even under non-hypoxic conditions, the prodrugs with acid-sensitive hydrazone bond were relatively stable under normal physiological conditions and released the drug rapidly in an environment mimicking the intracellular acidic microenvironment. Three prodrugs with different drug binding rates (DBRs) HESDOX1.7, HESDOX3.3 and HESDOX5.9 showed spherical morphologies with diameters around 60 nm. It is also noteworthy that the size of the HESDOX micelles decreased with the improvement of the DBR because of the expansion of the hydrophobic DOX moiety and compaction of the micellar core. The drug release could be accelerated by the decrease of pH. All DOX formulations demonstrated more efficient antitumor efficacy in comparison to free DOX, which resulted from their enhanced accumulation at the tumor site and improved DOX release in tumor cells due to the acidic intracellular microenvironment. On this basis, cyclic Arg-Gly-Asp peptides (cRGD) have been utilized. A targeted acid-sensitive polysaccharide prodrug (HESDOX/cRGD) was synthesized by Schiff base reaction between the aldehyde groups of oxidized HES and the amino groups of DOX and cyclo (Arg-Gly-Asp-D-Phe-Lys) peptide (c(RGDfK)), forming two imine linkers in the conjugate.62 HESDOX/cRGD exhibited a better inhibition effect on tumors and had a significant difference compared with HESDOX. That was because cRGD showed high affinity with αvβ3 integrin, which could selectively transport antitumor drugs to tumor tissues and stimulate the internalization process. In another work, HES and DOX were conjugated by a pH-sensitive hydrazine bond (Hyd). pH-Insensitive HES–SAD was also synthesized as a control.63 The different release profiles of the two polymers at various pH values verified the potential use of this pH-sensitive conjugate. In addition, a higher cellular proliferation inhibition efficacy was achieved in HES–Hyd–DOX group. Similarly, another pH-responsive luteinizing hormone-releasing hormone (LHRH)-conjugated prodrug (HES–DOX/LHRH) was facilely synthesized by conjugating oxidized HES (HES–CHO) with DOX and LHRH through an acid-sensitive Schiff base bond, which self-assembled into nanoscopic micelle with a radius of about 55 nm.64 Under the physiological condition of pH 7.4, the release rate of DOX after 72 h was only 30%. Under the conditions of pH 6.8/5.5, the release rate of HES–DOX increased significantly to 42.1%/62.5%, and the release rate of HES–DOX/LHRH was 40.1%/71.2%. This showed that the Schiff bond was stable under normal tissue physiological conditions, which can significantly reduce systemic toxicity, and acidic conditions in tumor sites triggered the cleavage of Schiff bond to promote drug release. More importantly, targeted HES–DOX/LHRH possessed better anti-tumor and anti-metastasis effects on a RM-1-xenografted C57BL/6 mouse model compared to free DOX and non-targeted HES–DOX, indicating that the LHRH-modified HES–DOX conjugate has great potential for treatment of metastatic prostate cancer with lower systemic toxicity. It is known that the concentration of glutathione (GSH) is higher in tumor than that in normal tissues. Taking advantage of this difference, a novel redox-sensitive conjugate HES–SS–DOX was developed.65 The disulfide bonds would be rapidly broken to trigger drug release under the environment of high reduction environment in tumor sites, while remained stable in blood circulation. Redox-insensitive HES–DOX was also synthesized as a control through the amide bonds. The drug release in vitro confirmed the GSH-triggered drug release of HES–SS–DOX. The drug release rate of HES–SS–DOX in PBS (pH 7.4) with 10 mM or 2 mM GSH was significantly higher than that in PBS (pH 7.4) with 2 uM or without GSH. The in vitro cytotoxicity of free DOX, HES–SS–DOX, and HES–DOX against H22, HepG-2, and Bel-7402 cells was studied. The results showed that at the concentration of DOX at 10 μg mL−1, the cytotoxicity of HES–SS–DOX conjugate was much higher than that of HES–DOX, which was close to free DOX. The in vivo study showed similar results. HES–SS–DOX possessed a longer plasma half-life and higher tumor site accumulation, bringing about higher tumor suppression efficiency with reduced toxicity. The HES in the above studies played an important role in relieving the toxicity of DOX to some extent. Especially the design of sensitive bonds made great achievements. Nevertheless, HESDOX exhibited better activity in vitro than HES–SS–DOX, thus, the release of DOX in a good manner is a concern.
DOX conjugates through an efficient Schiff base reaction between the aldehyde group (–CHO) in oxidized HES and the amino group (–NH2) of DOX.61 Based on the fact that both the intratumoral (pH 6.8) and intracellular microenvironments (endosome: pH 5.0–6.5, lysosome: pH 4.0–5.0) are acidic for that cancer cells produce lactic acid from glucose even under non-hypoxic conditions, the prodrugs with acid-sensitive hydrazone bond were relatively stable under normal physiological conditions and released the drug rapidly in an environment mimicking the intracellular acidic microenvironment. Three prodrugs with different drug binding rates (DBRs) HESDOX1.7, HESDOX3.3 and HESDOX5.9 showed spherical morphologies with diameters around 60 nm. It is also noteworthy that the size of the HESDOX micelles decreased with the improvement of the DBR because of the expansion of the hydrophobic DOX moiety and compaction of the micellar core. The drug release could be accelerated by the decrease of pH. All DOX formulations demonstrated more efficient antitumor efficacy in comparison to free DOX, which resulted from their enhanced accumulation at the tumor site and improved DOX release in tumor cells due to the acidic intracellular microenvironment. On this basis, cyclic Arg-Gly-Asp peptides (cRGD) have been utilized. A targeted acid-sensitive polysaccharide prodrug (HESDOX/cRGD) was synthesized by Schiff base reaction between the aldehyde groups of oxidized HES and the amino groups of DOX and cyclo (Arg-Gly-Asp-D-Phe-Lys) peptide (c(RGDfK)), forming two imine linkers in the conjugate.62 HESDOX/cRGD exhibited a better inhibition effect on tumors and had a significant difference compared with HESDOX. That was because cRGD showed high affinity with αvβ3 integrin, which could selectively transport antitumor drugs to tumor tissues and stimulate the internalization process. In another work, HES and DOX were conjugated by a pH-sensitive hydrazine bond (Hyd). pH-Insensitive HES–SAD was also synthesized as a control.63 The different release profiles of the two polymers at various pH values verified the potential use of this pH-sensitive conjugate. In addition, a higher cellular proliferation inhibition efficacy was achieved in HES–Hyd–DOX group. Similarly, another pH-responsive luteinizing hormone-releasing hormone (LHRH)-conjugated prodrug (HES–DOX/LHRH) was facilely synthesized by conjugating oxidized HES (HES–CHO) with DOX and LHRH through an acid-sensitive Schiff base bond, which self-assembled into nanoscopic micelle with a radius of about 55 nm.64 Under the physiological condition of pH 7.4, the release rate of DOX after 72 h was only 30%. Under the conditions of pH 6.8/5.5, the release rate of HES–DOX increased significantly to 42.1%/62.5%, and the release rate of HES–DOX/LHRH was 40.1%/71.2%. This showed that the Schiff bond was stable under normal tissue physiological conditions, which can significantly reduce systemic toxicity, and acidic conditions in tumor sites triggered the cleavage of Schiff bond to promote drug release. More importantly, targeted HES–DOX/LHRH possessed better anti-tumor and anti-metastasis effects on a RM-1-xenografted C57BL/6 mouse model compared to free DOX and non-targeted HES–DOX, indicating that the LHRH-modified HES–DOX conjugate has great potential for treatment of metastatic prostate cancer with lower systemic toxicity. It is known that the concentration of glutathione (GSH) is higher in tumor than that in normal tissues. Taking advantage of this difference, a novel redox-sensitive conjugate HES–SS–DOX was developed.65 The disulfide bonds would be rapidly broken to trigger drug release under the environment of high reduction environment in tumor sites, while remained stable in blood circulation. Redox-insensitive HES–DOX was also synthesized as a control through the amide bonds. The drug release in vitro confirmed the GSH-triggered drug release of HES–SS–DOX. The drug release rate of HES–SS–DOX in PBS (pH 7.4) with 10 mM or 2 mM GSH was significantly higher than that in PBS (pH 7.4) with 2 uM or without GSH. The in vitro cytotoxicity of free DOX, HES–SS–DOX, and HES–DOX against H22, HepG-2, and Bel-7402 cells was studied. The results showed that at the concentration of DOX at 10 μg mL−1, the cytotoxicity of HES–SS–DOX conjugate was much higher than that of HES–DOX, which was close to free DOX. The in vivo study showed similar results. HES–SS–DOX possessed a longer plasma half-life and higher tumor site accumulation, bringing about higher tumor suppression efficiency with reduced toxicity. The HES in the above studies played an important role in relieving the toxicity of DOX to some extent. Especially the design of sensitive bonds made great achievements. Nevertheless, HESDOX exhibited better activity in vitro than HES–SS–DOX, thus, the release of DOX in a good manner is a concern.
