
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew insight on the simultaneous H2 and HNO2 production in concentrated HNO3 aqueous solutions under alpha radiation†
Raluca M. Musat *a,
Jean-Luc Roujoua,
Vincent Dauvoisa,
Muriel Ferrya,
Carole Marchanda and
Gérard Baldacchinob
*a,
Jean-Luc Roujoua,
Vincent Dauvoisa,
Muriel Ferrya,
Carole Marchanda and
Gérard Baldacchinob
aDES – Service d'Étude du Comportement des Radionucleides (SECR), CEA, Université Paris Saclay, F-91191, Gif-sur-Yvette, France. E-mail: rmusat@gmail.com
bUniversité Paris-Saclay, CEA, CNRS, LIDYL, F-91191 Gif-sur-Yvette, France
First published on 25th March 2021
Abstract
Knowledge of hydrogen and nitrous acid yields (G(H2) and G(HNO2)) from α radiolysis of nitric acid solutions is of critical importance for the technological aspects of reprocessing of spent nuclear fuel (SNF). This study provides critical information on the G values for external alpha irradiation of concentrated HNO3 solutions. An investigation-specifically developed experimental setup allows performing this investigation without encountering issues related to extreme high local doses. In situ monitoring of the UV-visible induced absorption in irradiated HNO3 solutions permitted quantification of HNO2 production, and mass spectrometry was used to quantify H2. The influence of the dose rate and HNO3 concentration was investigated, and the primary yields of these two species were determined. It was found that dose rate increase leads to diminished production of HNO2 and H2, while HNO3 concentration increase leads to increased HNO2 formation and reduced H2 production. The values of the primary yields of these two species were determined and compared to the literature reported values. While the determined values show similar trends as those reported, this study provides accurate radiolytic yields for H2 and HNO2 that are radioelement-independent compared to the α radiolysis using radioisotope/HNO3 mixtures and provides the basis for perfecting numerical codes used for simulating the radiolytic processes associated with SNF reprocessing.
Introduction
The share of nuclear energy in the context of climate change and of the 2015 Paris agreement1 is dependent on many factors, the most important one being closing the nuclear fuel cycle, aiming at turning nuclear energy into a virtually zero waste energy source.The technology surrounding recycling of nuclear fuel has been developed and implemented on a reasonably large scale in several countries. Historically, the main process used for nuclear fuel recycling is the PUREX (Plutonium and Uranium Extraction) process that relies on separation of Pu and U from spent fuel dissolved in HNO3. The challenge however remains the recovery of all-long lived actinides and recycling them so as to produce short lived fission products, improving the proliferation resistance, reducing waste volume, and making this process cost-effective. For any large-scale separation process to be adopted and fulfil the above requirements, it must be robust under high dose-rate radiation. The effect of radiation on solvent (HNO3) extractions may result in decreased ligand concentrations, and accumulation of possibly dangerous degradation products. Among these, the most hazardous product is H2 that can be formed during the radiolytic degradation of HNO3,2–4 due to its flammability range, low ignition energy and high deflagration index5,6 inside a canister. Due to the multi-component aspect of spent nuclear fuel, a multidirectional approach is needed, taking into account the alpha, beta and gamma radiation induced degradation. Many studies were dedicated to the radiolytic degradation of HNO3 soon after the development of the PUREX process in the ‘50s, providing a wealth of information on the radiolytic mechanism and yields of H2, NO3˙ and NO2−/HNO2 in the γ3,7–25 or β radiolysis8,16,17,22,26–37 of aqueous nitrate and nitric acid solutions. However, fewer studies have been dedicated to the α radiolysis of these solutions, and the majority of these investigations were performed in the radiolysis of HNO3 in the presence of radionuclides (243Am, 241Am, 244Cm, 210Po, 240Pu, 238Pu).3,4,38–51 The formation of radiolytic products (H2, O2, H2O2, NO3˙ and NO2−/HNO2) is generally evaluated using direct methods such as spectrophotometry, gas chromatography, ion chromatography or mass spectrometry40 or indirect methods such as the classical or modified Shinn52,53 or Ghormley method.54 For the gas detection, all measurements are performed by sampling the headspace at the end of irradiations or at different residence times and replacing the sampled gas by laboratory air. These studies unanimously point to a decrease in the production of H2, and increased NO2−/HNO2 production with increasing HNO3 concentration. Elevated linear energy transfer (LET) associated with α irradiation implies generally higher primary yields (G values) than those corresponding to β and γ rays, due to increased second order processes as a result of closely spaced spurs and intra-track recombination favouring molecular products yields.55,56 The measured G-values of H2 range between 9.1 × 10−9 − 4.6 × 10−8 mol J−1 and 1.93 × 10−7 mol J−1 for pure water under γ55,57,58 and α57 irradiation respectively, decreasing to 2.1 × 10−9 mol J−1 for 6 mol dm−3 HNO3, under γ radiation. The vast majority of these studies has been performed using radionuclides as α sources, raising the problem of nuclides–specific yields and chemical interactions.
In this article we investigate, using energetic external helions, the α-radiation processes occurring in nitric acid solutions with concentrations ranging from 2 to 5 mol dm−3. For gaseous products analysis, we perform sampling of the headspace using volumes that do not disturb the system and/or do not require replacing them with laboratory air that may affect the investigated process. A specially designed quartz cell allowing fast sample circulation and dose evaluation, enables us to perform the in situ dosimetry and follow-up experiments. Comparison with the dose evaluated from the α particle flux and from in situ Fricke dosimetry shows agreement, ensuring that the extremely high doses generally associated with the use of particle accelerators are eliminated. At the same time, this study does not involve the use of radioelements as α-source, avoiding their chemical implication in the system and supplies independent radiolytic yields. The information provided by these measurements offers a comprehensive understanding of the radiolytic processes occurring in HNO3, creating a baseline for evaluations of future nuclear solvent extraction systems.
Experimental
HNO3 70% was purchased from Sigma-Aldrich and used without further purification. The investigated solutions are prepared from its dilution to reach 2, 3, 4 and 5 mol dm−3 HNO3 using ultrapure Milli-Q water with less than 5 ppb organic carbon and a resistivity of 18.2 MΩ cm. 100 ml of fresh solutions are prepared prior to irradiations. The physicochemical properties of the investigated solutions are presented in Table 1.| [HNO3] (mol dm3) | ρ (kg cm−3) | fs | fw | F (kg cm−3) |
|---|---|---|---|---|
| 0 | 1.00 | 0 | 1 | 1 |
| 2 | 1.06 | 0.11 | 0.89 | 1.053 |
| 3 | 1.09 | 0.16 | 0.84 | 1.08 |
| 4 | 1.1 | 0.21 | 0.79 | 1.1 |
| 5 | 1.13 | 0.26 | 0.74 | 1.13 |
For the irradiations, a special monoblock quartz cell was developed that allows continuous circulation of the solution, avoiding accumulation of degradation products and exalted local doses. The total volume of the irradiation cell is 200 ml, and the volume of the irradiated solution is 100 ml, keeping a liquid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :gas ratio of 1:1. Alpha particles delivered by the cyclotron enter the cell through a quartz window with a thickness of 500 μm, and a surface diameter of 6 mm. The beam diameter is fixed at the same size as the entrance window, so no gas phase irradiation occurs. Using a BVP-Z standard Ismatec pump (IDEX Corp., Cole-Palmer, DE) pump, the solution is continuously flown, at a rate of 3.6 dm3 min−1, so that the solution passes in a thin jet in front of the quartz window and is then sprayed onto the cell walls, favouring the liquid–gas exchange, and allowing the two phases to reach an equilibrium very efficiently. The irradiated solution is then injected into a 1 cm optical pathlength quartz cell (Hellma Analytics, DE), before returning to the irradiation loop. An optical fiber-connected spectrometer (AvaSpec, Avantes, NL) allows in situ monitoring of the UV-vis absorption inside the cells. The spectra are recorded for the entirety of the irradiation (5400 s for each sample), with spectra collected every 60 s as an average of 50 scans, in the wavelength range from 300 to 400 nm. To this irradiation cell, a loop is attached with a gas micro-pump (KNF, DE) that continuously circulates the gaseous phase. Periodically, the gas phase is sampled using a home-built remotely controlled electro-valves system. Each gas sample consists of a 2 ml volume. The gas samples are analysed post-irradiation using a Prisma Pro QMG 220 gas phase quadrupole mass spectrometer (Pfeiffer Vacuum, DE) to quantify the production of H2. The Prisma Pro QMG 220 has a quadrupole analyser QMA 200 and yttriated iridium filaments, allowing detection in the mass range 1–100 u, with a resolution of 0.5 u, and a minimum detection limit of 3 × 10−15 mbar. The sampled volume does not induce any disturbance in the measurement, as verified by irradiating 2 mol dm−3 solutions of HNO3, at a dose rate of 0.11 Gy s−1. These solutions were irradiated for 7200 s, and for 14400 s. In the first solution irradiation case the gaseous atmosphere is sampled every 1200 s, whereas for the second irradiated solution, the sampling started 7200 s after the beginning of the irradiation, every 1200 s. The coherence of data and correspondence of hydrogen concentration at 7200 s (832 Gy) – last point of the first solution and first point of the second solution – evidence that no perturbation was induced by sampling the gas atmosphere. Fig. 1S in ESI† presents these data.
:gas ratio of 1:1. Alpha particles delivered by the cyclotron enter the cell through a quartz window with a thickness of 500 μm, and a surface diameter of 6 mm. The beam diameter is fixed at the same size as the entrance window, so no gas phase irradiation occurs. Using a BVP-Z standard Ismatec pump (IDEX Corp., Cole-Palmer, DE) pump, the solution is continuously flown, at a rate of 3.6 dm3 min−1, so that the solution passes in a thin jet in front of the quartz window and is then sprayed onto the cell walls, favouring the liquid–gas exchange, and allowing the two phases to reach an equilibrium very efficiently. The irradiated solution is then injected into a 1 cm optical pathlength quartz cell (Hellma Analytics, DE), before returning to the irradiation loop. An optical fiber-connected spectrometer (AvaSpec, Avantes, NL) allows in situ monitoring of the UV-vis absorption inside the cells. The spectra are recorded for the entirety of the irradiation (5400 s for each sample), with spectra collected every 60 s as an average of 50 scans, in the wavelength range from 300 to 400 nm. To this irradiation cell, a loop is attached with a gas micro-pump (KNF, DE) that continuously circulates the gaseous phase. Periodically, the gas phase is sampled using a home-built remotely controlled electro-valves system. Each gas sample consists of a 2 ml volume. The gas samples are analysed post-irradiation using a Prisma Pro QMG 220 gas phase quadrupole mass spectrometer (Pfeiffer Vacuum, DE) to quantify the production of H2. The Prisma Pro QMG 220 has a quadrupole analyser QMA 200 and yttriated iridium filaments, allowing detection in the mass range 1–100 u, with a resolution of 0.5 u, and a minimum detection limit of 3 × 10−15 mbar. The sampled volume does not induce any disturbance in the measurement, as verified by irradiating 2 mol dm−3 solutions of HNO3, at a dose rate of 0.11 Gy s−1. These solutions were irradiated for 7200 s, and for 14400 s. In the first solution irradiation case the gaseous atmosphere is sampled every 1200 s, whereas for the second irradiated solution, the sampling started 7200 s after the beginning of the irradiation, every 1200 s. The coherence of data and correspondence of hydrogen concentration at 7200 s (832 Gy) – last point of the first solution and first point of the second solution – evidence that no perturbation was induced by sampling the gas atmosphere. Fig. 1S in ESI† presents these data.
