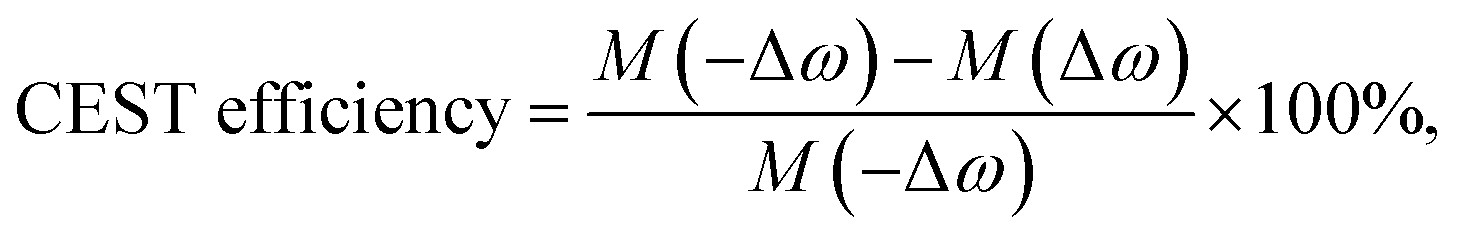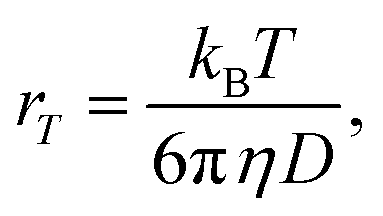DOI:
10.1039/D0RA10410H
(Paper)
RSC Adv., 2021,
11, 6526-6534
Paracetamol and other acetanilide analogs as inter-molecular hydrogen bonding assisted diamagnetic CEST MRI contrast agents†
Received
10th December 2020
, Accepted 29th January 2021
First published on 8th February 2021
Abstract
Paracetamol and a few other acetanilide derivatives are reported as a special class of diamagnetic Chemical Exchange Saturation Transfer (diaCEST) MRI contrast agents, that exhibit contrast only when the molecules form inter-molecular hydrogen bonding mediated molecular chains or sheets. Without the protection of the hydrogen bonding their contrast producing labile proton exchanges too quickly with the solvent to produce any appreciable contrast. Through a number of variable temperature experiments we demonstrate that under the conditions when the hydrogen bond network breaks and the high exchange returns back, the contrast drops quickly. The well-known analgesic drug paracetamol shows 12% contrast at a concentration of 15 mM at physiological conditions. With the proven safety track-record for human consumption and appreciable physiological contrast, paracetamol shows promise as a diaCEST agent for in vivo studies.
1. Introduction
In any diagnostic imaging technique such as Magnetic Resonance Imaging (MRI)1 or Computed Tomography (CT)2 it is always desired that the region of interest remains at a different contrast level from the surroundings. In MRI, differential water proton density or water content is the source of natural contrast that becomes useful in studying, for example, bones embedded in soft tissues. Also, in T1 or T2 weighted MRI imaging, the relaxation time constants of water produce natural contrast. However, in the absence of such natural contrast due to similar water content of the study region and the surroundings or due to unfavorable relaxation rates, artificial contrast is achieved through chemical MRI contrast agents3 following the seminal work by Young et al.4 Traditional paramagnetic metal-containing contrast agents bring contrast by altering either the longitudinal or the transverse relaxation rate constants of the water molecules.3a,5 In the year 2000, Ward et al. introduced a new class of compounds, that produce contrast by partially saturating the water through Chemical Exchange Saturation Transfer (CEST).6 In particular, the CEST contrast agents contain one or more labile protons that are saturated using low power continuous irradiation immediately prior to the MRI signal detection. The saturated labile protons then, in turn, transfer the saturation through multiple exchange events with the solvent during the long continuous irradiation. It is quite obvious that the CEST efficiency improves with increasing exchange rate (kex) of a labile proton.7 However, if the exchange becomes too fast on the NMR time scale, that is if kex becomes larger than the offset (Δω) of resonance frequency of a labile proton from the bulk water, then the CEST efficiency starts dropping again. For optimum CEST effect, the exchange rate should be as high as possible as long as it remains in the slow to medium exchange regime (kex ≤ Δω) to avoid coalescence.7a Naturally, constant efforts are being made in the search of molecules having labile protons with Δω as large as possible, so that faster-exchanging protons can take part in the CEST to improve the CEST efficiency.
