
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient access to 3′-O-phosphoramidite derivatives of tRNA related N6-threonylcarbamoyladenosine (t6A) and 2-methylthio-N6-threonylcarbamoyladenosine (ms2t6A)†
Katarzyna Debiec and
Elzbieta Sochacka*
and
Elzbieta Sochacka*
Institute of Organic Chemistry, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland. E-mail: elzbieta.sochacka@p.lodz.pl
First published on 7th January 2021
Abstract
An efficient method of ureido linkage formation during epimerization-free one-pot synthesis of protected hypermodified N6-threonylcarbamoyladenosine (t6A) and its 2-SMe analog (ms2t6A) was developed. The method is based on a Tf2O-mediated direct conversion of the N-Boc-protecting group of N-Boc-threonine into the isocyanate derivative, followed by reaction with the N6 exo-amine function of the sugar protected nucleoside (yield 86–94%). Starting from 2′,3′,5′-tri-O-acetyl protected adenosine or 2-methylthioadenosine, the corresponding 3′-O-phosphoramidite monomers were obtained in 48% and 42% overall yield (5 step synthesis). In an analogous synthesis, using the 2′-O-(tert-butyldimethylsilyl)-3′,5′-O-(di-tert-butylsilylene) protection system at the adenosine ribose moiety, the t6A-phosphoramidite monomer was obtained in a less laborious manner and in a remarkably better yield of 74%.
Introduction
Transfer RNAs (tRNAs) are known for having a substantial content of modified nucleoside units.1,2 To date, in tRNAs from all domains of life, more than 130 modified units have been identified, which differ in chemical structure,3–6 distribution within the tRNA molecules,7 and their biological activity.8–13The majority of modified units are present in the anticodon loop and stem domain of tRNAs, particularly at position 34 (the wobble position) and at position 37, i.e. adjacent to the anticodon at its 3′-side.3,7,14,15 Considering the latter modifications, special interest has been paid to several N6-threonylcarbamoyladenosines (depicted in Fig. 1), which are widely involved in the decoding of the A-starting codons (ANN).15,16 Among them, the most abundant N6-threonylcarbamoyladenosine (t6A)17 and its analogs containing either the –SMe group at the purine C2 atom (ms2t6A),18 or the methyl substituent at the N6-atom (m6t6A)19 have been known for many years and their diverse functions during the protein biosynthesis were intensively studied.6,9,15,16 Recently, next members of the t6A family have been identified in the tRNA anticodon loops, i.e. cyclic N6-threonylcarbamoyladenosine (ct6A)20,21 cyclic 2-methylthio-N6-threonylcarbamoyladenosine (ms2ct6A),22 and a t6A derivative having the threonine methyl group converted into a hydroxymetyl one (hydroxy-N6-threonylcarbamoyl-adenosine, ht6A).23
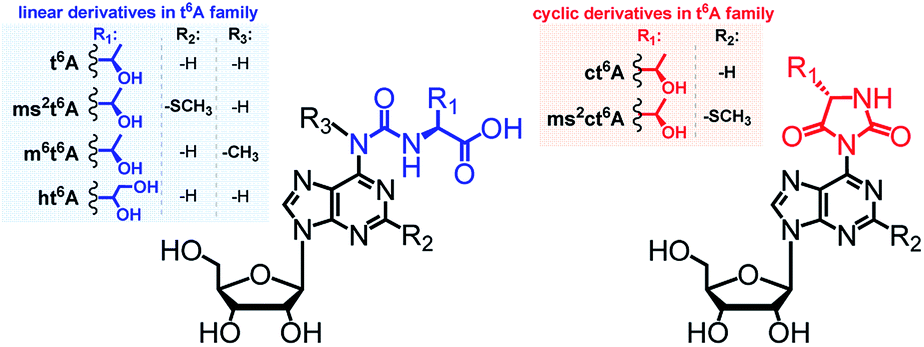 | ||
| Fig. 1 Abbreviations and structures of L-threonylcarbamoyl modified adenosines (the t6A37 family) located in tRNAs at the position 37. | ||
Recognition of the structural aspects and biological functions of the t6A nucleoside family is highly dependent on the synthetic availability of these nucleosides, as well as their 3′-O-phosphoramidite derivatives, which are essential for fast and efficient synthesis of model oligonucleotides with the sequence of the appropriate tRNA anticodon stems and loops (ASL of tRNAs). To date, several procedures have been developed to modify adenosine or 2-methylthioadenosine (ms2A) at the N6 position with a threonylcarbamoyl chain (a ureido system is formed) using either a carbamate or isocyanate approach (Scheme 1, paths A and B, respectively).
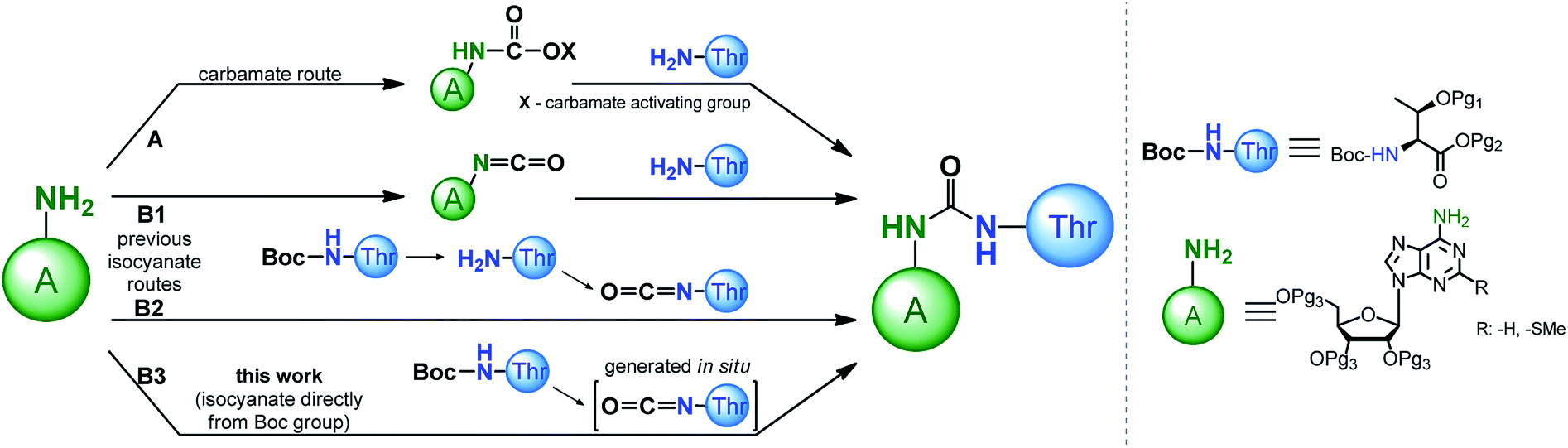 | ||
| Scheme 1 Approaches for the formation of the ureido linkage in t6A modified nucleoside. | ||
Adenosine N6-ethyl carbamate was used in the first syntheses of “free” t6A nucleoside,24–27 as well as in the preparation of its stereoisomers containing L, D, L-allo, and D-allo-Thr.28 Analogously, t6A derivatives suitable for synthesis of the corresponding 3′-O-phosphoramidite derivative (protected with tert-butyldimethylsilyl (TBDMS) on the OH and trimethylsilylethyl ester (TMSE) at the COOH of threonine residue) can be prepared.29 The carbamate method has been significantly improved by the use of more active phenyl carbamate derivatives of adenosine/2-methylthio-adenosine.30–40 This was possible with phenoxycarbonyl tetrazole31 or 1-N-methyl-3-phenoxycarbonyl-imidazolium chloride41 used as effective reagents introducing the carbamate functionality onto the weakly nucleophilic N6-amine function of A/ms2A nucleosides.