All irradiations are performed at the CEMHTI cyclotron (Orleans, France) that delivers α particles with an energy of 45 MeV. The delivered helions pass through titanium screen sheets and the quartz window before entering the solution, losing part of their energy. The energy loss was calculated using SRIM (The Stopping Power and Range of Ions in Matter), based on the TRIM code,59,60 with an evaluated energy inside the irradiation cell at 12.98 MeV, and the LET of the helions at 102 × 10−3 MeV μm−1.
The flux of particles within the irradiation cell is measured using a Faraday cup, and is set at 1, 2.5, 4, 5 and 10 nA before experiments. The irradiation time for each sample is of 5400 s. The dose rate is calculated considering the α particle flux and energy according to eqn (1):
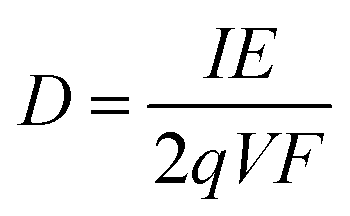 | (1) |
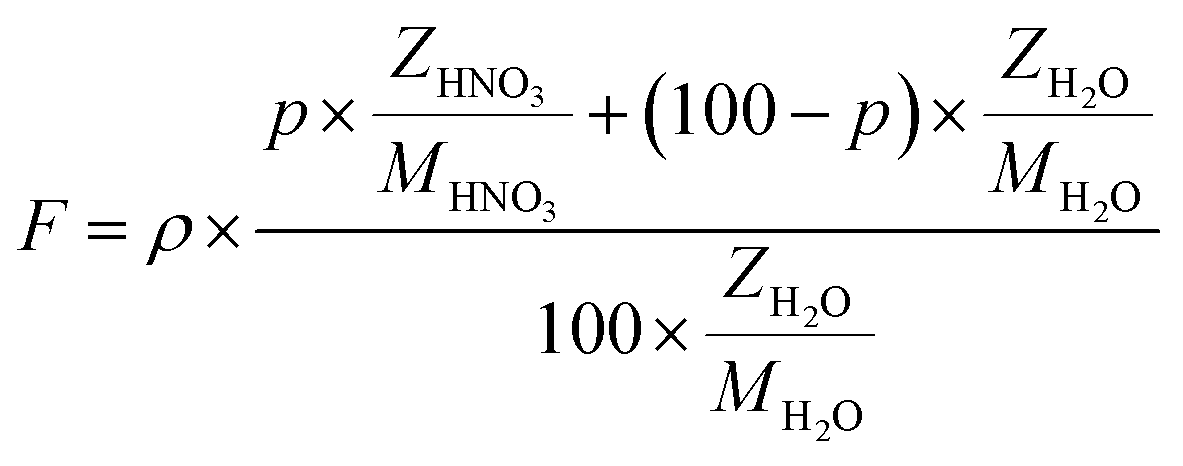 | (2) |
Because of the continuous circulation (flow rate = 60 cm3 s−1) of the target solutions inside the irradiation cell, the locally irradiated volume, Vi (Vi = S × l, S – beam facing cell surface, l – penetration depth of the α particles l = 163.4 μm from SRIM calculations) is constantly refreshed, so that in one second the irradiated volume is V = 60 cm3. The evaluated dose rates for the set currents are presented in Table 2.
| Current (nA) | D (Gy s−1) | Dmax (Gy s−1) | DFricke (Gy s−1) |
|---|---|---|---|
| 1 | 0.105 | 0.31 | 0.11 |
| 2.5 | 0.26 | 0.78 | 0.32 |
| 4 | 0.42 | 1.25 | 0.52 |
| 5 | 0.525 | 1.57 | 0.65 |
| 10 | 1.05 | 3.14 | 1.15 |
The maximum dose rate deposited was evaluated by looking at the Bragg curve (presented in Fig. 2S†) in our solution, determined using the SRIM software,59 according to:
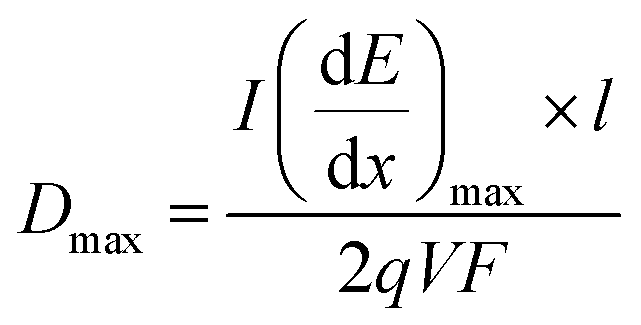 | (3) |
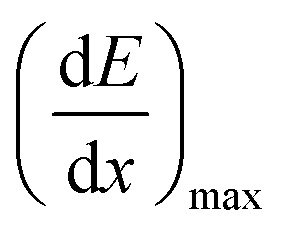 is the LET at Bragg peak (keV μm−1), l the penetration depth of the α particles (163.4 μm), q is the electron charge (1.6 × 10−19 C), V the irradiated volume (m3), and F the dose factor (kg m−3). The evaluated maximum dose rates are presented in Table 2.
is the LET at Bragg peak (keV μm−1), l the penetration depth of the α particles (163.4 μm), q is the electron charge (1.6 × 10−19 C), V the irradiated volume (m3), and F the dose factor (kg m−3). The evaluated maximum dose rates are presented in Table 2.
Prior to all irradiation, an in situ dosimetry is also performed using the super Fricke dosimeter, with the UV absorption of the irradiated solutions monitored in real-time using two 10 m optical fibers-coupled AvaSpec – dual channel spectrophotometer (Avantes. NL), attached to the quartz optical cell. The super Fricke dosimeter resides on the classical chemical dosimeter principles: oxidation of Fe2+ to Fe3+ by the radiolytically produced oxidizing species, but its limited dose range is extended by increasing the O2 and ferrous sulfate concentrations.61 Solutions of 10−2 mol dm−3 (NH4)2Fe(SO4)2 (>99%, AnalaR Normapur, UK) are prepared in 0.4 mol dm−3 H2SO4 (>98%, Carlo Erba, IT), with 10−3 mol dm−3 NaCl (Sigma-Aldrich, DE) added to suppress the effect of any impurities and bubbled with O2 prior to irradiations. The ferric ions’ concentration is monitored following the absorption of target solutions at 304 nm (ε = 2197 M−1 cm−1). According to the literature, the radiolytic yield of ferric ions for α particles of 12.98 MeV was extrapolated at 6.22 × 10−7 mol J−1.62–65 From the evolution of the absorbed dose in time, calculated according to eqn (4) and presented in Fig. 3S† in ESI,† we can evaluate the dose rate. The obtained values are presented in Table 2.
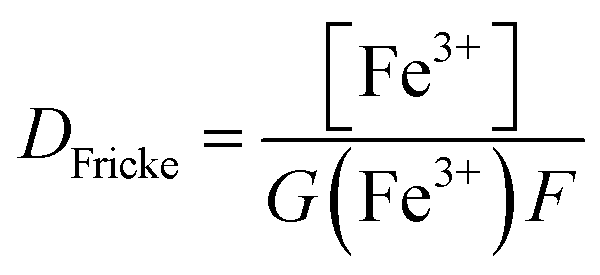 | (4) |
For the discussion herein, the dose rate we consider in all yield evaluations is the one measured using the super Fricke system, as it is determined under the same experimental conditions as the measurements in HNO3 solutions.
Results and discussion
Nitrous acid (HNO2) and H2 are the most important stable radiolytic products issued from exposure of HNO3 solutions to high ionizing radiation fields, and that can have deleterious effects on the reprocessing technology. HNO2 can react with actinides, changing their oxidation state, impacting their separation and extraction efficiency, while production and accumulation of H2 is associated with flammability hazards.The principal reactions occurring when ionizing radiation passes HNO3 aqueous solutions are listed in Table 3. Nitrite ions are produced indirectly via attack on nitrate ions by the water radiolysis primary species (reactions (13)–(31)) or directly by ionizing radiation (reaction (9)). Kazanjian et al.2 showed that the formation of HNO2 in HNO3 in solutions of concentrations higher than 1 mol dm−3 is a result of both direct and indirect action of ionizing radiation; Balcerzyk et al.35 showed that even in HNO3 solutions of concentrations of 1 mol dm−3, the direct effects of ionizing radiation have a non-negligible contribution to the formation of nitrate radicals or ions, which in turn can lead to formation of nitrite. Considering the acidity of the investigated solution and following its pKa value: pKa(HNO2/NO2−) = 3.24 at 25 °C,66 nitrite ions exist as nitrous acid.