A class of CEST agents known as the paraCEST8 agents, achieves Δω of the order of several kilohertz through paramagnetic downfield shift in the presence of a metal center. However, the presence of a metal center raises safety concerns.9 Diamagnetic CEST agents10 (diaCEST), on the other hand, are considered comparatively safer in absence of any metal center but produces a much lower Δω (typically less than 5 ppm) and hence inferior CEST efficiency. Recently, two classes of diaCEST agents were reported that produce comparatively larger offsets and hence much higher CEST contrast by utilizing, respectively, intramolecular hydrogen bonding and diatropic ring currents. Some benzoic acid derivatives (salicylic11 and anthranilic12 acids, in particular), phenols13 and modified imidazole14 compounds belong to the first class of molecules in which intramolecular hydrogen bonding (will be referred as intraMHB hereon for brevity) shifts the labile proton responsible for CEST contrast downfield (as large as 9.3 ppm for salicylic acid). Certain meso-substituted porphyrin derivatives, on the other hand, form the second class of diaCEST agents in which the diatropic ring current produced by the conjugated π-electrons brings a large upfield shift (∼8 ppm) to the labile inner-core amine protons.15
Herein, we introduce a few amide (acetanilide, in particular) derivatives as a new class of diaCEST agents, that produces comparable contrast as the previous two classes in spite of the fact that they do not show a similar large shift. Amide is already a popular choice12,16 for diaCEST agents as amides are found in various natural products, drugs and biomolecules and hence is considered in general non-toxic.17 The amide moiety however plays an additional role as the core structural motif in the present work. Amides are known to form intra as well inter-molecular hydrogen bonds.18 The compounds we present here as diaCEST agents do not exhibit contrast as an individual entity due to very fast exchange of their labile protons but do so only when they form ‘intermolecular hydrogen bonding (interMHB for brevity)’ mediated molecular chains or sheets. The interMHB helps to bring the exchange rate down to a favorable slow to medium exchange regime. In other words, the molecules act as a CEST contrast agent only as a group. We show this interesting behavior through a number of variable temperature experiments. We demonstrate that under mild heating above the physiological temperature when the interMHB chains start breaking, the isolated molecules cease to contribute to observable contrast as then the exchange rate breaches the slow to medium exchange regime (kex ≤ Δω). Although the weaker intrerMHB cannot produce as spectacular Δω as is produced by the much stronger intraMHB or the intense diatropic ring current, it slows down the exchange rate very optimally. For example, paracetamol (acetaminophen), a well-known analgesic drug19 and a notable member of this proposed class of CEST agents shows Δω of only 4.43 ppm, but yet it produces nearly similar contrast efficiency as compared to that shown by salicylic acid. The interMHB plays here the crucial role of optimizing the interplay between Δω and kex which is pivotal for any good CEST MRI contrast agent.
2. Experiments and methods
2.1 Synthesis and characterization
Acetanilide and substituted anilines were purchased from Sigma-Aldrich and were used without further purification. Other acetanilide derivatives (Table 1) were synthesized from the respective commercially available substituted aniline precursors following literature procedure.20 Acetic anhydride (12.7 mmol) was added to the aniline precursors (10 mmol) suspended in water (3 mL). The mixture was vigorously stirred in a warm water bath. Once the solid particles got dissolved, the reaction mixture was cooled. The solid product was then filtered and washed with cold water. The solid acetyl derivatives were finally recrystallized from hot water and dried at ambient conditions. All the synthesized compounds were characterized using 1H and 13C 1D NMR spectra acquired in DMSO-d6 as the solvent.