The isocyanate approach to the synthesis of t6A/ms2t6A (Scheme 1, path B1,2) was shown to have limited applicability in the preparation of “free” nucleosides.24 Because this method required a threonine derivative protected on the OH and COOH functions, it was considered inferior to the carbamate approach in which unprotected amino acid can be used.24–26 However, the isocyanate route was recently postulated by the Carell's group as a possible pathway for the formation of t6A under prebiotic conditions.42
In the synthesis of threonine protected t6A/ms2t6A derivatives for the subsequent preparation of the corresponding 3′-O-phosphoramidites, the isocyanate approach39,43 is much less explored than the carbamate procedures.29,34–40 Initially, the isocyanate derivative was generated from the N6-amine function of sugar protected adenosine (Scheme 1, path B1), but its condensation with the free amine function of L-threonine was ineffective and the ureido-nucleoside product was obtained in a low 19% yield.43 Noticeably better results were obtained in our recently published method (Scheme 1, path B2), based on the reaction of isocyanate derivative of the amino acid substrate (prepared by removing of Boc-protection and phosgene treatment of the free amine function of L-threonine appropriately blocked on the OH and COOH functions) with the sugar protected nucleoside (overall yield of this three steps procedure ∼55%).39 This result of isocyanate procedure turned our attention to the methods of synthesis of unsymmetrical ureas involving the formation of the isocyanate functionality directly from the carbamate type protecting groups of amino acids (e.g. N-Boc protecting group).44–53 Most likely, such variant of the isocyanate method (Scheme 1, path B3) applied in the synthesis of the 3′-O-phosphoramidite derivatives of t6A/ms2t6A would be greatly advantageous in comparison to our previous isocyanate route (Scheme 1, path B2) owing to a smaller number of synthesis steps in the preparation of threonine derivative (the removal of N-Boc protection is unnecessary) and escaping the use of toxic phosgene.
Here we report a new one-pot procedure for the introduction of an ureido linkage into t6A/ms2t6A using a Tf2O-mediated generation of the isocyanate derivative directly from N-Boc-protecting group of L-threonine, followed by its straight reaction with the N6 exo-amine function of the sugar protected nucleoside. We have also showed that this approach is compatible with the use of the recently introduced 2′-O-(tert-butyldimethylsilyl)-3′,5′-O-(di-tert-butylsilylene) ribonucleoside sugar protection system,54–56 that allows to prepare the 3′-O-phosphoramidite monomeric unit more effectively and in a less laborious manner.
Results and discussion
Search for the best conditions leading to the formation of the ureido compound 4a was performed using trimethylsilylethyl (TMSE) ester of N-Boc-O-tert-butyldimethylsilyl (TBDMS) protected L-threonine39 (1) and 2′,3′,5′-tri-O-acetyladenosine (3a) (Table 1, see ESI for spectroscopic data of 1, 2, 3a, Fig. S1–S7†). In all cases, the final condensation of isocyanate 2 with 3a was performed in the presence of Et3N (2-fold molar excess over the Tf2O activator used for isocyanate formation) in boiling toluene for 16 h. It was reported that addition of Et3N, which is an effective scavenger of trifluoromethanesulfonic acid (generated in the step of isocyanate formation), helps to maintain a concentration of the unprotonated amine component sufficient for effective nucleophilic attack on the isocyanate moiety.48,50
|
||||
|---|---|---|---|---|
| Entry | Boc-Thr 1b (equiv.) | Tf2O/basec (equiv.) | Solvent, time, temp. (°C) | Yield of 4ad (%) |
| a All reactions were performed in a 0.2 mmol scale in 6 mL of the corresponding solvent.b The number of equivalents was calculated in respect to the nucleoside reagent 3a.c The ratios of 1/Tf2O = 1.5 and Tf2O/base = 2 were applied.d Isolated yield after column chromatography.e The reaction was carried out also in 0 °C and after stirring for 3 h no consumption of 1 was observed according to TLC analysis. | ||||
| 1 | 1.0 | Tf2O (1.5)/2-Cl-Py (3.0) | CH2Cl2, rt, 15 min | 19% |
| 2 | 1.0 | Tf2O (1.5)/2-Cl-Py (3.0) | CH2Cl2, 0 °C, 5 min | 46% |
| 3 | 1.0 | Tf2O (2.0)/2-Cl-Py (4.0) | CH2Cl2, 0 °C, 5 min | 42% |
| 4 | 1.0 | Tf2O (1.5)/Py (3.0) | CH2Cl2, rte, 3 h | — |
| 5 | 1.0 | Tf2O (1.5)/DMAP (3.0) | CH2Cl2, rte, 3 h | — |
| 6 | 1.0 | Tf2O (1.5)/Et3N (3.0) | CH2Cl2, rte, 3 h | — |
| 7 | 1.0 | Tf2O (1.5)/2,6-lutidine (3.0) | CH2Cl2, rte, 30 min | 16% |
| 8 | 1.5 | Tf2O (2.25)/2-Cl-Py (4.5) | CH2Cl2, 0 °C, 5 min | 71% |
| 9 | 2.0 | Tf2O (3.0)/2-Cl-Py (6.0) | CH2Cl2, 0 °C, 5 min | 80% |
| 10 | 2.5 | Tf2O (3.75)/2-Cl-Py (7.5) | CH2Cl2, 0 °C, 5 min | 92% |
| 11 | 2.5 | Tf2O (3.75)/2-Cl-Py (7.5) | Toluene, rt, 15 min | 92% |
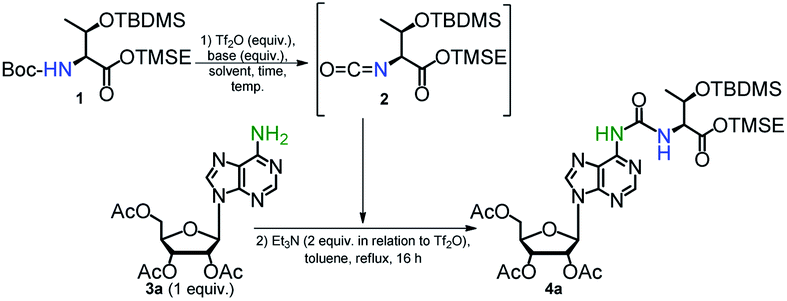
To optimize the triflic anhydride (Tf2O) mediated conversion of N-Boc-protected threonine 1 (a dichloromethane solution) into the isocyanate derivative 2 (the first step of the one-pot synthesis of 4a) we were changing the amount of Tf2O activator, basicity of amine, temperature and reaction time (entries 1–7). When the amount of 2 reached the plateau (TLC monitoring) the reaction mixture was concentrated, the residue was dissolved in toluene and Et3N and the nucleoside substrate 3a was added. The reaction 1 → 2 for 15 min at room temp. (entry 1), followed by reaction with 3a, afforded the final product 4a in a low 19% yield and several by-products were detected by TLC analysis. An experiment conducted at lower temperature (0 °C) for much shorter time (5 min) (entry 2) was more productive (46% yield) but the yield did not further increase when higher concentration of Tf2O (2 equiv.) was used (entry 3). Compound 1 did not react when more common bases such as pyridine, 4-dimethylaminopyridine or triethylamine were used (entries 4–6). In the case of 2,6-lutidine, some isocyanate 2 was generated after 30 min at rt, but the final product 4a was formed in only 16% yield (entry 7). Neither acetic anhydride nor trifluoroacetic anhydride were able to promote the formation of isocyanate 2 regardless of the temperature applied.
