DOI:
10.1039/D0RA09811F
(Paper)
RSC Adv., 2021,
11, 2783-2792
Tungsten-substituted molybdophosphoric acid impregnated with kaolin: effective catalysts for the synthesis of 3,4-dihydropyrimidin-2(1H)-ones via biginelli reaction†
Received
19th November 2020
, Accepted 4th January 2021
First published on 13th January 2021
Abstract
A series of highly reusable heterogeneous catalysts (10–25 wt% PMo7W5/kaolin), consisting of tungsten-substituted molybdophosphoric acid, H3PMo7W5O40·24H2O (PMo7W5) impregnated with acid treated kaolin clay was synthesized by the wetness impregnation method. The newly synthesized catalyst was fully characterized using inductively coupled plasma-atomic emission spectroscopy (ICP-AES), Fourier transform infrared (FT-IR), powder X-ray diffraction (XRD), scanning electron microscopy (SEM), energy dispersive X-ray analysis (EDX), transmission electron microscopy (TEM), Brunauer–Emmett–Teller (BET) analysis and thermal analysis (TG-DTA). The synthesized materials were shown to be efficient in the synthesis of 3,4-dihydropyrimidin-2(1H)-ones via Biginelli reaction under solvent-free conditions. The obtained results indicate that 20% PMo7W5/kaolin catalyst showed remarkably enhanced catalytic activity compared to the bulk PMo7W5 catalyst, and also the (10 and 15%) PMo7W5 catalyst supported on kaolin clay.
1. Introduction
Heteropoly acids (HPA) are well known as environmentally benign and economically feasible alternatives to traditional acid catalysts due to their Brönsted acidity, high proton mobility and relatively better stability.1–4 One of the important structural subclass of HPAs is the Keggin anion, which is typically represented by the general formula XM12O40,x−8 where X is the central atom (Si, P, B, Zr etc.), x is its oxidation state, and M is the metal ion (Mo6+or W6+).5–7 The M6+ ions can be replaced by many other metal ions, e.g., V5+, Co2+, Zn2+, Ni2+ etc.8–12 The Keggin anion is composed of a central tetrahedral XO4 surrounded by 12 edge and corner sharing metal-oxygen octahedra MO6. The octahedra are arranged in four M3O13 groups. Each group is formed by three octahedra sharing edges and having a mutual oxygen atom which is also shared with the central tetrahedral XO4.13,14 The catalytic properties of Keggin type heteropoly acids can be tuned by changing their central heteroatoms, framework poly-atoms, and charge-compensating cations because the substitution of these atoms changes their acid and redox properties.15–17 It is well known that the acid strength of tungstophosphoric acid (H3PW12O40·nH2O) is greater than that of molybdophosphoric acid (H3PMo12O40·nH2O). Therefore, it can be projected that replacement of molybdenum (Mo) by tungsten (W) in the peripheral metal atom positions of the anions can increase the acid strength of the heteropoly acids, giving high catalytic activity for acid-catalyzed reactions.18,19 Extremely low surface area, poor stability and rapid deactivation are the major problems associated with heteropoly acids.20 Hence, it is imperative to employ an appropriate support to distribute the heteropoly acid.21–25
Kaolin, a clay mineral which is abundant on the earth, it is composed of abundantly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 clay mineral Al2Si2O5(OH)4 structure per alumina-silicate producing bulky congested particles of SiO4 tetrahedral sheets and AlO2(OH)4 octahedral sheets.26,27 Kaolin clay are promising supports due to their common fascinating features, such as their inherent acidity, excellent thermal stability and easily controlled structural morphology.28 Therefore, the acidified-kaolin with larger specific surface area is widely used as a very good catalyst carrier.29 Actually, heteropoly acid-impregnated solid acids have caused great interests in many fields.30,31
:1 clay mineral Al2Si2O5(OH)4 structure per alumina-silicate producing bulky congested particles of SiO4 tetrahedral sheets and AlO2(OH)4 octahedral sheets.26,27 Kaolin clay are promising supports due to their common fascinating features, such as their inherent acidity, excellent thermal stability and easily controlled structural morphology.28 Therefore, the acidified-kaolin with larger specific surface area is widely used as a very good catalyst carrier.29 Actually, heteropoly acid-impregnated solid acids have caused great interests in many fields.30,31
3,4-Dihydropyrimidin-2-(1H)-ones (DHPM) are of significant interest in industry as well as in academia because of their promising biological and pharmacological activities such as antitumor, antibacterial, antiviral and anti-inflammatory activities.32,33 DHPM also have some other interesting pharmacological properties of being calcium channel modulators, anti-HIV in some natural products containing the DHPM skeleton and anti-cancer by inhibiting kinesin motor protein.34,35 Therefore, development of new, efficient and convenient protocols that lead to substituted DHPMs is of considerable attention. This has led to the recent disclosure of several one-pot methodologies for the synthesis of DHPM derivatives such as [bmim] [FeCl4],36 [bmim]BF4-immobilized Cu(II) acetylacetonate,37 piperidinium triflate38 and ammonium carbonate.39 However, some of existing methods associated with certain limitations such as environmental pollution caused by utilization of organic solvents, long reaction time, exotic reaction conditions and expensive catalysts. Therefore, it is crucial to further develop an efficient and convenient method to construct such significant scaffold (Scheme 1).
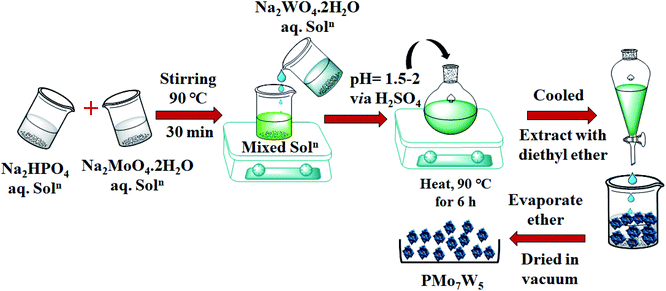 |
| | Scheme 1 Synthesis of tungsten-substituted molybdophosphoric acid, H3[PMo7W5O40]·24H2O. | |
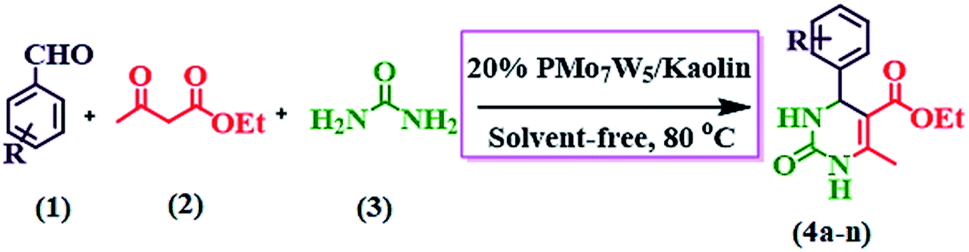
Encouraged by the intense ongoing research activity in the field of heterogeneous acid catalysis and in pursuit of our continuous interest in the area of catalysis by supported heteropoly acids.40 Herein, we wish to report a simple, green and efficient protocol for the synthesis of 3,4-dihydropyrimidin-2(1H)-ones using series of tungsten-substituted molybdophosphoric acid (H3PMo7W5O40·24H2O) impregnated with kaolin clay catalysts through Biginelli reaction (Scheme 2).
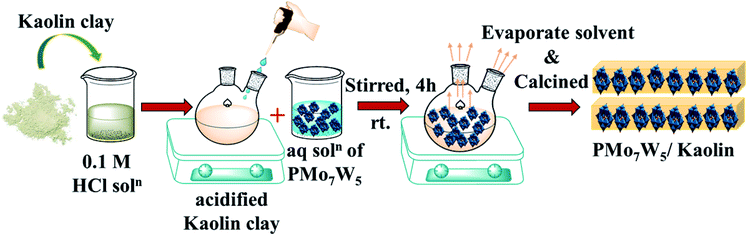 |
| | Scheme 2 Preparation of the series of catalysts having 10–25% loading of H3[PMo7W5O40]·24H2O on kaolin clay support. | |
2. Experimental section
2.1 Materials and general characterization
Disodium phosphate (Na2HPO4), Sodium molybdate (Na2MoO4·2H2O) sodium tungstate (Na2WO4·2H2O) and kaolin clay (kaolin-product code: 15160) were purchased from MOLYCHEM in India and used without further purification. All the chemicals and solvents involved in the organic synthesis were purchased from Merck, Sigma Aldrich and Alfa Aesar.
Element content was measured on an ARCOS, Simultaneous ICP Spectrometer inductively coupled plasma atomic emission spectroscopy (ICP-AES). The Fourier-transform infrared spectroscopy (FTIR) spectrum was performed on a Bruker ALPHA (Eco-ATR) spectrophotometer. The materials were characterized by X-ray powder diffraction (XRD) using a Bruker AXS Company, D8 ADVANCE diffractometer (Germany). Scanning electron microscopy (SEM) images were obtained using a FEI Nova NanoSEM 450 combined with a Bruker XFlash 6I30 instrument for energy-dispersive X-ray spectroscopy (EDX), with a scanning electron electrode at 15 kV. Transmission electron microscopy (TEM) images were collected using a (HR-TEM: Jeol/JEM 2100) operated at an accelerating voltage of 200 kV. Nitrogen adsorption–desorption isotherms were measured with a NOVA Station A instrument at 77 K. The surface area calculated by the Brunauer–Emmett–Teller (BET) method and pore size distribution derived from adsorption branches of the isotherms using the distribution Barrett–Joyner–Halenda (BJH) method. The thermal stability of the sample was carried out using simultaneous thermogravimetry (TG) and differential thermal analysis (DTA) technique, measurements were performed using a SHIMADZU, DTG-60H simultaneous DTA-TG apparatus. The progress of the reaction monitored by thin-layer chromatography on Merck's silica plates and imagining accomplished by iodine/ultraviolet light. Melting points of all the synthesized analogues were resolute in open capillary tube and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker Avance 400 Spectrometer in DMSO and CDCl3. Chemical shifts are expressed in δ parts per million relative to tetramethylsilane (TMS) as the internal standard. All yields refer to the isolated products.