H2O ![[long arrow, wavy then straight]](https://www.rsc.org/images/entities/char_e0f6.gif) e−pre, e−sol, H2O˙+, H3O+, OH˙, H˙ e−pre, e−sol, H2O˙+, H3O+, OH˙, H˙ |
(5) | |
| H2O H2O* |
(6) | |
| H2O* → H˙ + OH˙ | (7) | |
| H2O* → H2 + O˙ | (8) | |
| NO3− NO−3* → NO−2 + O˙ |
(9) | |
| NO3− NO3˙ + e− |
(10) | |
| HNO3 NO3˙ + H˙ |
(11) | |
| HNO3 HNO3* → HNO2 + O˙ |
(12) | |
| O˙(1D) + H2O → H2O2 | (13) | |
| O˙(3P) + NO3− → NO2− + O2 | 2.2 × 108;69 | (14) |
| NO3− + H+ ↔ HNO3 | pKa15 = 1.4;66 ; k15 = 6 × 108;32 k−15 = 2 × 1010 s−1;32 | (15) |
| NO2− + H+ ↔ HNO2 | pKa16 = 3.24; k16 = 5 × 1010;70 k−16 = 3 × 107 s−1;70 | (16) |
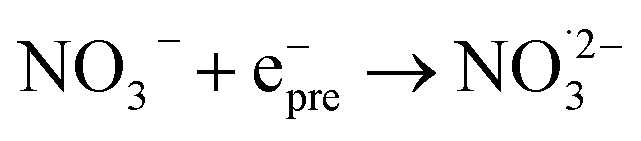 |
4.5 × 1012;71 | (17) |
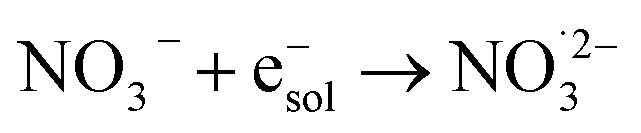 |
9.7 × 109;72 | (18) |
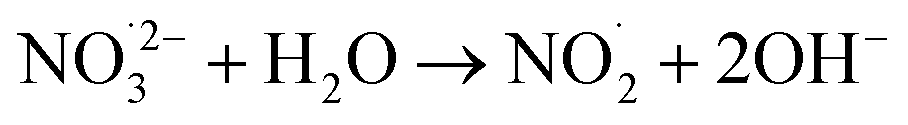 |
1 × 103;73 | (19) |
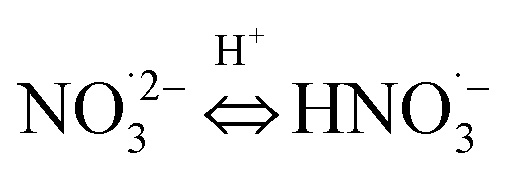 |
pKa20 = 4.8;74 5 × 108;74 | (20) |
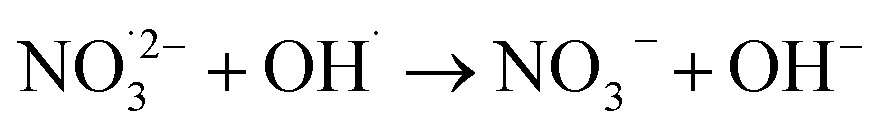 |
2.5 × 109;73 | (21) |
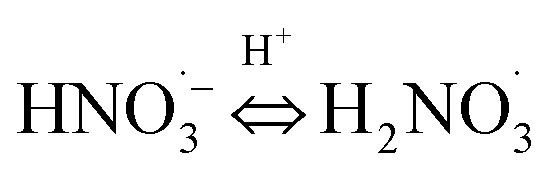 |
pKa22 = 7.5;74 | (22) |
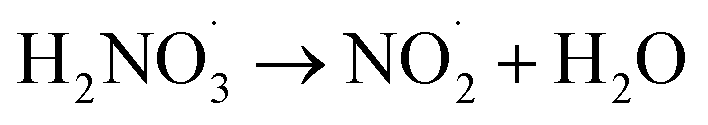 |
7 × 105;73 | (23) |
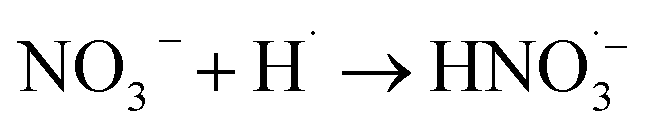 |
1 × 107;73 | (24) |
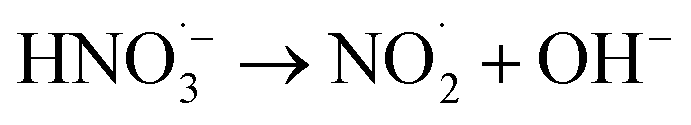 |
2.31 × 105 s−1;74 | (25) |
| H2O+ + H2O → OH˙ + H3O+ | ∼1013;75,76 | (26) |
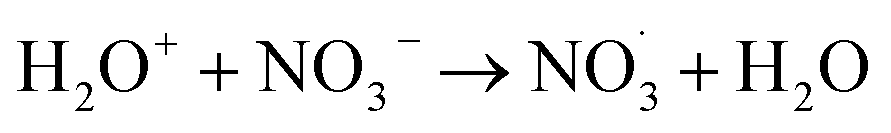 |
1 × 1012;35 | (27) |
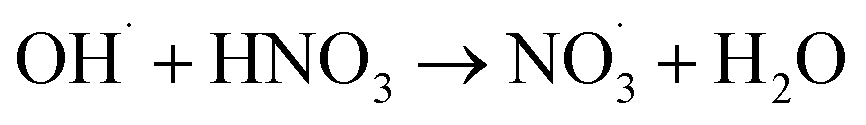 |
5.3 × 107;37 | (28) |
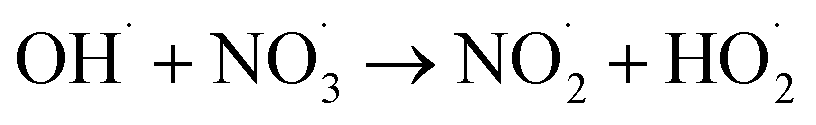 |
1.1 × 1010;36 | (29) |
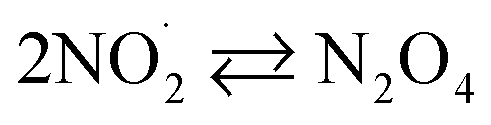 |
k30 = 4.5 × 108;73,77 k−30 = 6.9 × 103 s−1;77,78 | (30) |
| N2O4 + H2O → HNO3 + HNO2 | 18;77 1 × 103 s−1;77 | (31) |
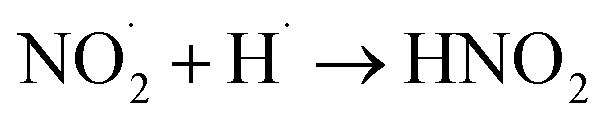 |
1 × 1010 s−1;73 | (32) |
| H+ + e−pre → H˙ | (33) | |
| H+ + e−sol → H˙ | 2.3 × 1010;79 | (34) |
| e−sol + e−sol → H2 + OH− + OH− | 7.3 × 109;80 | (35) |
| e−sol + H˙ → H2 + OH− | 2.7 × 1010;80 | (36) |
| e−sol + OH˙ → OH− | 3.5 × 1010;80 | (37) |
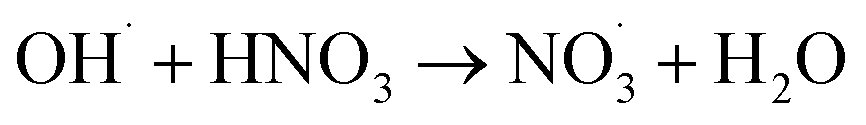 |
5.3 × 107;37,81 | (38) |
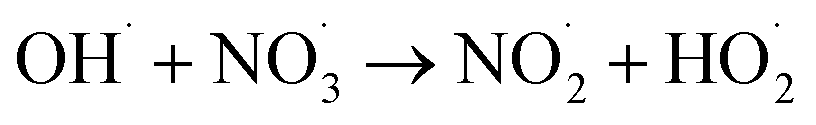 |
1.1 × 1010;36 | (39) |
| H˙ + H˙ → H2 | 5.1 × 109;80 | (40) |
| H˙ + OH˙ → H2O | 1.1 × 1010;80 | (41) |
| OH˙ + OH˙ → H2O2 | 4.8 × 109;80 | (42) |
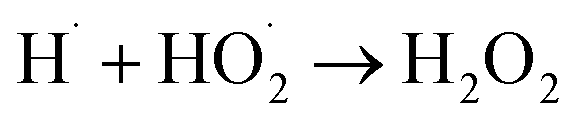 |
1.3 × 1010;80 | (43) |
The time evolution of the transient absorbance spectra recorded up to 5400 s with the AvaSpec fiber-coupled spectrometer is presented in Fig. 1. The 3D plot and time slices of the recorded data show the well-known spectrum of HNO2 with three absorbance maxima at 348, 358 and 372 nm.33,67 From Fig. 1, we extracted the time evolution of the absorbance at 358 nm, and converted it into HNO2 formation (ε358nm = 57.05 M−1 cm−1;68).
 | ||
| Fig. 1 (Top) Recorded image of the induced absorbance in a solution of 3 mol dm−3 HNO3 by α radiation at a dose rate of 0.64 Gy/s. (Bottom) Same data visualized as transient spectra at different absorbed doses. | ||
While for low concentrations of HNO3 (<1 mol dm−3), the indirect effects represent the exclusive nitrite formation mechanism, this is not the case for the investigated solutions in this study: even for the lowest concentration investigated, 2 mol dm−3 HNO3 solution, as we can see from the electron fraction of NO3− relative to water (Table 1), 11% of the deposited dose is absorbed by nitrate, leading to the formation of nitrite ions and nitrate radicals (reactions (9) and (10)). The direct action of ionizing radiation can result in the formation of excited O˙ atom, in the singlet (1D) or triplet state (3P), via reaction (9),69 leading to H2O2 (ref. 82–84) or HNO2 (ref. 69) formation, respectively via reactions (13) and (14). When considering the radiolytic yield of HNO2, we need to also take into account its depletion via reactions with species resulting from the radiolysis of water 44–47, or from NO3˙ radicals' consumption of nitrites (reactions (48) and (49)):
 | (44) |
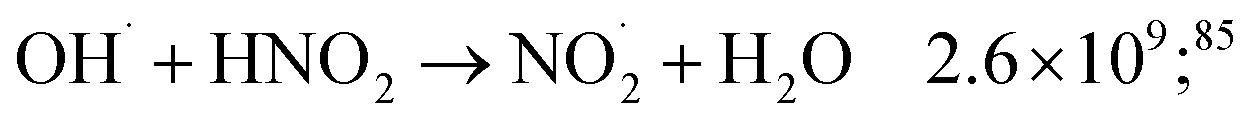 | (45) |
| H2O2 + HNO2 → NO3− + H+ + H2O k46 = k′[H+] = 2.5 × 103 M−1 s−1 for [H+] = 1 mol dm−3;86 | (46) |
| H2O2 + HNO2 → HOONO + H2O 1.68 × 1010;87 7.2 × 105;88 | (47) |
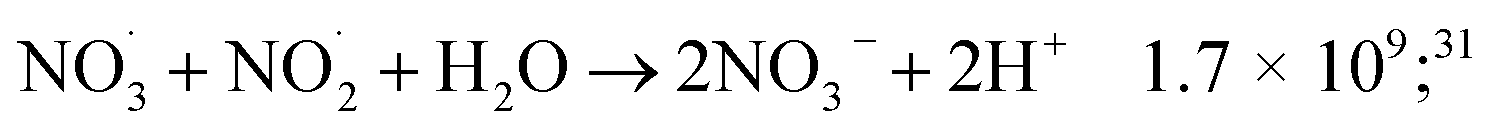 | (48) |
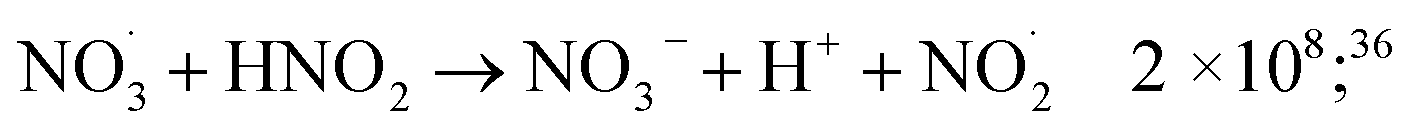 | (49) |
Fig. 2 presents the production of HNO2 in solutions of 2 mol dm−3 HNO3, irradiated at dose rates from 0.11 to 1.14 Gy s−1 in the upper panel, and in solutions of several HNO3 concentrations in the middle panel. As previously observed in α self-radiolysis, the formation of nitrite in HNO3 is proportional to the absorbed dose. Kazanjian et al.89 observed an increase of nitrite, that reaches a maximum concentration in γ radiolysis for HNO3 solutions with concentrations lower than 1 mol dm−3 while at concentrations higher than 1 mol dm−3, the nitrite increase is linear. The authors suggest that a similar maximum concentration of nitrites for higher doses and β and α radiation, is not excluded. Our measurements show that a new regime (break in the slope), or possibly a maximum of the HNO2 production is reached at the lowest dose rate and lowest concentration. As Fig. 2 shows, such slope-breaking could be expected for the other solutions at higher doses, and for more concentrated HNO3 solutions at increased dose rates. This new regime of HNO2 production could correspond to a steady state regime where accumulation of HNO2 is equilibrated by its degradation and consumption by products resulting from the radiochemical transformations of water (reactions (46)–(49)).