In so far reported procedures for the one-pot syntheses of ureas from carbamates, the use of an excess of amine substrate, usually up to 3 equivalents (or more for less nucleophilic amines) is recommended to obtain the higher efficiency of the process.48,50,51 However, in the case of t6A/ms2t6A synthesis, the amine nucleoside substrate, especially non-native 2-methylthioadenosine (ms2A) is a very costly reagent. Therefore, in the second step of optimizations we examined an excess of N-Boc protected L-threonine derivative 1 to nucleoside 3a in a range 1.5–2.5 (entries 8–10), yet the concentrations of Tf2O and 2-Cl-Py against 1 were kept as determined previously (entry 2). We were glad to see that 1.5 molar excess of 1 to 3a led to a significantly better yield of 4a (71%, entry 8), while very high conversion of 3a to 4a was observed when 2.5 equivalents of 1 was applied (92%, entry 10). Unfortunately, further increase in the excess of 1 (3 equiv. or more) did not lead to a higher isolated yield of product 4a. Finally, the use of toluene instead of dichloromethane for the formation of 2 allowed us to carry out the whole process in the same solvent (92% yield, entry 11) which facilitate the preparative procedure for the one-pot synthesis of t6A derivative 4a.
The optimized method described above was used in synthesis of the phosphoramidite derivatives of t6A and ms2t6A (6a, and 6b, respectively; Scheme 2). Starting from 2.5 mmol of appropriately protected Boc-L-threonine 139 and 1 mmol of adenosine derivative 3a57 or 3b,39 the modified nucleosides 4a and 4b were obtained in 92% and 86% yield, respectively. Next, the acetyl groups in 4a/4b were removed under conditions safe for the installed N6-threonylcarbamoyl chain (Et3N/MeOH, rt, 24 h) and the resultant 5a/5b were appropriately protected and phosphitylated according to the previously reported procedures39 to give t6A/ms2t6A-phosphoramidites (6a/6b) in 48% and 42% overall yield, respectively (see ESI† for details).
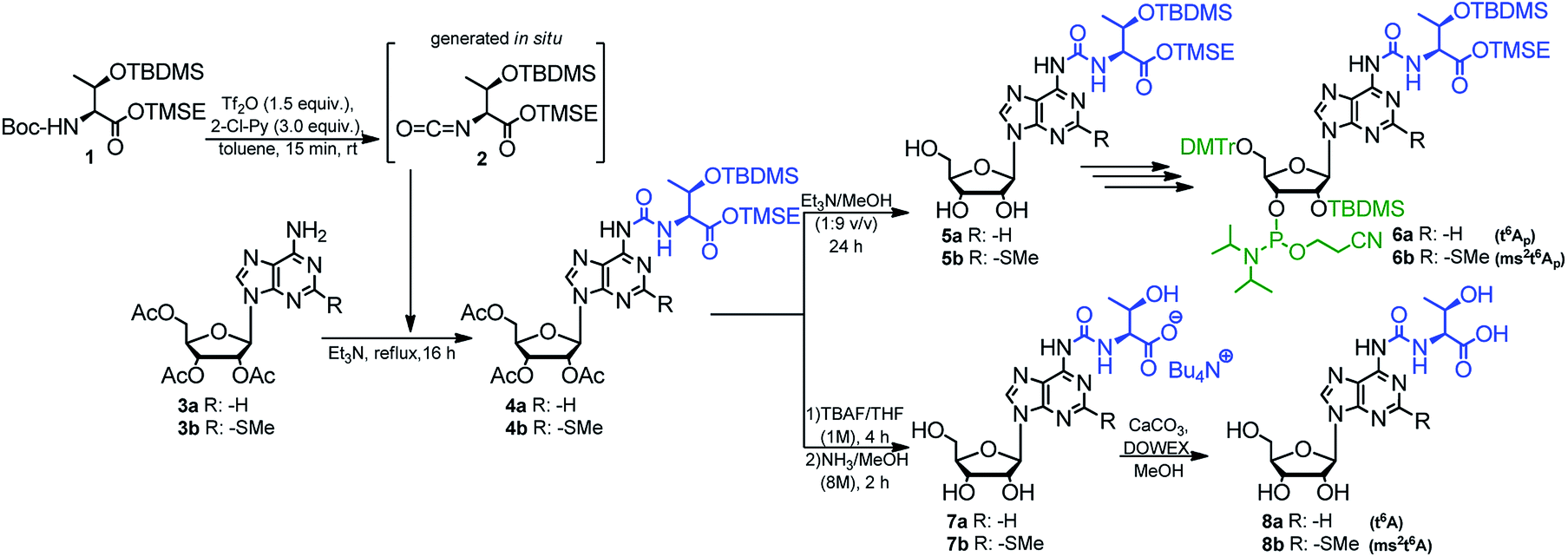 | ||
| Scheme 2 Preparation of t6A and ms2t6A 3′-O-phosphoramidities and samples of modified nucleosides t6A and ms2t6A. | ||
Also, the nucleosides 4a/4b were deprotected to yield 8a/8b (Scheme 2), to be used as standards in analysis of enzymic hydrolysates of t6A- or ms2t6A-containing oligomers. The silyl protecting groups (TBDMS, TMSE) were removed with excess 1 M tetrabutylammonium fluoride (TBAF) in THF (4 h, rt), and the acetyl groups were cleaved off with NH3/MeOH (2 h, rt) (see experimental details in ESI†). The reactions were virtually quantitative and the HPLC profiles recorded for the reaction mixtures (Fig. 2) contained single, slightly tailing peaks (profiles in panels (A), part I for t6A and part II for ms2t6A). The tailing was not observed, when the highly lipophilic tetrabutylammonium cations were replaced with H+ ions using DOWEX, H+/CaCO3 treatment.58 The resultant acidic forms 8a/8b had the same HPLC mobility as genuine L-t6A/L-ms2t6A standards21,22,38,39 (compare profiles in panels (B) and (C)). The profiles recorded for 8a co-injected with D-allo-t6A21,38 and for 8b co-injected with D-allo-ms2t6A22,39 (panels (E)) indicate that the new procedure for ureido linkage formation is safe in terms of the stereochemistry at the Cα of the amino acid component. The profiles for the D-allo nucleoside standards are shown in panels (D).