2.2 Catalyst preparation
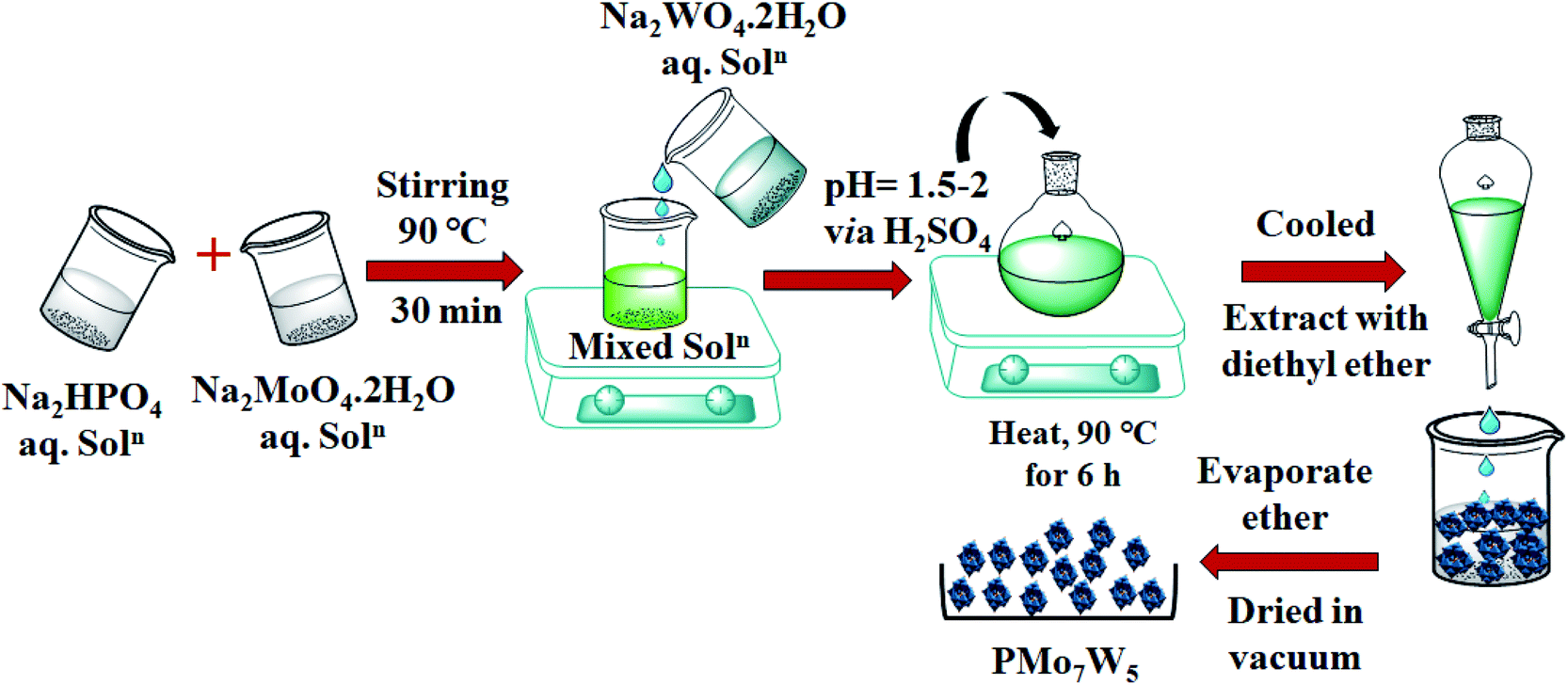
2.2.1 Preparation of H3[PMo7W5O40]·24H2O (PMo7W5). HPA with general formula H3[PMo7W5O40]·24H2O, was synthesized using the procedure reported by Huixiong.18 Briefly, Disodium phosphate (0.63 g, Na2HPO4) and desired amount of Sodium molybdate (7.48 g, Na2MoO4·2H2O) were dissolved in distilled water. The obtained solution was stirred at 90 °C. After being stirred for 30 min, aqueous solution of sodium tungstate (7.28 g, Na2WO4·2H2O) was added to the above heated solution. Subsequently, sulfuric acid (H2SO4) solution was added drop wise until the solution pH value reached about 1.5–2. The resulting mixture was heated at 90 °C for 6 h. Finally, the solution was cooled and extracted with diethyl ether in sulfuric acid environment. The powder H3[PMo7W5O40]·24H2O was obtained after concentrated etherate solution was dried in vacuum.
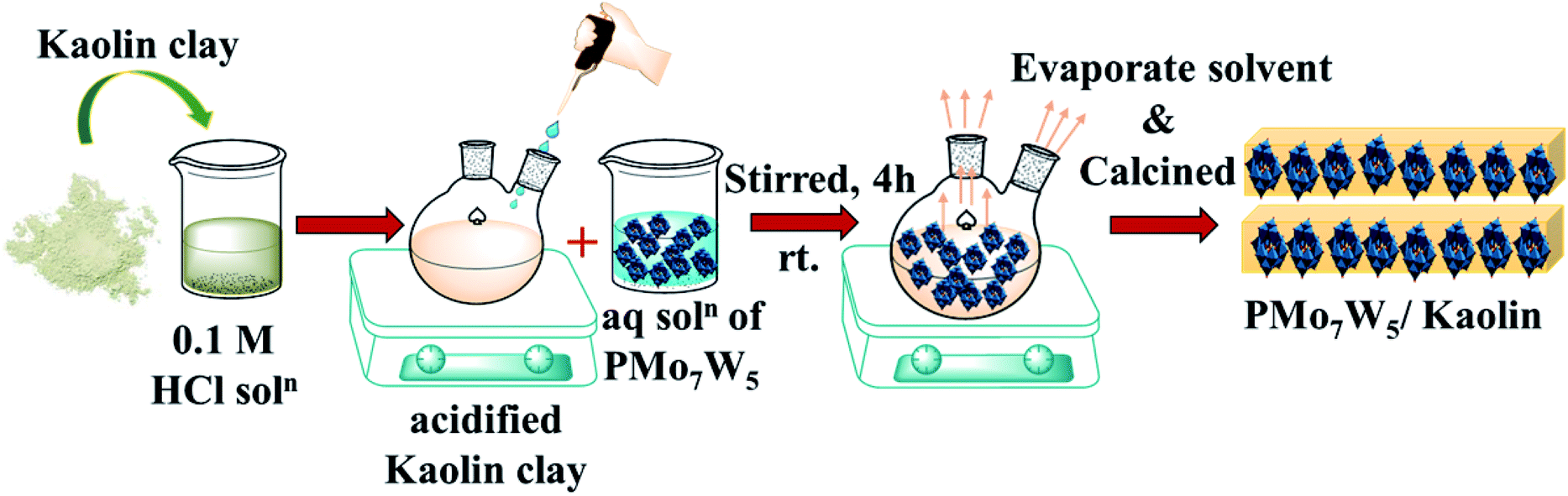
2.2.2 Preparation of PMo7W5/kaolin. PMo7W5 impregnated-kaolin catalysts were prepared according to literature procedures with small modifications.21 In a typical synthesis of 10% PMo7W5/kaolin, 0.5 g of PMo7W5 dissolved in 0.1 mol L−1 HCl solution was added into the flask containing 4.5 g of kaolin and stirred for 4 hours at room temperature. The resulting mixture was heated to 80 °C until complete evaporation of the liquid part. Then solid residue calcined in an oven at 200 K for 5 h. A series of PMo7W5/kaolin (10, 15, 20, 25 wt%) were prepared using the same method.
2.2.3 General procedure of the synthesis of 3,4-dihydropyrimidin-2(1H)-ones. In a typical experiment, a mixture of aromatic aldehyde (3 mmol), ethyl acetoacetate (3 mmol), urea (3.2 mmol) and 20% PMo7W5/kaolin (0.1 g) was stirred at 80 °C under solvent-free conditions for suitable time as indicated by thin-layer chromatography. After completion of reaction, the reaction mixture was diluted using hot ethanol (10.0 mL) and filtered for catalyst separation. The crude product was obtained by solvent evaporation under reduced pressure and recrystallized from ethanol. The recovered catalyst was washed with ethanol (10 mL) and dried overnight for further reuse. The physical data (Melting point, IR and H1 & C13NMR) of known compounds (see S5†) were found to be identical with those reported in the various literature.