5-Fluorouracil (5-Fu) is a small molecule drug that is widely used in the treatment of many tumors, especially solid tumors. Luo et al. developed a sustained-release drug delivery system for 5-FU to improve its short half-life.66 5-Fluorouracil-1-acetic acid (FUAC), which is transformed from 5-FU was firstly prepared and then conjugated to HES through ester bonds to afford the HES–FUAC conjugate. The conjugate was very stable in an acidic buffer with a pH value of 5.8. The release rate of FUAC was slow. As the pH value increased (pH 7.0–10.0), the hydrolysis was significantly accelerated. The conjugate was also hydrolyzable in human and rat plasma, releasing FUAC with half-lives of 20.4 h and 24.6 h, respectively. This significantly increased half-life of FUAC in the blood circulation confirmed that this conjugation can realize long-term circulation and sustained release function of FUAC in vivo. However, its antitumor efficacy in vivo needs to be further verified.
For developing a sustained-release drug delivery system with enhanced solubility, stability, and bioavailability, the anticancer drug curcumin (CUR), as a food-derived natural product with low toxicity, was conjugated to HES via an acid-labile ester linker.67 The amphiphilic HES–CUR conjugate could self-assemble into micellar nanoparticles (HES–CUR NPs) with a diameter below 100 nm. The HES–CUR NPs kept good stability as well as anti-oxidative ability during the storage. Compared with free CUR solution, HES–CUR NPs enhanced the solubility and improved the cytotoxicity to HeLa cells and Caco-2 human colorectal adenocarcinoma cells. Similarly, employing HES to conjugate with cis-platinum was a promising way to potentiate the water solubility and anti-cancer efficacy of cis-platinum, which attacks DNA, blocks replication and induces cell apoptosis. HES–Pt (IV) was firstly synthesized via esterification reaction, and then modified with galactose moieties by the reaction between hydroxyl group of HES 200/0.5 and carboxyl group of lactobionic acid (LA). The resulting LA–HES–Pt can specifically bind to the asialoglycoprotein receptors (ASGPR) overexpressed on the surfaces of hepatoma cells HepG-2 to promote the cellular uptake of LA–HES–Pt by hepatoma cells. The platinum conjugated on the LA–HES–Pt was reduced to cis-platinum after cellular uptake. The modification of cis-platinum with HES significantly improved the water solubility of cis-platinum. In vitro experiment verified that LA–HES–Pt had the higher cytotoxicity to HepG-2 cells compared with HES–Pt or free cis-platinum due to the ASGPR–LA-mediated endocytosis. Both the reported HES–CUR NPs and LA–HES–Pt have significant clinical translation potential and warrant further in vivo investigation.28
10-Hydroxy camptothecin (10-HCPT) is a camptothecin derivative with strong cytotoxicity. But the application is inhibited for its low water solubility and stability in vitro and in vivo.68 Under physiological environment, 10-HCPT can regulate the balance of its lactone structure and carboxylate structure through pH. Among them, lactone structure is an important form of antitumor effect, while carboxylate structure does not have antitumor activity. Studies have shown that coupling of the hydroxyl group at position 20 of the HCPT molecule with a water-soluble macromolecular material can significantly avoid ring-opening reactions of lactones and improve water solubility.69 Li et al. prepared a 10-HCPT–HES conjugate to deliver 10-HCPT.70,71 10-HCPT and HES were conjugated by ester bonds. 10-HCPT–HES has a water solubility of 0.72 mg mL−1 as 10-HCPT (approximately 100 times than that of the free 10-HCPT). The release of 10-HCPT from 10-HCPT–HES was increased with the decrease of pH value (4.0–7.4), suggesting prolonged circulation and favorable cancer treatment. As for the Hep-3B and SMMC-7721 cell lines, the 10-HCPT–HES conjugates exhibited higher cytotoxicity in comparison with free 10-HCPT. The half-life of 10-HCPT was extended to 4.38 h from 10 min. The 10-HCPT–HES conjugate effectively suppressed tumor growth accordingly. When 10-HCPT–HES conjugate was continuously modified with lysine (Ly), the prepared micelles showed satisfactory anti-tumor effect due to the existence of active targeting between Ly and amino acid (AA) transporters on the surface of liver cancer cells.72
Li et al. have synthesized a HES-based α-amylase and redox-responsive HES–SS–PTX NPs.73 The nanoparticle has a hydrophobic core (PTX) and a hydrophilic shell structure (HES). During blood circulation, α-amylase will specifically broke the α-1, 4 glycosidic bond on the HES linear chain, which gradually reduced the diameter of the nanoparticle and this was beneficial for deep penetration. After being taken up by the tumor cells, the nanoparticles disintegrated and released PTX to kill the tumor cells under the action of GSH. HES–SS–PTX NPs were firstly prepared by a modified emulsification method. When it comes to α-amylase concentration of 100 U L−1, size reduction took place within 10 min from 160 nm to 120 nm, indicating that HES can be rapidly degraded by α-amylase. Compared with commercially available Taxol, the half-life of HES–SS–PTX nanoparticles can be as long as 15 h, which was about 7 times longer than that of Taxol. The antitumor effect was also enhanced, accompanied with lower toxicity. These results indicated that nanoparticles with dual response of α-amylase and redox based on HES have great application potential in clinical tumor treatment. The degradation of HES by α-amylase achieved sustained and controlled release of drugs, and this was the advantage over PEG.
In addition to the small molecule drugs mentioned above, metal nanoparticles have attracted enormous interest in recent times. Among them, gold nanoparticles have a wide range of applications, which also have an antibacterial effect.74 However, the conventional chemical methods to prepare the gold nanoparticles have limitations.75 Das et al. developed a green synthesis of gold nanoparticles using a novel biodegradable graft copolymer, where methylacrylate (MA) was grafted on HES bone.76 Then, the partially hydrolyzed graft copolymer was used for synthesizing gold nanoparticles (GgNPS). The synthesized GgNPS with a mean diameter of 16–20 nm have shown excellent antibacterial activity on two Gram positive bacteria, but the effect on negative bacteria was not observed. This was because that the graft copolymer hindered the interaction of gold nanoparticles with the outer membrane component of the Gram negative bacteria, which has to be solved.