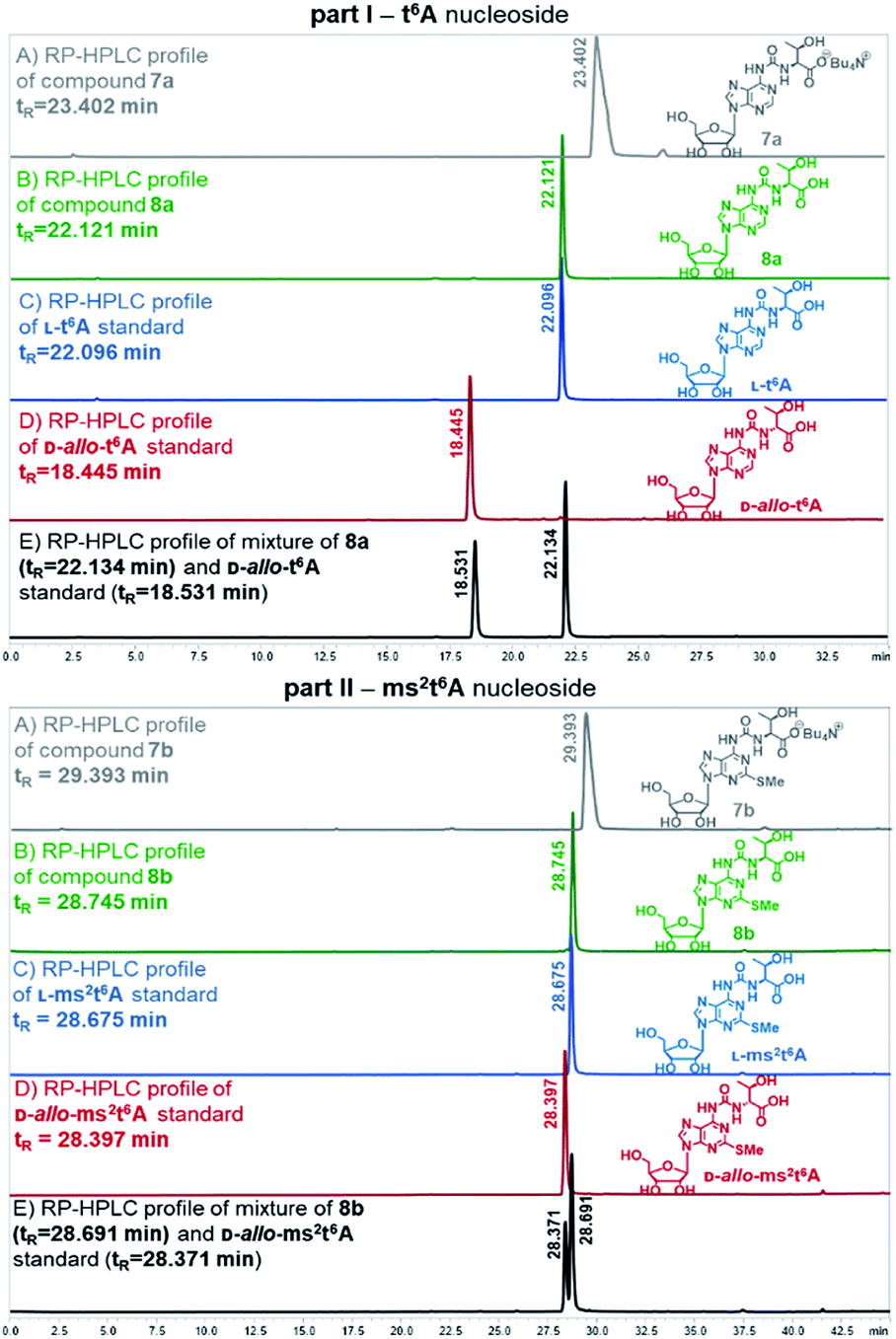 | ||
| Fig. 2 HPLC profiles recorded for L-t6A and L-ms2t6A nucleosides and the corresponding D-allo nucleoside standards (D-allo-t6A, D-allo-ms2t6A). | ||
The phosphoramidite derivative of t6A (6a) was also synthesized using 2′-O-(tert-butyldimethylsilyl)-3′,5′-O-(di-tert-butylsilylene) protected adenosine 954,59 as the nucleoside substrate (Scheme 3). The one-pot conversion 9 → 10 proceeded in 94% yield. Subsequent selective removal of the cyclic silyl protecting group (HF in pyridine, 0 °C) furnished compound 11 (96% yield), further converted into the 5′-O-DMTr derivative 12 (90% yield). The reaction of 12 with 2-cyanoethyl N,N-diisopropylchloro-phosphoramidite gave finally the target t6A-phosphoramidite in 91% yield (combined yield 74% for 9 → 6a).
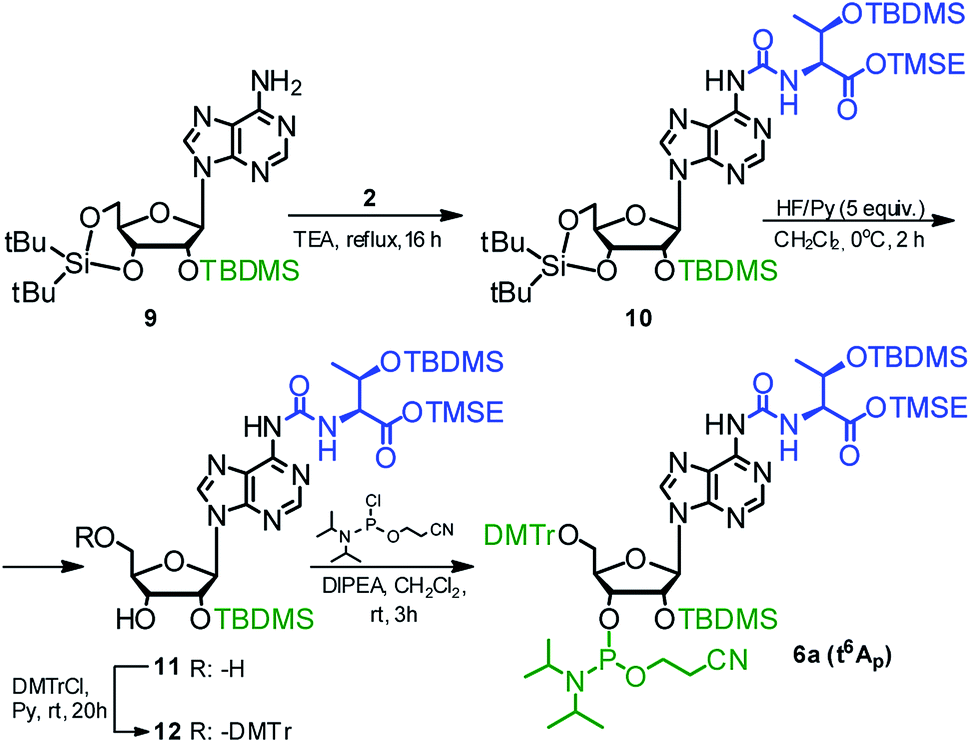 | ||
| Scheme 3 Synthesis of t6A phosphoramidite (6a) using 2′-O-(tert-butyldimethylsilyl)-3′,5′-O-(di-tert-butylsilylene)adenosine as the nucleoside substrate. | ||
Conclusions
The presented here modification of the isocyanate method of formation of the ureido linkage between adenosine and threonine greatly facilitates synthesis of fully protected L-threonylcarbamoyl modified adenosines 4a,b rendering subsequent preparation of the t6A/ms2t6A phosphoramidite monomers 6a,b much more efficient. The developed one-pot procedure for 4a,b synthesis, consisting in the epimerization-free formation of L-threonine isocyanate directly from the N-Boc-Thr upon activation with Tf2O in the presence of 2-Cl-Py, followed by its straight reaction with the N6 exo-amine function of the sugar protected nucleoside, eliminates the use of toxic phosgene and provides a shorter protocol for the preparation of the protected t6A/ms2t6A derivatives compared to the previously reported isocyanate and carbamate routes. In addition, the protected nucleosides 4a,b were efficiently deprotected yielding free nucleosides 8a,b to be used as the standards, e.g. in HPLC analysis of enzymatically digested oligomers bearing t6A/ms2t6A units. Moreover, the in situ formed threonine isocyanate reacted efficiently with 2′-O-(tert-butyldimethyl-silyl)-3′,5′-O-(di-tert-butylsilylene)adenosine and the resultant conjugate was conveniently transformed into the t6A-phosphoramidite in a very good overall yield 74%. Developed procedures for the synthesis of t6A/ms2t6A 3′-O-phosphoramidities will significantly facilitate the availability of monomeric units for the chemical synthesis of various model tRNA fragments suitable for the structure–activity-relationship and biological studies of the t6A family nucleosides.Experimental
General remarks
Commercial reagents and analytical grade solvents were used without additional purification unless otherwise stated. Analytical thin layer chromatography (TLC) was done on silica gel coated plates (60 F254, Supelco) with UV light (254 nm) or the ninhydrin test (for amino acids) detection. The products were purified by chromatography on a silica gel 60 (mesh 230–400, Fluka) column eluted with the indicated solvent mixtures. NMR spectra were recorded using a 700 MHz (for 1H) instrument, 176 MHz for 13C and 283 MHz for 31P. Chemical shifts (δ) are reported in ppm relative to residual solvent signals CDCl3: 7.26 ppm for 1H NMR, 77.16 ppm for 13C NMR; DMSO-d6: 2.50 ppm for 1H NMR, 39.52 ppm for 13C NMR. The signal multiplicities are described as s (singlet), d (doublet), dd (doublet of doublets), ddd (doublets of doublets of doublets), dq (dublet of quartets), t (triplet), td (triplet of doublets), q (quartet), qd (quartet of doublets), m (multiplet), and br s (broad singlet). High-resolution mass spectra were recorded on Synapt G2Si mass spectrometer (Waters) equipped with an ESI source and quadrupole-time-of-flight mass analyzer. HPLC analysis of nucleosides was performed on a Shimadzu Prominence HPLC system equipped with an SPD-M20A spectral photodiode array detector using a Kinetex® column (RP, C18, 5 μm, 4.6 × 250 mm, 100 Å, Phenomenex). Analyses were run at 30 °C and the elution profiles were UV monitored at λ = 254 nm.General procedure for the one-pot synthesis of 4a, 4b and 10 from Boc-L-threonine 1