3. Result and discussion
3.1 Catalyst characterization
3.1.1 ICP-AES analysis. The results of ICP-AES elemental analysis revealed that the atomic ratio of P/Mo/W is nearly maintained 1.05:6.68:5.18 which corresponds to the formula H3[PMo7W5O40]·24H2O (PMo7W5).18,40
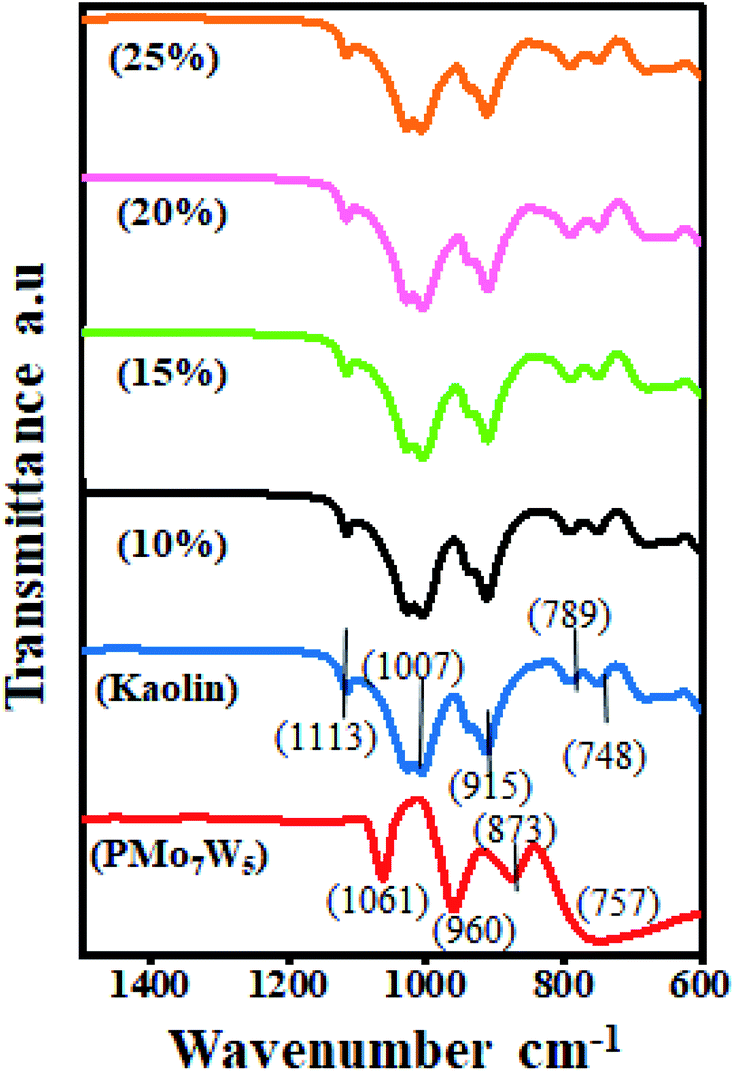
3.1.2 FT-IR spectroscopy. Primarily, FT-IR spectroscopy was used to confirm the successful functionalization of the PMo7W5/kaolin catalyst (Fig. 1). For bulk PMo7W5, characteristic bands exhibiting at 1061 (P–Oa in central tetrahedral), 960 (terminal M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Od), 873 (M–Ob–M), and 757 cm−1 (M–Oc–M) are accorded with asymmetric vibrations in Keggin unit.17 In the FT-IR spectrum of kaolin, absorptions at 748 cm−1 and 789 cm−1 are attributed to Si–O–Al vibrations and the band at 915 cm−1 is assigned to Al-OH bending vibrations. The peak at 1007 cm−1 is assigned to Si–O–Si in-plane vibrations and at 1113 cm−1 is assigned to asymmetric Si–O–Si stretching vibrations.26 The spectrum of PMo7W5/kaolin with different percentages of PMo7W5 showed two bands at 938 and 910 cm−1, which might be attributed to the (terminal MOd) and (M–Ob–M), respectively. However, the bands at 1061 and 757 cm−1 were not prominent due to overlapping with the strong bands of silica in the kaolin support.21
Od), 873 (M–Ob–M), and 757 cm−1 (M–Oc–M) are accorded with asymmetric vibrations in Keggin unit.17 In the FT-IR spectrum of kaolin, absorptions at 748 cm−1 and 789 cm−1 are attributed to Si–O–Al vibrations and the band at 915 cm−1 is assigned to Al-OH bending vibrations. The peak at 1007 cm−1 is assigned to Si–O–Si in-plane vibrations and at 1113 cm−1 is assigned to asymmetric Si–O–Si stretching vibrations.26 The spectrum of PMo7W5/kaolin with different percentages of PMo7W5 showed two bands at 938 and 910 cm−1, which might be attributed to the (terminal MOd) and (M–Ob–M), respectively. However, the bands at 1061 and 757 cm−1 were not prominent due to overlapping with the strong bands of silica in the kaolin support.21
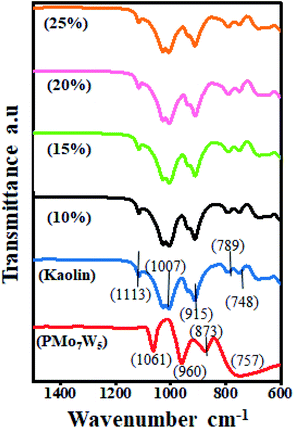 |
| | Fig. 1 FT-IR analysis of bulk PMo7W5, kaolin and PMo7W5/kaolin composites. | |
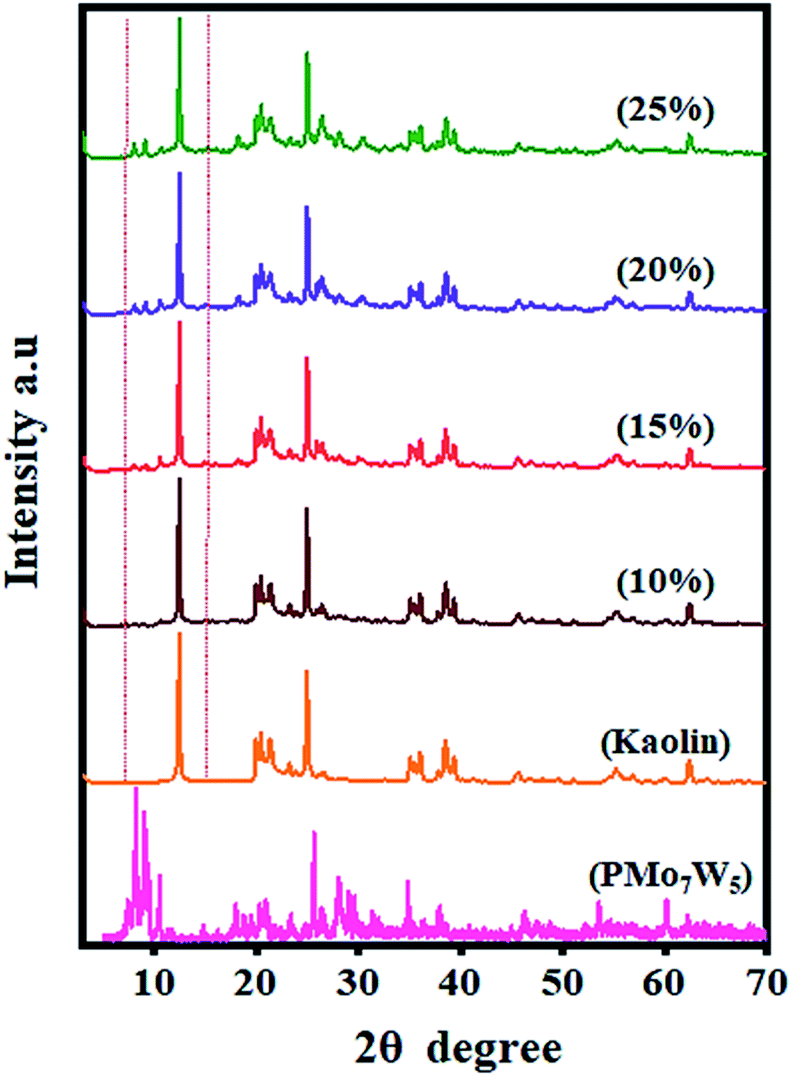
3.1.3 XRD analysis. Fine dispersion of PMo7W5 on the kaolin support was confirmed by XRD analysis. Fig. 2 shows the XRD patterns of PMo7W5, kaolin support and PMo7W5/kaolin composites (with PMo7W5 loading from 10, 15, 20 and 25%). Unsupported PMo7W5 showed the characteristic XRD peaks of the Keggin structure mainly exist in four ranges of 2θ = 7–10°, 18–23°, 25–30° and 31–37° (ref. 17) On the other hand, several prominent peaks of kaolin detected at about 2θ = 12.36°, 20.44°, 21.42°, 25.10°, 35.03°, 38.48° and 62.38°. The peak at 2θ = 12.36° is the distinctive XRD form of kaolin.41 Interestingly, 10, 15, 20 and 25% PMo7W5/kaolin catalyst exhibited no characteristic peaks of PMo7W5 but showed almost the same XRD pattern as kaolin, indicating the retention of the original characteristics of kaolin clay.42 The above results strongly supports that PMo7W5 was finely and molecularly dispersed on the kaolin support in the PMo7W5/kaolin catalyst.28,43
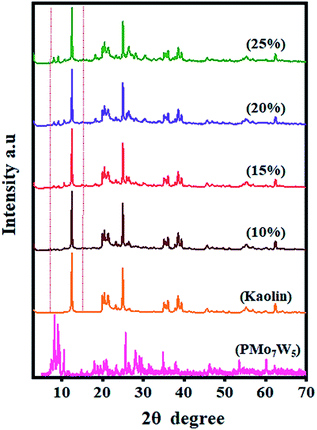 |
| | Fig. 2 XRD patterns of bulk PMo7W5, kaolin and PMo7W5/kaolin composites. | |
3.1.4 SEM and TEM analysis. The surface morphology and texture of produced samples was investigated using SEM and TEM analysis (Fig. 3a–d). As seen in Fig. 3a, the irregularly shaped particles with rough, flaky edges were observed for bulk PMo7W5 sample. The SEM images of kaolin and 20% PMo7W5/kaolin are shown in Fig. 3b and c respectively. These images clearly show that the surface morphology of supported catalyst is almost identical to that of pure kaolin. No change in surface morphology of the catalyst demonstrate that PMo7W5 species are well dispersed inside the hexagonal pores.41 Further, no separate crystallites of bulk phase of PMo7W5 were found in 20% PMo7W5/Kaolin.
 |
| | Fig. 3 FE-SEM images of bulk PMo7W5 (a), kaolin (b), 20% PMo7W5/kaolin (c) and TEM images of 20% PMo7W5/kaolin (d). | |
The TEM images of 20% PMo7W5/kaolin (Fig. 3d) shows that most of the hexagonal pores covered with dark colored fine particles. This indicates uniform dispersion of PMo7W5 inside the hexagonal pores of kaolin clay support.