Polymer–drug and polymer–protein conjugates are promising candidates for the delivery of therapeutic agents. Kessler et al. have reported a novel compound named AGEM400 (HES) and evaluated its activity in vitro, where AGEM400 is a novel dimeric erythropoietin mimetic peptide.77 The molecular weight of HES used in AGEM400 (HES) was about 130 kDa. About up to 5 peptide molecules were coupled to one HES molecule. By the conjugation to HES, not only the circulating half-life of AGEM400 was prolonged, but also AGEM400 (HES) had effect on inducing normal in vitro erythropoiesis and stimulating survival of EPO-dependent cell line UT7/EPO. However, it displayed weak effects on three different EPO-responsive cell lines. Similarly, the covalent coupling of HES with anakinra (a 17.26 kDa protein) by reductive amination reaction resulted in increased molecular size, which greatly extended the half-life of anakinra in vivo without change of anakinra' structure and stability.78 These works only evaluated the half-life extension of protein drugs. The in vivo therapeutic effects and potential side effects need to be further studied.
DOX released the free DOX in response to acidic pH while HES–SS–DOX released DOX–SH in response to GSH. The activity of DOX–SH is reduced compared with free DOX. This is one of the reasons why HES–SS–DOX exhibited reduced in vitro activity than HESDOX. However, HESDOX also has apparent limitations. HES was oxidized for the conjugation of DOX in HESDOX, which disrupts the backbone of HES and may lead to unfavorable effects. In addition to stimuli-responsive bonds, ester bond and amide bond are also proven to be effective for some HES–small molecule conjugates such as HES–Pt, HES–FUAC, HES–CUR and 10-HCPT–HES, which may ascribed to the enzyme-mediated drug release. Compared to the above mentioned HES-small molecules, HES–DFO and HES–protein conjugates do not need to enter into cells to exert their effects and could take full advantage of long-circulation of HES, showing more promising clinic translation potential. However, HES–protein conjugates are difficult to synthesize and control the quality due to the presence of multiple functional groups on proteins. Although polyhydroxyl sites on HES provide targets for covalent attachment of drugs, how to achieve precise and controllable conjugation is crucial to the successful translation of HES–drug conjugates.
| HES kind | Drug | Type of bond | In vivo studies (models) | Key findings | Ref. | |
|---|---|---|---|---|---|---|
| Small agents | Not available | Deferoxamine (DFO) | Imine | Murine model of acute iron toxicity | Prevent the mortality | 57 |
| HES 130/0.4 | Methotrexate (MTX) | Ester | P388 murine leukaemia, MV-4-11 human leukaemia | Higher antitumor efficacy | 58 | |
| HES 130/0.4 | Doxorubicin (DOX) | Imine | B16F10 melanoma | Acid-sensitive, enhanced antitumor activity and security | 61 | |
| HES 130/0.4 | Doxorubicin (DOX) | Imine | A375 human malignant melanoma | Targeted acid-sensitive and improved antitumor efficacy | 62 | |
| HES 130/0.4 | Doxorubicin (DOX) | Hydrazone | No | Acid responsiveness, better proliferation inhibition in HepG2 cells | 63 | |
| HES 130/0.4 | Doxorubicin (DOX) | Imine | RM-1-xenografted C57BL/6 mouse | pH-responsive, improved distribution, anti-tumor and anti-metastasis | 64 | |
| HES 200/0.5 | Doxorubicin (DOX) | Amide | H22-tumor mice model | Redox-sensitive, targeted drug delivery and better antitumor efficacy | 65 | |
| HES 130/0.4 | 5-Fluorouracil (5-Fu) | Ester | SD rats | Sustained-release of FUAC | 66 | |
| HES 200/0.5 | Curcumin (CUR) | Ester | No | Improved solubility and stability of CUR, better anticancer activity | 67 | |
| HES 200/0.5 | cis-Platinum (Pt) | Ester | No | Improved solubility of Pt, targeted delivery to HepG-2 cancer cells | 28 | |
| HES 200/0.5, HES 130/0.4 | 10-Hydroxy camptothecin (10-HCPT) | Amide | Hep-3B solid tumor in nude mice | Improved solubility and stability of 10-HCPT, higher cytotoxicity | 70 and 71 | |
| HES 130/0.4 | Paclitaxel (PTX) | Disulfide bond | 4T1-tumor mice model | α-Amylase- and redox-responsive, increased in vivo half life time | 73 | |
| Biomolecules | HES 130/0.5 | Erythropoietin (AGEM400) | Thioether | No | Prolonged half-life, superior efficacy | 77 |
| HES 85 kDa | Anakinra | Imine | Male Wistar rats | Extended half-life | 78 |
3.2 HES based drug carriers
Although great progresses have actually been made in the field of polymer-conjugates among different nanocarriers, such conjugation is not the perfect way. Many conjugations pose a hazard to other normal cells because of their poor stability and premature drug release. Hu et al. developed HES–SS–DOX conjugate in their previous study, which showed a certain ability to suppress the growth of tumors. Nevertheless, the therapeutic effect of the HES–SS–DOX conjugate needs to be essentially improved since they were unable to eradicate tumors in the treatment period after multiple injections.65 In view of the good in vivo biocompatibility but limited efficiency of HES–SS–DOX conjugates, Yu et al. once again developed a new type of HES–SS–DOX@ICG nanoparticles (NPs) for fully eradicating tumors.85 Indocyanine green (ICG), as a photothermal agent, also has cytotoxicity to the targeted cancer cells, which was directly coated onto HES–SS–DOX conjugates. These HES–SS–DOX@ICG NPs were stable and able to eradicate tumors with one single injection and single near infrared (NIR) laser irradiation. This nanoplatform exploits the well-known instability of FDA approved ICG for enhanced tumor penetration and can also encapsulate other components via supramolecular self-assembly for combination therapies. This combination has promising potential for chemo-photothermal cancer therapy in clinic.
To continuously solve ICG's instability and expand its application, ICG was encapsulated in HES–oleic acid conjugate nanoparticles (HES–OA NPs). Then ICG@HES–OA NPs was combined with β-phenylethyl isothiocyanate (PEITC) for potent PDT.86 PEITC has a role in depleting GSH efficiently and selectively after conjugation with intracellular GSH by the action of GSH S-transferases inside the tumor cells. ICG@HES–OA NPs enhanced the stability of ICG greatly. PDT based on the combination of ICG@HES–OA NPs and PEITC exhibited synergistic antitumor effect. Similarly, DOX and ICG were co-loaded by galactose-functionalized HES–PCL nanocolloidosomes (Gal-HES–PCL NCs) for in vivo chemo/photothermal combination therapies.87 The resultant DOX/ICG@Gal-HES–PCL NCs possessed selective drug release behaviors due to the distinctive structure of nanocolloidosomes.
