
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of H2SO4/H2O2 pre-treatment on electrochemical properties of exfoliated graphite prepared by an electro-exfoliation method†
Oktaviardi Bityasmawan Abdillaha,
Octia Flowerib,
Tirta Rona Mayangsari c,
Sigit Puji Santosad,
Takashi Ogie and
Ferry Iskandar*abd
c,
Sigit Puji Santosad,
Takashi Ogie and
Ferry Iskandar*abd
aDepartment of Physics, Faculty of Mathematics and Natural Sciences, Institut Teknologi Bandung, Jl. Ganesha 10, Bandung 40132, Indonesia. E-mail: ferry@fi.itb.ac.id
bResearch Center for Nanosciences and Nanotechnology (RCNN), Institut Teknologi Bandung, Jl. Ganesha 10, Bandung 40132, Indonesia
cDepartment of Chemistry, Universitas Pertamina, Jl. Teuku Nyak Arief, Simprug, Jakarta 12220, Indonesia
dNational Center for Sustainable Transportation Technology (NCSTT), Institut Teknologi Bandung, Jl. Ganesha 10, Bandung 40132, Indonesia
eChemical Engineering Program, Graduate School of Advanced Science and Engineering, Hiroshima University, 1-4-1 Kagamiyama, Hiroshima 739-8527, Japan
First published on 15th March 2021
Abstract
The effect of pre-treating graphite sheets in a H2SO4/H2O2 solution before electro-exfoliation is reported. It was revealed that the volume fraction of H2SO4 to H2O2 during pre-treatment could control the degree of exfoliation of the resulting exfoliated graphite (EG). X-ray diffraction (XRD), Raman, and Fourier transform infrared (FTIR) spectroscopy analyses have suggested that EG produced by first pre-treating the graphite sheet in H2SO4/H2O2 solution with the H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O2 volume fraction of 95:5 demonstrates the highest exfoliation degree. This sample also demonstrated excellent electrochemical properties with good electrical conductivity (36.22 S cm−1) and relatively low charge transfer resistance (Rct) of 21.35 Ω. This sample also showed the highest specific capacitance of all samples, i.e., 71.95 F g−1 at 1 mV s−1 when measured at a voltage range of −0.9 to 0 V. Further measurement at an extended potential window down to −1.4 V revealed the superior specific capacitance value of 150.69 F g−1. The superior morphology characteristics and the excellent electrical properties of the obtained EG are several reasons behind its exceptional properties. The pre-treatment of graphite sheets in H2SO4/H2O2 solution allegedly leads to easier and faster exfoliation. The faster exfoliation is allegedly able to prevent massive oxidation, which frequently induces the formation of graphite/graphene oxide (GO) in a prolonged process. However, too large H2O2 volume fraction involved during pre-treatment seems to cause excessive expansion and frail structure of the graphite sheets, which leads to an early breakdown of the structure during electrochemical exfoliation and prohibits layer by layer exfoliation.
:H2O2 volume fraction of 95:5 demonstrates the highest exfoliation degree. This sample also demonstrated excellent electrochemical properties with good electrical conductivity (36.22 S cm−1) and relatively low charge transfer resistance (Rct) of 21.35 Ω. This sample also showed the highest specific capacitance of all samples, i.e., 71.95 F g−1 at 1 mV s−1 when measured at a voltage range of −0.9 to 0 V. Further measurement at an extended potential window down to −1.4 V revealed the superior specific capacitance value of 150.69 F g−1. The superior morphology characteristics and the excellent electrical properties of the obtained EG are several reasons behind its exceptional properties. The pre-treatment of graphite sheets in H2SO4/H2O2 solution allegedly leads to easier and faster exfoliation. The faster exfoliation is allegedly able to prevent massive oxidation, which frequently induces the formation of graphite/graphene oxide (GO) in a prolonged process. However, too large H2O2 volume fraction involved during pre-treatment seems to cause excessive expansion and frail structure of the graphite sheets, which leads to an early breakdown of the structure during electrochemical exfoliation and prohibits layer by layer exfoliation.
Introduction
Exfoliated graphite (EG) is a type of graphene-like material formed when stacked graphitic layers undergo partial separation.1,2 Even though the produced material is not as thin as graphene, it is considered a good deal as its properties resemble that of graphene and it is easier to produce and is scalable for industry.3 Like graphene, EG can be applied in various applications such as catalyst supports, gaskets, the anode of lithium-ion batteries, supercapacitors, etc., in which materials with large surface area and superior electrochemical properties are needed.1,4–6 Among various feasible applications, graphene and EG applied as supercapacitors have been highly projected and widely studied.7,8 The supercapacitor is a type of energy storage device with several advantages such as high power density, relatively long lifetime, and simple operation.9 This superiority makes it suitable for the applications which require high power output and fast charge–discharge process such as load-levelling in various energy sources and energy recovery from regenerative braking in vehicles.10 Compared to pristine graphene, EG is expected to show better long-term properties as a supercapacitor because it normally contains enough oxygen functional groups bound to its edges and basal planes.11 The presence of oxygen-containing functional groups is beneficial as they can prevent the re-stacking of graphene layers, thus increasing the specific capacitance.11–13 However, the excessive oxygen functional groups are not good for supercapacitor applications since they can also disrupt the electron transport in the materials, and thus decreasing the specific capacitance.14,15Various methods have been frequently used to fabricate EG, including micromechanical exfoliation,16 chemical exfoliation through reduction of exfoliated graphite oxide,17 and electro-exfoliation.18 Among them, electro-exfoliation has several advantages such as cost-effective, easy, and high scalability.19 This method offers controllable properties of EG products by selecting applied voltages, aqueous or non-aqueous electrolytes, and types of graphite source.19,20 Besides, this method also produces EG with lower oxygen content, and higher electrical conductivity than that produced via chemical exfoliation route.18,21 For those positive reasons, the electro-exfoliation method is considered highly suitable to be applied in producing EG that fits various applications in which EG with excellent electrochemical properties is required, including supercapacitor.21
Based on the applied bias, the electro-exfoliation process could be classified into two types, i.e., cathodic and anodic exfoliation. Cathodic exfoliation is a process where a negative potential is applied to the graphite electrode. This process requires organic electrolytes or solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N-methyl-2-pyrrolidone (NMP), or propylene carbonate (PC). On the other hand, in anodic exfoliation, a positive potential is applied to the graphite precursor, and it can be easily conducted in an aqueous solution of inorganic electrolytes.20 The needs for organic solvents make the application of cathodic exfoliation limited because the organic solvents are relatively expensive, and their utilisation may cause pollution to the environment. Therefore, anodic exfoliation is preferred as it can be carried out in an aqueous solution using less expensive electrolyte materials.
However, despite its advantages, the conventional anodic exfoliation tends to produce a rather low yield.22,23 To improve the exfoliation efficiency, Munuera et al. involved a simple pre-treatment before the electrochemical process by immersing a graphite precursor in concentrated sulfuric acid (H2SO4) for 48 h.24 They could improve the yield of the graphene product up to five times produced without the pre-treatment, i.e., 50 wt%. The pre-treatment is believed to improve the exfoliation of graphite during the electrochemical process by letting H2SO4 molecules fill the voids and interstitial spaces within the graphite, turning the graphite become hygroscopic materials. The change in the graphite properties is supposed to allow easier intercalation of the anions and water from electrolytes into the graphite during the electrochemical process, resulting in a more extensive exfoliation of graphene layers. The high yield of EG obtained via H2SO4 pre-treatment has made the anodic exfoliation process of graphite more efficient. However, modification to the procedure is still needed as the pre-treatment time up to 48 h is still rather long. Therefore, an improved pre-treatment procedure is needed to produce EG with good properties and high yield in a relatively shorter time.
The initial expansion of graphite has become one powerful way to increase efficiency and reduce the required time for graphite exfoliation.25–27 This method involves an interlayer gas evolution caused by a chemical reaction between graphite intercalant molecules (such as FeCl3, CrO3, and H2SO4) and reactive species (such as (NH4)2S2O8, and H2O2). Lin et al. have studied the initial expansion of graphite by treating the graphite in the mixture of CrO3 and H2O2.27 They have successfully obtained the highly expanded graphite (1000 folds of initial volume), which could be easily exfoliated to produce few-layer graphene. The utilisation of H2O2 as reactive species is interesting because it could be carried out at room temperature without producing harmful byproduct.27 Besides, Gu et al. have also reported the utilisation of H2O2 and H2SO4 to open up the spacing of graphite layers into expandable graphite.28 The expandable graphite was then subjected to thermal shock to produce worm-like expanded graphite (WEG), which was used as the precursor for graphene synthesis via ultrasonication.