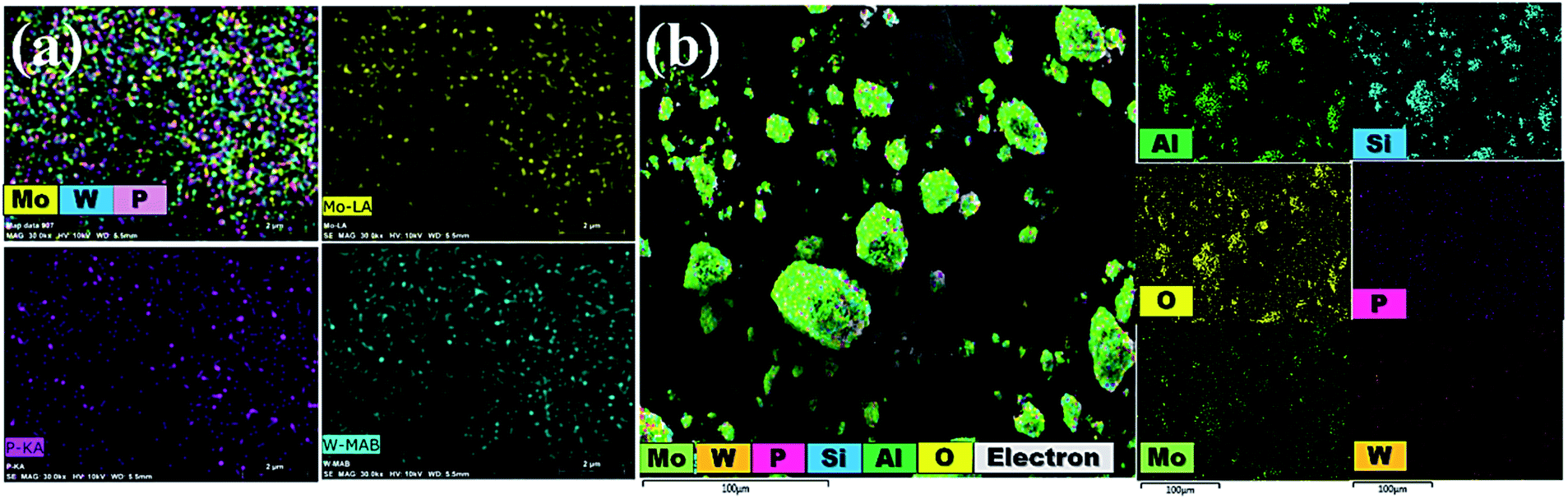
3.1.5 EDX analysis and mapping images. The chemical constitution of bulk PMo7W5 and 20% PMo7W5/kaolin were evidenced by EDX analysis as depicted in Fig. S2.† The results of newly prepared PMo7W5 confirms the presence of P, Mo and W (Fig. S1-a†). Moreover, the EDX results of 20% PMo7W5/kaolin (Fig. S2-b†) showed the presence of P, Mo and W elements of PMo7 W5 and Si, Al and O of kaolin clay. Which strongly indicates the successfully formation of 20% PMo7W5/kaolin.42Additionally, EDS mapping images proved a uniform distribution of W, Mo and P in the desired PMo7W5 catalyst system (Fig. 4a). The elemental mapping of Fig. 4b depicted the construction of a well-dispersed composite material of P, Mo, W, Si, Al and O in the synthesized 20% PMo7W5/kaolin catalyst, which is in moral contract with FT-IR, XRD and SEM results.
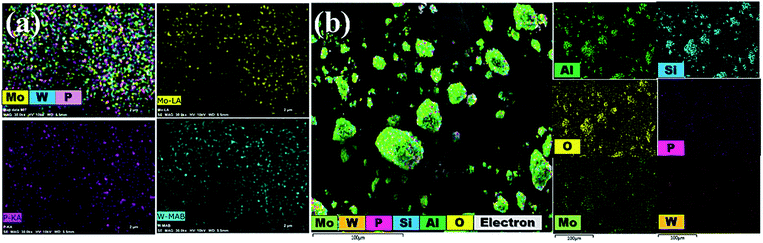 |
| | Fig. 4 Elemental mapping images of (a) bulk PMo7W5 (b) 20% PMo7W5/kaolin catalyst. | |
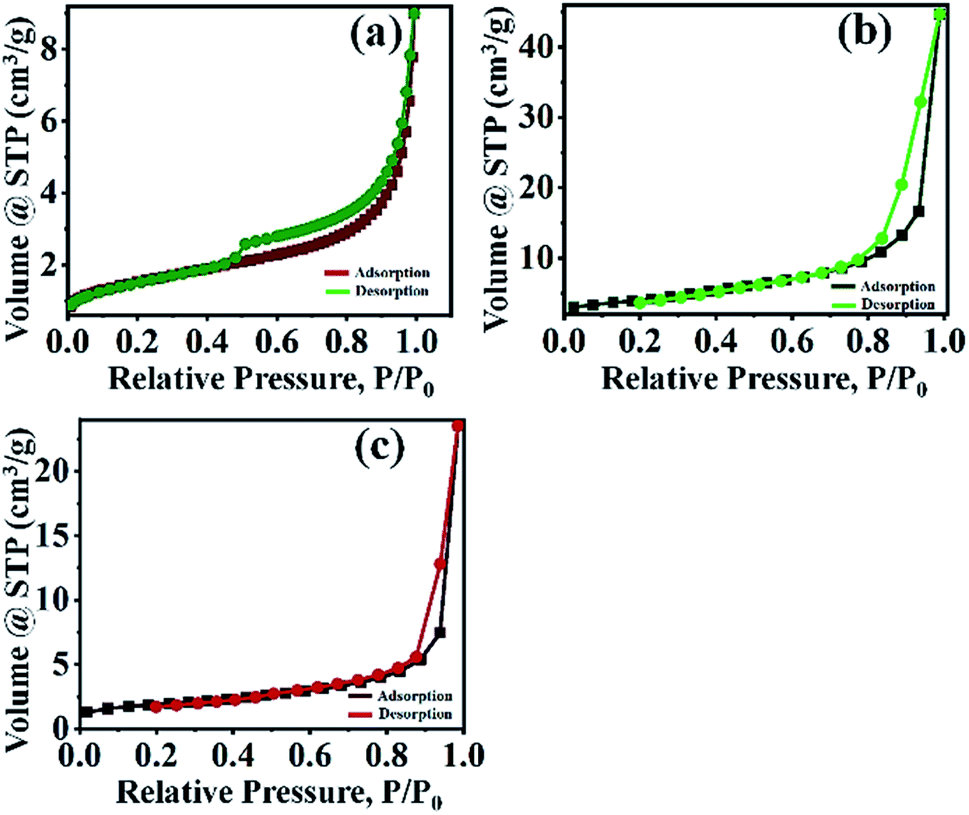
3.1.6 BET analysis. The specific surface area is an important representative of a catalyst. The specific surface area, pore diameter, and pore volume of the catalysts were determined by BET and BJH methods. The specific surface area of the PMo7W5, kaolin support and 20% PMo7W5/kaolin catalysts were 5.3801, 16.093 and 6.199 m2 g−1, respectively (Table 1). The porosities of these catalysts was investigated by N2 adsorption/desorption measurement. N2 adsorption/desorption isotherm are the type IV in nature according to the IUPAC classification. The bulk PMo7W5 exhibit a well expressed H4 hysteresis loop (Fig. 5a), whereas, parent kaolin clay and 20% PMo7W5/kaolin catalyst exhibit H3 hysteresis loop (Fig. 5b and c) in the range of 0.4–1.0 P/P0 at high relative pressure, which is typical for mesoporous materials. As demonstrated in Table 1, the pore diameter of the PMo7W5, kaolin support and 20% PMo7W5/kaolin catalyst were 10.409, 15.850, and 3.882 nm respectively. Also pore volume (0.014–0.070 cm3 g−1) of the catalysts were calculated using the BJH method (see Fig. S3-a–c†).26,44,45
Table 1 Textural properties of the prepared catalyst
| Entry |
Samples |
SBET (m2 g−1) |
Dpore (nm) |
Vpore (cm3 g−1) |
| 1 |
PMo7W5 |
5.3801 |
10.409 |
0.014 |
| 2 |
Kaolin clay |
16.093 |
15.850 |
0.070 |
| 3 |
20% PMo7W5/kaolin |
6.199 |
3.882 |
0.036 |
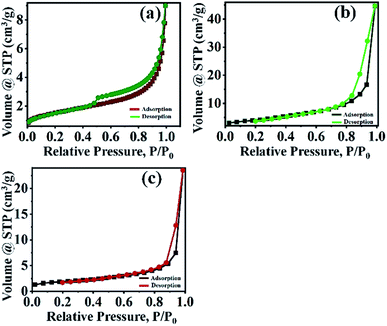 |
| | Fig. 5 N2 adsorption–desorption isotherms of (a) PMo7W5 (b) kaolin support (c) 20% PMo7W5/kaolin. | |
The study proven that the surface area of 20% PMo7W5/kaolin catalyst slightly increased but not more than that of kaolin support.46 This could primarily due to deposition and incorporation of PMo7W5 catalyst into the pores of kaolin support.47
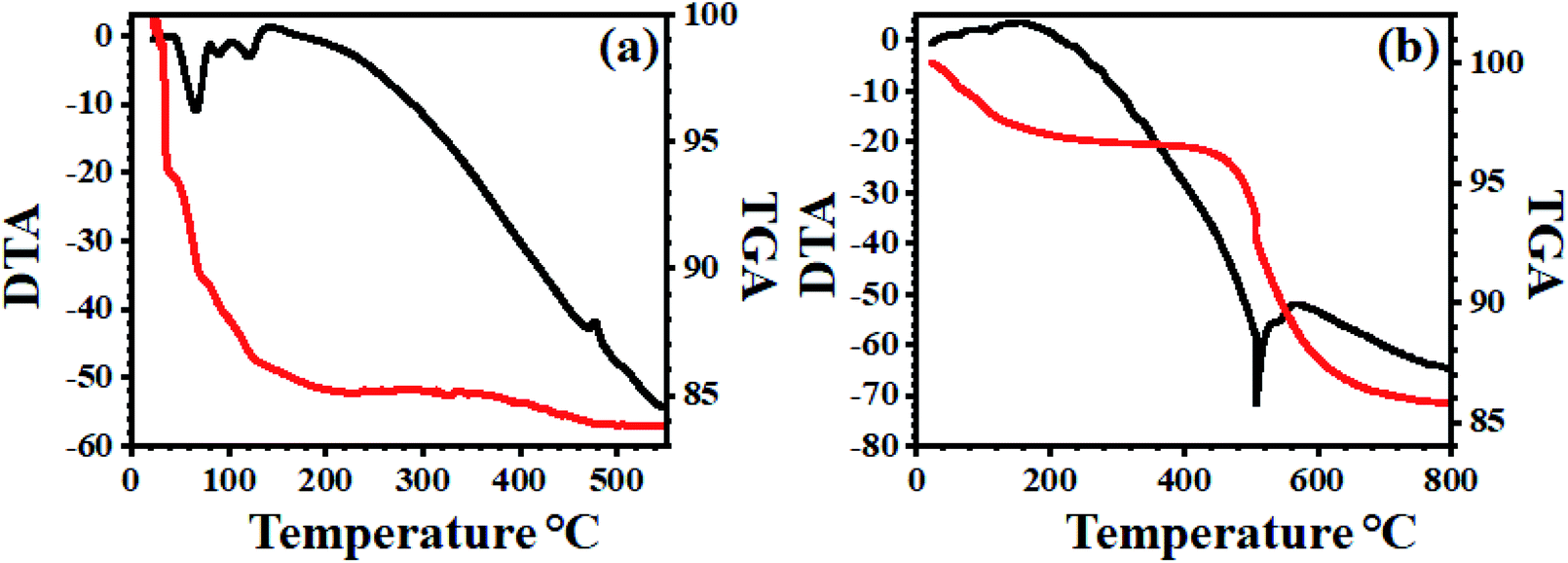
3.1.7 TG-DT analysis. The thermal stability of bulk PMo7W5 and 20% PMo7W5/kaolin examined by the thermogravimetric (TG) and differential thermal analysis (DTA). The hydrated solids usually obtained with a large amount of water of crystallization. Generally, three types of crystallographic water molecule could be distinguishes in the hydrated solid (Fig. 6a).48,49 The TG curve for bulk PMo7W5 shows the total weight loss of 16.01% below 460 °C indicating that 24 water molecules calculated were lost. The first mass loss stage, free crystalized water, 10.20% of the total sample mass was lost and an endothermic peak appeared at 95 °C for 15-water molecule. The second mass lost for H+ -combined water, about 3.27% of the total sample mass was loss and endothermic peak appeared at 147 °C for 4.9 water molecule. The structure water, 2.5% of the total sample mass loss and DTA curve shows the exothermic peak at 467 °C, which demonstrate stability of Keggin unit and these third mass losses for oxide mixture.