DOX is used as one of effective chemotherapeutic drugs, but the low specificity toward tumor and severe side effects limit its clinical use. As a substitute for traditional PEGylation, different nanocarriers based on HES have sprung up. Wu et al. presented the preparation of HES-coated PDA (HES–PDA) NPs as a nanocarrier for DOX.88 Not only could it be a straightforward method to improve the stability of the PDA (bio-inspired polydopamine) NPs, but also reinforced the application of DOX. DOX-loaded HES–PDA NPs were studied in comparison with PEG-coated PDA NPs (DOX@PEG–PDA NPs). Collectively, DOX@HES–PDA NPs can implement the same function, with some advantages, compared with DOX@PEG–PDA NPs for cancer chemotherapy. Yu et al. designed HES-grafted-polylactide (HES-g-PLA) copolymers, which assembled into two kinds of NPs that were used as partner nanocarriers for delivering DOX.89 As results showed, these HES-g-PLA NPs indeed enhanced DOX delivery with no impairment to the body. The HES-g-PLA NPs, as a reticuloendothelial system (RES)-blocking agent, can temporarily block up the RES for a suitable period of time, and they were suitable for delivering various kinds of anticancer drugs toward tumors if the adopted drug could be efficiently loaded into the small-size HES-g-PLA NPs. Docetaxel (DTX) as loaded drug in Liu et al.‘s study has come to same conclusion.90 To enhance the tumor targeting and penetration ability of chemotherapy-related drugs, Hu et al. reported a novel kind of polymeric reduction responsive vehicle, nanoclusters (NCs). The NCs were self-assembled from hydrophobically modified HES (HES–SS–C18). A tumor-penetrating peptide IRGD was coated on the surface to enhance their tumor cell internalization.91 Because of interaction between iRGD and integrin αv, DOX@iRGD–HES–SS–C18 NCs can specifically bind to the cell membranes. Then, DOX was released in the tumor's reductive environment. The intelligent nanoclusters realized more accurate targeting. In the process of seeking to improve the targeting of drugs, heterogeneous distribution of drug inside tumor was found to causing regional insufficient chemotherapy, which resulted in drug resistance and tumor metastasis. Conscious of this problem, there was a co-delivery nanoparticle (HES–PLA), where DOX and TGF-β receptor inhibitor, LY2157299 (LY), were administered together.92 In vitro and in vivo studies, DOX/LY@HES–PLA nanoparticle demonstrated great effect on suppressing primary tumor and distant metastasis (nearly no nodule on the lung), which provided implications for clinical applications. If the targeting molecule can be modified on the surface of this carrier, it will greatly increase the uptake of tumor cells.
Folic acid (FA) has a high affinity to folate receptors (mainly FRα) which are overexpressed on the surface of various tumor carcinoma cells. When covalently linked (via γ-carboxyl group) to the NH2 terminated conjugate, which was used for coupling with the carboxylic groups on the HES nanocapsules surface, the resultant HES–FA conjugate remained its physiological properties and binding affinity to FRα.93 After fractionation, HES–FA–F, with a small diameter (174 nm) showed a better uptake into HeLa cells. However, the uptake of nanoparticles in vivo is not yet known.
Jiang et al. also introduced a microspheres delivery system AcHES–PLGA, composed of poly(D, L-lactide-co-glycolide) (PLGA) and poly(acryloyl hydroxyethyl starch) (acryloyl derivatized HES; AcHES), which was loaded with bovine insulin.96 The composite microspheres showed more favorable and complete in vitro release than conventional PLGA microspheres. In vivo evaluation, when treated with insulin-loaded AcHES–PLGA, the glucose suppression could be easily seen in diabetic animals. Then, HES microcapsules with encapsulated bovine serum albumin (BSA) were synthesized by crosslinking with terephthaloyl chloride at the emulsion droplet interface. The loading of BSA in HES microparticles can induce both a T helper 1/T helper 2 immune response, which was critical for the control of malignant cells as well as infectious pathogens.97 The HES vehicle may provide a suitable delivery and presentation system in the development of vaccine.
To investigate physicochemical characterization, the in vitro cytocompatibility and hemotoxicity of HES-decorated polyplexes, Noga et al. prepared a series of HES–PEI conjugates coupled to a 22 kDa linear polyethylenimine (LPEI22).98 It is noteworthy that various HES-decorated polyplexes had hydrodynamic diameters of approximately 70–80 nm, which meant that the amount of HES in the polyplexes didn't play an important role in the particle diameter. At the same time, the HES-mediated particle shielding effect was manifested by a decrease in surface charge that was strongly correlated to the molar mass of HES, where the charge decreases linearly with increasing molar mass. HES–PEI conjugates showed lower cytotoxicity, no aggregation, and much lower hemolysis compared to unmodified PEI, providing the potential for gene delivery.
Frick et al. linked human IL-2 to the surface of HES nanocapsules via copper-free click reaction to obtain “HES-D-IL-2” nanocapsules.99 The uptake experiment in vitro revealed that HES-D-IL-2 capsules could lead to vigorous T cell proliferation compared with only IL-2. In addition, by controlling the degree of functionalization as well as by assessing the amounts of surface-bound IL-2, HES-D-IL-2 with different ratios of IL-2 were synthesized. This nanocapsule-mediated technique is a promising strategy for T cell-based immunotherapies and may be translated to other cytokine-related targeting systems.
HES nanocapsules (NCs) themselves do not show any non-specific cell interactions. Only the presence of a targeting agent on their surface allows them to target specifically to the desired site. Freichels et al. described the synthesis of nanocapsules (NCs) covalently attached with mannose residues.100 Amine groups on the surface of HES NCs were reacted either directly with the isothiocyanate group (ITC-Man) or through reductive amination (di-Man and tri-Man). The uptake of NCs by dendritic cells was significantly enhanced by the modification of mannose on the surface of NCs. However, the targeting function of mannosylated HES NCs in vivo is absent.
| HES kind | Nanocarrier | Drug | In vivo studies (models) | Key findings | Ref. | |
|---|---|---|---|---|---|---|
| Small agents | HES 200/0.5 | HES polymer | Dexamethasone (DXM) | No | Targeted transport of DXM to NPCs | 81 |
| HES 200 | HES polymer | Monophosphoryl lipid A (MPLA) | No | Enhanced the uptake and phagocytosis | 82 | |
| Not precisely | HES polymer | Diclofenac sodium (DS) | SD rats | Reduced dosage and frequency of administration of DS | 83 | |
| HES 200/0.5 | HES–PLLA | Octenidine | No | Inhibited the enzymatic degradation | 84 | |
| HES 20/0.5 | HES–SS–DOX conjugate | Indocyanine green (ICG) | H22 tumor-bearing mice | Highly efficient anti-tumor performance with ICG | 85 | |
| HES 130/0.4 | HES–OA conjugate | Indocyanine green (ICG) | H22 tumor-bearing mice | Enhanced ICG stability, PDT synergistic antitumor effect | 86 | |
| HES 130/0.4 | Gal-HES–PCL | Doxorubicin (DOX) and indocyanine green (ICG) | H22 tumor-bearing mice | Chemo/photothermal combination therapies | 87 | |
| HES 25/0.5 | HES–PDA | Doxorubicin (DOX) | H22 tumor-bearing mice | Improved antitumor efficacy | 88 | |
| HES 70/0.5 | HES-g-PLA copolymers | Doxorubicin (DOX) | H22 tumor-bearing mice | Effectively delivery of DOX | 89 | |
| HES 130/0.4 | iRGD–HES–SS–C18 | Doxorubicin (DOX) | No | Enhanced cellular uptake and antitumor efficacy | 91 | |
| HES 70/0.5 | HES–PLA | Doxorubicin (DOX) and LY2157299 (LY) | 4T1 cell in zebrafish | Simultaneously suppress primary tumor and distant metastasis | 92 | |
| HES 200 | HES NCs | Folic acid (FA) | No | Specific cellular uptake into HeLa cells | 93 | |
| Biomolecules | HES 130/0.4 | HES–(P(EG)6 MA) polymer | No | No | A delivery system for the controlled release of proteins | 94 |
| HES 130 | HES–HEMA | Lysozyme | No | Controlled release | 95 | |
| HES 422/0.76 | AcHES–PLGA | Insulin | Diabetic animal model | Sustained release of proteins | 96 | |
| HES 130/0.4 | HES | Bovine serum albumin (BSA) | No | Enhanced immune responses | 97 | |
| HES 70/0.5 | HES–PEI copolymers | Plasmid pCMVluc (pDNA) | No | The shielding and controlled deshielding of DNA polyplexes | 98 | |
| HES 200/0.5 | HES NCs | Cytokine interleukin-2 (IL-2) | Wild-type C57BL/6 mice | Direct and specifc target to human and murine T cells | 99 | |
| HES 200/0.45 | HES NCs | (Oligo)mannose | No | Targeted delivery to dendritic cells | 100 |
4. Conclusion and outlook
At the end of the last century, the discovery and confirmation of EPR effect have greatly promoted the development of nano-drug delivery systems.102 As summarized in Fig. 2, there are mainly two common drug-loading strategies: one is loading drug on the carrier through non-covalent bond interaction, the other is covalently conjugating to the carrier. No matter which way, the choice of carrier, such as PEG, N-(2-hydroxypropyl)-methacrylamide (pHPMA), polypeptide and polysaccharides is crucial. However, the non-degradability of PEG and pHPMA polymers in vivo inevitably limits the applications of these two materials. Polypeptides including α,β-polyasparthydrazide (PAHy), polyglutamic acid (PGA), polylysine (PLL) and poly(L-aspartic acid) (PAA) exhibit good biocompatibility, biodegradability and abundant reaction groups.103–105 In addition to the above advantages of poly (amino acids), HES, as a polysaccharide, leads to low incidence of immune reaction due to molecular structure similar to human glycogen and is easy to get at low price.106–108 Compared with resistant starch which is indigestible by body enzymes, HES has slightly worse anti-digestion physiological characteristics, but it can be completely metabolized and has better biological safety.109,110The compounds, depending on different intrinsic characteristic, were covalently conjugated with HES or loaded onto nanocarriers to improve the drug's solubility, enhance the therapeutic efficacy, and reduce side effects. The nano drug delivery systems will have a better effect on maintaining the stability and efficacy of drug molecules. A variety of non-covalent bonding nanomedicine has been approved for clinical useages.3 There still exists challenge to balance the contradiction between drug loading system stability and rapid drug release at the target sites, while the conjugation strategy is preferable because conjugating drugs onto polysaccharides can produce more stable prodrugs with only two ingredients, which can minimize the possible toxicity induced by foreign materials. Apart from the compatibility, the drug loading capability of the polymer is crucial. The abundant reaction groups (hydroxyl groups) on HES greatly improve the limitations of low drug-loading ability of PEG for that it has only two terminal groups for covalent attachment with the drug molecules, thus exhibiting a clinical prospect.