To a stirred solution of Boc-L-threonine 1 (1.08 g, 2.5 mmol) in dry toluene (30 mL) 2-chloropyridine (2-Cl-Py, 0.7 mL, 7.5 mmol) was added, followed by trifluoromethanesulfonic anhydride (Tf2O, 0.64 mL, 3.75 mmol) and after stirring for 15 min at room temperature triethylamine (Et3N, 1.04 mL, 7.5 mmol) and sugar-protected adenosine (3a, 3b or 9, 1.0 mmol) were added. The reaction mixture was stirred under reflux for 16 h. Then the solvent was evaporated under reduced pressure and 4a,b or 10 were isolated by silica gel column chromatography.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v).
:5 v/v).1H NMR (700 MHz, DMSO-d6) δ: 9.98 (s, 1H, NH-6), 9.86 (d, 1H, 3J = 9.2 Hz, NH Thr), 8.65 (s, 1H, H-8), 8.42 (s, 1H, H-2), 6.30 (d, 1H, 3J = 5.4 Hz, H-1′), 6.07–5.99 (m, 1H, H-2′), 5.64 (dd, 1H, 3J = 5.9 Hz, 3J = 4.5 Hz, H-3′), 4.49 (qd, 1H, 3J = 6.2 Hz, 3J = 1.8 Hz, CH-β Thr), 4.44–4.37 (m, 3H, CH-α Thr, H-4′, H-5′), 4.30–4.24 (m, 1H, H-5′′), 4.18 (ddd, 1H, 2J = 11.0 Hz, 3J = 10.0 Hz, 3J = 6.8 Hz, O–CH TMSE), 4.12 (ddd, 1H, 3J = 10.9 Hz, 3J = 10.0 Hz, 3J = 6.5 Hz, O–CH TMSE), 2.13 (s, 3H, CH3–CO Ac), 2.04 (s, 3H, CH3–CO Ac), 2.01 (s, 3H, CH3–CO Ac), 1.19 (d, 3J = 6.3 Hz, 3H, CH3 Thr), 1.01–0.92 (m, 2H, Si–CH2 TMSE), 0.89 (s, 9H, Si–C(CH3)3 TBDMS), 0.08 (s, 3H, Si–CH3 TBDMS), 0.01 (s, 3H, Si–CH3 TBDMS), −0.00 (s, 9H, Si(CH3)3 TMSE); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C32H53N6O11Si2 753.3311; found 753.3307 (see Fig. S8 and S26 in the ESI†).
:5 v/v).1H NMR (700 MHz, DMSO-d6) δ: 9.98 (s, 1H, NH-6), 9.24 (d, 1H, 3J = 8.6 Hz, NH Thr), 8.45 (s, 1H, H-8), 6.24 (d, 1H, 3J = 4.3 Hz, H-1′), 6.07 (dd, 1H, 3J = 6.0 Hz, 3J = 4.2 Hz, H-2′), 5.69 (t, 1H, 3J = 6.1 Hz, H-3′), 4.46–4.44 (m, 2H, CH-α Thr, CH-β Thr), 4.42 (dd, 1H, 2J = 12.0 Hz, 3J = 3.7 Hz, H-5′), 4.40–4.35 (m, 1H, H-4′), 4.24–4.16 (m, 2H, 2H, H-5′′, O–CH TMSE), 4.11 (td, 1H, 2J = 10.7 Hz, 3J = 6.2 Hz, O–CH TMSE), 2.58 (s, 3H, S–CH3), 2.11 (s, 3H, CH3–CO Ac), 2.07 (s, 3H, CH3–CO Ac), 1.95 (s, 3H, CH3–CO Ac), 1.19 (d, 3H, 3J = 6.3 Hz, CH3 Thr), 1.04–0.93 (m, 2H, Si–CH2 TMSE), 0.85 (s, 9H, Si–C(CH3)3 TBDMS), 0.08 (s, 3H, Si–CH3 TBDMS), 0.03 (s, 3H, Si–CH3 TBDMS), 0.01 (s, 9H, Si(CH3)3 TMSE); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C33H55N6O11SSi2 799.3188; found 799.3177 (see Fig. S14 and S27 in the ESI†).
Preparation of nucleoside standards 8a and 8b
Fully-protected adenosine 4a or 4b (0.02 g, 0.03 mmol) was dissolved in 1 M solution of TBAF in THF (0.4 mL, 0.40 mmol) and the reaction mixture was stirred for 4 h at room temperature. After this time NH3 in dry MeOH (8 M solution, 0.2 mL) was added for deprotection of all acetyl groups from ribose moiety. The reaction was carried out for 2 h and then NH3 was removed under reduced pressure to obtain tetrabutylammonium salts 7a/7b. To exchange Bu4N+ counterion to H+, CaCO3 (0.28 g), dry DOWEX 50WX8 H+ form (0.84 g) and distilled methanol (0.6 mL) were added and the reaction mixture was stirred for 1 h at room temperature.58 After this time the resulting mixture was filtered through Celite plug and washed with MeOH. The filtrate was analysed by HPLC and the presence of fully-deprotected only one isomer of 8a/8b with natural L-threonine residue was confirmed (for 8a Rt = 22.121 min, for 8b Rt = 28.745 min, see Fig. 2 panel (B)).RP-HPLC conditions for analysis of t6A derivatives: C18 column with linear gradient of buffer A (0.1% AcOH in H2O) and buffer B (ACN) with a flow of 1 mL min−1 as follows: 0–15 min from 2% to 8% B, 15–30 min from 8% to 25% B, 30–35 min 2% B. RP-HPLC conditions for analysis of ms2t6A derivatives: C18 column with linear gradient of buffer A (0.1% AcOH in H2O) and buffer B (ACN) with a flow of 1 mL min−1 as follows: 0–30 min from 2% B to 15% B, 30–40 min from 15% B to 30% B, 40–45 min 2% B.