A simple modification of the pre-treatment process was carried out by involving H2O2 in the H2SO4 pre-treatment of graphite in the present work. During this process, the graphite is expected to experience initial expansion due to the evolution of oxygen gases caused by H2O2 decomposition, which is probably beneficial to ensure a more efficient electro-exfoliation process. To the best of our knowledge, none has reported the pre-treatment process of graphite precursor in the H2SO4/H2O2 solution before the electro-exfoliation procedure. The effect of graphite pre-treatment in H2SO4/H2O2 solution on the electro-exfoliation process and the electrochemical properties (e.g., charge transfer resistance, specific capacitance, and potential window) of the EG products were investigated. This study is beneficial for the development of exceptional EG-based supercapacitors via a facile and efficient electro-exfoliation method.
Materials and methods
Materials
Graphite sheet (Hi-tech Carbon, Ltd.), concentrated sulfuric acid (H2SO4 98%, Smartlab Indonesia), hydrogen peroxide (H2O2 50%, Bratachem), ammonium sulfate ((NH4)2SO4, Pure Analysis, Merck), polyvinylidene fluoride (PVDF, MTI), conductive carbon black (MTI), N-methyl-2-pyrrolidone (NMP, Pure Analysis, Merck), potassium hydroxide (KOH, Pure Analysis, Merck), deionised water (DI water), and ethanol.Pre-treatment of the graphite sheet
Graphite sheets were cut into a rectangular shape with an approximate size of 5.5 × 2.5 cm. H2SO4/H2O2 solutions with the various volume fraction of H2SO4:H2O2, i.e., 100:0, 95:5, 93:7, and 91:9, were freshly prepared by mixing the desired amount of concentrated sulfuric acid (H2SO4 98%) and hydrogen peroxide (H2O2 50%) solution to a volume total of 40 mL. Pre-treatment was conducted by immersing a ply of graphite sheet in the H2SO4:H2O2 solution by leaving about 1.5 cm part unsubmerged for alligator clamp. After pre-treatment for 3 min, the graphite sheet was carefully moved to the different chamber for the electro-exfoliation process. The graphite sheet (GS) samples after pre-treatment were named after the variation of H2SO4:H2O2 volume ratio used during pre-treatment, i.e., GS 100:0, GS 95:5, GS 93:7, and GS 91:9.
Preparation of electrochemically exfoliated graphite
In the electro-exfoliation process, the pre-treated graphite sheet and Pt wire were set as the working electrode and counter electrode, respectively. As for electrolyte, 100 mL of ammonium sulfate ((NH4)2SO4) 0.1 M solution was prepared by dissolving 1.32 g of (NH4)2SO4 in DI water. During the electrochemical exfoliation process, the electrodes were given a constant potential bias of 10 V until no more current could be observed, which means the end of the process. After that, the exfoliated product was washed repeatedly using DI water until a pH of 7.0 was reached. This action was carried out to remove the remaining ions attached to the sample. After being dried in an oven at a temperature of 40 °C for 6 h, the product was dispersed in DI water (1 mg mL−1) using an ultrasonic homogeniser for an hour (pulsed 480 W). The resulting dispersion was centrifuged at 1000 rpm for 20 min to separate large particles from the dispersion. Finally, the top part of the dispersion was poured onto a Petri dish and oven-dried at 80 °C for 18 h. The resulting exfoliated graphite (EG) samples were named after the variation of H2SO4:H2O2 volume ratio used during pre-treatment, i.e., EG 100:0, EG 95:5, EG 93:7, and EG 91:9.
Characterisations
The structural ordering was characterised using an X-ray diffractometer (XRD, Bruker D8 Advance, Bruker) with Cu Kα radiation at 1.5406 Å. Meanwhile, Raman spectroscopy (Modular Horiba Jobin Yvon type iHR320, Horiba) with 532 nm laser excitation was utilised to study the crystal defect and exfoliation of the samples. Morphology observation and C/O elemental composition analysis of the samples was carried out using a scanning electron microscope and energy dispersive X-ray, respectively (SEM and EDX, SU3500, Hitachi). Fourier transform infrared spectroscopy (FT-IR, Prestige 21, Shimadzu) characterisation was employed to observe the functional group contained in the samples.The electrical conductivity of pelletised samples was measured using a four-point probe connected to a direct current (DC) source (R6240A, Advantest) and multimeter (2100 Series: 61/2-digit USB multimeter, Keithley). Meanwhile, their electrochemical impedance spectroscopy (EIS) measurement and cyclic voltammetry (CV) was carried out using potentiostat/galvanostat (PARSTAT 3000A, Princeton Applied Research) with Ag/AgCl as reference electrode and Pt wire as a counter electrode in a three-electrode system. Before the measurement, the EG samples were first mixed with PVDF binder and conductive carbon black ratio of 8:1:1 using NMP as a dispersant to form slurries. The resulting slurries were deposited on a stainless-steel foil and dried in a vacuum oven at 100 °C for 12 h to make electrodes. The electrodes were then cut into a size of ca. 1 × 1 cm. EIS measurement was carried out with 10 mV AC voltage at a frequency range of 100000 to 0.1 Hz. Meanwhile, CV measurement was conducted with a scan rate of 1–200 mV s−1 in 6 M KOH aqueous electrolyte.
Results and discussion
Fig. 1(a)–(e) shows the photographed images of the graphite sheet before and after the pre-treatment process carried out by immersing the pristine graphite sheet (p-GS) in H2SO4/H2O2 solution for 3 min. It can be seen that after pre-treatment, the graphite sheet pre-treated in a solution containing H2O2 underwent expansion. The expansion was most likely caused by the presence of H2O2 in the pre-treatment solution, as H2O2 may be self-decomposed following chemical reaction shown in eqn (1), which has been reported to occur more vigorously in the presence of concentrated sulfuric acid.29| 2H2O2(aq) → 2H2O(l) + O2(g) | (1) |
 | ||
| Fig. 1 Photographed images (a–e) and optical microscope images (f–j) of p-GS (a and f), and GS pre-treated in H2SO4/H2O2 solution with various volume fraction of H2SO4:H2O2, i.e. GS 100:0 (b and g), GS 95:5 (c and h), GS 93:7 (d and i), and GS 91:9 (e and j). Yellow arrows show several macro-cracking formed in the sample pre-treated in solution with a higher volume fraction of H2O2. | ||
As a result, oxygen gases (O2) are generated within the graphite slabs, and forces expansion of the interlayer graphite.27,30,31 The proposed mechanisms are in line with the fact that the expansion of the graphite sheet is greater with the increasing H2O2 volume fraction involved during pre-treatment, as shown by the photographed and optical microscope images of all samples in Fig. 1(a)–(j). This judgment is also in line with the visible evidence witnessed during the pre-treatment of the graphite sheets; more bubbles were generated when more H2O2 was involved, indicating more oxygen produced during the process. Morphology of the pre-treated samples observed using an optical microscope revealed several macro cracking seen from the samples pre-treated using a higher proportion of H2O2 (H2SO4:H2O2 93:7 and 91:9) as indicated by the yellow arrows. This phenomenon suggests the more extensive expansion due to a larger amount of oxygen bubbles generated within the graphite slabs.