Polymer micelles have many advantages over other nano-drug delivery systems. For example, the size of polymer micelles is smaller than liposomes, which is very important for transcutaneous lymphatic administration or intravascular drug extravasation into tumor tissues.111 Many types of amphiphilic polymers can form micellar structures, where the hydrophobic part is usually composed of a biodegradable hydrophobic polymer, such as PLA or PCL and the hydrophilic part is usually PEG.112 After PEG–PLA and PEG–PCL form micelles, the hydrophilic shell composed of PEG can reduce the adsorption of plasma proteins and opsonins on the micelles, thus prolonging the circulation time of the micelles in vivo.113 However, the hydrophilic layer formed by PEG also prevents the nanoparticle from contacting the cell membrane composed of the phospholipid bilayer, thereby affecting the endocytosis efficiency of the nanoparticle by tumor cells.114 Even the nano-formulations that have been approved for clinical use like still have drug efficacy and security issues.115,116 In addition, studies have shown that PEG–PLA can inhibit the efflux function of P-glycoprotein at the cellular level.117 Similar HES–PLA or HES–PCL not only has the advantages of PEGylation modification, but also solves the dilemma caused by PEG. Under the action of α-amylase in the blood, the HES hydration layer on the periphery of the nanoparticles is continuously trimmed, which is conducive to the incorporation and release of drugs. It was reported that the amount of amylase decreased with the decrease of HES.118 Therefore, the nanoparticles coated with HES possess a property of α-amylase-responsive. The degradation by α-amylase leads to the decrease of particle size,73 which would be beneficial for nanoparticles tumor targeting, accumulation, and penetration.119 It is speculated that the targeted degradation may be realized at the tumor site for the reason that the expression of starch hydrolase may increase according with rapidly metabolism of tumor cells. As a result, the increase of local drug concentration at the tumor could be realized. It is known that α-amylase is produced mainly in pancreas. Thus, the more α-amylase may accelerate such a degradation process, so as to achieve better effect on pancreatic cancer.
Cytokines are a heterogeneous group of soluble small polypeptides or glycoproteins, playing an important regulatory role in the immune system.120 The cytokine therapy that has emerged in recent years can convert an immunological “cold” microenvironment to “hot” in immunotherapy. Intratumoral injection or targeting antibodies can relieve dose-limiting systemic toxicities to some extent associated with cytokines themself.121–123 HES which is completely degraded without residue, can also increase the stability and extend the half-life during the circulation to avoid the side effects. The poly-hydroxyl groups on HES provide coupling of targeted substances or chemotherapeutics in addition to cytokines, realizing tumor both targeting and immuno-chemotherapy at the same time. At present, PEG has been applied to the modification of a variety of protein drugs, such as ADAGEN in market, PEG-modified asparaginase, interferon α2a and 2b and other protein drugs have been approved by the FDA for clinical use, verifying the feasibility of coupling this macromolecule to protein or factor.124,125 But how to achieve optimal concentrations in the tumor microenvironment has not been addressed yet.
Apart from the anticancer drugs, there are also other drugs accompanied with low solubility, stability, bioavailability or side effects.126–128 Nanomedicine can reduce these shortcomings. The CS-MnFe2O4 NPs as carriers of ofloxacin realized a controlled release over 3 days, compared with that half-life of free is 8 to 9 h129 The resveratrol with cyclodextrin-based nanosponges displayed significantly better permeation, stability and cytotoxicity.130 As mentioned above, HES as a carrier has good application in improving these shortcomings. In addition, as HES also showed a tremendous decrease of the other pro-inflammatory cytokines (IL-1, IL-6, IL-8, and TNF) in human peripheral blood mononuclear cells, it may play an anti-inflammatory effect other than the carrier in inflammation-related diseases.131 The application of Morin/HES–DOCA-NPs remarkably extended the half-life of Morin and had a superior serum urate lowering effect in vivo. The success of this formulation provided a solid proof for novel self-assembled HES NPs can be applied to additional therapeutic agents to improve the treatment of many other diseases.132 Last but not least, targeting ligands, molecular transporting moieties, or fluorescence probes will be introduced to nano drug delivery system based on HES in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by grants from the National Key Research and Development Program of China (2018YFA0208900, 2020YFA0211200), the National Science Foundation of China (31972927 and 31700867), Scientific Research Foundation of Huazhong University of Science and Technology (3004170130), Program for HUST Academic Frontier Youth Team (2018QYTD01), and the HCP Program of HUST.References
- S. K. Nitta and K. Numata, Int. J. Mol. Sci., 2013, 14, 1629–1654 CrossRef CAS