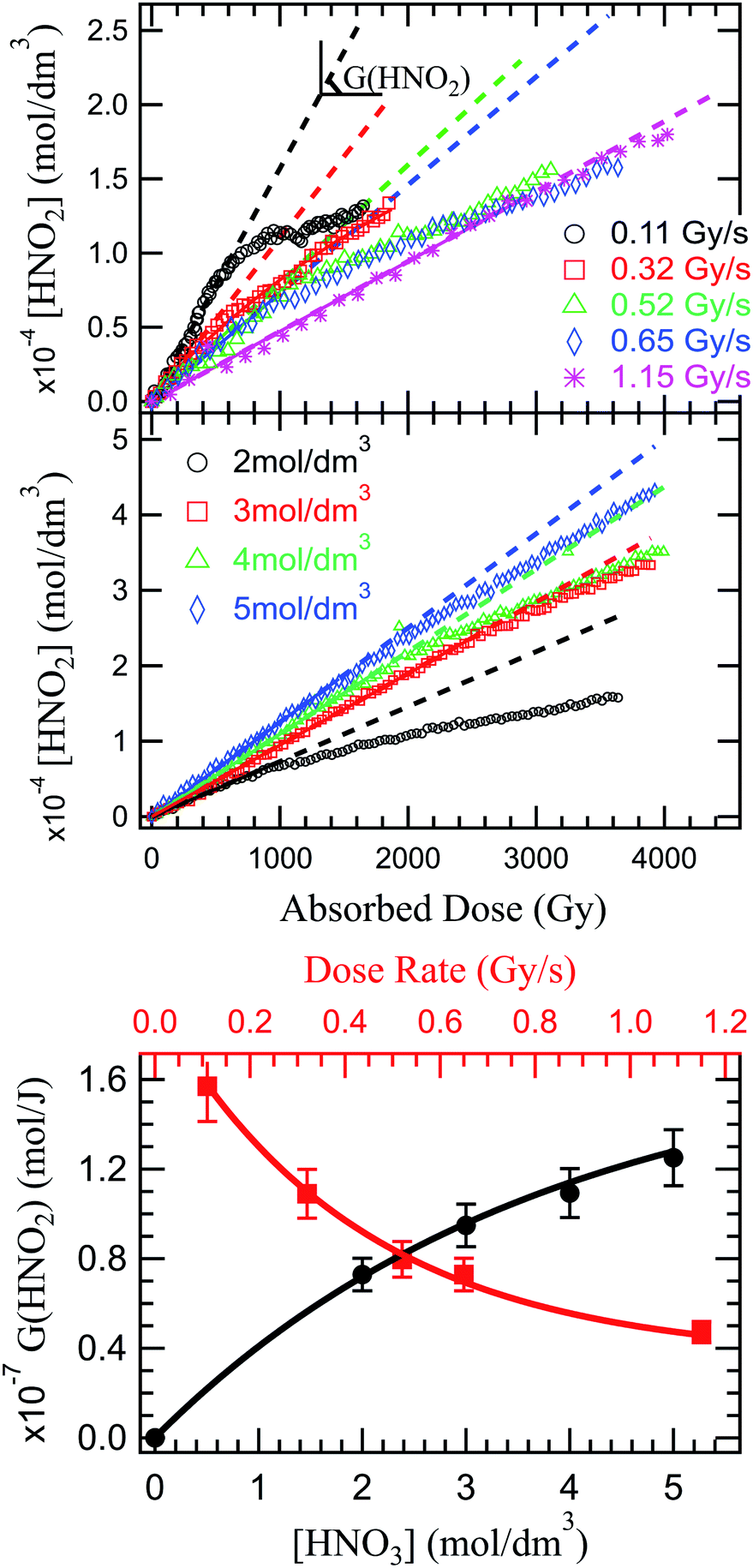 | ||
| Fig. 2 Top image: dose dependence of the HNO2 concentration in solutions of 2 mol dm−3 HNO3 under α irradiation at various dose rates. Middle image: HNO2 concentration as a function of the absorbed dose in solutions of 2, 3, 4 and 5 mol dm−3 at 0.64 Gy s−1. Lower image: HNO2 radiolytic yield dependence on HNO3 concentration (black) and G(HNO2) dependence on the dose rate in a 2 mol dm−3 HNO3 solution (red). | ||
The observed nonlinear dose dependency of HNO2 concentration is a result of the complex processes involved in its production: both inhomogeneous (intra-track) and homogeneous chemistry are impacting HNO2 formation.23 As listed in Table 3, the capture of presolvated and solvated electron by NO3− leads to formation of NO32−, that can in turn lead to the HNO2 precursor, NO2˙ (reaction (18)), or can be back oxidized by O2 or H2O2 to NO3−:
 | (50) |
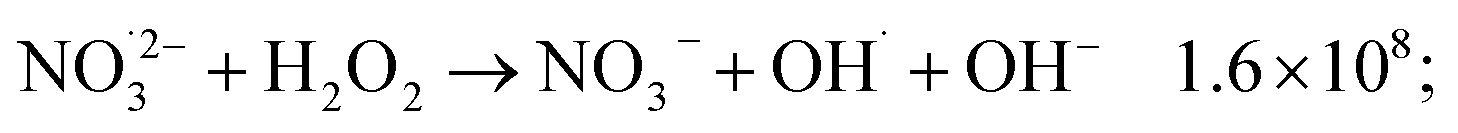 | (51) |
H2O2 can progressively accumulate in the system, and in time, reactions ((46) and (47)) and (51) start playing a more important role, which may lead to the levelling observed at high doses. Further investigations are required to see if such a stationary regime is also observed for other dose rates and concentrations.
Unlike previous observations that reported no dose rate effect for HNO2 production in 1 mol dm−3 and higher concentrations of HNO3 at dose rates ranging from 0.93 to 1.5 Gy s−1, but rather assigned observed yield differences as a result of LET effects,3 a distinct dose rate effect can be observed in the present investigation: an increase of the dose rate leads to a decrease in G(HNO2) (Fig. 2 lower panel). An increase in the dose rate will lead to an overlap of the heterogenous zones, and to an increase of biradical reactions, favouring formation of molecular products (H2O2 via reactions (42) and (43), H2 via (35)−(37)).90 If the concentrations of HNO3 solutions is high enough, HNO3 can interfere in this mechanism. Otherwise, HNO3 will only react with the molecular products resulting from H2O radiolysis. Considering the concentration of the studied solutions, the molecular products formed in the radiolysis of HNO3, can react with the molecular products issued from radiolysis of H2O. H2O2 (H2 having a low reactivity) is therefore responsible for the decrease in the HNO2 production, either by consumption of its precursor (reaction (51)) or its decomposition (reaction (46)/(47)). Dose rate also affects the solvated electron yield91 which in turn is critical in the formation of HNO2 precursors.
NO3− is effectively converted into precursors of NO2˙ (NO32−) through capture of presolvated and solvated electrons. Therefore we expect an increase of HNO3 concentration to lead to an increase of HNO2 production. Another contribution to this increase could be due to less important destruction of HNO2 by H2O2. Moisy et al.92 showed that in concentrated solutions of HNO3, the G(H2O2) decreases linearly with increasing HNO3 concentrations. H2O2 is mainly formed via recombination of ˙OH radicals, and increased concentrations of HNO3 will lead to ˙OH scavenging via reaction (21) and (28). As the concentration of HNO3 increases, the dissociation degree decreases, as shown in Fig. 4S (see ESI†). Therefore, the ˙OH reaction with the undissociated HNO3 molecules becomes more important with the increase in HNO3 concentration, leading to a decrease in H2O2, and ultimately to an increase of HNO2. This is confirmed experimentally in measurement of HNO2 production in solutions of 2, 3, 4 and 5 mol dm3 HNO3 irradiated at the same dose rate (0.64 Gy s−1). The results are presented in Fig. 2 (middle panel).
The radiolytic yields were determined from the slope at origin of these curves, and are presented in Fig. 2 (lower panel). Moisy et al.92 reported that the radiolytic yield rises with the acidity until it reaches a plateau for concentrations of H+ higher than 1 mol dm−3. However, these measurements were performed in NaNO3–HNO3 mixtures, and both the NO3− concentration and H+ concentration impact the HNO2 production. Probably due to the lower NO3− concentration, our measurements show no plateau in the investigated HNO3 concentration range, but we do not exclude its existence at higher concentrations of HNO3.
In the production of molecular hydrogen in the radiolysis of water, two main mechanisms have been suggested: intra-track chemistry, where the role of the H2 precursors (reactions (33)–(36)) has been identified and underlined by several authors,20,50,93 and dissociative chemistry of excited water molecules (reactions (7) and (8)) formed by direct excitation or recombination reaction of the water radical cation with the presolvated electron. In HNO3 solutions, competition reactions leading to production of H atom through direct effects (reaction (11), and consumption of H2 precursors (reactions (17) and (18)) occur; but the overall reported trend is a decrease of H2 in concentrated HNO3 solutions. As for HNO2, we looked at what impact of the dose rate and the solution concentration has on H2 production in the external α radiolysis of HNO3.
The dose rate effect on the production of solvated electron has been already shown in the literature,90,91,94 indicating an increased recombination of solvated electron with H˙ and ˙OH radicals in the complicated process of spur overlap (reactions (36) and (37)). Higher quantities of H2O2 produced favorably at higher dose rates and under aerated conditions as is the case of these investigations can also play a role in scavenging this H2 precursors:95,96
| e−sol + H2O2 → OH˙ + OH− + H2O 1.4 × 1010;97 | (52) |
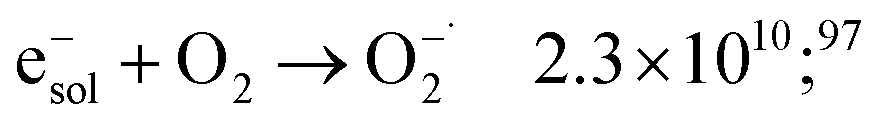 | (53) |
As the solvated electrons recombinations (reactions (34)–(36)) are a great contributor to H2 production in the radiolysis of water,20,98,99 a dose rate increase implying a reduced solvated electron production, will ultimately lead to a decrease in hydrogen production. This behavior is observed experimentally and reported in Fig. 3 (upper panel). The production of H2 is proportional to the absorbed dose, and inversely proportional to the dose rate. No stationary regime is observed in the production of H2 under the investigated conditions, leaving room for debate whether this regime exists or not.