Synthesis of t6A 3′-O-phoshoramidite 6a from 9
:5 v/v).1H NMR (700 MHz, CDCl3) δ: 10.08 (d, 1H, 3J = 9.1 Hz, NH Thr), 8.49 (s, 1H, H-2), 8.43 (br s, 1H, NH-6), 8.14 (s, 1H, H-8), 5.97 (br s, 1H, H-1′), 4.61–4.56 (m, 3H, H-2′, CH-β Thr, CH-α Thr), 4.52–4.47 (m, 2H, H-3′, H-5′), 4.29–4.21 (m, 2H, H4′, O–CH TMSE), 4.21–4.15 (m, 1H, O–CH2 TMSE), 4.06 (dd, 2J = 10.5 Hz, 3J = 9.3 Hz, 1H, H5′′), 1.26 (d, 3H, 3J = 6.3 Hz, CH3 Thr), 1.09 (s, 9H, Si–C(CH3)3 tBu2Si), 1.05 (s, 9H, Si–C(CH3)3 tBu2Si), 1.02–0.99 (m, 2H, Si–CH2 TMSE), 0.95 (s, 9H, Si–C(CH3)3 TBDMS), 0.94 (s, 9H, Si–C(CH3)3 TBDMS), 0.17 (s, 3H, Si–CH3 TBDMS), 0.15 (s, 3H, Si–CH3 TBDMS), 0.10 (s, 3H, Si–CH3 TBDMS), 0.05 (s, 3H, Si–CH3 TBDMS), 0.02 (s, 9H, Si(CH3)3 TMSE); 13C NMR (176 MHz, CDCl3) δ: 171.16 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O Thr), 154.44 (NH–
O Thr), 154.44 (NH–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O–NH), 151.48 (C-2), 150.61 (C-6), 149.82 (C-4), 141.31 (C-8), 121.25 (C-5), 92.45 (C-1′), 76.01 (C-3′), 75.74 (C-2′), 74.93 (C-4′), 68.97 (Cα Thr), 67.91 (C-5′), 63.81 (O–CH2 TMSE), 59.75 (Cβ Thr), 27.64 (Si–C(H3)3 tBu2Si), 27.19 (Si–C(H3)3 tBu2Si), 26.05 (Si–C(H3)3 TBDMS), 25.75 (Si–C(H3)3 TBDMS), 22.88 (Si–(CH3)3 tBu2Si), 21.32 (CH3 Thr), 20.51 (Si–(CH3)3 tBu2Si), 18.47 (Si–(CH3)3 TBDMS), 18.03 (Si–(CH3)3 TBDMS), 17.51 (Si–CH2 TMSE), −1.41(Si(CH3)3 TMSE), −4.07 (Si–CH3 TBDMS), −4.15 (Si–CH3 TBDMS), −4.84 (Si–CH3 TBDMS), −5.15 (Si–CH3 TBDMS); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C40H77N6O8Si4 881.4880; found 881.4868 (see Fig. S20, S21 and S28 in the ESI†).
O–NH), 151.48 (C-2), 150.61 (C-6), 149.82 (C-4), 141.31 (C-8), 121.25 (C-5), 92.45 (C-1′), 76.01 (C-3′), 75.74 (C-2′), 74.93 (C-4′), 68.97 (Cα Thr), 67.91 (C-5′), 63.81 (O–CH2 TMSE), 59.75 (Cβ Thr), 27.64 (Si–C(H3)3 tBu2Si), 27.19 (Si–C(H3)3 tBu2Si), 26.05 (Si–C(H3)3 TBDMS), 25.75 (Si–C(H3)3 TBDMS), 22.88 (Si–(CH3)3 tBu2Si), 21.32 (CH3 Thr), 20.51 (Si–(CH3)3 tBu2Si), 18.47 (Si–(CH3)3 TBDMS), 18.03 (Si–(CH3)3 TBDMS), 17.51 (Si–CH2 TMSE), −1.41(Si(CH3)3 TMSE), −4.07 (Si–CH3 TBDMS), −4.15 (Si–CH3 TBDMS), −4.84 (Si–CH3 TBDMS), −5.15 (Si–CH3 TBDMS); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C40H77N6O8Si4 881.4880; found 881.4868 (see Fig. S20, S21 and S28 in the ESI†).
:5 v/v).1H NMR (700 MHz, DMSO-d6) δ: 9.92 (s, 1H, NH-6), 9.88 (d, 1H, 3J = 9.0 Hz, NH Thr), 8.71 (s, 1H, H-8), 8.39 (s, 1H, H-2), 6.03 (d, 1H, 3J = 5.6 Hz, H-1′), 5.18 (dd, 1H, 3J = 6.3 Hz, 3J = 5.1 Hz, 5′OH), 5.13 (d, 1H, 3J = 5.3 Hz, 3′OH), 4.71 (dd, 1H, 3J = 5.7 Hz, 3J = 4.8 Hz, H2′), 4.48 (qd, 1H, 3J = 6.2 Hz, 3J = 1.9 Hz, CH-β Thr), 4.41 (dd, 1H, 3J = 9.0 Hz, 3J = 2.0 Hz, CH-α Thr), 4.20–4.15 (m, 2H, H3′, O–CH TMSE), 4.12 (ddd, 1H, 2J = 11.0 Hz, 3J = 9.9 Hz, 3J = 6.6 Hz, O–CH TMSE), 4.02 (q, 1H, 3J = 3.7 Hz, H4′), 3.74 (ddd, 1H, 2J = 12.1 Hz, 3J = 5.1 Hz, 3J = 4.0 Hz, H5′), 3.74 (ddd, 1H, 2J = 12.1 Hz, 3J = 6.3 Hz, 3J = 3.6 Hz, H5′′), 1.19 (d, 3J = 6.3 Hz, 3H, CH3 Thr), 0.99–0.91 (m, 2H, Si–CH2 TMSE), 0.88 (s, 9H, Si–C(CH3)3 TBDMS), 0.72 (s, 9H, Si–C(CH3)3 TBDMS), 0.07 (s, 3H, Si–CH3 TBDMS), 0.00 (s, 3H, Si–CH3 TBDMS), −0.02 (s, 9H, Si–(CH3)3 TMSE), −0.07 (s, 3H, Si–CH3 TBDMS), −0.18 (s, 3H, Si–CH3 TBDMS); 13C NMR (176 MHz, DMSO-d6) δ: 170.62 (CO Thr), 153.68 (NH–O–NH), 150.34 (C-6), 150.13 (C-4), 150.10 (C-2), 142.27 (C-8), 120.55 (C-5), 87.90 (C-1′), 85.98 (C-4′), 75.66 (C-2′), 70.12 (C-3′), 68.36 (Cβ Thr), 62.85 (O–CH2 TMSE), 61.06 (C-5′), 58.90 (Cα Thr), 25.47 (Si–C(H3)3 TBDMS), 25.34 (Si–C(H3)3 TBDMS), 20.86 (CH3 Thr), 17.73 (Si–(CH3)3 TBDMS), 17.47 (Si–(CH3)3 TBDMS), 16.69 (Si–CH2 TMSE), −1.62 (Si(H3)3 TMSE), −4.35 (Si–CH3 TBDMS), −4.94 (Si–CH3 TBDMS), −5.42 (Si–CH3 TBDMS), −5.59 (Si–CH3 TBDMS); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C32H61N6O8Si3 741.3859; found 741.3860 (see Fig. S22, S23 and S29 in the ESI†).