.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS
- S. Marchal, A. El Hor, M. Millard, V. Gillon and L. Bezdetnaya, Drugs, 2015, 75, 1601–1611 CrossRef CAS
- C. P. Hollis, H. L. Weiss, M. Leggas, B. M. Evers, R. A. Gemeinhart and T. Li, J. Controlled Release, 2013, 172, 12–21 CrossRef CAS
- Y. Tsukioka, Y. Matsumura, T. Hamaguchi, H. Koike, F. Moriyasu and T. Kakizoe, Jpn. J. Cancer Res., 2002, 93, 1145–1153 CrossRef CAS
- J. Zheng, L. Y. Li, H. K. Tsao, Y. J. Sheng, S. F. Chen and S. Y. Jiang, Biophys. J., 2005, 89, 158–166 CrossRef CAS
- G. Pasut and F. M. Veronese, J. Controlled Release, 2012, 161, 461–472 CrossRef CAS
- S. Mishra, P. Webster and M. E. Davis, Eur. J. Cell Biol., 2004, 83, 97–111 CrossRef CAS
- H. Hatakeyama, H. Akita and H. Harashima, Adv. Drug Delivery Rev., 2011, 63, 152–160 CrossRef CAS
- B. Stella, S. Arpicco, M. T. Peracchia, D. Desmaele, J. Hoebeke, M. Renoir, J. D'Angelo, L. Cattel and P. Couvreur, J. Pharm. Sci., 2000, 89, 1452–1464 CrossRef CAS
- H. P. Ramesh and R. N. Tharanathan, Crit. Rev. Biotechnol., 2003, 23, 149–173 CrossRef CAS
- G. Saravanakumar, D. G. Jo and J. H. Park, Curr. Med. Chem., 2012, 19, 3212–3229 CrossRef CAS
- A. Abed, N. Assoul, M. Ba, S. M. Derkaoui, P. Portes, L. Louedec, P. Flaud, I. Bataille, D. Letourneur and A. Meddahi-Pelle, J. Biomed. Mater. Res., Part A, 2011, 96A, 535–542 CrossRef CAS
- S. Rodrigues, L. Cardoso, A. M. Rosa da Costa and A. Grenha, Materials, 2015, 8, 5647–5670 CrossRef CAS
- G. Tripodo, A. Trapani, M. L. Torre, G. Giammona, G. Trapani and D. Mandracchia, Eur. J. Pharm. Biopharm., 2015, 97, 400–416 CrossRef CAS
- K. Divya, V. Smitha and M. S. Jisha, Int. J. Biol. Macromol., 2018, 114, 572–577 CrossRef CAS
- A. B. Sieval, M. Thanou, A. F. Kotze, J. E. Verhoef, J. Brussee and H. E. Junginger, Carbohydr. Polym., 1998, 36, 157–165 CrossRef CAS
- A. Passi and D. Vigetti, Adv. Drug Delivery Rev., 2019, 146, 83–96 CrossRef CAS
- J. M. Mishler, Clin. Haematol., 1984, 13, 75–92 CAS
- M. Westphal, M. F. M. James, S. Kozek-Langenecker, R. Stocker, B. Guidet and H. Van Aken, Anesthesiology, 2009, 111, 187–202 CrossRef CAS
- J. Boldt and S. Suttner, Minerva Anestesiol., 2005, 71, 741–758 CAS
- S. A. Kozek-Langenecker, Anesthesiology, 2005, 103, 654–660 CrossRef
- N. E. Kuz'mina, S. V. Moiseev, V. I. Krylov, V. A. Yashkir and V. A. Merkulov, J. Anal. Chem., 2015, 70, 32–38 CrossRef
- J. Treib, J. F. Baron, M. T. Grauer and R. G. Strauss, Intensive Care Med., 1999, 25, 258–268 CrossRef CAS
- J. Treib, A. Haass, G. Pindur, U. T. Seyfert, W. Treib, M. T. Grauer, F. Jung, E. Wenzel and K. Schimrigk, Thromb. Haemostasis, 1995, 74, 1452–1456 CrossRef CAS
- W. M. Kulicke, D. Roessner and W. Kull, Starch-Starke, 1993, 45, 445–450 CrossRef CAS
- H. Kohler, H. Zschiedrich, A. Linfante, F. Appel, H. Pitz and R. Clasen, Klin. Wochenschr., 1982, 60, 293–301 CrossRef CAS
- C. Xiao, H. Hu, H. Yang, S. Li, H. Zhou, J. Ruan, Y. Zhu, X. Yang and Z. Li, Nanoscale Adv., 2019, 1, 1002–1012 RSC
- S. Maier, C. Holz-Hoelzl, W. Pajk, H. Ulmer, C. Hengl, M. Duenser, T. Haas, C. Velik-Salchner, D. Fries, A. Greiner, W. Hasibeder and H. Knoter, J. Trauma: Inj., Infect., Crit. Care, 2009, 66, 337–345 CrossRef CAS
- W. A. Osthaus, D. Huber, C. Baeumker, L. Witt, M. Metzelder, J. Kuebler and R. Suempelmann, Pediatr. Anesth., 2008, 18, 922–928 CrossRef
- B. E. Ickx, F. Bepperling, C. Melot, C. Schulman and P. J. Van der Linden, Br. J. Anaesth., 2003, 91, 196–202 CrossRef CAS
- J. Waitzinger, F. Bepperling, G. Pabst, J. Opitz, A. Fackelmayer and J. Boldt, Clin. Drug Invest., 1999, 17, 119–125 CrossRef CAS
- P. Meybohm, E. Cavus, V. Doerges, B. Weber, K.-H. Stadlbauer, V. Wenzel, J. Scholz, M. Steffen and B. Bein, Resuscitation, 2008, 76, 449–456 CrossRef CAS
- R. Woessner, M. T. Grauer, H. J. Dieterich, F. Bepperling, D. Baus, T. Kahles, S. Georgi, O. Bianchi, M. Morgenthaler and J. Treib, Pathophysiol. Haemostasis Thromb., 2003, 33, 121–126 CrossRef CAS
- J. Tian, X. Lin, R. Guan and J. G. Xu, Anesth. Analg., 2004, 98, 768–774 CrossRef CAS
- J. Tian, X. Lin, W. Zhou and J. G. Xu, Ann. Clin. Lab. Sci., 2003, 33, 451–458 CAS
- R. Lv, Z.-Q. Zhou, H.-W. Wu, Y. Jin, W. Zhou and J.-G. Xu, Anesth. Analg., 2006, 103, 149–155 CrossRef CAS
- J. L. Pascual, L. E. Ferri, P. Chaudhury, A. J. Seely, G. Campisi, B. Giannias, D. C. Evans and N. V. Christou, Surg. Infect., 2001, 2, 275–287 CrossRef CAS
- R. Lv, W. Zhou, L. D. Zhang and J. G. Xu, Acta Anaesthesiol. Scand., 2005, 49, 635–642 CrossRef CAS
- K. Lang, S. Suttner, J. Boldt, B. Kumle and D. Nagel, Can. J. Anaesth., 2003, 50, 1009–1016 CrossRef
- G. Scharbert, E. Deusch, H. G. Kress, M. Greher, B. Gustorff and S. A. Kozek-Langenecker, Anesth. Analg., 2004, 99, 823–827 CrossRef CAS
- S. H. Hwang, Y. S. Won, J. S. Yu, J. Y. Yang and C. S. Choi, J. Korean Neurosurg. Soc., 2007, 42, 377–381 CrossRef CAS
- C. Jungheinrich, W. Sauermann, F. Bepperling and N. H. Vogt, Drugs R&D, 2004, 5, 1–9 CrossRef CAS
- C. A. Volta, V. Alvisi, M. Campi, E. Marangoni, R. Alvisi, M. Castellazzi, E. Fainardi, M. C. Manfrinato, F. Dallocchio and T. Bellini, Anesthesiology, 2007, 106, 85–91 CrossRef CAS
- K. Kessenbrock, V. Plaks and Z. Werb, Cell, 2010, 141, 52–67 CrossRef CAS