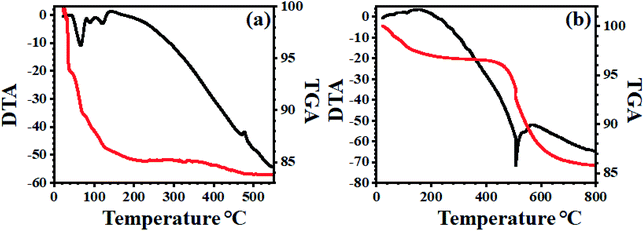 |
| | Fig. 6 TG-DT analysis of (a) PMo7W5 and (b) 20% PMo7W5/kaolin catalyst. | |
The TG-DTA of supported PMo7W5 on kaolin showed the mass loss of about 3.51% up to 430 °C temperature corresponds to dehydration of surface and interlayer water molecules (Fig. 6b). The gradual mass loss about 7.19% within the temperature range 430 °C to 750 °C, which indicates an increase in the thermal stability of bulk PMo7W5 on kaolin clay support.40 This might be due to formation of intermolecular bonding interaction between kaolin and bulk PMo7W5. The above result reveals that strong chemical interaction between kaolin and PMo7W5 catalyst.
3.2 Catalytic activity
Our initial studies were focused on the optimization of the reaction conditions for the synthesis of ethyl 6-methyl-2-oxo-4-aryl-1,2,3,4-tetrahydropyrimidine-5-carboxylate derivatives. Benzaldehyde (3 mmol), ethyl acetoacetate (3 mmol) and urea (3.2 mmol) were chosen as substrates for model reaction (Scheme 3).
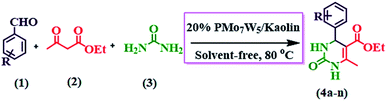 |
| | Scheme 3 Synthesis of ethyl 6-methyl-2-oxo-4-aryl-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4a–n) by using 20% PMo7W5/kaolin catalyst. | |
Various amounts of PMo7W5/kaolin were used to study the effect of the composition of the catalyst on the conversion and obtained results are summarized in Table 2. Pure kaolin showed extremely low catalytic activity in terms of the reaction time and the yield of the desired product (Table 2, entry 1). Bulk PMo7W5 gave a moderate yield of the product but only after an extended reaction time (Table 2, entry 2). It was observed that both yield of the product and reaction time were improved upon increasing the catalyst loading up to 20% w/w (Table 2, entries 3, 4, and 5). Further, an increase in PMo7W5 catalyst loading above 20% w/w on kaolin support have no effect on yield of product and reaction time due to leaching of catalyst from the support (Table 2, entry 6). The increase in activity of the 20% PMo7W5/kaolin catalyst was due to high dispersion of PMo7W5 catalyst on kaolin support. This would lead to an increase in its surface area and the number of active sites compared with the bulk PMo7W5 catalyst. Hence, 20% PMo7W5/kaolin catalyst was found to be optimal amount and adequate to push the reaction forward.
Table 2 Effect of H3PMo7W5O40·24H2O loading on support kaolin for the synthesis of ethyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4a)a
| Entry |
Catalyst |
Timeb (min) |
Yieldc (%) |
| Reaction conditions: benzaldehyde (3 mmol), ethyl acetoacetate (3 mmol), urea (3.2 mmol) and PMo7W5/kaolin catalyst. Reaction progress monitored by TLC. Isolated yields. |
| 1 |
Pure kaolin |
120 |
45 |
| 2 |
Bulk H3PMo7W5O40·24H2O |
55 |
60 |
| 3 |
10% PMo7W5/kaolin |
35 |
68 |
| 4 |
15% PMo7W5/kaolin |
20 |
85 |
| 5 |
20% PMo7W5/kaolin |
08 |
95 |
| 6 |
25% PMo7W5/kaolin |
08 |
95 |
The determination of appropriate amount of the catalyst for catalyzing the reaction is another critical parameter in terms of reaction efficiency. To find appropriate amount of catalyst, the model reaction was carried out in the presence of different amounts (25, 50, 70, 100 and 125 mg) of 20% PMo7W5/kaolin and obtained results are summarized in Table 3. When the amount of 20% PMo7W5/kaolin increases gradually, product yield also increases (Table 3, entries 1–5). The obtained results demonstrates that 100 mg amount of 20% PMo7W5/kaolin catalyst gave 95% yield of product at 80 °C (Table 3, entry 5). Additional increase in the amount (125 mg) of 20% PMo7W5/kaolin does not increase in the yield of the product (Table 3, entry 5). This may be due to overtiredness of the catalytic site or accomplishment of the maximum conversion efficiency of the catalyst. It is also important to note that at 60 and 70 °C low to moderate conversion observed, whereas at 80 °C excellent conversion of starting material were seen (Table 3, entry 5 versus entries 7 and 8). The effectiveness of model reaction also studied without using any catalyst. Where, trace amount of the product obtained after a long period (Table 3, entry 1).
Table 3 Effect of temperature and amount of catalyst on ethyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4a)a
| Entry |
Catalyst (mg) |
Temp. °C |
Timeb |
Yieldc (%) |
| Reaction conditions: benzaldehyde (3 mmol), ethyl acetoacetate (3 mmol), urea (3.2 mmol) and 20% PMo7W5/kaolin catalyst. Reaction progress monitored by TLC. Isolated yields. |
| 1 |
0 |
80 |
24 h |
Trace |
| 2 |
25 |
80 |
8 min |
65 |
| 3 |
50 |
80 |
8 min |
70 |
| 4 |
70 |
80 |
8 min |
82 |
| 5 |
100 |
80 |
8 min |
95 |
| 6 |
125 |
80 |
8 min |
95 |
| 7 |
100 |
70 |
8 min |
65 |
| 8 |
100 |
60 |
8 min |
45 |
Further, the efficiency of 20% PMo7W5/kaolin was investigated by using 100 mg of 20% PMo7W5/kaolin in various organic solvents (Table 4). Initially, the model reaction was carried out in ethanol and water at reflux temperature the moderate yield were obtained (Table 4, entries 1, 2). When the methanol, acetonitrile, dichloromethane were used as solvent at reflux temperature and DMF at 80 °C, it gave lower yields of product (Table 4, entries 3–6). None of the above solvents demonstrates the advantage of time and yield instead of solvent-free condition (Table 4, entry 7). Therefore, solvent-free condition was superior in terms of cost and it is environmentally benign to promote further derivatives (Table 5).