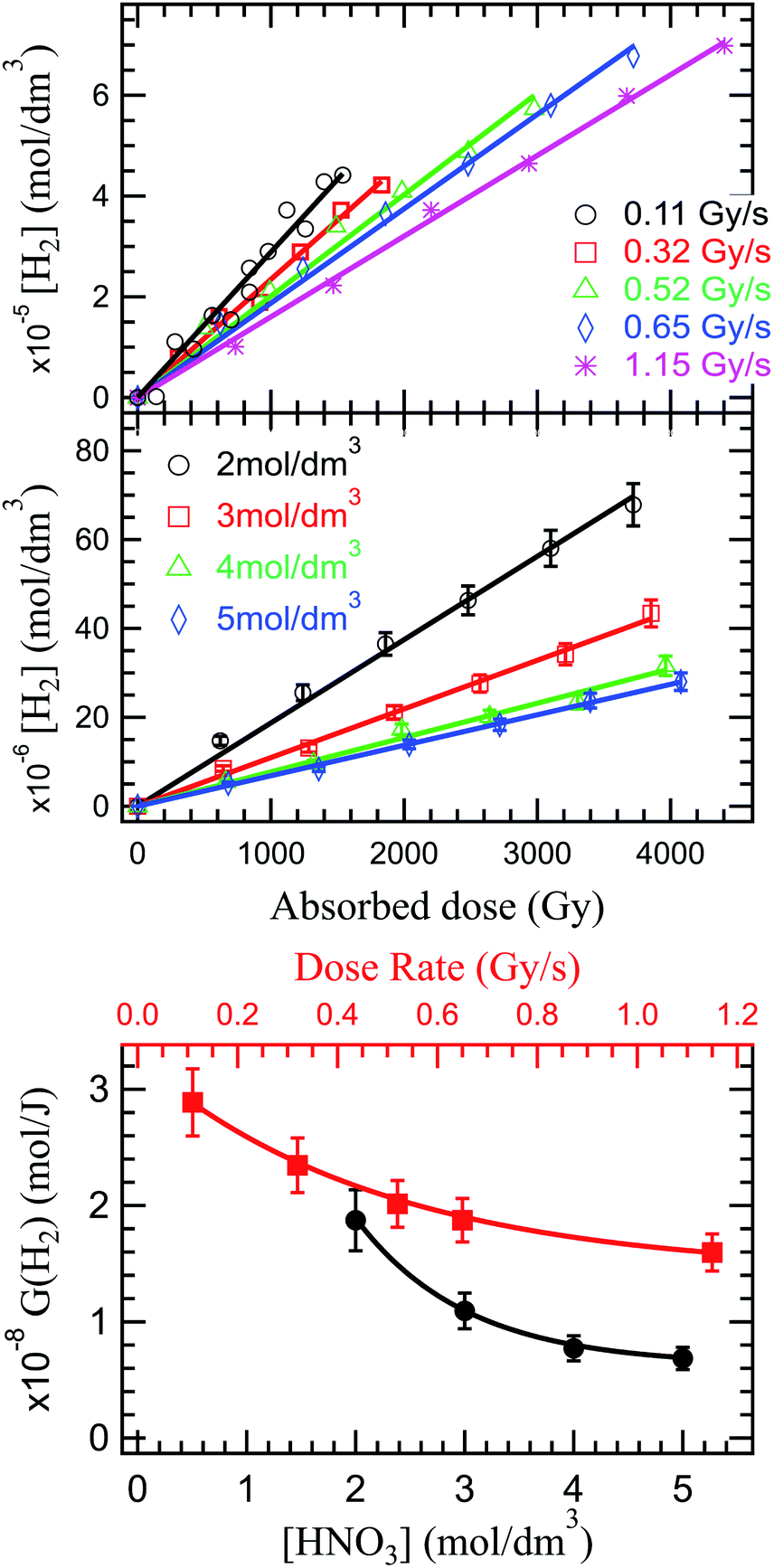 | ||
| Fig. 3 Top image: dose dependence of the H2 concentration (top) in solutions of 2 mol dm−3 HNO3 under α irradiation at various dose rates. Middle image: H2 concentration as a function of the absorbed dose in solutions of HNO3 for several concentrations indicating a decrease in H2 with increasing concentrations. Lower image: H2 radiolytic yield dependence on the concentration of HNO3 (black) and G(H2) dependence on the dose rate in a 2 mol dm−3 HNO3 solution (red). | ||
It has been shown that the excited water molecule decomposition is the greatest contributor to H2 formation (∼70%) in the radiolysis of water, and the role of the presolvated electron has been underlined.93 This latter species can undergo dissociative recombination with the parent water cation:
| e−pre + H2O+ → H2O* → H2 + O˙ 4.3 × 1012;93 | (54) |
In HNO3 solutions, these precursors are scavenged by nitrate ions (reaction (17)). Horne et al.24 showed that these reactions are not sufficient to account for the decrease of H2 yield in high HNO3 concentration, and identified the excited water molecule quenching by nitrate ion as responsible for the observed decrease:
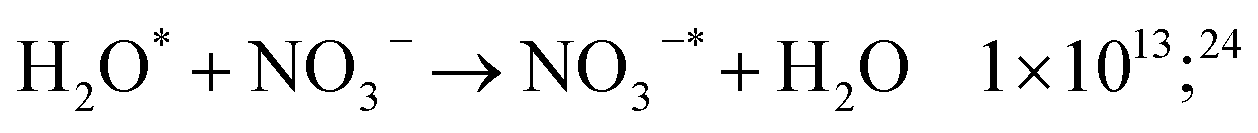 | (55) |
H˙ atoms, which are also produced can at their turn be scavenged by nitrate and nitrite ions and radicals as follows:
| NO3− + H˙ → HNO3− 1.0 × 107;78 5.56 × 106;100 | (56) |
| NO2− + H˙ → HNO2− 1.61 × 109;100 | (57) |
| HNO2 + H˙ → H2O + NO˙ 3.85 × 108;100 | (58) |
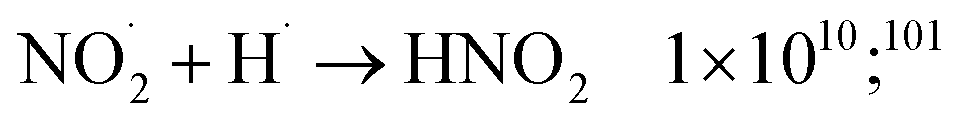 | (59) |
Previous reports indicated a decreased H2 production with increased nitrate concentration, due to the scavenging of H2 precursors (epre− and esol−) by nitrate (reaction (17) and (18)). But, as shown above, a complex series of reactions occurs in HNO3 solutions that results in a diminished H2 production decrease. This trend is observed in our investigations in Fig. 3 (middle and lower panel).
Fig. 4 shows a compilation of literature reported values of radiolytic yields of H2 and HNO2 measured in the α radiolysis of HNO3 solutions.
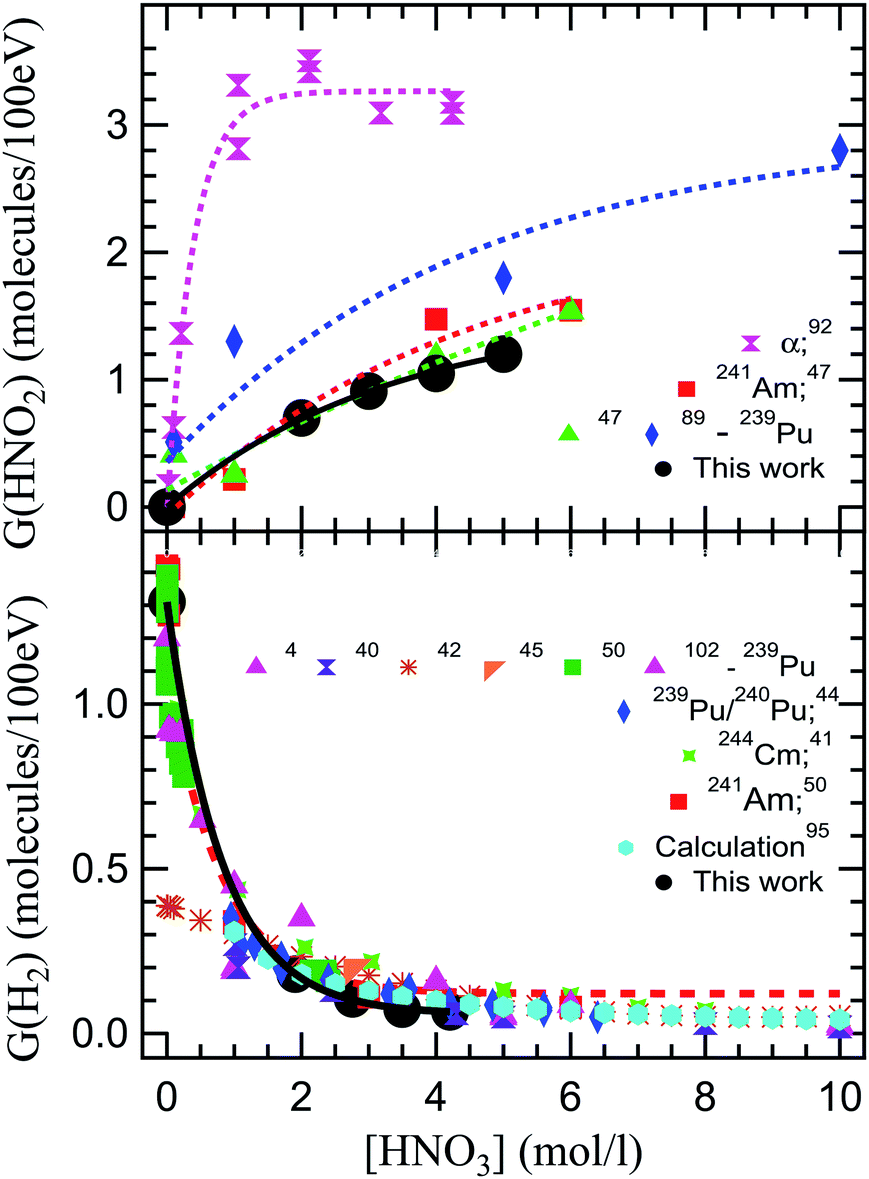 | ||
| Fig. 4 Comparison of our radiolytic yields of HNO2 (top image) and H2 (lower image) with the values reported in the literature. The G(HNO2) values are extracted from alpha radiolysis of HNO3 using helions,92 241Am,47 and 239Pu.47,89 G(H2) are the values reported in the alpha radiolysis of HNO3 solutions using 239Pu,4,40,42,45,50,102 mixtures of 239Pu/240Pu,44 244Cm,41 241Am,50 and calculated values.51,103 | ||
Looking at the previously reported G(HNO2), our results are closest to the ones obtained for α radiolysis using plutonium nitrate in HNO3,47 and rather inconsistent with measurements using helions.92 However, as mentioned, the external α irradiations were performed in NaNO3–HNO3 mixtures of constant NO3− concentration, by varying the H+ concentration. These results are obtained from measurements using hydrazinium as an HNO2 scavenger and following the decay of N2H5+ as well as the formation of NH3. This reaction occurs in competition with H2O2 consumption of HNO2, explaining the observed differences. Pu and Cm self-radiolysis of HNO3 provide higher values for G(H2), while Am yielded closer values to these external α investigations. No external α irradiation measurements on the radiolytic yield of H2 have been reported before this work. Compared to previous reports, we observe that our measurement indicate a slightly lower production of H2 in the α radiolysis of HNO3, but careful consideration needs to be taken when such comparisons are made, as the experimental conditions are different. For the α radiolysis investigations, there is a long standing debate over the use of external helions versus internal α particle emitters. This concerns the homogenous dose that is usually measured by performing a dosimetry versus the very high local dose deposited in solution in the case of external helions, and the localized dose deposition versus the calculation of the average deposited doses from the activities of the different actinides used as internal alpha sources. The discrepancies between the local dose deposited by external helions, evaluated from the ion current densities, and the dosimetry evaluated deposited dose have lead authors to the use of radionuclides in the detriment of cyclotrons for α sources. However, this is not applicable for these investigations. Continuous intense flow of our solutions insures (see Experimental section – dosimetry) that the irradiated volume is constantly refreshed, avoiding accumulation of degradation products and the mentioned enormous local doses, while insuring the rapid liquid/gas exchange. Moreover, when using internal α particle emitters, radionuclides-specific yields have been recorded due to the progressive disproportionation as a function of the nitric acid concentration induced by the complexing nature of HNO3 and its chemical involvement in the investigated processes.46,49,104–113 Simultaneously, investigations using radionuclides may pose exposure and contamination risks to the manipulators and require special equipment for safe handling that render them cumbersome. At the same time, questions are imposed about loss of products during handling. Working in a closed circuit allows us to avoid sampling of the headspace and replacing the sampled volume with laboratory air that can dilute the gaseous volume or induce leaks of H2 produced, considering the fugacity of H2 gas. Radioisotope/HNO3 mixtures result in self-radiolysis and high locally deposited energy, issue partially alleviated by stirring the solutions. Our experimental set-up allowed us to perform experiments, sampling the gas phase without inducing perturbations of the system. To the best of our knowledge, this is the first time that this type of experiment has been performed using external α irradiation.