Fig. 2(a) shows the Raman spectra of the p-GS and EG samples. All spectra reveal the presence of three peaks, which are typically seen in Raman spectra of graphitic carbon materials, i.e., D, G, and 2D peaks at Raman shift of ca. 1350 cm−1, 1580 cm−1, and 2650 cm−1, respectively. Sourced from the stretching of C–C bonds (sp3), D peak is often associated with the structural defect and oxidation of graphitic carbon.32,33 On the contrary, the G peak is attributed to the structural perfection of graphitic carbon as it is originated from the stretching of sp2 hybridised carbons (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C).34 Thus, the ratio of D to G peak intensity (ID/IG) is often used to assess the extent of damage in carbonaceous materials.35 It can be seen from Fig. 2(a) that the intensity of D peak and ID/IG value of all EG samples both are higher than that of the pristine graphite sheet, indicating more defective and oxidised structure was formed after electrochemical exfoliation. However, the ID/IG value of EG decreases with the increasing of H2O2 volume fraction used during the pre-treatment, suggesting better crystallinity. The shoulder peak, seen at Raman shift of around 1620 cm−1, corresponds to D′ peak, are originated from a single phonon intra-valley scattering process. The intensity of the D′ peak is known to be proportional to the average number of defects in the unit cell of samples.36 Fig. 2(a) shows that the intensity of this peak decreases with the increasing H2O2 amount used during pre-treatment. This result is consistent with the ID/IG value of EG that samples subjected to pre-treatment in higher H2O2 volume fraction demonstrated a lower structural disorder.
C).34 Thus, the ratio of D to G peak intensity (ID/IG) is often used to assess the extent of damage in carbonaceous materials.35 It can be seen from Fig. 2(a) that the intensity of D peak and ID/IG value of all EG samples both are higher than that of the pristine graphite sheet, indicating more defective and oxidised structure was formed after electrochemical exfoliation. However, the ID/IG value of EG decreases with the increasing of H2O2 volume fraction used during the pre-treatment, suggesting better crystallinity. The shoulder peak, seen at Raman shift of around 1620 cm−1, corresponds to D′ peak, are originated from a single phonon intra-valley scattering process. The intensity of the D′ peak is known to be proportional to the average number of defects in the unit cell of samples.36 Fig. 2(a) shows that the intensity of this peak decreases with the increasing H2O2 amount used during pre-treatment. This result is consistent with the ID/IG value of EG that samples subjected to pre-treatment in higher H2O2 volume fraction demonstrated a lower structural disorder.
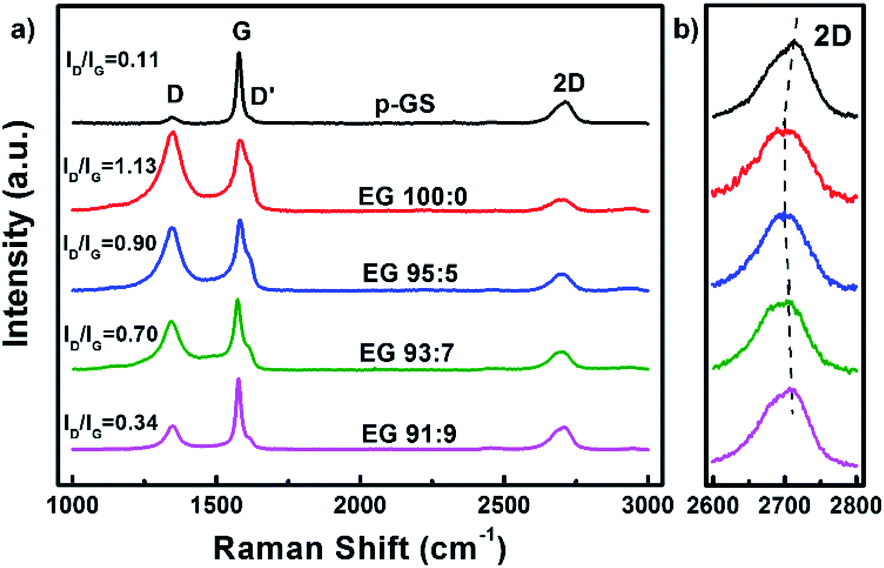 | ||
| Fig. 2 Raman spectra of the EG obtained after pre-treatment in H2SO4/H2O2 solution with various volume fractions of H2SO4:H2O2 (EG 100:0, EG 95:5, EG 93:7, and EG 91:9) at Raman shift of 1000–3000 cm−1, and (b) their enlarged spectra at 2600–2800 cm−1. Raman spectrum of pristine graphite sheet (p-GS) is also provided as a comparison. | ||
While D and G peaks are mainly considered to assess the damage and structural perfection, a 2D peak is often analysed to assess the layered characteristic of graphene. Several studies have reported the dependency of shape and position of the 2D peak on the graphene thickness.37–39 The 2D peak of single-layer graphene is often observed as a symmetric peak at ca. 2680 cm−1 under the exposure of 532 nm laser excitation.36 Whereas, graphite and multilayers graphene typically show a wide asymmetric peak at Raman shift of 2600–2800 cm−1, which can be deconvoluted into several smaller peaks. The enlarged Raman spectra of all samples along 2600–2800 cm−1 (2D) depicted in Fig. 2(b) shows that none of the samples resembles the characteristic of a single graphene layer. However, it can be seen that the position of 2D peaks in EG samples shifted to the left as compared to the pristine graphite sheet. The 2D peak gradually red-shifted in the spectra of samples pre-treated with H2SO4/H2O2 solution with volume fraction H2SO4:H2O2 100:0 and 95:5, indicating the thinner graphene layers in these samples. However, increment of H2O2 volume fraction did not shift the peak further. Instead, it pushed the 2D peak back nearing that of the pristine graphite sheet, advising that the thicker EG was obtained when the graphite sheet is pre-treated with an excessive amount of H2O2. Another sign was the resemblance of the 2D peak shape of these samples with that of the graphite sheet shown in Fig. 2(b). This phenomenon allegedly caused by the extreme early expansion of the graphite sheet during the pre-treatment, especially with the large H2O2 volume fraction (EG 93:7 and EG 91:9), marked by several macro cracking that was visible within the samples (Fig. 1(i) and (j)). The extreme early expansion and macro cracking made the samples so friable that they were broken into thick graphite chunks during the electrochemical exfoliation process, as was observed using Scanning Electron Microscopy (SEM) shown in Fig. 3.
 | ||
| Fig. 3 Scanning electron microscope (SEM) images of the (a) p-GS, (b) EG 100:0, (c) EG 95:5, (d) EG 93:7, and (e) EG 91:9. | ||
The SEM images of the pristine graphite sheet and the obtained EG after centrifugation are depicted in Fig. 3. It can be seen that the thick graphite sheet samples look more delaminated, forming thinner layers of EG. The SEM images of EG 100:0 (Fig. 3(b)) and EG 95:5 (Fig. 3(c)) show the presence of thin layers of graphene. Meanwhile, the SEM images of EG 93:7 (Fig. 3(d)) and EG 91:9 (Fig. 3(e)) reveal a thick fragment of multilayers graphene, which resembles the SEM image of graphite (Fig. 3(a)). The result of SEM observation is in line with Raman characterisation, indicating that delamination of the graphite sheet could not be conducted well in the sample pre-treated with a large volume fraction of H2O2. As noted earlier, the extreme expansion of the graphite sheet during pre-treatment with a large amount of H2O2 apparently caused the structure of the graphite sheet to crumble easily.
Fig. 4(a) shows the normalised X-ray diffraction (XRD) patterns of all samples. They show the existence of graphitic materials, indicated by the peak around 2θ of 26° and 55°, which correspond to diffraction plane (002) and (004), respectively. However, small bumps can be seen at 2θ of ca. 12° and 43° in the sample prepared without H2O2. This phenomenon likely corresponds to the diffraction peak (001) and (101) of graphene oxide (GO), respectively.40 The enlarged XRD pattern along 2θ of 10–14° shown in Fig. S1(a)† indicates that only sample 100:0 revealed a (001) peak of GO. Whereas, the enlarged pattern along 2θ of 41–47° (Fig. S1(b)†) reveals that all EG samples which previously subjected to pre-treatment show very small bumps at 2θ of 43°, which correspond to (101) plane of GO.40 However, it can be seen that the intensity of the bumps decreases gradually with the increase of the H2O2 amount.