- C. M. Thorson, R. M. Van Haren, M. L. Ryan, E. Curia, D. Sleeman, J. U. Levi, A. S. Livingstone and K. G. Proctor, Surgery, 2014, 155, 134–144 CrossRef
- H. Liang, C. Yang, B. Zhang, H. Wang, H. Liu, Z. Zhao, Z. Zhang, X. Wen and X. Lai, Med. Oncol., 2015, 32, 151 CrossRef
- T. Tamada, K. Okada, R. Ishida, K. Kamishita and T. Irikura, Chem. Pharm. Bull., 1971, 19, 286–291 CrossRef CAS
- A. Stolzing, Y. Naaldijk, V. Fedorova and S. Sethe, Transfus. Apher. Sci., 2012, 46, 137–147 CrossRef CAS
- F. J. Lionetti and S. M. Hunt, Cryobiology, 1975, 12, 110–118 CrossRef CAS
- H. Kim, S. Tanaka, S. Une, M. Nakaichi, S. Sumida and Y. Taura, J. Vet. Med. Sci., 2004, 66, 1543–1547 CrossRef CAS
- A. Sputtek, E. P. Horn, J. S. A. Esch and P. Kuhnl, Anasthesiologie Intensivmedizin Notfallmedizin Schmerztherapie, 2001, 36, S162–S164 CrossRef
- N. Neveux, J. P. DeBandt, C. Charrueau, E. Savier, J. C. Chaumeil, L. Hannoun, J. Giboudeau and L. A. Cynober, Hepatology, 1997, 25, 678–682 CrossRef CAS
- J. Y. Mabrut, M. Adham, J. P. Bourgeot, A. Eljaafari, E. DelaRoche, C. Ducerf, J. Baulieux and D. Rigal, Transplant. Proc., 2001, 33, 609–611 CrossRef CAS
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS
- M. E. Brecher, H. G. Owen and N. Bandarenko, J. Clin. Apher., 1997, 12, 146–153 CrossRef CAS
- J. R. Mahoney Jr, P. E. Hallaway, B. E. Hedlund and J. W. Eaton, J. Clin. Invest., 1989, 84, 1362–1366 CrossRef CAS
- T. M. Goszczynski, B. Filip-Psurska, K. Kempinska, J. Wietrzyk and J. Boratynski, Pharmacol. Res. Perspect., 2014, 2, e00047 CAS
- O. M. Abo-Salem, J. Biochem. Mol. Toxicol., 2012, 26, 1–9 CrossRef CAS
- S. Yilmaz, A. Atessahin, E. Sahna, I. Karahan and S. Ozer, Toxicology, 2006, 218, 164–171 CrossRef CAS
- D. Li, J. Ding, X. Zhuang, L. Chen and X. Chen, J. Mater. Chem. B, 2016, 4, 5167–5177 RSC
- D. Li, X. Peng, L. Chen, J. Ding and X. Chen, ACS Biomater. Sci. Eng., 2018, 4, 539–546 CrossRef CAS
- Y. Zhu, X. Yao, X. Chen and L. Chen, J. Appl. Polym. Sci., 2015, 132, 42778 Search PubMed
- K. Zhao, D. Li, W. Xu, J. Ding, W. Jiang, M. Li, C. Wang and X. Chen, Biomaterials, 2017, 116, 82–94 CrossRef CAS
- H. Hu, Y. Li, Q. Zhou, Y. Ao, C. Yu, Y. Wan, H. Xu, Z. Li and X. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 30833–30844 CrossRef CAS
- Q. Luo, P. Wang, Y. Miao, H. He and X. Tang, Carbohydr. Polym., 2012, 87, 2642–2647 CrossRef CAS
- S. Chen, J. Wu, Q. Tang, C. Xu, Y. Huang, D. Huang, F. Luo, Y. Wu, F. Yan, Z. Weng and S. Wang, Carbohydr. Polym., 2020, 228, 115398 CrossRef CAS
- N. Subramanian, N. Sundaraganesan, S. Sudha, V. Aroulmoji, G. D. Sockalingam and M. Bergamin, Spectrochim. Acta, Part A, 2011, 78, 1058–1067 CrossRef CAS
- H. G. Lerchen, J. Baumgarten, K. von dem Bruch, T. E. Lehmann, M. Sperzel, G. Kempka and H. H. Fiebig, J. Med. Chem., 2001, 44, 4186–4195 CrossRef CAS
- G. Li, C. Cai, Y. Qi and X. Tang, Drug Delivery, 2016, 23, 277–284 CrossRef CAS
- G. Li, Y. Li, Y. Tang, Y. Zhang, Y. Zhang, T. Yin, H. Xu, C. Cai and X. Tang, Int. J. Pharm., 2014, 471, 234–244 CrossRef CAS
- G. Li, M. Zhao and L. Zhao, Drug Delivery, 2020, 27, 519–529 CrossRef CAS
- Y. Li, H. Hu, Q. Zhou, Y. Ao, C. Xiao, J. Wan, Y. Wan, H. Xu, Z. Li and X. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 19215–19230 CrossRef CAS
- V. D. Badwaik, L. M. Vangala, D. S. Pender, C. B. Willis, Z. P. Aguilar, M. S. Gonzalez, R. Paripelly and R. Dakshinamurthy, Nanoscale Res. Lett., 2012, 7, 623 CrossRef
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS
- S. Das, A. Pandey, S. Pal, H. Kolya and T. Tripathy, J. Mol. Liq., 2015, 212, 259–265 CrossRef CAS
- C. Kessler, A. Greindl, B. Breuer, U. Haberl, A. Rybka, M. Emgenbroich, H.-G. Frank and A. J. G. Poetgens, Cytokine, 2012, 57, 226–237 CrossRef CAS
- R. Liebner, R. Mathaes, M. Meyer, T. Hey, G. Winter and A. Besheer, Eur. J. Pharm. Biopharm., 2014, 87, 378–385 CrossRef CAS
- A. W. Meikle and F. H. Tyler, Am. J. Med., 1977, 63, 200–207 CrossRef CAS
- L. W. Doyle, R. A. Ehrenkranz and H. L. Halliday, Neonatology, 2010, 98, 217–224 CrossRef CAS
- M. Fichter, G. Baier, M. Dedters, L. Pretsch, A. Pietrzak-Nguyen, K. Landfester and S. Gehring, Nanomedicine, 2013, 9, 1223–1234 CrossRef CAS
- M. Fichter, M. Dedters, A. Pietrzak-Nguyen, L. Pretsch, C. U. Meyer, S. Strand, F. Zepp, G. Baier, K. Landfester and S. Gehring, Vaccine, 2015, 33, 838–846 CrossRef CAS
- D. Narayanan, G. J. Pillai, S. V. Nair and D. Menon, Drug Delivery Transl. Res., 2019, 9, 867–878 CrossRef CAS
- G. Baier, A. Cavallaro, K. Friedemann, B. Mueller, G. Glasser, K. Vasilev and K. Landfester, Nanomedicine, 2014, 10, 131–139 CrossRef CAS
- C. Yu, C. Liu, S. Wang, Z. Li, H. Hu, Y. Wan and X. Yang, Cancers, 2019, 11, 207 CrossRef CAS
- H. Hu, J. Chen, H. Yang, X. Huang, H. Wu, Y. Wu, F. Li, Y. Yi, C. Xiao, Y. Li, Y. Tang, Z. Li, B. Zhang and X. Yang, Nanoscale, 2019, 11, 6384–6393 RSC
- H. Hu, C. Xiao, H. Wu, Y. Li, Q. Zhou, Y. Tang, C. Yu, X. Yang and Z. Li, ACS Appl. Mater. Interfaces, 2017, 9, 42225–42238 CrossRef CAS
- H. Wu, H. Hu, J. Wan, Y. Li, Y. Wu, Y. Tang, C. Xiao, H. Xu, X. Yang and Z. Li, Chem. Eng. J., 2018, 349, 129–145 CrossRef CAS
- C. Yu, Q. Zhou, F. Xiao, Y. Li, H. Hu, Y. Wan, Z. Li and X. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 10481–10493 CrossRef CAS
- Q. Liu, X. Yang, H. Xu, K. Pan and Y. Yang, Eur. Polym. J., 2013, 49, 3522–3529 CrossRef CAS