:2 v/v).1H NMR (700 MHz, DMSO-d6) δ: 9.92–9.91 (m, 2H, NH-6, NH Thr), 8.59 (s, 1H, H-8), 8.28 (s, 1H, H-2), 7.42–7.38 (m, 2H, HAr DMTr), 7.29–7.22 (m, 6H, HAr DMTr), 7.22–7.16 (m, 1H, HAr DMTr), 6.84–6.79 (m, 4H, HAr DMTr), 6.02 (d, 1H, 3J = 5.3 Hz, H-1′), 5.17 (d, 1H, 3J = 5.7 Hz, 3′OH), 5.01 (t, 1H, 3J = 5.2 Hz, H-2′), 4.48 (qd, 1H, 3J = 6.3 Hz, 3J = 2.0 Hz, CH-β Thr), 4.41 (dd, 1H, 3J = 9.0 Hz, 3J = 1.9 Hz, CH-α Thr), 4.29–4.23 (m, 1H, H-3′), 4.18–4.15 (m, 1H, O–CH TMSE), 4.15–4.08 (m, 2H, H-4′, O–CH TMSE), 3.71 (s, 6H, 2× O–CH3 DMTr), 3.32 (dd, 1H, 2J = 10.6 Hz, 3J = 3.9 Hz, H-5′), 3.25 (dd, 1H, 2J = 10.5 Hz, 3J = 5.1 Hz, H-5′′), 1.19 (d, 3H, 3J = 6.2 Hz, CH3 Thr), 1.00–0.89 (m, 2H, Si–CH2 TMSE), 0.85 (s, 9H, Si–C(CH3)3 TBDMS), 0.73 (s, 9H, Si–C(CH3)3 TBDMS), 0.06 (s, 3H, Si–CH3 TBDMS), 0.00 (s, 3H, Si–CH3 TBDMS), −0.02 (s, 9H, Si(CH3)3 TMSE), −0.05 (s, 3H, Si–CH3 TBDMS), −0.16 (s, 3H, Si–CH3 TBDMS); 13C NMR (176 MHz, DMSO-d6) δ: 171.17 (CO), 158.51 (CAr DMTr), 154.23 (NH–CO–NH), 150.86 (C-6), 150.59 (C-4), 150.47 (C-2), 145.35 (CAr DMTr), 143.44 (C-8), 135.95 (CAr DMTr), 130.18 (CAr DMTr), 128.17 (CAr DMTr), 128.15 (CAr DMTr), 127.05 (CAr DMTr), 121.26 (C-5), 113.52 (CAr DMTr), 88.89 (C-1′), 86.02 (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) DMTr), 84.38 (C-4′), 74.84 (C-2′), 70.69 (C-3′), 68.86 (C-β), 63.84 (C-5′), 63.36 (O–CH2 TMSE), 59.40 (C-α), 55.44 (O–CH3 DMTr), 25.99 (C–Si–(CH3)3 TBDMS), 25.79 (C–Si–(CH3)3 TBDMS), 21.35 (CH3), 18.25 (C–Si–(CH3)3 TBDMS), 17.93 (C–Si–(CH3)3 TBDMS), 17.16 (Si–CH2 TMSE), −1.11 (Si–(CH3)3 TMSE), −3.90 (Si–CH3 TBDMS), −4.36 (Si–CH3 TBDMS), −4.86 (Si–CH3 TBDMS), −5.10 (Si–CH3 TBDMS); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C53H79N6O10Si3 1043.5165; found 1043.5170 (see Fig. S24, S25 and S30 in the ESI†).
DMTr), 84.38 (C-4′), 74.84 (C-2′), 70.69 (C-3′), 68.86 (C-β), 63.84 (C-5′), 63.36 (O–CH2 TMSE), 59.40 (C-α), 55.44 (O–CH3 DMTr), 25.99 (C–Si–(CH3)3 TBDMS), 25.79 (C–Si–(CH3)3 TBDMS), 21.35 (CH3), 18.25 (C–Si–(CH3)3 TBDMS), 17.93 (C–Si–(CH3)3 TBDMS), 17.16 (Si–CH2 TMSE), −1.11 (Si–(CH3)3 TMSE), −3.90 (Si–CH3 TBDMS), −4.36 (Si–CH3 TBDMS), −4.86 (Si–CH3 TBDMS), −5.10 (Si–CH3 TBDMS); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C53H79N6O10Si3 1043.5165; found 1043.5170 (see Fig. S24, S25 and S30 in the ESI†).
:1 v/v) to obtain pure product 6a in 92% yield (0.64 g, 0.52 mmol). TLC: Rf = 0.52 (CHCl3/acetone, 95:5 v/v).31P NMR: (283 MHz, C6H6) δ: 149.89, 148.04 (see Fig. S12 in the ESI†).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Center, Poland, UMO-2017/25/B/ST5/00971 to E. S. Thanks are directed to Dr Piotr Guga for critical reading of the manuscript.Notes and references
- P. Boccaletto, M. A. Machnicka, E. Purta, P. Piatkowski, B. Baginski, K. Wirecki, V. de Crecy-Lagard, R. Ross, P. A. Limbach, A. Kotter, M. Helm and J. M. Bujnicki, Nucleic Acids Res., 2018, 46, D303 CrossRef CAS.
- W. A. Cantara, P. F. Crain, J. Rozenski, J. A. McCloskey, K. A. Harris, X. Zhang, F. A. Vendeix, D. Fabris and P. F. Agris, Nucleic Acids Res., 2011, 39, D195 CrossRef CAS.
- P. J. McCown, A. Ruszkowska, C. N. Kunkler, K. Berger, J. P. Hulewicz, M. C. Wang, N. A. Springer and J. A. Brown, Wiley Interdiscip. Rev.: RNA, 2020, 11, e1595 CAS.
- V. Väre, E. Eruysal, A. Narendran, K. Sarachan and P. Agris, Biomolecules, 2017, 7, 29 CrossRef.
- M. Helm and J. D. Alfonzo, Chem. Biol., 2014, 21, 174 CrossRef CAS.
- G. R. Björk and T. G. Hagervall, EcoSal Plus, 2014, 6, ESP-0007-2013 CrossRef.
- M. A. Machnicka, A. Olchowik, H. Grosjean and J. M. Bujnicki, RNA Biol., 2014, 11, 1619 CrossRef.
- L. Pollo-Oliveira and V. de Crécy-Lagard, Biochemistry, 2019, 58, 355 CrossRef CAS.
- B. El Yacoubi, M. Bailly and V. de Crécy-Lagard, Annu. Rev. Genet., 2012, 46, 69 CrossRef CAS.
- F. Tuorto and F. Lyko, Open Biol., 2016, 6, 160287 CrossRef.
- M. Duechler, G. Leszczyńska, E. Sochacka and B. Nawrot, Cell. Mol. Life Sci., 2016, 73, 3075 CrossRef CAS.
- L. Endres, P. C. Dedon and T. J. Begley, RNA Biol., 2015, 12, 603 CrossRef.
- A. G. T. Torres, E. Batlle and L. Ribas de Pouplana, Trends Mol. Med., 2014, 20, 306 CrossRef CAS.
- M. A. Machnicka, A. Olchowik, H. Grosjean and J. M. Bujnicki, RNA Biol., 2014, 11, 1619 CrossRef.
- A. Pichard-Kostuch, M.-C. Daugeron, P. Forterre and T. Basta, in RNA metabolism and gene expression in Archaea, ed. B. Clouet d'Orval, Springer International Publishing AG, 2017, ch. 8, pp. 177–200 Search PubMed.
- P. C. Thiaville, B. El Yacoubi, C. Köhrer, J. J. Thiaville, C. Deutsch, D. Iwata-Reuyl, J. M. Bacusmo, J. Armengaud, Y. Bessho, C. Wetzel, X. Cao, P. A. Limbach, U. L. RajBhandary and V. de Crecy-Lagard, Mol. Microbiol., 2015, 98, 1199 CrossRef CAS.