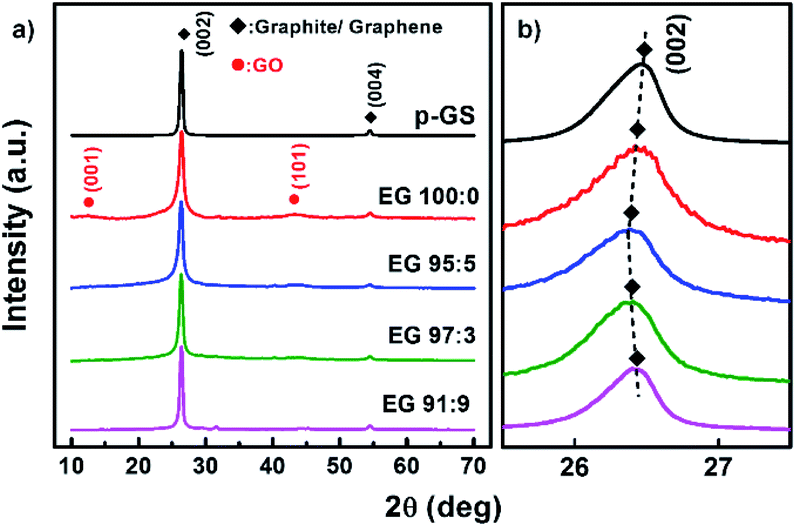 | ||
| Fig. 4 (a) Normalised X-ray diffraction (XRD) patterns of exfoliated graphite (EG) obtained after pre-treatment in H2SO4/H2O2 solution with various volume fractions of H2SO4:H2O2 (EG 100:0, EG 95:5, EG 93:7, and EG 91:9) along 2θ of 10–70 degree, and (b) their enlarged patterns at 2θ of 25.5–27.5 degree. XRD pattern of pristine graphite sheet (p-GS) is also provided as a comparison. | ||
The formation of GO in the sample pre-treated with only H2SO4 is supposedly triggered by the formation of graphite intercalation compound (GIC). GIC is widely known as an important stage in GO production via Hummer's and other chemical exfoliation methods.41,42 The intercalation of H2SO4 and HSO4− compounds in the graphite interlayer may facilitate oxidising agents (e.g., HNO3 and KMnO4) enter the graphite slab, which may further lead to oxidation of carbon atoms on the basal plane.43,44 Cao et al. have reported that the electrochemical exfoliation of the GIC-graphite with low stage index (n) leads to the formation of highly oxidised GO.44 GIC formation facilitates the entrance of water during the electrochemical process. The reaction between the nucleophilic water with the positively charged graphite results in the generation of oxygen-containing functional groups.44,45 GIC with lower n facilitates more water entering the graphite slabs, increasing the possibility of water attacking the positively charged graphite layers to form GO during the electrochemical process. However, in the case of less intercalation (high n), the product of this process will be a mixture of GO and graphene layers.44
Fig. 4(b) shows the enlarged diffraction pattern of all samples along the 2θ of 25–28°. It can be seen that (002) graphitic peaks shifted to lower 2θ, in the EG 100:0 and EG 95:5, and shifted back to higher 2θ in the samples EG 93:7 and 91:9. This trend agrees well with the shifting of 2D peak shown in Raman spectra (Fig. 2(b)), as the shifting of (002) peaks to lower 2θ is also known as a sign of the expansion of graphite interlayer spacing along the c-axis (d002).46 The expansion can be caused by at least two reasons, including the delamination of graphite layers and oxidation of graphite edge and basal plane.
During the electrochemical exfoliation process, positive electric potential causes the insertion of nucleophilic water molecules into graphite interlayer then forms oxygen-containing functional groups. After that, the water molecules in the graphite slabs are slowly oxidised to form oxygen gases. The gases are then accumulated and caused the generation of pressure inside the graphite slab, which may overcome van der Waals interactions between the graphite layers and cause the delamination of graphene layers.18,47
Fig. 5 shows the proposed mechanism of electrochemical exfoliation that involves pre-treatment in H2SO4, with and without the addition of H2O2. The graphite sheet sample pre-treated in the H2SO4/H2O2 solution experienced early expansion due to the decomposition reaction of H2O2 that releases O2 gases. This process reduces the delamination time, which is considerably longer in the electrochemical exfoliation of graphite sheet that is only pre-treated using H2SO4. Besides, the less time-consuming procedure can prevent the massive attack of nucleophilic water to the graphite sheet that often leads to the graphene oxide formation in a prolonged time. In order to get information related to the functionality of the obtained EG, Fourier transform-infrared (FT-IR) spectra of the samples were collected and compared with the FT-IR spectrum of the pristine graphite sheet (Fig. 6). All spectra show an absorbance peak at ca. 1620 cm−1, attributed to vibration of CC bending from sp2 hybridised carbons.48,49 Whereas the peaks related to oxygen functional groups originated from alcohol and water, such as O–H stretching, CO–H bending, and C–O stretching is visible at ca. 3400 cm−1, 1380 cm−1, 1090 cm−1, respectively.50,51 While the peak at 3429 cm−1 (O–H stretching) looks similar for all samples, the C–O stretching peak at 1116 cm−1 looks more prominent in the EG samples, suggesting oxidation of the graphite sheet during the electro-exfoliation process. However, it can also be seen that the peak decreases gradually with the increasing amount of H2O2 added during pre-treatment, proposing less oxidation of the graphite pre-treated with more amount of H2O2. As mentioned previously, this phenomenon can be caused by a very fast breakdown of the fragile graphite sheet pre-treated with a higher dose of H2O2.