Conclusions
The two most important species resulting from the radiolysis of HNO3 are HNO2 and H2. When considering the use of HNO3 in the retreatment of spent nuclear fuel, and therefore its radiolysis due to the high radiation fields it is exposed to, reliable information on the radiolytic yield of G(HNO2) and G(H2) is essential. If not scavenged, HNO2 can be responsible for partitioning failure as it plays on the oxidation state of U, Pu and minor actinides. H2 concentrations have to be closely monitored in order to avoid its accumulation in the retreatment units and the associated explosion risk. The main scope of these experiments was to gain insight into the α-radiolytic production of these two species, and identify any dose rate and concentration effects. UV-visible spectroscopy and gas mass spectrometry are versatile and extremely sensitive tools for performing quantitative analysis of desired products. Continuously monitoring HNO2 and H2 allowed us to perform measurements and quantify their production. These productions show dose rate effects that have been neglected up to now in the production of HNO2, as well as in the production of H2. Increasing concentrations of HNO3 have opposite effects on these products, leading to HNO2 increase and H2 decrease.Table 4 summarizes the determined radiolytic yields as a function of the HNO3 concentration and dose rate. Our measurements provide accurate values for the radiolytic yields of these species that are chemically independent of any radioisotope that could be used as an α radiation source. The information provided by this study is the first of this kind and starts shedding light on the complexity of the HNO3 chemical system under irradiation.
| [HNO3] (mol dm−3) | G(H2) (mol J−1) | G(HNO2) (mol J−1) | Dose rate (Gy s−1) | G(H2), 2 mol dm−3 HNO3 (mol J−1) | G(HNO2), 2 mol dm−3 HNO3 (mol J−1) |
|---|---|---|---|---|---|
| 0 | 1.9 × 10−7;57 | 0 | 0.11 | 2.4 × 10−8 | 1.6 × 10−7 |
| 2 | 1.8 × 10−8 | 7.2 × 10−8 | 0.32 | 2.3 × 10−8 | 1.1 × 10−7 |
| 3 | 1.1 × 10−8 | 9.6 × 10−8 | 0.52 | 2.1 × 10−8 | 7.6 × 10−8 |
| 4 | 7.6 × 10−9 | 1.1 × 10−7 | 0.65 | 1.9 × 10−8 | 7.2 × 10−8 |
| 5 | 7.1 × 10−9 | 1.3 × 10−7 | 1.15 | 1.7 × 10−8 | 4.6 × 10−8 |
From both fundamental and practical aspects, this study paves the way for further investigations on the radiolytic processes occurring in HNO3 mixtures relevant to SNF retreatment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to extend their gratitude to the entire CEMHTI team (Orléans, France. CNRS Campus) without whose help these time consuming and challenging experiments could not have been possible. We address special thanks to Thierry Sauvage and Dominique Baux for their constructive assistance in using the cyclotron installation. The authors acknowledge the financial funding from ORANO, EDF and CEA that made this project happen. The authors would like to thank Virginie Blin, COSTO project (ORANO/EDF/CEA) coordinator for her continuous support.Notes and references
- Paris Agreement (Dec. 13, 2015), in UNFCCC, COP Report No. 21, Addendum, at 21, U.N. Doc. FCCC/CP/2015/10/Add, 1 (Jan. 29, 2016) Search PubMed.
- F. J. Miner, A. R. Kazanjian, A. K. Brown, P. G. Hagan and J. W. Berry, Radiation Chemistry of Nitric Acid Solutions, The Dow Chemical Company, United States, 1969 Search PubMed.
- A. R. Kazanjian, F. J. Miner, A. K. Brown, P. G. Hagan and J. W. Berry, Radiolysis of Nitric Acid Solution : L.E.T. Effects, Trans. Faraday Soc., 1970, 66, 2192–2198 RSC.
- A. R. Kazanjian and D. R. Horrell, Radiolytically Generated Gases in Plutonium–Nitric Acid Solutions, Radiation Effects: Incorporating Plasma Science and Plasma Technology, 1972, 13, 277–280 CrossRef CAS.
- D. A. Crowl and Y.-D. Jo, The hazards and risks of hydrogen, J. Loss Prev. Process Ind., 2007, 20, 158–164 CrossRef CAS.
- F. L. Dryer, M. Chaos, Z. Zhao, J. N. Stein, J. Y. Alpert and C. J. Homer, Spontaneous Ignition of Pressurized Releases of Hydrogen and Natural Gas into Air, Combust. Sci. Technol., 2007, 179, 663–694 CrossRef CAS.
- H. A. Mahlman, Activity Concept in Radiation Chemistry, J. Chem. Phys., 1959, 31, 993–995 CrossRef CAS.
- L. L. Burger and M. D. Money, Nitrous Acid Behavior in Purex Systems, Hanford Laboratories Operation, United States, 1959 Search PubMed.
- H. A. Mahlman, Hydrogen Formation in the Radiation Chemistry of Water, J. Chem. Phys., 1960, 32, 601–603 CrossRef CAS.
- H. A. Mahlman, The OH Yield in the Co60 γ Radiolysis of HNO3, J. Chem. Phys., 1961, 35, 936–939 CrossRef CAS.
- H. A. Mahlman, The “Direct Effect” in the Radiolysis of Aqueous Sodium Nitrate Solutions, J. Phys. Chem., 1963, 67, 1466–1469 CrossRef CAS.
- M. L. Hyder, The Radiolysis of Aqueous Nitrate Solutions, J. Phys. Chem., 1965, 69, 1858–1865 CrossRef CAS.
- M. Daniels and E. E. Wigg, Radiation Chemistry of the Aqueous Nitrate System. I. γ-Radiolysis of Dilute Solutions, J. Phys. Chem., 1967, 71, 1024–1033 CrossRef CAS.
- M. Daniels and E. E. Wigg, Radiation Chemistry of the Aqueous Nitrate System. II. Scavenging and pH Effects in the Cobalt-60 Gamma Radiolysis of Concentrated Sodium Nitrate Solutions, J. Phys. Chem., 1969, 73, 3703–3709 CrossRef CAS.
- Z. D. Draganic and I. G. Draganic, Studies on the Formation of Primary Yields of Hydrogen Peroxide and Molecular Hydrogen (GH2O2 and GH2) in the Radiolysis of Neutral Aqueous Solutions, J. Phys. Chem., 1971, 75, 3950–3957 CrossRef CAS.
- R. W. Matthews, H. A. Mahlman and T. J. Sworski, Elementary Processes in the Radiolysis of Aqueous Nitric Acid Solutions. Determination of Both GOH and GNO3, J. Phys. Chem., 1972, 76, 2680–2684 CrossRef CAS.
- P. K. Bhattacharyya and R. D. Saini, Radiolytic Yields G(HNO2) and G(H2O2) in the Aqueous Nitric Acid System, Int. J. Radiat. Phys. Chem., 1973, 5, 91–99 CrossRef CAS.
- L. G. Rodenas, R. F. Prini and S. J. Liberman, Radiolysis of Aqueous Solutions of Gadolinium Nitrate, J. Radioanal. Nucl. Chem., 1990, 139, 277–286 CrossRef CAS.
- N. Nakagiri and T. Miyata, Evaluation of Value for Hydrogen Release from High-level Liquid Waste, (I). Gamma-Ray Radiolysis of Aqueous Nitric Acid Solutions., J. At. Energy Soc. Jpn., 1994, 36, 744–751 CrossRef CAS.
- B. Pastina, J. A. LaVerne and S. M. Pimblott, Dependence of Molecular Hydrogen Formation in Water on Scavengers of the Precursor to the Hydrated Electron, J. Phys. Chem. A, 1999, 103, 5841–5846 CrossRef CAS.
- K. Yoshida, H. Abe, Y. Yamane, S. Tashiro and K. Muramatsu, Research on the State-of-the-art of Accident Consequence Analysis Method for Non-reactor Nuclear Facilities, Japan Atomic Energy Agency, Japan, 2007 Search PubMed.
- G. Elias, PhD thesis, University of Idaho, 2010.
- G. P. Horne, T. A. Donoclift, H. E. Sims, R. M. Orr and S. M. Pimblott, Multi-Scale Modeling of the Gamma Radiolysis of Nitrate Solutions, J. Phys. Chem. B, 2016, 120, 11781–11789 CrossRef CAS PubMed.