- M. P. Schweizer, G. B. Chheda, L. Baczynskyj and R. H. Hall, Biochemistry, 1969, 8, 3283 CrossRef CAS.
- Z. Yamaizumi, S. Nishimura, K. Limburg, M. Raba, H. J. Gross, P. F. Crain and J. M. McCloskey, J. Am. Chem. Soc., 1979, 101, 2224 CrossRef CAS.
- F. Kimura-Harada, D. L. von Minden, J. A. McCloskey and S. Nishimura, Biochemistry, 1972, 11, 3910 CrossRef CAS.
- K. Miyauchi, S. Kimura and T. Suzuki, Nat. Chem. Biol., 2013, 9, 105 CrossRef CAS.
- M. Matuszewski, J. Wojciechowski, K. Miyauchi, Z. Gdaniec, W. M. Wolf, T. Suzuki and E. Sochacka, Nucleic Acids Res., 2017, 45, 2137 CrossRef CAS.
- B. Kang, K. Miyauchi, M. Matuszewski, G. S. D'Almeida, M. A. T. Rubio, J. D. Alfonzo, K. Inoue, Y. Sakaguchi, T. Suzuki, E. Sochacka and T. Suzuki, Nucleic Acids Res., 2017, 45, 2124 CrossRef CAS.
- A. Nagao, M. Ohara, K. Miyauchi, S. I. Yokobori, A. Yamagishi, K. Watanabe and T. Suzuki, Nat. Struct. Mol. Biol., 2017, 24, 778 CrossRef CAS.
- G. B. Chheda and C. I. Hong, J. Med. Chem., 1971, 14, 748 CrossRef CAS.
- C. I. Hong and G. B. Chheda, in Nucleic Acid Chemistry, ed. L. B. Towsend and R. S. Tipson, John Wiley & Sons, New York, 1972, pp. 661–664 Search PubMed.
- C. I. Hong, G. B. Chheda, S. P. Dutta, A. O. Grady-Curtis and G. L. Tritsch, J. Med. Chem., 1973, 16, 139 CrossRef CAS.
- R. W. Adamiak and M. Wiewiórowski, Bull. Acad. Pol. Sci., Ser. Sci. Chim., 1974, 23, 241 Search PubMed.
- D. Martin and E. Schlimme, Z. Naturforsch., C: J. Biosci., 1994, 49, 834 CAS.
- M. Sundaram, P. F. Crain and D. R. Davis, J. Org. Chem., 2000, 65, 5609 CrossRef CAS.
- P. A. Lyon and C. B. Reese, J. Chem. Soc., Perkin Trans. 1, 1978, 131 RSC.
- R. W. Adamiak and J. Stawinski, Tetrahedron Lett., 1977, 18, 1935 CrossRef.
- R. W. Adamiak, E. Biala, K. Grzeskowiak, R. Kierzek, A. Kraszewski, W. T. Markiewicz, J. Okupniak, J. Stawinski and M. Wiewiorowski, Nucleic Acids Res., 1978, 5, 1889 CrossRef CAS.
- E. Sochacka, Nucleosides Nucleotides, 1998, 17, 327 CrossRef CAS.
- G. Leszczynska, J. Pieta, B. Sproat and A. Małkiewicz, Tetrahedron Lett., 2011, 52, 4443 CrossRef CAS.
- A. C. Bajji and D. R. Davis, J. Org. Chem., 2002, 67, 5352 CrossRef CAS.
- C. Baiji, M. Sundaram, D. G. Myszka and D. R. Davis, J. Am. Chem. Soc., 2002, 124, 14302 CrossRef.
- D. R. Davis and A. C. Bajji, Methods Mol. Biol., 2005, 288, 187 CAS.
- M. Matuszewski, K. Debiec and E. Sochacka, Chem. Commun., 2017, 53, 7945 RSC.
- K. Debiec, M. Matuszewski, K. Podskoczyj, G. Leszczynska and E. Sochacka, Chem.–Eur. J., 2019, 25, 13309 CrossRef CAS.
- M. Nainyte, F. Muller, G. Ganazzoli, Ch.-Y. Chan, A. Crisp, D. Globisch and T. Carell, Chem.–Eur. J., 2020, 26, 14856 CrossRef CAS.
- F. Himmelsbach, B. S. Schultz, T. Trichtinger, R. Charubala and W. Pfleiderer, Tetrahedron, 1984, 40, 59 CrossRef CAS.
- Ch. Schneider, S. Becker, H. Okamura, A. Crisp, T. Amatov, M. Stadlmeier and T. Carell, Angew. Chem., Int. Ed., 2018, 57, 5943 CrossRef CAS.
- V. Boudou, J. Langridge, A. V. Aerschot, C. Hendrix, A. Millar, P. Weiss and P. Herdewijn, Helv. Chim. Acta, 2000, 83, 152 CrossRef CAS.
- M. Lamothe, M. Perez, V. Colovray-Gotteland and S. Halazy, Synlett, 1996, 507 CrossRef CAS.
- P. Y. Chong, S. Z. Janicki and P. A. Petillo, J. Org. Chem., 1998, 63, 8515 CrossRef CAS.
- S. Gastaldi, S. M. Weinreb and D. Stien, J. Org. Chem., 2000, 65, 3239 CrossRef CAS.
- J. In, S. Hwang, C. Kim, J. H. Seo and S. Kim, Eur. J. Org. Chem., 2013, 965 CrossRef CAS.
- C. Spyropoulos and C. G. Kokotos, J. Org. Chem., 2014, 79, 4477 CrossRef CAS.
- H. Cho, J. O. Lee, S. Hwang, J. H. Seo and S. Kim, Asian J. Org. Chem., 2016, 5, 287 CrossRef CAS.
- H.-K. Kim and A. Lee, Tetrahedron Lett., 2016, 57, 4890 CrossRef CAS.
- P. Bana, A. Lako, N. Z. Kiss, Z. Beni, A. Szigetvari, J. Koti, G. I. Turos, J. Eles and I. Greiner, Org. Process Res. Dev., 2017, 21, 611 CrossRef CAS.
- S. Kang and H.-K. Kim, Tetrahedron, 2018, 74, 4036 CrossRef CAS.
- M. Wang, J. Han, X. Si, Y. Hu, J. Zhu and X. Sun, Tetrahedron Lett., 2018, 59, 1614 CrossRef CAS.
- V. Serebryany and L. Beigelman, Tetrahedron Lett., 2002, 43, 1983 CrossRef CAS.
- V. Serebryany and L. Beigelman, Nucleosides, Nucleotides Nucleic Acids, 2003, 22, 1007 CrossRef CAS.
- Y. Saito, A. Nyilas and L. A. Agrofoglio, Carbohydr. Res., 2001, 331, 83 CrossRef CAS.
- C. B. Reese, L. H. K. Shek and Z. Zhao, J. Chem. Soc., Perkin Trans. 1, 1995, 3077 RSC.
- Y. Kaburagi and Y. Kishi, Org. Lett., 2007, 9, 723 CrossRef CAS.
- S. Shishodia, D. Zhang, A. H. El-Sagheer, T. Brown, T. D. W. Claridge, C. J. Schofield and R. J. Hopkinson, Org. Biomol. Chem., 2018, 16, 4021 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for synthesis 6a/6b from 4a/4b and spectroscopic data (NMR, IR, HRMS). See DOI: 10.1039/d0ra09803e |
| This journal is © The Royal Society of Chemistry 2021 |