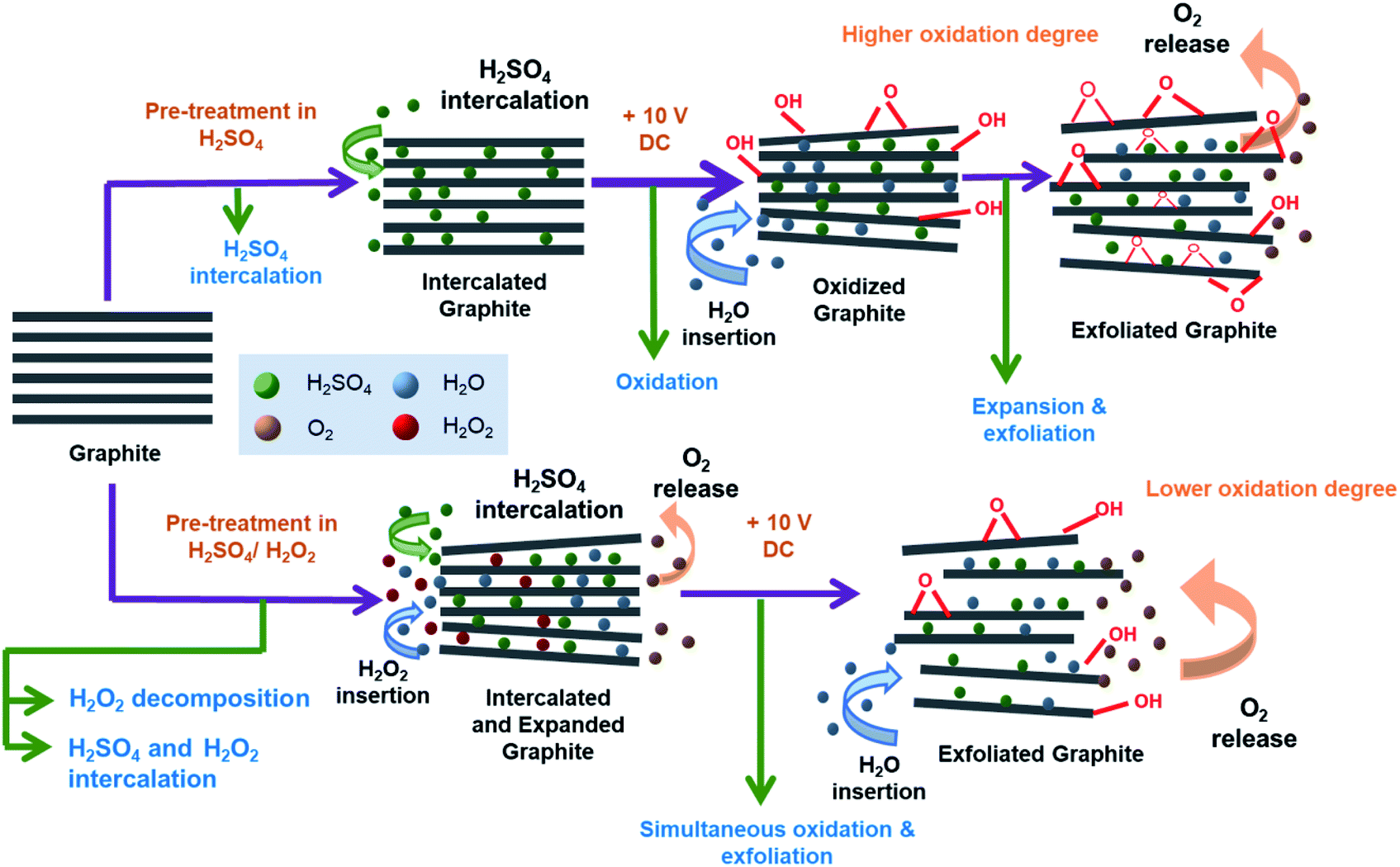 | ||
| Fig. 5 The proposed mechanism of graphite sheet electrochemical exfoliation after pre-treatment in H2SO4 with and without the addition of H2O2. | ||
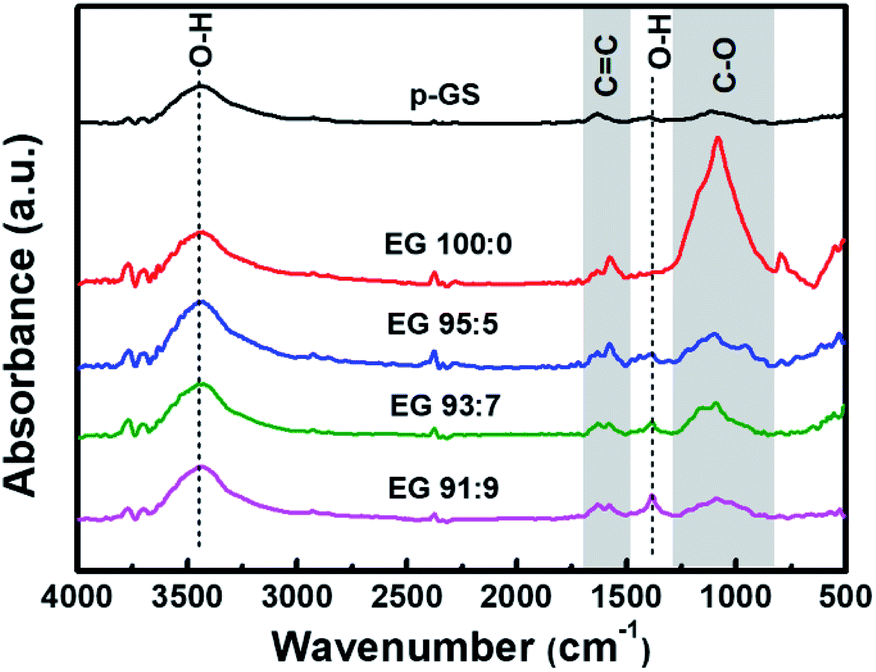 | ||
| Fig. 6 Fourier transform-infrared (FT-IR) spectra of EG obtained with previously pre-treated p-GS in H2SO4/H2O2 solution with various volume fractions of H2SO4:H2O2 (EG 100:0, EG 95:5, EG 93:7, and EG 91:9) along wavenumber of 4000–500 cm−1. FT-IR spectrum of pristine graphite sheet (p-GS) is also provided as a comparison. | ||
The oxidation mechanism of graphite during electrochemical exfoliation is often explained as the nucleophilic substitution reaction between the positively charged graphite sheets and water molecules. The reaction causes C–OH group formation and may lead to the formation of C–O–C epoxide groups at a prolonged time.44 Meanwhile, further oxidation of C–OH may form –CO (carbonyl) or OCO (carbon dioxide). However, FT-IR spectra of EG shown in Fig. 6 reveal no peak indicating the presence of CO carbonyl groups, suggesting the low to moderate oxidation of the obtained EGs. The spectra show that the peak attributed to C–O stretching decreases, and CO–H bending increases with the increase of H2O2 used during pre-treatment. It seems that –COH groups transformed into the –COC epoxide group at the longer electrochemical reaction in samples pre-treated with a small amount of H2O2.
The oxidation level of all samples represented as a relative percentage of oxygen-containing functional group (RPOCFG) has been determined by comparing the integrated area of peaks attributed to oxygen functional groups (1398 cm−1 and 1116 cm−1) to the integrated area of all peaks along 900–1850 cm−1, as proposed by Kumar et al.48 Table 1 shows the calculated RPOCFG value of each EG sample. The RPOCFG value tends to decrease with the increasing amount of H2O2 involved during the pre-treatment. The lowest oxidation degree is demonstrated by the EG 91:9, with an RPOCFG of 87.49%. The same trend was also revealed from the elemental analysis using an energy dispersive X-ray spectrometer (EDS), represented as the O/C ratio and shown in Table 1. The O/C ratio of EG samples is higher than that of the pristine graphite sheet, suggesting the oxidation of graphite sheets during the electrochemical exfoliation. It can be seen that the O/C ratio decreases with the increase of H2O2 amount involved during pre-treatment, which are in line with the result of the RPOCFG calculation.
| Sample | RPOCFG (%) | C (at%) | O (at%) | O/C |
|---|---|---|---|---|
| p-GS | 83.50 | 96.07 | 3.93 | 0.04 |
| EG 100:0 |
98.04 | 66.64 | 33.36 | 0.50 |
| EG 95:5 |
91.38 | 85.36 | 14.64 | 0.17 |
| EG 93:7 |
88.87 | 85.81 | 14.19 | 0.16 |
| EG 91:9 |
87.49 | 88.51 | 11.49 | 0.13 |
The reason behind the less oxidation of EG 91:9 is the friable structure of the pre-treated graphite sheets in a solution containing more H2O2. The sample allegedly prematurely broke into large pieces of graphite fragment during the early stage of the electrochemical process, preventing the continuous delamination of the graphite layer and evolution of oxygen functional groups from –COH to –COC. This proposed mechanism agrees well with the SEM observation shown in Fig. 3. The EG prepared with a higher amount of H2O2 during pre-treatment (EG 93:7 and EG 91:9) seemed less exfoliated. The samples apparently broken into chunks of graphite instead of exfoliated as a thin layer of EG.