- G. P. Horne, S. M. Pimblott and J. A. LaVerne, Inhibition of Radiolytic Molecular Hydrogen Formation by Quenching of Excited State Water, J. Phys. Chem. B, 2017, 121, 5385–5390 CrossRef CAS PubMed.
- D. Watanabe, Y. Wada, A. Sasahira, M. Itori and T. Ebina, Nitrous Acid Generation From Radiolysis of Nitric Acid Aqueous Solution Under Gas Flow Condition, J. Nucl. Sci. Technol., 2017, 54, 182–187 CrossRef CAS.
- R. K. Broszkiewicz, The Radiation-Induced Formation of NO3 in Aqueous Solutions, Int. J. Appl. Radiat. Isot., 1966, 18, 25–32 CrossRef.
- M. Daniels, Radiation Chemistry of the Aqueous Nitrate System. III. Pulse Electron Radiolysis of Concentrated Sodium Nitrate Solutions, J. Phys. Chem., 1969, 73, 3710–3717 CrossRef CAS.
- E. Kozłowska-Milner and R. K. Broszkiewicz, Pulse Radiolysis of HNO3 and HNO3(aq), Radiat. Phys. Chem., 1977, 1978(11), 253–260 Search PubMed.
- M. V. Vladimirova, I. A. Kulikov, O. A. Sosnovskii and A. A. Ryabova, Reduction of PuO22+ in y Radioysis in Aqueous HNO3, At. Energ., 1981, 51, 55–57 CAS.
- P. Neta and R. E. Huie, Rate constants for reactions of nitrogen oxide (NO3) radicals in aqueous solutions, J. Phys. Chem., 1986, 90, 4644–4648 CrossRef CAS.
- Y. Katsumura, P. Y. Jiang, R. Nagaishi, T. Oishi, K. Ishigure and Y. Yoshida, Pulse Radiolysis Study of Aqueous Nitric Acid Solutions: Formation Mechanism, Yield, and Reactivity of NO3 Radical, J. Phys. Chem., 1991, 95, 4435–4439 CrossRef CAS.
- G. A. Poskrebyshev, P. Neta and R. E. Huie, Equilibrium constant of the reaction ˙OH + HNO3 ⇆ H2O + NO3. in aqueous solution, J. Geophys. Res.: Atmos., 2001, 106, 4995–5004 CrossRef CAS.
- M. Precek, A. Paulenova, P. Tkac and N. Knapp, Effect of Gamma Irradiation on the Oxidation State of Neptunium in Nitric Acid in the Presence of Selected Scavengers, Sep. Sci. Technol., 2010, 45, 1699–1705 CrossRef CAS.
- M. Precek, A. Paulenova and B. J. Mincher, Reduction of Np(VI) in Irradiated Solutions of Nitric Acid, Procedia Chem., 2012, 7, 51–58 CrossRef CAS.
- A. Balcerzyk, A. K. El Omar, U. Schmidhammer, P. Pernot and M. Mostafavi, Picosecond Pulse Radiolysis Study of Highly Concentrated Nitric Acid Solutions: Formation Mechanism of NO3˙ Radical, J. Phys. Chem. A, 2012, 116, 7302–7307 CrossRef CAS PubMed.
- G. Garaix, G. P. Horne, L. Venault, P. Moisy, S. M. Pimblott, J. Marignier and M. Mostafavi, Decay Mechanism of NO3˙ Radical in Highly Concentrated Nitrate and Nitric Acidic Solutions in the Absence and Presence of Hydrazine, J. Phys. Chem. B, 2016, 120, 5008–5014 CrossRef CAS PubMed.
- R. Musat, S. A. Denisov, J.-L. Marignier and M. Mostafavi, Decoding the Three-Pronged Mechanism of NO3˙ Radical Formation in HNO3 Solutions at 22 and 80 °C Using Picosecond Pulse Radiolysis, J. Phys. Chem. B, 2018, 122, 2121–2129 CrossRef CAS PubMed.
- M. Lefort, Radiochimie des solutions aqueuses: Remarques particulières à l’action des rayons α, J. Chim. Phys., 1954, 51, 351–353 CrossRef CAS.
- M. V. Vladimirova, Alpha Radiolysis of Aqueous Solutions, Russ. Chem. Rev., 1964, 33, 212–220 CrossRef.
- J. C. Sheppard, Alpha Radiolysis of Plutonium (IV): Nitric Acid Solutions, Battelle-Northwest, Richland, Washington Pacific Northwest Lab, United States, 1968 Search PubMed.
- N. E. Bibler, Curium-244 alpha Radiolysis of Nitric Acid. Oxygen Production from Direct Radiolysis of Nitrate Ions, J. Phys. Chem., 1974, 78, 211–215 CrossRef CAS.
- A. Maimoni, Density and Radiolytic Decomposition of Plutonium Nitrate Solutions, Lawrence Livermore Laboratory, United States, 1979 Search PubMed.
- S. Tachimori, Numerical Simulation for Chemical Reactions of Actinide Elements in Aqueous Nitric Acid Solution, J. Nucl. Sci. Technol., 1991, 28, 218–227 CrossRef CAS.
- Y. Kuno, T. Hina and J. Masui, Radiolytically Generated Hydrogen and Oxygen from Plutonium Nitrate Solutions, J. Nucl. Sci. Technol., 1993, 30, 919–925 CrossRef CAS.
- J. R. Smith, Radiolysis Gases from Nitric Acid Solutions Containing HSA and HAN (U), Westinghouse Savannah River Company, 1994 Search PubMed.
- M. V. Vladimirova and A. V. Khaperskaya, Mechanism and Kinetics of Rh(IV) Radiation-Chemical Reduction in HNO3 Solutions, Radiochemistry, 2003, 45, 33–39 CrossRef CAS.
- G. P. Horne, C. R. Gregson, H. E. Sims, R. M. Orr, R. J. Taylor and S. M. Pimblott, Plutonium and Americium Alpha Radiolysis of Nitric Acid Solutions, J. Phys. Chem. B, 2017, 121, 883–889 CrossRef CAS PubMed.
- Z. Liu, Z. Fang, L. Wang, H. He and M.-Z. Lin, Alpha Radiolysis of Nitric Acid Aqueous Solution Irradiated by 238Pu Source, Nucl. Sci. Tech., 2017, 28, 54 CrossRef.
- T. S. Grimes, G. P. Horne, C. J. Dares, S. M. Pimblott, S. P. Mezyk and B. J. Mincher, Kinetics of the Autoreduction of Hexavalent Americium in Aqueous Nitric Acid, Inorg. Chem., 2017, 56, 8295–8301 CrossRef CAS PubMed.
- C. R. Gregson, G. P. Horne, R. M. Orr, S. M. Pimblott, H. E. Sims, R. J. Taylor and K. J. Webb, Molecular Hydrogen Yields from the α-Self-Radiolysis of Nitric Acid Solutions Containing Plutonium or Americium, J. Phys. Chem. B, 2018, 122, 2627–2634 CrossRef CAS.
- R. Nagaishi, A Model for Radiolysis of Nitric Acid and its Application to the Radiation Chemistry of Uranium Ion in Nitric Acid Medium, Radiat. Phys. Chem., 2001, 60, 369–375 CrossRef CAS.
- M. B. Shinn, Colorimetric Method for Determination of Nitrate, Ind. Eng. Chem., Anal. Ed., 1941, 13(1), 33–35 CrossRef CAS.
- K. Bendschneider and R. J. Robinson, New Spectrophotometric Method for the Determination of Nitrite in Sea Water, University of Washington Oceanographic Laboratories, Seattle, WA, USA, 1952 Search PubMed.
- A. O. Allen, C. J. Hochanadel, J. A. Ghormley and T. W. Davis, Decomposition of Water and Aqueous Solutions under Mixed Fast Neutron and γ-Radiation, J. Phys. Chem., 1952, 56, 575–586 CrossRef.
- J. W. T. Spinks and R. J. Woods, An introduction to radiation chemistry, Wiley, New York, 3rd edn, 1990 Search PubMed.
- F. Crumière, J. Vandenborre, R. Essehli, G. Blain, J. Barbet and M. Fattahi, LET effects on the hydrogen production induced by the radiolysis of pure water, Radiat. Phys. Chem., 2013, 82, 74–79 CrossRef.
- R. Essehli, F. Crumière, G. Blain, J. Vandenborre, F. Pottier, B. Grambow, M. Fattahi and M. Mostafavi, H2 Production by γ and He Ions Water Radiolysis, Effect of Presence TiO2 Nanoparticles, Int. J. Hydrogen Energy, 2011, 36, 14342–14348 CrossRef CAS.
- J. A. LaVerne and L. Tandon, H2 Production in the Radiolysis of Water on CeO2 and ZrO2, J. Phys. Chem. B, 2002, 106, 380–386 CrossRef CAS.
- J. F. Ziegler and J. P. Biersack, in Treatise on Heavy-Ion Science: Volume 6: Astrophysics, Chemistry, and Condensed Matter, Springer US, Boston, MA, 1985, pp. 93–129 Search PubMed.
- J. F. Ziegler, M. D. Ziegler and J. P. Biersack, SRIM – The stopping and range of ions in matter, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 2010(268), 1818–1823 CrossRef.
- R. W. Matthews, Aqueous Chemical Dosimetry, Int. J. Appl. Radiat. Isot., 1982, 33, 1159–1170 CrossRef CAS.
- R. D. Saini and P. K. Bhattacharyya, Radiolytic Oxidation of U(IV) Sulphate in Aqueous Solution by Alpha Particles from Cyclotron, Int. J. Radiat. Appl. Instrum. C Radiat. Phys. Chem., 1987, 29, 375–379 CAS.
- J. A. LaVerne and R. H. Schuler, Radiation Chemical Studies with Heavy Ions: Oxidation of Ferrous Ion in the Fricke Dosimeter, J. Phys. Chem., 1987, 91, 5770–5776 CrossRef CAS.
- M. Matsui, H. Seki, T. Karasawa and M. Imamura, Radiation Chemical Studies with Cyclotron Beams, (I): Fricke Solution, J. Nucl. Sci. Technol., 1970, 7, 97–104 CrossRef CAS.
- C. Costa, J. Vandenborre, F. Crumière, G. Blain, R. Essehli and M. Fattahi, Chemical Dosimetry during Alpha Irradiation: A Specific System for UV-vis in situ Measurement, Am. J. Anal. Chem., 2012, 03, 6–11 CrossRef CAS.
- R. M. Smith and A. E. Martell, Critical Stability Constants: Inorganic Complexes, Springer-Verlag New York, 1976 Search PubMed.
- J. Chlistunoff, K. J. Ziegler, L. Lasdon and K. P. Johnston, Nitric/Nitrous Acid Equilibria in Supercritical Water, J. Phys. Chem. A, 1999, 103, 1678–1688 CrossRef CAS.