Table 4 Effect of different solvents on the ethyl 6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4a)a
| Entry |
Catalyst (mg) |
Temp. °C |
Solvent |
Timeb |
Yieldc (%) |
| Reaction conditions: benzaldehyde (3 mmol), ethyl acetoacetate (3 mmol), urea (3.2 mmol) and 20% PMo7W5/kaolin catalyst. Reaction progress monitored by TLC. Isolated yields. |
| 1 |
100 |
Reflux |
EtOH |
15 min |
72 |
| 2 |
100 |
Reflux |
H2O |
10 min |
68 |
| 3 |
100 |
Reflux |
MeOH |
25 min |
62 |
| 4 |
100 |
Reflux |
CH3CN |
45 min |
45 |
| 5 |
100 |
80 |
DMF |
65 min |
55 |
| 6 |
100 |
Reflux |
CH2Cl2 |
30 min |
59 |
| 7 |
100 |
80 |
Solvent-free |
8 min |
95 |
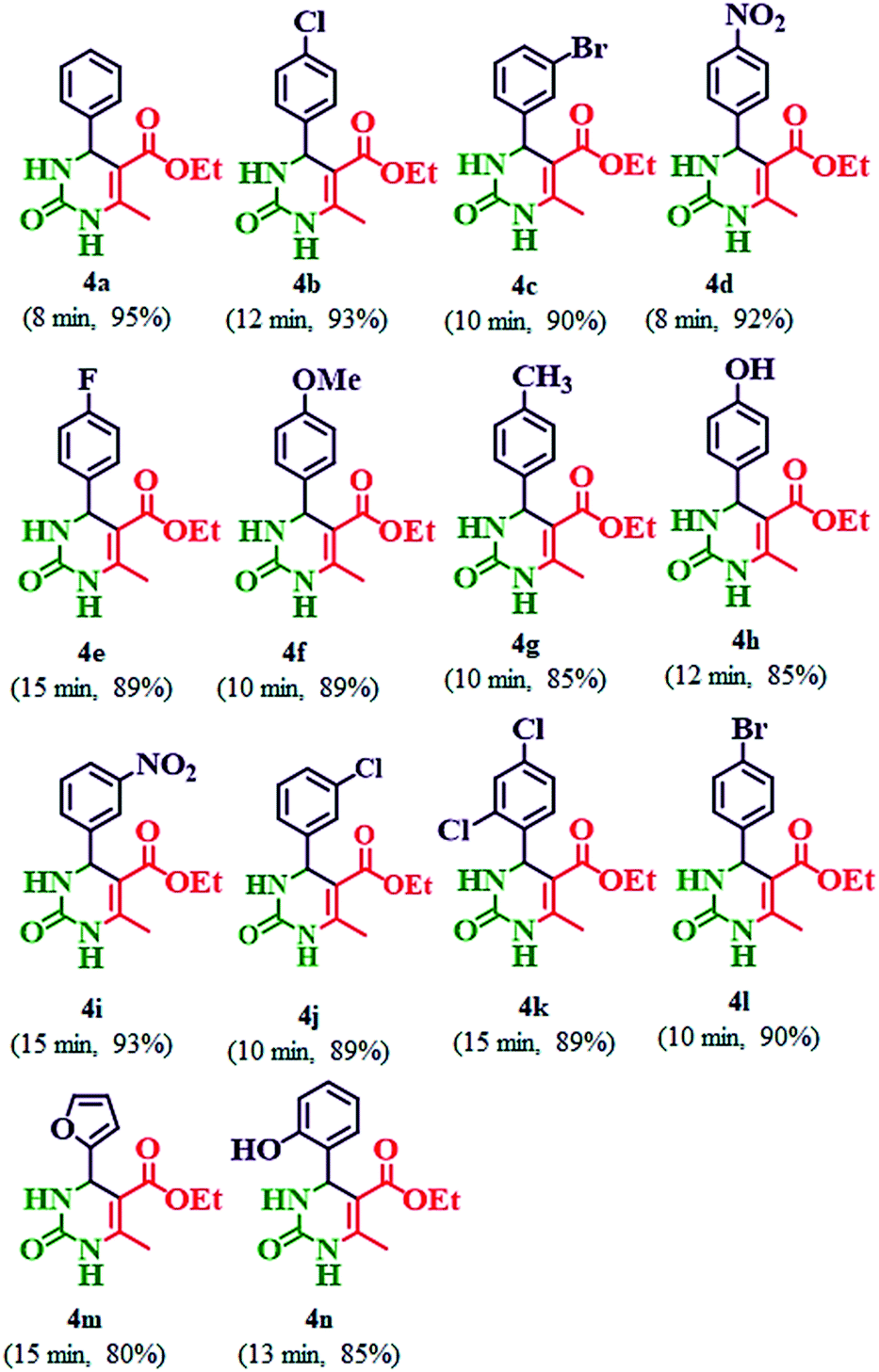
Table 5 Synthesis of ethyl 6-methyl-2-oxo-4-aryl-1,2,3,4-tetrahydropyrimidine-5-carboxylate (4a–n) by using 20% PMo7W5/kaolin catalysta,b
| Reaction conditions: aldehydes (4a–n) (3 mmol), ethyl acetoacetate (3 mmol), and urea (3.2 mmol) in 20% PMo7W5/kaolin (0.1 g) stirred at 80 °C. Isolated yields. |
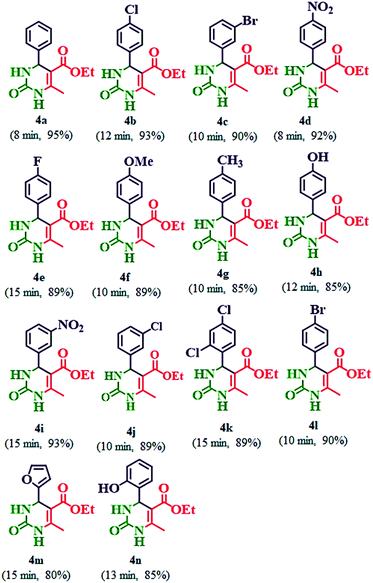 |
A comparative study was performed for the use of 20% PMo7W5/kaolin with some of the reported catalysts for the synthesis of 6-methyl-2-oxo-4-aryl-1,2,3,4-tetrahydropyrimidine-5-carboxylate (Table 6). Reaction with different catalysts required a higher amount of catalyst and longer reaction times compared with 20% PMo7W5/kaolin in solvent-free systems. In most methods, the reaction was achieved in solvent such as ethanol, acetic acid and dioxane. Thus, 20% PMo7W5/kaolin encouraged the reactions more effectively than the other catalysts and should be considered as one of the top choices for selecting an economically convenient, user-friendly catalyst.
Table 6 Biginelli reaction of benzaldehyde, ethyl acetoacetate and urea with different catalystsa
| Entry |
Catalyst |
Condition |
Time |
Yieldb (%) Ref |
| Reaction conditions: benzaldehydes (3 mmol), ethyl acetoacetate (3 mmol), and urea (3.2 mmol) in 20% PMo7W5/kaolin (0.1 g) stirred at 80 °C. Isolated yields. |
| 1 |
p-Sulfonic acid calixarenes |
EtOH, reflux |
8 h |
69 (ref. 50) |
| 2 |
NH4H2PO4/MCM-41 |
Solvent-free, 100 °C |
6 h |
72 (ref. 51) |
| 3 |
[Et3NH] [HSO4] |
Solvent-free, 100 °C |
1 h |
75 (ref. 52) |
| 4 |
12-Tungstophosphoric acid |
Reflux, acetic acid |
6 h |
98 (ref. 53) |
| 5 |
[Btto][p-TSA] |
Solvent-free, 90 °C |
30 min |
96 (ref. 54) |
| 6 |
Fe3O4/PAA-SO3H |
Solvent-free, rt |
2 h |
90 (ref. 55) |
| 7 |
SnCl2/nano SiO2 |
EtOH, reflux |
40 min |
94 (ref. 56) |
| 8 |
Bentonite/PS-SO3H |
Solvent-free, 120 °C |
30 min |
89 (ref. 57) |
| 9 |
Fe3O4@SBA-15 |
EtOH, 90 °C |
6 h |
85 (ref. 58) |
| 10 |
PS-PEG-SO3H |
Dioxane & 2-propanol, 80 °C |
10 h |
80 (ref. 59) |
| 11 |
20% PMo7W5/kaolin |
Solvent-free, 80 °C |
8 min |
95 (this work) |
3.3 Recycling of the catalyst
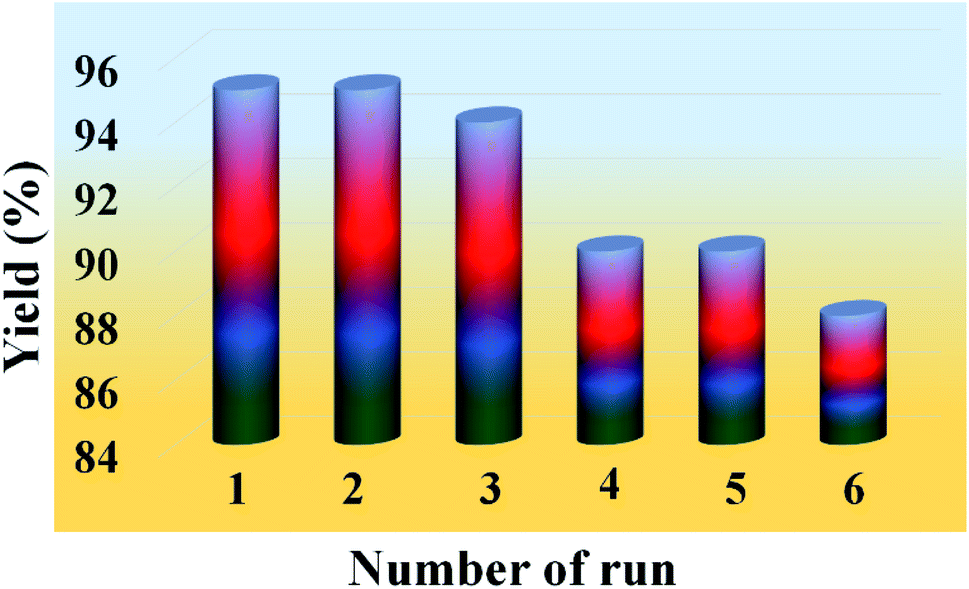
One of the most important features of the present methodology is the recyclability of the catalyst. It was observed that the 20% PMo7W5/kaolin catalyst could be reused multiple times. For this purpose, the same model reaction was performed again studied under the optimized conditions (Table 7). After reaction completion, the reaction mixture was diluted using hot ethanol and filtered for catalyst separation, the solid catalyst was washed with ethanol several times, dried and calcined at 200 °C for 5 h and reused for subsequent reaction. The results (Fig. 8) revealed that the catalyst exhibited good catalytic activity up to six consecutive cycles.