The electrical conductivity of the obtained EG in a function of the H2O2 volume fraction involved during pre-treatment is displayed in Fig. 7. The electrical conductivity tends to increase with the increase of H2O2 amount involved during pre-treatment, and the highest was demonstrated by sample EG 91:9 with electrical conductivity of 103.72 ± 6.59 S cm−1. This trend is inversely proportional to the RPOCFG and O/C because electrical conductivity is affected negatively by the presence of defects and functional groups. Covalent functionalisation of graphene may halt the delocalisation of π electron from the carbon atoms due to the transformation of planar sp2 hybridisation into tetrahedral sp3 hybridisation.52 Besides, oxygen content in the sample could disrupt electron transport, causing a decrease in electrical conductivity.52
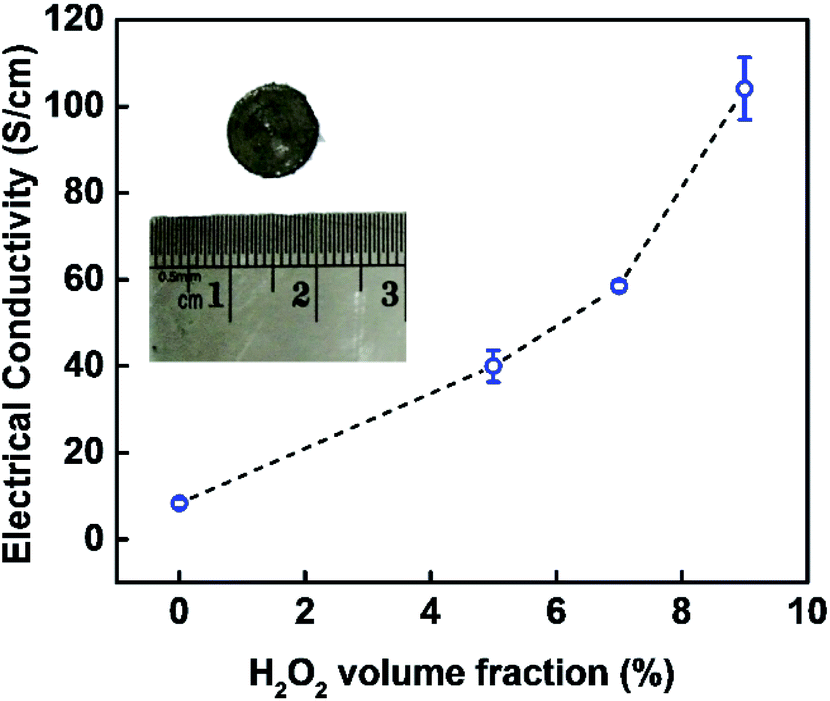 | ||
| Fig. 7 The electrical conductivity of EG in a function of H2O2 volume fraction involved during pre-treatment. Data are the average ± standard deviation (SD) of three independent measurements. | ||
Fig. 8 shows the Nyquist plots obtained from the electrochemical impedance spectroscopy (EIS) measurement of the EG samples. The plots were fitted using an equivalent circuit depicted in Fig. S2.† The semicircle in the plot corresponds to the resistance between graphene sheets and contact resistance between the electrode and current collector,53,54 of which diameter represents the value of charge transfer resistance (Rct) at the interface of electrolyte and electrode.55 Among all samples, EG 91:9, the least oxidised sample, shows the lowest Rct, i.e., 2.59 Ω (Table 2). This result suggests that the oxidation degree of graphitic materials is equally proportional to the Rct value. The reason behind it is that the oxygen functional group in the EG samples will act as an insulating layer that inhibits interfacial charge transfer and increases the Rct value.56 The Rct value corresponds with electrical conductivity measurement that the sample with the lowest Rct has the highest electrical conductivity.57
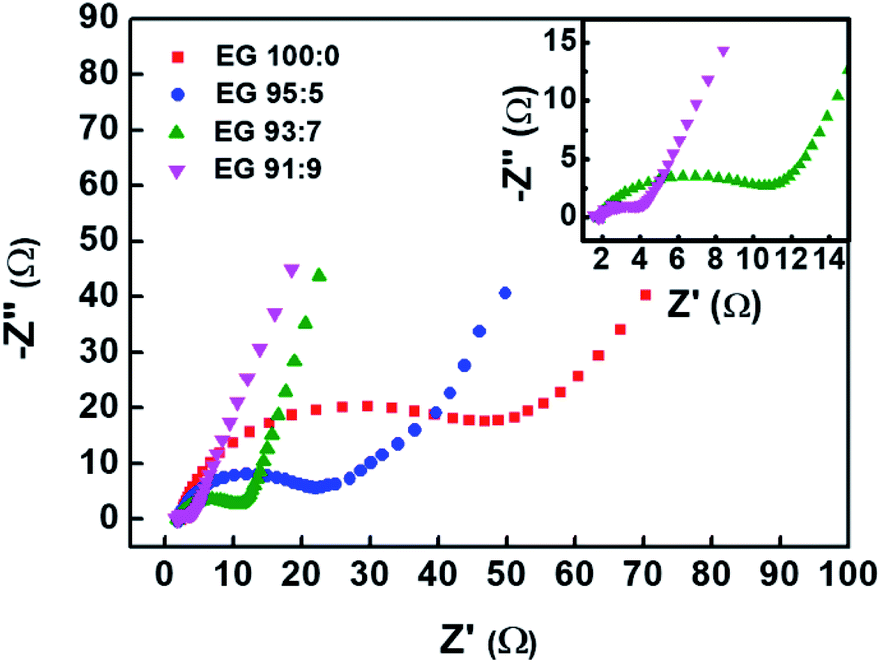 | ||
| Fig. 8 Nyquist plots of EG samples prepared using previously pre-treated graphite sheets in H2SO4/H2O2 solution with various volume fractions of H2SO4:H2O2 (EG 100:0, EG 95:5, EG 93:7, and EG 91:9). | ||
| Sample | Rct (Ω) |
|---|---|
| EG 100:0 |
50.21 |
| EG 95:5 |
21.35 |
| EG 93:7 |
10.47 |
| EG 91:9 |
2.59 |
The capacitive behaviour of all samples was analysed by conducting cyclic voltammetry (CV) measurement. Fig. 9(a) shows the cyclic voltammograms of pristine graphite and various EG samples, measured at a scan rate of 1 mV s−1 along −0.9 to 0 V. All samples, except EG 100:0, revealed electric double layer capacitive (EDLC) properties indicated by their pseudo-rectangular-shape CV profiles.9,12 CV profile of EG 100:0 demonstrates oblique-shape CV profile, which allegedly caused by the high electrical resistivity of this sample. Table 3 shows the specific capacitance of samples measured at a scan rate of 1 mV s−1. The specific capacitance of all samples was calculated from their cyclic voltammogram using the following formula:58
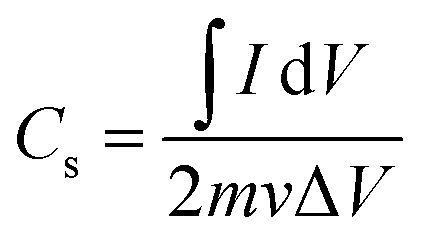 | (2) |
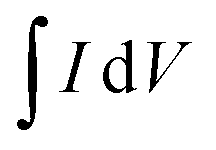 is an integrated area of cyclic voltammogram, m is mass of active material (g), v is scan rate (V s−1), and ΔV is a potential window (V).
is an integrated area of cyclic voltammogram, m is mass of active material (g), v is scan rate (V s−1), and ΔV is a potential window (V).
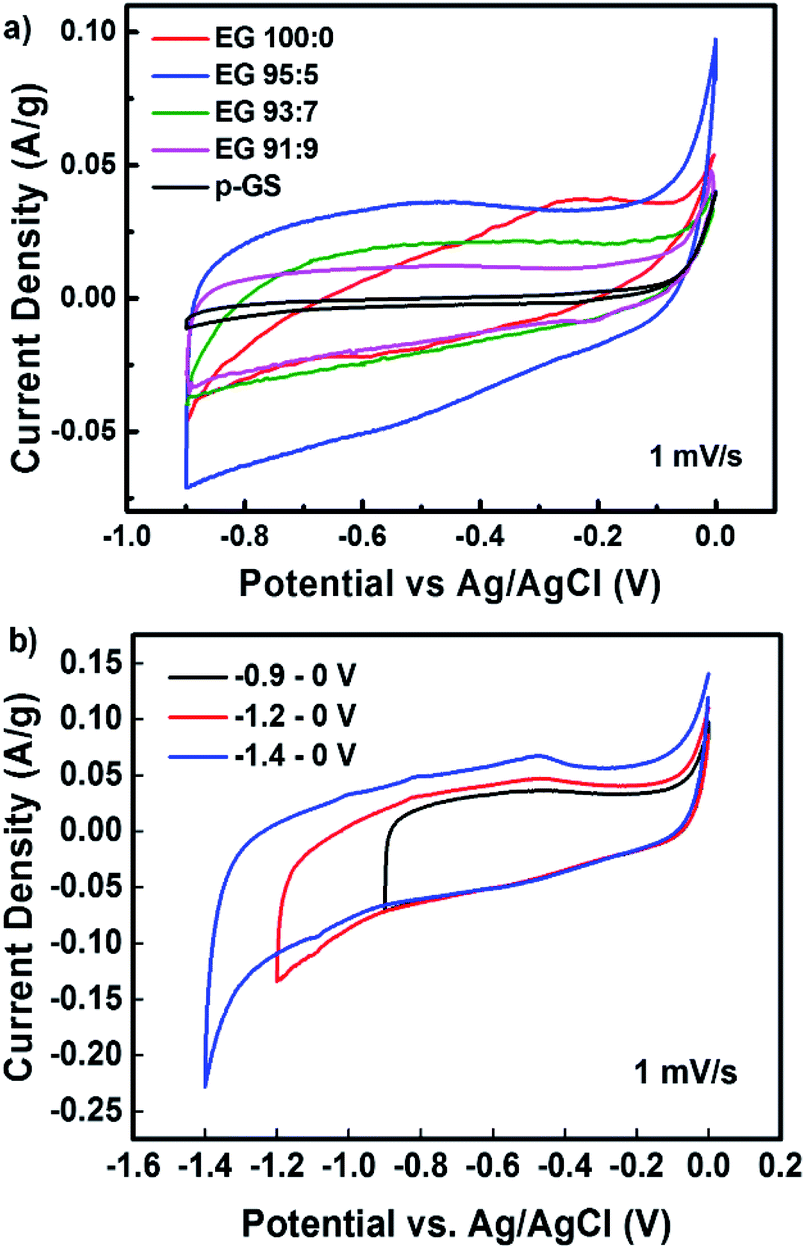 | ||
| Fig. 9 Cyclic voltammogram of (a) EG samples prepared using previously pre-treated graphite sheet in H2SO4 with a various volume fraction of H2O2, compared with that of the pristine graphite sheet (at 1 mV s−1), (b) EG 95:5 at various potential windows using a scan rate of 1 mV s−1. | ||
| Sample | Cs (F g−1) |
|---|---|
| p-GS | 11.77 |
| EG 100:0 |
51.05 |
| EG 95:5 |
71.95 |
| EG 93:7 |
40.54 |
| EG 91:9 |
35.50 |
All EG samples exhibited higher specific capacitance than the pristine graphite sheet because of their large surface area caused by successful electro-exfoliation. The large surface area is an essential factor determining the specific capacitance of carbon-based supercapacitor, as samples with a large surface area can adsorb more ions on their surface.59 Among all samples, EG 95:5 sample, showed the highest specific capacitance of 71.95 F g−1, which is higher than electrochemically exfoliated graphene prepared without any pre-treatment as reported by Parvez et al.18 The reason behind the excellent capacitive properties is the high degree of exfoliation, as confirmed by Raman spectroscopy measurement and SEM observation. Even though EG 95:5 and EG 100:0 showed a similar degree of exfoliation, more electrically conductive EG 95:5 enabled higher electron mobility on the electrode surface, leading to the higher specific capacitance of this sample.14,59,60