- E. Riordan, N. Minogue, D. Healy, P. O'Driscol and J. R. Sodeau, Spectroscopic and Optimization Modeling Study of Nitrous Acid in Aqueous Solution, J. Phys. Chem. A, 2005, 109, 779–786 CrossRef CAS.
- P.-Y. Jiang, R. Nagaishi, T. Yotsuyanagi, Y. Katsumura and K. Ishigure, γ-Radiolysis study of concentrated nitric acid solutions, J. Chem. Soc., Faraday Trans., 1994, 90, 93–95 RSC.
- Z. B. Alfassi, N-Centered Radicals, Wiley, Chichester , New York, 1998 Search PubMed.
- S. M. Pimblott and J. A. LaVerne, On the Radiation Chemical Kinetics of the Precursor to the Hydrated Electron, J. Phys. Chem. A, 1998, 102, 2967–2975 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (˙OH/˙O−) in Aqueous Solution, J. Phys. Chem. Ref. Data, 1988, 17, 2 CrossRef.
- T. Loegager and K. Sehested, Formation and Decay of Peroxynitrous Acid: a Pulse Radiolysis Study, J. Phys. Chem., 1993, 97, 6664–6669 CrossRef CAS.
- M. Grätzel, A. Henglein and S. Taniguchi, Pulsradiolytische Beobachtungen über die Reduktion des NO3− -Ions und über Bildung und Zerfall der persalpetrigen Säure in wäßriger Lösung, Berichte der Bunsengesellschaft für physikalische Chemie, 1970, 74, 292–298 CrossRef.
- A. Furuhama, M. Dupuis and K. Hirao, Reactions associated with ionization in water: A direct ab initio dynamics study of ionization in (H2O)17, J. Chem. Phys., 2006, 124, 3–13 CrossRef.
- O. Marsalek, C. G. Elles, P. A. Pieniazek, E. Pluhařová, J. VandeVondele, S. E. Bradforth and P. Jungwirth, Chasing charge localization and chemical reactivity following photoionization in liquid water, J. Chem. Phys., 2011, 135, 224510 CrossRef PubMed.
- M. Grätzel, A. Henglein, J. Lilie and G. Beck, Pulsradiolytische Untersuchung einiger Elementarprozesse der Oxydation und Reduktion des Nitritions, Berichte der Bunsengesellschaft für physikalische Chemie, 1969, 73, 646–653 Search PubMed.
- T. Loegager and K. Sehested, Formation and Decay of Peroxynitric Acid: A Pulse Radiolysis Study, J. Phys. Chem., 1993, 97, 10047–10052 CrossRef CAS.
- C. D. Jonah, J. R. Miller and M. S. Matheson, The reaction of hydrated electron + oxonium. Concentration effects of acid or salts, J. Phys. Chem., 1977, 81, 931–934 CrossRef CAS.
- A. J. Elliott and D. M. Bartels, The Reaction Set, Rate Constants and g-Values for the Simulation of the Radiolysis of Light Water over the Range 20° to 350 °C Based on Information Available in 2008, Atomic Energy of Canada Limited, Canada, 2009 Search PubMed.
- P. Y. Jiang, Y. Katsumura, K. Ishigure and Y. Yoshida, Reduction Potential of the Nitrate Radical in Aqueous Solution, Inorg. Chem., 1992, 31, 5135–5136 CrossRef CAS.
- H. Taube, Photochemical Reactions of Ozone in Solution, Trans. Faraday Soc., 1957, 53, 656 RSC.
- P. G. Sennikov, S. K. Ignatov and O. Schrems, Complexes and Clusters of Water Relevant to Atmospheric Chemistry: H2O Complexes with Oxidants, ChemPhysChem, 2005, 6, 392–412 CrossRef CAS.
- S. Xu, V. Jirasek and P. Lukes, Molecular Dynamics Simulations of Singlet Oxygen Atoms Reactions with Water Leading to Hydrogen Peroxide, J. Phys. D: Appl. Phys., 2020, 53, 275204 CrossRef CAS.
- M. Fischer and P. Warneck, Photodecomposition of Nitrite and Undissociated Nitrous Acid in Aqueous Solution, J. Phys. Chem., 1996, 100, 18749–18756 CrossRef CAS.
- S. I. Nikitenko, P. Moisy, L. Venault and C. Madic, Kinetics of Nitrous Acid Formation in a Two-Phase Tri-n-Butylphosphate–Diluent/Aqueous Nitric Acid Extraction System Under the Effect of Power Ultrasound, Ultrason. Sonochem., 2000, 7, 135–144 CrossRef CAS.
- D. Vione, V. Maurino, C. Minero, D. Borghesi, M. Lucchiari and E. Pelizzetti, New Processes in the Environmental Chemistry of Nitrite. 2. The Role of Hydrogen Peroxide, Environ. Sci. Technol. Libr., 2003, 37, 4635–4641 CrossRef CAS.
- B. J. Mincher, M. Precek, S. P. Mezyk, G. Elias, L. R. Martin and A. Paulenova, The Redox Chemistry of Neptunium in γ-Irradiated Aqueous Nitric Acid, Radiochim. Acta, 2013, 101, 259–266 CrossRef CAS.
- F. J. Miner, A. R. Kazanjian, A. K. Brown, P. G. Hagan and J. W. Berry, Radiation Chemistry of Nitric Acid Solutions, The Dow Chemical Company, United States, 1969 Search PubMed.
- C. Ferradini and J.-P. Jay-Gerin, La radiolyse de l'eau et des solutions aqueuses : historique et actualité, Can. J. Chem., 1999, 77, 1542–1575 CrossRef CAS.
- J. E. Fanning, C. N. Trumbore, P. G. Barkley, D. R. Short and J. H. Olson, Preliminary Report of a Spur Model Including Spur Overlap, J. Phys. Chem., 1977, 81, 1026–1029 CrossRef CAS.
- G. Garaix, L. Venault, A. Costagliola, J. Maurin, M. Guigue, R. Omnee, G. Blain, J. Vandenborre, M. Fattahi, N. Vigier and P. Moisy, Alpha Radiolysis of Nitric Acid and Sodium Nitrate with 4He2+ Beam of 13.5 MeV Energy, Radiat. Phys. Chem., 2015, 106, 394–403 CrossRef CAS.
- J. A. LaVerne and S. M. Pimblott, New Mechanism for H2 Formation in Water, J. Phys. Chem. A, 2000, 104, 9820–9822 CrossRef CAS.
- C. N. Trumbore, D. R. Short, J. E. Fanning and J. H. Olson, Effects of Pulse Dose on Hydrated Electron Decay Kinetics in the Pulse Radiolysis of Water. A Computer Modeling Study, J. Phys. Chem., 1978, 82, 2762–2767 CrossRef CAS.
- A. Mozumder, Fundamentals of radiation chemistry, Academic Press, San Diego, 1999 Search PubMed.
- B. J. Mincher, G. Elias, L. R. Martin and S. P. Mezyk, Radiation Chemistry and the Nuclear Fuel Cycle, J. Radioanal. Nucl. Chem., 2009, 282, 645–649 CrossRef CAS.
- A. J. Elliot and D. M. Bartels, The Reaction Set, Rate Constants and g-Values for the Simulation of the Radiolysis of Light Water over the Range 20° to 350°C Based on Information Available in 2008, Atomic Energy of Canada Limited, Canada, 2009 Search PubMed.
- E. Peled and G. Czapski, Molecular Hydrogen Formation (GH2) in the Radiation Chemistry of Aqueous Solutions, J. Phys. Chem., 1970, 74, 2903–2911 CrossRef CAS.
- H. A. Schwarz, Applications of the Spur Diffusion Model to the Radiation Chemistry of Aqueous Solutions, J. Phys. Chem., 1969, 73, 1928–1937 CrossRef CAS.
- S. P. Mezyk and D. M. Bartels, Temperature Dependence of Hydrogen Atom Reaction with Nitrate and Nitrite Species in Aqueous Solution, J. Phys. Chem. A, 1997, 101, 6233–6237 CrossRef CAS.
- B. Smaller, E. C. Avery and J. R. Remko, EPR Pulse Radiolysis Studies of the Hydrogen Atom in Aqueous Solution. I. Reactivity of the Hydrogen Atom, J. Chem. Phys., 1971, 55, 2414–2418 CrossRef CAS.
- J. I. Savel'ev, Z. V. Ersova and M. V. Vladimirova, Radiolyse Alpha des Solutions Aqueuses d'Acide Nitrique, Radiochimie, 1967, 9, 244–249 Search PubMed.
- N. E. Bibler, J. M. Pareizs, T. L. Fellinger and C. J. Bannochie, in INIS-US–09-WM-07162, Tucson, Arizona, 2007, vol. 41 Search PubMed.
- K. W. Bagnall, The chemistry of polonium, Q. Rev., Chem. Soc., 1957, 11, 30 RSC.
- P. I. Artyukhin, V. I. Medvedovskii and A. D. Gel’man, Disproportionation of Pu(IV) and Pu(V) in Nitric Acid Solutions, Zh. Neorg. Khim., 1959, 4, 1324–1331 CAS.
- H. Escure, D. Gourisse and J. Lucas, Dismutation du neptunium pentavalent en solution nitrique—I, J. Inorg. Nucl. Chem., 1971, 33, 1871–1876 CrossRef CAS.
- V. S. Koltunov and M. F. Tikhonov, Kinetics of Neptunium (5) Disproprotionation in Nitric Acid Solution, Radiokhimiya, 1975, 17, 560–563 CAS.
- Z.-M. Zhou, Y.-J. Zhang and H.-F. Du, Kinetic Studies on the Oxidation of Uranium (IV) in Nitric Acid Solution, J. Radioanal. Nucl. Chem., 1994, 188, 177–187 CrossRef CAS.
- V. S. Koltunov, V. I. Marchenko, G. I. Zhuravleva and O. A. Savilova, Kinetics of Redox Reactions of U, Pu, and Np in TBP Solutions: VII. Kinetics of Reduction of Pu(IV) and Np(VI) with Butanal Oxime in Undiluted TBP, Radiochemistry, 2001, 43, 334–337 CrossRef CAS.
- C. Gregson, C. Boxall, M. Carrott, S. Edwards, M. Sarsfield, R. Taylor and D. Woodhead, Neptunium (V) Oxidation by Nitrous Acid in Nitric Acid, Procedia Chem., 2012, 7, 398–403 CrossRef CAS.
- M. Precek, PhD thesis, Oregon State University, 2013.
- H. Chen, R. J. Taylor, M. Jobson, D. A. Woodhead, C. Boxall, A. J. Masters and S. Edwards, Simulation of Neptunium Extraction in an Advanced PUREX Process—Model Improvement, Solvent Extr. Ion Exch., 2017, 35, 1–18 CrossRef CAS.
- M. Chotkowski, Redox interactions of technetium with neptunium in acid solutions, J. Radioanal. Nucl. Chem., 2018, 317, 527–533 CrossRef CAS PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/d0ra10061g |
| This journal is © The Royal Society of Chemistry 2021 |