- H. Hu, J. Wan, X. Huang, Y. Tang, C. Xiao, H. Xu, X. Yang and Z. Li, Nanoscale, 2018, 10, 10514–10527 RSC
- Q. Zhou, Y. Li, Y. Zhu, C. Yu, H. Jia, B. Bao, H. Hu, C. Xiao, J. Zhang, X. Zeng, Y. Wan, H. Xu, Z. Li and X. Yang, J. Controlled Release, 2018, 275, 67–77 CrossRef CAS
- G. Baier, D. Baumann, J. M. Siebert, A. Musyanovych, V. Mailaender and K. Landfester, Biomacromolecules, 2012, 13, 2704–2715 CrossRef CAS
- A. Bertz, S. Woehl-Bruhn, S. Miethe, B. Tiersch, J. Koetz, M. Hust, H. Bunjes and H. Menzel, J. Biotechnol., 2013, 163, 243–249 CrossRef CAS
- A. D. A. Schwoerer, S. Harling, K. Scheibe, H. Menzel and R. Daniels, Eur. J. Pharm. Biopharm., 2009, 73, 351–356 CrossRef CAS
- G. Jiang, W. Qiu and P. P. DeLuca, Pharm. Res., 2003, 20, 452–459 CrossRef CAS
- E. Balasse, J. Odot, G. Gatouillat, M.-C. Andry and C. Madoulet, Int. J. Pharm., 2008, 353, 131–138 CrossRef CAS
- M. Noga, D. Edinger, E. Wagner, G. Winter and A. Besheer, J. Biomater. Sci., Polym. Ed., 2014, 25, 855–871 CrossRef CAS
- S. U. Frick, M. P. Domogalla, G. Baier, F. R. Wurm, V. Mailaender, K. Landfester and K. Steinbrink, ACS Nano, 2016, 10, 9216–9226 CrossRef CAS
- H. Freichels, M. Wagner, P. Okwieka, R. G. Meyer, V. Mailaender, K. Landfester and A. Musyanovych, J. Mater. Chem. B, 2013, 1, 4338–4348 RSC
- A. Besheer, J. Vogel, D. Glanz, J. Kressler, T. Groth and K. Maeder, Mol. Pharm., 2009, 6, 407–415 CrossRef CAS
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS
- H. Lu and J. Cheng, J. Am. Chem. Soc., 2007, 129, 14114–14115 CrossRef CAS
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS
- C. Deng, J. Wu, R. Cheng, F. Meng, H.-A. Klok and Z. Zhong, Prog. Polym. Sci., 2014, 39, 330–364 CrossRef CAS
- F. Schortgen, N. Deye, L. Brochard and C. S. Grp, Intensive Care Med., 2004, 30, 2222–2229 CrossRef
- C. M. Munsch, E. Macintyre, S. J. Machin, I. J. Mackie and T. Treasure, Br. J. Surg., 1988, 75, 675–678 CrossRef CAS
- C. M. Paleos, Z. Sideratou and D. Tsiourvas, Bioconjugate Chem., 2017, 28, 1611–1624 CrossRef CAS
- M. G. Sajilata, R. S. Singhal and P. R. Kulkarni, Compr. Rev. Food Sci. Food Saf., 2006, 5, 1–17 CrossRef CAS
- M. Yu, N. Ji, Y. Wang, L. Dai, L. Xiong and Q. Sun, Compr. Rev. Food Sci. Food Saf., 2020, 1–26 Search PubMed
- G. Kore, A. Kolate, A. Nej and A. Misra, J. Nanosci. Nanotechnol., 2014, 14, 288–307 CrossRef CAS
- K. Letchford and H. Burt, Eur. J. Pharm. Biopharm., 2007, 65, 259–269 CrossRef CAS
- Y. Sanada, I. Akiba, K. Sakurai, K. Shiraishi, M. Yokoyama, E. Mylonas, N. Ohta, N. Yagi, Y. Shinohara and Y. Amemiya, J. Am. Chem. Soc., 2013, 135, 2574–2582 CrossRef CAS
- G. Akner, Age Ageing, 2005, 34, 320–321 CrossRef
- D. L. Stirland, J. W. Nichols, S. Miura and Y. H. Bae, J. Controlled Release, 2013, 172, 1045–1064 CrossRef CAS
- D. W. Kim, S. Y. Kim, H. K. Kim, S. W. Kim, S. W. Shin, J. S. Kim, K. Park, M. Y. Lee and D. S. Heo, Ann. Oncol., 2007, 18, 2009–2014 CrossRef
- W. Li, X. Li, Y. Gao, Y. Zhou, S. Ma, Y. Zhao, J. Li, Y. Liu, X. Wang and D. Yin, Mol. Pharm., 2014, 11, 71–80 CrossRef CAS
- J. Treib, A. Haass, G. Pindur, U. T. Seyfert, W. Treib, M. T. Grauer, F. Jung, E. Wenzel and K. Schimrigk, Thromb. Haemostasis, 1995, 74, 1452–1456 CrossRef CAS
- L. A. Lane, X. Qian, A. M. Smith and S. Nie, in Annual Review of Physical Chemistry, ed. M. A. Johnson and T. J. Martinez, 2015, vol. 66, pp. 521–547 Search PubMed
- L. C. Borish and J. W. Steinke, J. Allergy Clin. Immunol., 2003, 111, S460–S475 CrossRef CAS
- D. Neri and P. M. Sondel, Curr. Opin. Immunol., 2016, 40, 96–102 CrossRef CAS
- N. Momin, N. K. Mehta, N. R. Bennett, L. Ma, J. R. Palmeri, M. M. Chinn, E. A. Lutz, B. Kang, D. J. Irvine, S. Spranger and K. D. Wittrup, Sci. Transl. Med., 2019, 11, eaaw2614 CrossRef
- J. Ishihara, A. Ishihara, K. Sasaki, S. S.-Y. Lee, J.-M. Williford, M. Yasui, H. Abe, L. Potin, P. Hosseinchi, K. Fukunaga, M. M. Raczy, L. T. Gray, A. Mansurov, K. Katsumata, M. Fukayama, S. J. Kron, M. A. Swartz and J. A. Hubbell, Sci. Transl. Med., 2019, 11, eaau3259 CrossRef CAS
- O. Kinstler, G. Molineux, M. Treuheit, D. Ladd and C. Gegg, Adv. Drug Delivery Rev., 2002, 54, 477–485 CrossRef CAS
- A. Basu, K. Yang, M. Wang, S. Liu, R. Chintala, T. Palm, H. Zhao, P. Peng, D. Wu, Z. Zhang, J. Hua, M.-C. Hsieh, J. Zhou, G. Petti, X. Li, A. Janjua, M. Mendez, J. Liu, C. Longley, Z. Zhang, M. Mehlig, V. Borowski, M. Viswanathan and D. Filpula, Bioconjugate Chem., 2006, 17, 618–630 CrossRef CAS
- P. Vitaglione, S. Sforza, G. Galaverna, C. Ghidini, N. Caporaso, P. P. Vescovi, V. Fogliano and R. Marchelli, Mol. Nutr. Food Res., 2005, 49, 495–504 CrossRef CAS
- M.-L. Yang and Y.-M. Song, RSC Adv., 2015, 5, 17824–17833 RSC
- S. B. Nia, M. Pooresmaeil and H. Namazi, Int. J. Biol. Macromol., 2020, 155, 1401–1409 CrossRef
- A. Taavoni-Gilan, J. Chin. Chem.
Soc., 2019, 66, 600–607 CrossRef CAS
- K. A. Ansari, P. R. Vavia, F. Trotta and R. Cavalli, AAPS PharmSciTech, 2011, 12, 279–286 CrossRef CAS
- S. Hoffmann, H. Caysa, J. Kuntsche, P. Kreideweiss, A. Leimert, T. Mueller and K. Maeder, Carbohydr. Polym., 2013, 95, 404–413 CrossRef CAS
- J. Li, Y. Yang, L. Lu, Q. Ma and J. Zhang, Int. J. Nanomed., 2018, 13, 2129–2141 CrossRef CAS
Footnote |
| † These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2021 |