Table 7 Recycling and reuse of the catalysta
| Entry |
Number of recycle |
Yield (%)b |
| Reaction conditions: benzaldehyde (3 mmol), ethyl acetoacetate (3 mmol), urea (3.2 mmol) and 20% PMo7W5/kaolin catalyst, time 8 min. Isolated yields. |
| 1 |
1 |
95 |
| 2 |
2 |
95 |
| 3 |
3 |
94 |
| 4 |
4 |
90 |
| 5 |
5 |
90 |
| 6 |
6 |
88 |
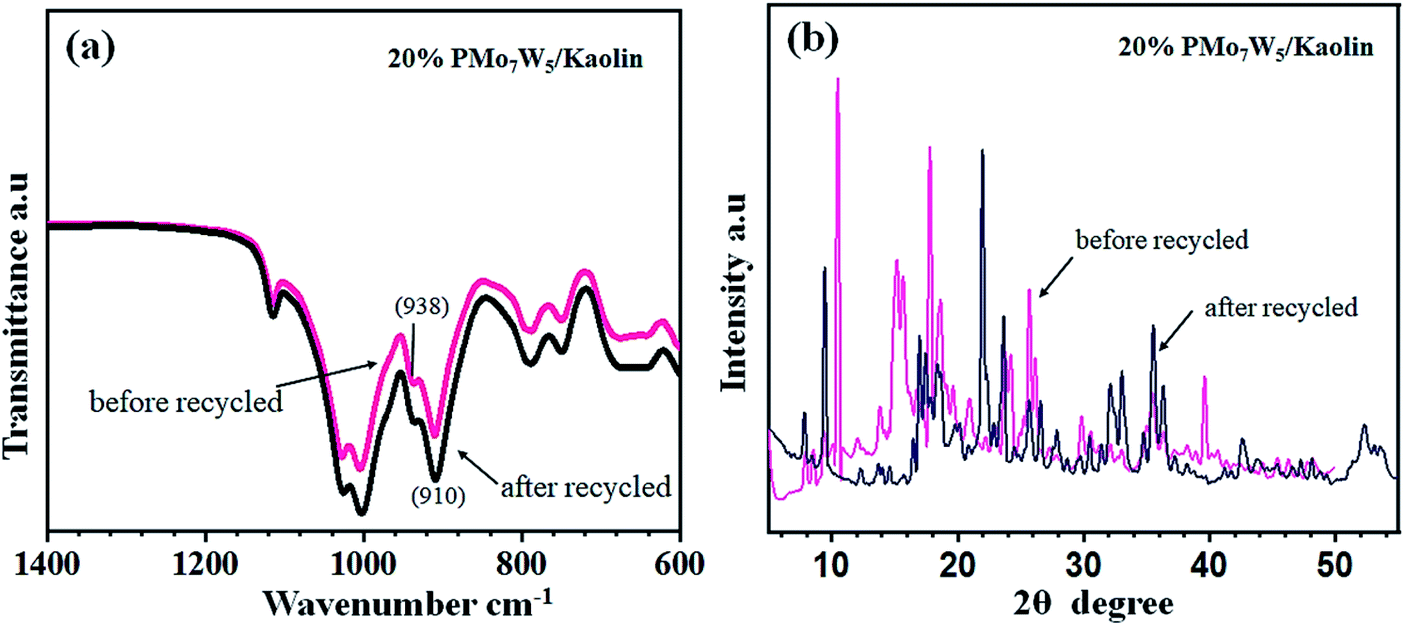
The FT-IR and XRD spectra of the recovered 20% PMo7W5/kaolin (after six cycles) were matched with those of the fresh sample. As documented in Fig. 7, the FT-IR showed two bands at 938 and 910 cm−1 were found to similar of fresh 20% PMo7W5/kaolin (Fig. 7a verses Fig. 1). XRD spectra displayed by the recovered 20% PMo7W5/kaolin catalyst at 2θ = 20.12°, 21.97°, 23.65, 25.63°, 35.53° and 59.32° were found to be almost similar to the fresh one (Fig. 7b verses Fig. 2).
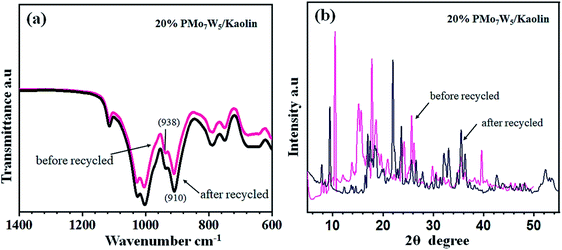 |
| | Fig. 7 FT-IR (a) and XRD (b) analysis of recovered 20% PMo7W5/kaolin catalyst. | |
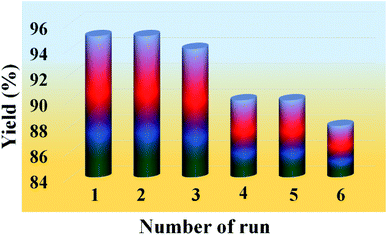 |
| | Fig. 8 Recyclability test of 20% PMo7W5/kaolin catalyst on model reaction. | |
3.4 Plausible reaction mechanism
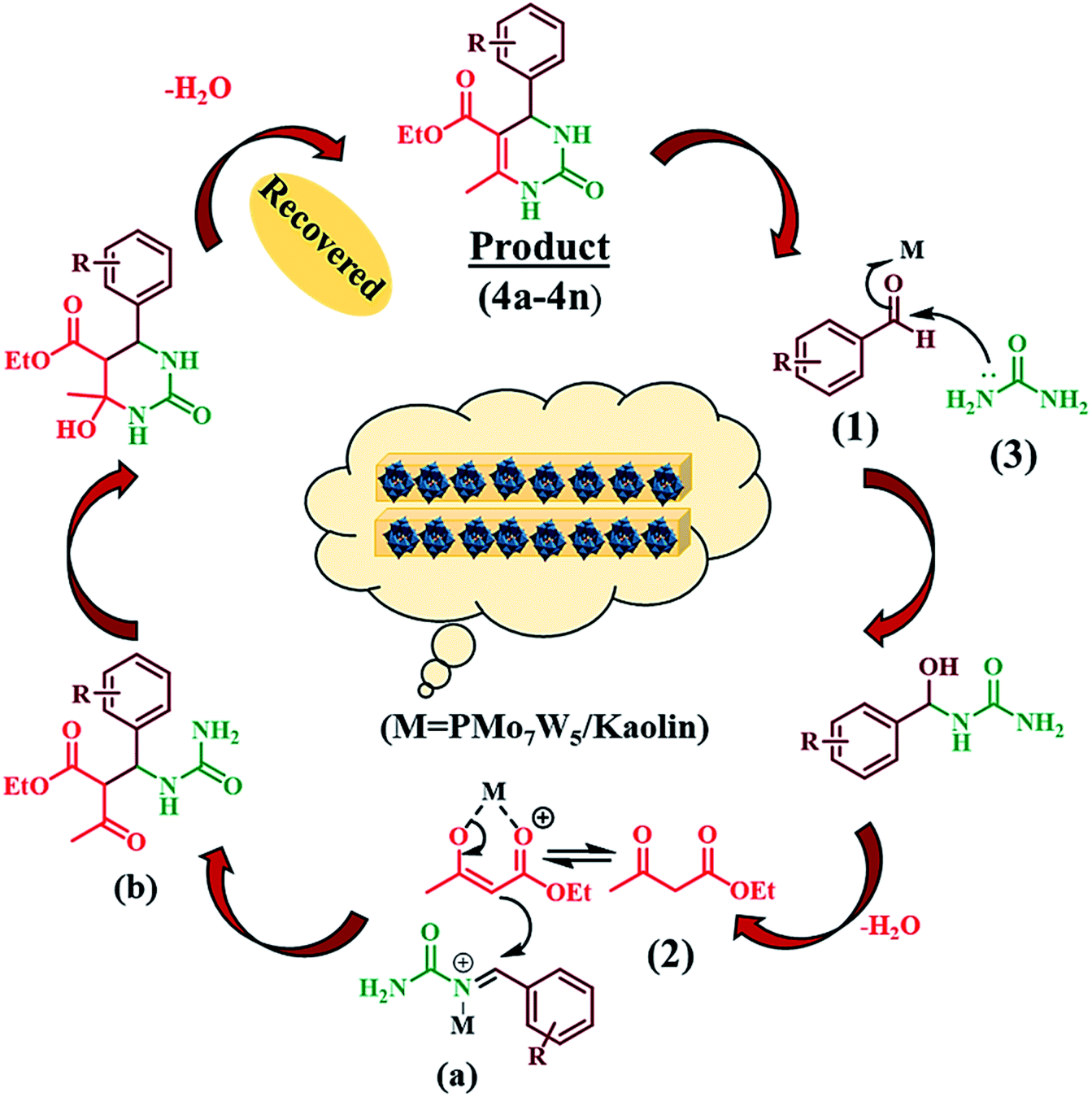
The possible reaction pathway for the Biginelli three-component condensation mediated by 20%PMo7W5/kaolin was depicted in Scheme 4. The first step involves nucleophilic attack of urea (3) on the electron deficient carbon of aldehyde (1). Here, electrophilicity of carbonyl group of aldehyde increased due to Brönsted and Lewis acidic nature of 20% PMo7W5/kaolin, which leads to formation of N-acyliminium ion intermediate (a). Interception of this iminium ion intermediate by activated 1,3-dicarbonyl compound (2) produces an open-chain ureide (b) which subsequently undergoes cyclization and dehydration to afford the corresponding dihydropyrimidinones.
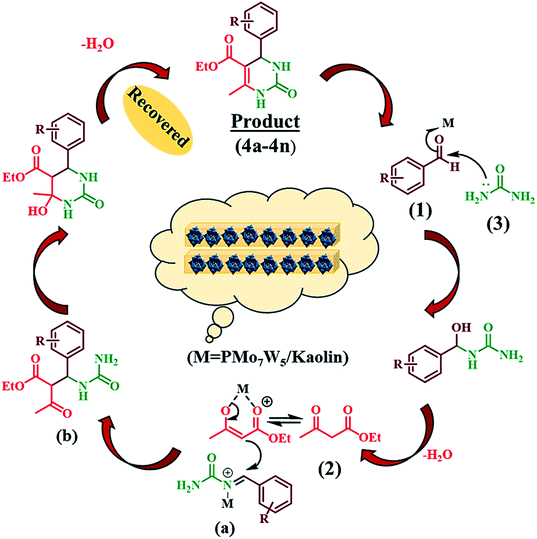 |
| | Scheme 4 Proposed mechanism for the synthesis of 3,4-dihydropyrimidin-2(1H)-ones. | |
4. Conclusion
In conclusion, we have successfully synthesis a series of tungsten-substituted molybdophosphoric acid (H3PMo7W5O40·24H2O) catalyst impregnated with acidified kaolin clay. The catalytic activity of PMo7W5/kaolin was probed through one-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones via Biginelli reaction. The 20% PMo7W5/kaolin showed higher catalytic activity than the bulk PMo7W5 catalyst, as well as the 10% and 15% PMo7W5/kaolin. The effects of various parameters such as catalyst loading, amount of catalyst, effect of solvents, influence of temperature on the rate of reaction, comparison of different catalyst was discussed in detail. The 20% PMo7W5/kaolin catalyst shows very high conversion rates in short reaction times. This catalyst was recovered easily from reaction mixture and reused at least six times without significant loss of its catalytic activity. Therefore, the newly synthesised 20% PMo7W5/kaolin could be used as a promising heterogeneous catalyst for a wide range of multifunctional applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The author DSA is gratefully acknowledges the University Grant Commission (UGC), New Delhi (India) for senior research fellowship (SRF). SGS is thankful to Dr Babasaheb Ambedkar Marathwada University, Aurangabad (MS), India (STAT/VI/RG/DEPT/2019-20/337-38) and UGC-DST SAP for financial assistance. We are also thankful to Department of Chemistry, Dr Babasaheb Ambedkar Marathwada University, Aurangabad (MS), India for providing laboratory facility.