Fig. 9(b) shows the CV plots of EG 95:5 sample measured at three different voltage windows, i.e., −0.9 to 0 V, −1.2 to 0 V, and −1.4 to 0 V. It can be seen that the pseudo-rectangular form of the CV plots could be maintained down to −1.4 V, suggesting that this sample owns the high potential to be applied as electrode materials for the wide voltage window supercapacitor. The voltammogram measured along the −1.4 to 0 V voltage window showed a hump at around −0.5 V. The hump probably corresponds to the redox reaction of oxygen-containing functional groups in the sample, which also has been reported by Oh et al.61 Table 4 lists the specific capacitances of EG 95:5 sample measured at different voltage windows. The data reveals the increase of capacitance value with the widening of the voltage window. More charges stored along the wider voltage window produced a higher current response, thus increasing the capacitance. Fig. S3† shows the CV profiles of EG 95:5 sample measured at various scan rates from 1 to 200 mV s−1 along −1.4 to 0 V. All CVs demonstrate the rectangular-like curves observed even at a high scan rate of 200 mV s−1, revealing the ideal EDLC performance.21
:5 at various voltage windows measured using a scan rate of 1 mV s−1 (Fig. 9(b))
| Voltage window (V) | Cs (F g−1) |
|---|---|
| −0.9 to 0 | 71.95 |
| −1.2 to 0 | 120.84 |
| −1.4 to 0 | 150.69 |
Aside from focusing on the improvement of power and energy density of supercapacitors, lowering the fabrication costs and ensuring the involvement of environmentally friendly processes are several points of interest in the fabrication of supercapacitor materials.62,63 Although several groups have reported the fabrication of EG with higher specific capacitance, the utilisation of additional organic substances in the exfoliation and dispersion process is not economically and environmentally favorable.21,64,65 Our method offers low a cost and easy process using a more environmentally friendly water-based process. Besides, it also demonstrates excellent exfoliation of graphene layers with outstanding capacitive properties.
Conclusions
Pre-treatment of graphite sheet in H2SO4/H2O2 solution before electrochemical exfoliation, as expected, could affect the exfoliation and oxidation degree of the resulting EG samples. The exfoliation process can be improved thanks to the early expansion of graphite sheets caused by H2O2 decomposition. However, the excessive corporation of H2O2 in the pre-treatment solution allegedly caused the extreme expansion of the graphite sheets, leading to fragile structures and hampers the electrochemical delamination process. Moderate expansion during the pre-treatment has successfully assisted the electrochemical exfoliation process producing high yielded well-exfoliated graphite sheets with a low oxidation degree in a relatively short time. The optimum exfoliation was demonstrated by the EG samples obtained by pre-treating graphite sheets in H2SO4/H2O2 solution with a volume fraction H2SO4:H2O2 of 95:5 before electro-exfoliation. This sample showed the best exfoliation degree among all the samples. It also demonstrated excellent capacitive properties with high specific capacitance and wide voltage range, making this sample suitable to be applied as the high energy density and high-power density supercapacitor materials.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was funded by Ministry of Research and Technology/National Research and Innovation Agency, Republic of Indonesia, under “Penelitian Dasar Unggulan Perguruan Tinggi (PDUPT)” Research Program Fiscal Year 2021. We would like to thank for the research facility supported by USAID through Sustainable Higher Education Research Alliances (SHERA) Program. Octia Floweri would like to thank ITB World Class University (WCU) Post-doctoral program.References
- D. D. L. Chung, J. Mater. Sci., 2016, 51, 554–568 CrossRef CAS.
- A. Bianco, H.-M. Cheng, T. Enoki, Y. Gogotsi, R. H. Hurt, N. Koratkar, T. Kyotani, M. Monthioux, C. R. Park, J. M. D. Tascon and J. Zhang, Carbon, 2013, 65, 1–6 CrossRef CAS.
- M. Cai, D. Thorpe, D. H. Adamson and H. C. Schniepp, J. Mater. Chem., 2012, 22, 24992–25002 RSC.
- Q. Zhao, J. Wang, Z. Li, Y. Qiao, C. Jin and Y. Guo, Ceram. Int., 2016, 42, 13273–13277 CrossRef CAS.
- M. Agostini, S. Brutti and J. Hassoun, ACS Appl. Mater. Interfaces, 2016, 8, 10850–10857 CrossRef CAS PubMed.
- A. Raghunandanan, M. Yeddala, P. Padikassu and R. Pitchai, ChemistrySelect, 2018, 3, 5032–5039 CrossRef CAS.
- A. Ambrosi and M. Pumera, Chem.–Eur. J., 2016, 22, 153–159 CrossRef CAS PubMed.
- J. Paredes and J. Munuera, J. Mater. Chem. A, 2017, 5, 7228–7242 RSC.
- R. Maharsi, A. F. Arif, T. Ogi, H. Widiyandari and F. Iskandar, RSC Adv., 2019, 9, 27896–27903 RSC.
- A. Izadi-Najafabadi, S. Yasuda, K. Kobashi, T. Yamada, D. N. Futaba, H. Hatori, M. Yumura, S. Iijima and K. Hata, Adv. Mater., 2010, 22, E235–E241 CrossRef CAS PubMed.
- A. Hamra, H. Lim, W. Chee and N. Huang, Appl. Surf. Sci., 2016, 360, 213–223 CrossRef CAS.
- L. W. Le Fevre, J. Cao, I. A. Kinloch, A. J. Forsyth and R. A. Dryfe, ChemistryOpen, 2019, 8, 418–428 CrossRef CAS PubMed.
- M. P. Down, S. J. Rowley-Neale, G. C. Smith and C. E. Banks, ACS Appl. Energy Mater., 2018, 1, 707–714 CrossRef CAS.
- N. Morimoto, T. Kubo and Y. Nishina, Sci. Rep., 2016, 6, 21715 CrossRef CAS PubMed.
- B. Zhao, P. Liu, Y. Jiang, D. Pan, H. Tao, J. Song, T. Fang and W. Xu, J. Power Sources, 2012, 198, 423–427 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- F. Iskandar, O. B. Abdillah, E. Stavila and A. H. Aimon, New J. Chem., 2018, 42, 16362–16371 RSC.
- K. Parvez, Z.-S. Wu, R. Li, X. Liu, R. Graf, X. Feng and K. Müllen, J. Am. Chem. Soc., 2014, 136, 6083–6091 CrossRef CAS PubMed.
- S. Yang, M. R. Lohe, K. Müllen and X. Feng, Adv. Mater., 2016, 28, 6213–6221 CrossRef CAS PubMed.
- W. Wu, C. Zhang and S. Hou, J. Mater. Sci., 2017, 52, 10649–10660 CrossRef CAS.
- I.-L. Tsai, J. Cao, L. Le Fevre, B. Wang, R. Todd, R. A. Dryfe and A. J. Forsyth, Electrochim. Acta, 2017, 257, 372–379 CrossRef CAS.
- J. Munuera, J. Paredes, S. Villar-Rodil, M. Ayán-Varela, A. Martínez-Alonso and J. Tascón, Nanoscale, 2016, 8, 2982–2998 RSC.
- C.-Y. Su, A.-Y. Lu, Y. Xu, F.-R. Chen, A. N. Khlobystov and L.-J. Li, ACS Nano, 2011, 5, 2332–2339 CrossRef CAS PubMed.
- J. Munuera, J. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. Tascón, Carbon, 2017, 115, 625–628 CrossRef CAS.
- A. M. Dimiev, G. Ceriotti, A. Metzger, N. D. Kim and J. M. Tour, ACS Nano, 2016, 10, 274–279 CrossRef CAS PubMed.
- X. Geng, Y. Guo, D. Li, W. Li, C. Zhu, X. Wei, M. Chen, S. Gao, S. Qiu, Y. Gong, L. Wu, M. Long, M. Sun, G. Pan and L. Liu, Sci. Rep., 2013, 3, 1134 CrossRef PubMed.
- S. Lin, L. Dong, J. Zhang and H. Lu, Chem. Mater., 2016, 28, 2138–2146 CrossRef CAS.
- W. Gu, W. Zhang, X. Li, H. Zhu, J. Wei, Z. Li, Q. Shu, C. Wang, K. Wang and W. Shen, J. Mater. Chem., 2009, 19, 3367–3369 RSC.
- L. K. Wu, K. Y. Chen, S. Y. Cheng, B. S. Lee and C. M. Shu, J. Therm. Anal. Calorim., 2008, 93, 115 CrossRef CAS.
- M. Salvatore, G. Carotenuto, S. De Nicola, C. Camerlingo, V. Ambrogi and C. Carfagna, Nanoscale Res. Lett., 2017, 12, 167 CrossRef PubMed.
- J. M. Skowroński, J. Mater. Sci., 1988, 23, 2243–2246 CrossRef.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 CrossRef CAS PubMed.
- H. Wang, C. Wei, K. Zhu, Y. Zhang, C. Gong, J. Guo, J. Zhang, L. Yu and J. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 34456–34466 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- B. Krauss, T. Lohmann, D. H. Chae, M. Haluska, K. von Klitzing and J. H. Smet, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 165428 CrossRef.
- J.-B. Wu, M.-L. Lin, X. Cong, H.-N. Liu and P.-H. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC.
- A. C. Ferrari, J. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. Novoselov and S. Roth, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- A. Gupta, G. Chen, P. Joshi, S. Tadigadapa and P. Eklund, Nano Lett., 2006, 6, 2667–2673 CrossRef CAS PubMed.
- L. Niu, M. Li, X. Tao, Z. Xie, X. Zhou, A. P. Raju, R. J. Young and Z. Zheng, Nanoscale, 2013, 5, 7202–7208 RSC.
- R. Gupta, Z. Alahmed and F. Yakuphanoglu, Mater. Lett., 2013, 112, 75–77 CrossRef CAS.
- A. M. Dimiev and J. M. Tour, ACS Nano, 2014, 8, 3060–3068 CrossRef CAS PubMed.
- N. Yadav and B. Lochab, FlatChem, 2019, 13, 40–49 CrossRef CAS.
- F. Kang, Y. Leng and T.-Y. Zhang, J. Phys. Chem. Solids, 1996, 57, 889–892 CrossRef CAS.
- J. Cao, P. He, M. A. Mohammed, X. Zhao, R. J. Young, B. Derby, I. A. Kinloch and R. A. Dryfe, J. Am. Chem. Soc., 2017, 139, 17446–17456 CrossRef CAS PubMed.
- F. Beck, J. Jiang and H. Krohn, J. Electroanal. Chem., 1995, 389, 161–165 CrossRef.
- Y. Htwe, W. Chow, Y. Suda, A. Thant and M. Mariatti, Appl. Surf. Sci., 2019, 469, 951–961 CrossRef CAS.
- S. T. Hossain and R. Wang, Electrochim. Acta, 2016, 216, 253–260 CrossRef CAS.
- N. Kumar and V. C. Srivastava, ACS Omega, 2018, 3, 10233–10242 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- R. Gao, N. Hu, Z. Yang, Q. Zhu, J. Chai, Y. Su, L. Zhang and Y. Zhang, Nanoscale Res. Lett., 2013, 8, 32 CrossRef PubMed.
- H. Yu, B. Zhang, C. Bulin, R. Li and R. Xing, Sci. Rep., 2016, 6, 36143 CrossRef CAS PubMed.
- T. Sreeprasad and V. Berry, Small, 2013, 9, 341–350 CrossRef CAS PubMed.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L.-C. Qin, Phys. Chem. Chem. Phys., 2011, 13, 17615–17624 RSC.
- X. Liu, X. Qi, Z. Zhang, L. Ren, G. Hao, Y. Liu, Y. Wang, K. Huang, X. Wei and J. Li, RSC Adv., 2014, 4, 13673–13679 RSC.
- S. S. Karade, D. P. Dubal and B. R. Sankapal, ChemistrySelect, 2017, 2, 10405–10412 CrossRef CAS.
- H.-L. Guo, X.-F. Wang, Q.-Y. Qian, F.-B. Wang and X.-H. Xia, ACS Nano, 2009, 3, 2653–2659 CrossRef CAS PubMed.
- F. Iskandar, B. Setiawan, T. Mayangsari, R. Maharsi, A. Purwanto and A. Aimon, Mater. Res. Express, 2018, 6, 025514 CrossRef.
- A. Sayah, F. Habelhames, A. Bahloul, B. Nessark, Y. Bonnassieux, D. Tendelier and M. El Jouad, J. Electroanal. Chem., 2018, 818, 26–34 CrossRef CAS.
- Y. Gong, D. Li, Q. Fu and C. Pan, Prog. Nat. Sci., 2015, 25, 379–385 CrossRef CAS.
- H. H. Shi, S. Jang, A. Reza-Ugalde and H. E. Naguib, ACS Appl. Energy Mater., 2020, 3, 987–997 CrossRef CAS.
- Y. J. Oh, J. J. Yoo, Y. I. Kim, J. K. Yoon, H. N. Yoon, J.-H. Kim and S. B. Park, Electrochim. Acta, 2014, 116, 118–128 CrossRef CAS.
- Z. Fan, J. Yan, T. Wei, L. Zhi, G. Ning, T. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366–2375 CrossRef CAS.
- Y.-G. Wang, H.-Q. Li and Y.-Y. Xia, Adv. Mater., 2006, 18, 2619–2623 CrossRef CAS.
- L. Wu, W. Li, P. Li, S. Liao, S. Qiu, M. Chen, Y. Guo, Q. Li, C. Zhu and L. Liu, Small, 2014, 10, 1421–1429 CrossRef CAS PubMed.
- Y. Zhang, Y. Xu, J. Zhu, L. Li, X. Du and X. Sun, Carbon, 2018, 127, 392–403 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Enlarged X-ray diffraction (XRD) pattern of graphite sheet precursor (p-GS) and exfoliated graphite (EG) obtained after pre-treatment in H2SO4/H2O2 with various volume fractions; the equivalent circuit used to fit the Nyquist plots from Electrochemical Impedance Spectroscopy (EIS) measurement; cyclic voltammetry (CV) curves of EG 95:5 measured at various scan rate (1–200 mV s−1) with potential window of 1.4 V. See DOI: 10.1039/d0ra10115j |
| This journal is © The Royal Society of Chemistry 2021 |