Notes and references
- H. R. Gurav, K. Y. Nandiwale and V. V. Bokade, J. Phys. Org. Chem., 2014, 27, 121–127 CrossRef CAS.
- R. Chen, J. Xin, D. Yan, H. Dong, X. Lu and S. Zhang, ChemSusChem, 2019, 12, 2715–2724 CrossRef CAS.
- J. L. Horan, A. Genupur, H. Ren, B. J. Sikora, M.-C. Kuo, F. Meng, S. F. Dec, G. M. Haugen, M. A. Yandrasits, S. J. Hamrock, M. H. Frey and A. M. Herring, ChemSusChem, 2009, 2, 226–229 CrossRef CAS.
- J. Li, X. Wang, W. Zhu and F. Cao, ChemSusChem, 2009, 2, 177–183 CrossRef CAS.
- N. L. Mulik, P. S. Niphadkar, K. V. Pandhare and V. V. Bokade, ChemistrySelect, 2018, 3, 832–836 CrossRef CAS.
- R. Chaudhary and M. Datta, J. Anal. Sci., Methods Instrum., 2013, 03, 193–201 Search PubMed.
- M. M. Heravi, M. Mirzaei, S. Y. S. Beheshtiha, V. Zadsirjan, F. Mashayekh Ameli and M. Bazargan, Appl. Organomet. Chem., 2018, 32, 1–10 Search PubMed.
- E. G. Zhizhina, Y. A. Rodikova, O. Y. Podyacheva and Z. P. Pai, Z. Anorg. Allg. Chem., 2018, 644, 869–876 CrossRef CAS.
- N. C. Coronel, M. J. da Silva, S. O. Ferreira, R. C. da Silva and R. Natalino, ChemistrySelect, 2019, 4, 302–310 CrossRef CAS.
- W.-S. Che, H.-H. Gai, W.-B. Hao and R.-H. Ma, Synth. React. Inorg., Met.-Org., Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2014, 44, 649–655 CrossRef CAS.
- S. Bencedira, O. Bechiri, M. Djenouhat and M. Boulkra, Arabian J. Sci. Eng., 2020, 45, 4669–4681 CrossRef CAS.
- S. Tang, W. Wu, Z. Fu, S. Zou, Y. Liu, H. Zhao, S. R. Kirk and D. Yin, ChemCatChem, 2015, 7, 2637–2645 CrossRef CAS.
- C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407–455 CrossRef CAS.
- J. Xu, R. W. Gable and C. Ritchie, Acta Crystallogr., 2018, 74, 1384–1389 CrossRef CAS.
- M. Chamack, A. R. Mahjoub and H. Aghayan, Chem. Eng. J., 2014, 255, 686–694 CrossRef CAS.
- H. Fakhri, A. R. Mahjoub and H. Aghayan, J. Radioanal. Nucl. Chem., 2019, 321, 449–461 CrossRef CAS.
- H. Aghayan, A. R. Khanchi, T. Yousefi and H. Ghasemi, J. Nucl. Mater., 2017, 496, 207–214 CrossRef CAS.
- H. Wu, M. Zhou, Y. Qu, H. Li and H. Yin, Chin. J. Chem. Eng., 2009, 17, 200–206 CrossRef CAS.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS.
- A. D. Newman, D. R. Brown, P. Siril, A. F. Lee and K. Wilson, Phys. Chem. Chem. Phys., 2006, 8, 2893 RSC.
- L. H. O. Pires, A. N. de Oliveira, N. Alex Jr, R. S. Angelica, C. E. F. da Costa, J. R. Zamian, L. A. S. do Nascimento and G. N. R. Filho, Appl. Catal., B, 2014, 160–161, 122–128 CrossRef CAS.
- T. Suppan, M. K. Kunjunni, A. Barik and R. R. Bhattacharjee, ChemistrySelect, 2018, 3, 1275–1281 CrossRef CAS.
- L. Muraleedharan, Bellundagere, M. Chandrashekara, Bangalore, S. J. Prakash, Yajnavalkya and S. Bhat, ChemistrySelect, 2018, 3, 801–808 CrossRef CAS.
- A. E. R. S. Khder, H. M. A. Hassan and M. S. El-Shall, Appl. Catal., A, 2012, 411–412, 77–86 CrossRef CAS.
- D. S. Park, B. K. Kwak, N. D. Kim, J. R. Park, J. H. Cho, S. Oh and J. Yi, ChemCatChem, 2012, 4, 836–843 CrossRef CAS.
- A. G. Olaremu, J. Miner. Mater. Charact. Eng., 2015, 03, 353–361 CAS.
- S. Yahaya, S. S. Jikan, N. A. Badarulzaman and A. D. Adamu, Path Sci., 2017, 3, 1001–1004 CrossRef.
- S. Attique, M. Batool, M. I. Jalees, K. Shehzad, U. Farooq, Z. Khan, F. Ashraf and A. T. Shah, Turk. J. Chem., 2018, 42, 684–693 CAS.
- J. P. Nguetnkam, R. Kamga, F. Villiéras, G. E. Ekodeck, A. Razafitianamaharavo and J. Yvon, J. Colloid Interface Sci., 2005, 289, 104–115 CrossRef CAS.
- S. K. Kundu, J. Mondal and A. Bhaumik, Dalton Trans., 2013, 42, 10515–10524 RSC.
- L. Chen, B. Nohair, D. Zhao and S. Kaliaguine, ChemCatChem, 2018, 10, 1918–1925 CrossRef CAS.
- K. S. Atwal, B. N. Swanson, S. E. Unger, D. M. Floyd, S. Moreland, A. Hedberg and B. C. O'Reilly, J. Med. Chem., 1991, 34, 806–811 CrossRef CAS.
- J. Azizian, M. K. Mohammadi, O. Firuzi, B. Mirza and R. Miri, Chem. Biol. Drug Des., 2010, 75, 375–380 CrossRef CAS.
- A. T. Khan, M. Lal, S. Ali and M. M. Khan, Tetrahedron Lett., 2011, 52, 5327–5332 CrossRef CAS.
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043–1052 CrossRef CAS.
- X. Chen and Y. Peng, Catal. Lett., 2008, 122, 310–313 CrossRef CAS.
- S. L. Jain, J. K. Joseph and B. Sain, Catal. Lett., 2007, 115, 52–55 CrossRef CAS.
- C. Ramalingan, S. J. Park, I. S. Lee and Y. W. Kwak, Tetrahedron, 2010, 66, 2987–2994 CrossRef CAS.
- F. Tamaddon, Z. Razmi and A. A. Jafari, Tetrahedron Lett., 2010, 51, 1187–1189 CrossRef CAS.
- D. S. Aher, K. R. Khillare, L. D. Chavan and S. G. Shankarwar, ChemistrySelect, 2020, 5, 7320–7331 CrossRef CAS.
- O. D. S. Lacerda, R. M. Cavalcanti, T. M. de Matos, R. S. Angélica, G. N. da Rocha Filho and I. D. C. L. Barros, Fuel, 2013, 108, 604–611 CrossRef CAS.
- L. V. Chopda and P. N. Dave, ChemistrySelect, 2020, 5, 2395–2400 CrossRef CAS.
- D. S. Aher, K. R. Khillare, L. D. Chavan and S. G. Shankarwar, ChemistrySelect, 2020, 5, 7320–7331 CrossRef CAS.
- L. Rozic, B. Grbic, S. Petrovic, N. Radic, L. Damjanovic and Z. Vukovic, Mater. Chem. Phys., 2015, 167, 42–48 CrossRef CAS.
- A. B. Gawade, M. S. Tiwari and G. D. Yadav, ACS Sustainable Chem. Eng., 2016, 4, 4113–4123 CrossRef CAS.
- V. V. Bokade and G. D. Yadav, Appl. Clay Sci., 2011, 53, 263–271 CrossRef CAS.
- G. Yadav, J. Catal., 2003, 217, 88–99 CAS.
- Z. Xie, H. Wu, Q. Wu and L. Ai, RSC Adv., 2018, 8, 13984–13988 RSC.
- H. Cai, X. Wu, Q. Wu and W. Yan, Dalton Trans., 2016, 45, 14238–14242 RSC.
- D. L. Da Silva, S. A. Fernandes, A. A. Sabino and A. De Fa, Tetrahedron Lett., 2011, 52, 6328–6330 CrossRef CAS.
- R. Tayebee and M. Ghadamgahi, Arabian J. Chem., 2017, 10, S757–S764 CrossRef CAS.
- H. Khabazzadeh, E. T. Kermani and T. Jazinizadeh, Arabian J. Chem., 2012, 5, 485–488 CrossRef CAS.
- M. M. Heravi, F. Derikvand and F. F. Bamoharram, J. Mol. Catal. A: Chem., 2005, 242, 173–175 CrossRef CAS.
- Y. Zhang, B. Wang, X. Zhang, J. Huang and C. Liu, Molecules, 2015, 20, 3811–3820 CrossRef CAS.
- F. Zamani and E. Izadi, Catal. Commun., 2013, 42, 104–108 CrossRef CAS.
- J. Safaei Ghomi, R. Teymuri and A. Ziarati, Monatsh. Chem., 2013, 144, 1865–1870 CrossRef CAS.
- R. J. Kalbasi, A. R. Massah and B. Daneshvarnejad, Appl. Clay Sci., 2012, 55, 1–9 CrossRef CAS.
- J. Mondal, T. Sen and A. Bhaumik, Dalton Trans., 2012, 41, 6173 RSC.
- Z.-J. Quan, Y.-X. Da, Z. Zhang and X.-C. Wang, Catal. Commun., 2009, 10, 1146–1148 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09811f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kiran R. Khillarea,
Laxmikant D. Chavanb and
Sunil G. Shankarwar
a,
Kiran R. Khillarea,
Laxmikant D. Chavanb and
Sunil G. Shankarwar