Table 1 Frequency offset, CEST contrast and exchange rates for acetanilide derivatives. All experiments were performed at 15 mM sample concentration in 10 mM PBS at the physiological condition of pH 7.4 and 37 °C
| Compound |
Δω [ppm] |
CEST contrast |
kex (ks−1) |
 |
5.21 |
12.4% |
1.84 |
 |
5.27 |
11.9% |
2.7 |
 |
5.07 |
6.8% |
1.7 |
 |
5.06 |
8.2% |
1.6 |
 |
5.53 |
0.5% |
1.07 |
 |
4.41 |
7.5% |
3.08 |
 |
4.61 |
8.1% |
4.18 |
 |
4.60 |
10.5% |
2.1 |
 |
4.43 |
12% |
1.2 |
2.2 Sample preparation and instrumentation
15 mM solutions of acetanilide and its derivatives were prepared using 10 mM (1×) PBS buffer. For the pH dependent studies seven samples were prepared with pH values ranging between 6.8 and 8.1 by using 0.5 M HCl and 0.5 M NaOH solutions. pH measurements were performed using a Hanna HI2210 pH-meter. All experiments were performed at the physiological temperature of 37 °C unless mentioned otherwise. For CEST studies in the biological media, 15 mM solutions at pH 7.4 were prepared by dissolving the necessary compounds in 1 mL of either 4% Fetal Bovine Serum (FBS) or 4% Horse Serum (HS) solution. All NMR experiments were performed on a 9.4 T Bruker AVANCE-III Nanobay NMR spectrometer equipped with a BBFO broadband probe, except for the exchange constant determination experiments. Exchange constants were calculated from experiments acquired on a 16.4 T Bruker AVANCE-III AV700 Ascend spectrometer equipped with a triple resonance room temperature probe. D2O filled capillaries were placed coaxially inside the NMR tube for deuterium lock. Methanol-d4 was used for temperature calibration using standard procedure.
2.3 Preparation of CEST z-spectra
CEST experiments were carried out with r.f. irradiation at 213 Hz (5 μT) field strength for 3 s. Data were collected at 1 ppm interval and normalized water peak intensity (100 × MZ/M0) was plotted against irradiation offset frequency to obtain the CEST z-spectra. MZ and M0 denote the water peak intensities, respectively, with and without on-resonance (on the solute exchangeable peak) saturation. CEST contrast or efficiency was calculated from the asymmetric magnetization transfer ratio (MTRassym) given by,7c| |
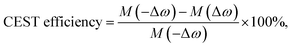 | (1) |
M(Δω) and M(−Δω) being, respectively, intensities of the water peak after on-resonance saturation (at the site of the exchangeable peak, offset Δω from water resonance frequency) and off-resonance saturation (equal offset on the other side of water peak, −Δω). In-house MATLAB (R2014b) scripts were used for post-acquisition processing and plotting.
2.4 Exchange rate calculation
Exchange rates were calculated using the method proposed by Dixon et al.21 The method uses a computationally simple linear regression method where the quantity MZ/(M0 − MZ) is fitted linearly against 1/ω12. ω1 is related to the r.f. power B1 as ω1 = γB1, γ being the gyromagnetic ratio of proton. The negative x-axis intercept of the linear fit gives the exchange rate as [1/ω12]intercept = −1/(kex)2. The method gives the best result under the condition that direct saturation (DS) of water is negligible, saturation powers (in Hz) used are not larger than the offset Δω and saturation time is long enough to ensure complete saturation. To satisfy these conditions, exchange rates were measured at the highest magnetic field available in the laboratory, with long saturation duration and using a sinc pulse-train that produces low DS. In the pulse train, 50 ms sinc pulses were used with 4 μs inter-pulse delay. Maximum saturation power was adjusted by taking the chemical shift offset of the compound under study. All fittings were performed in Microsoft Excel.
2.5 Diffusion measurements
Pulse field Gradient Stimulated Echo (PGSE)22 method was employed to determine the translational diffusion coefficient. To reduce the effect of Eddy current bipolar gradients were used. Attenuation of all 1H NMR signals with a reasonable signal to noise was measured as a function of gradient strength. Gradient strength was varied between 2% and 95% of the peak gradient value of 48.15 gauss per cm. The normalized peak intensity (I/I0) for any particular gradient strength (g) follows a relationship I/I0 = exp(−Dζg2), I0 being the equilibrium intensity and ζ is a constant that depends on a number of experimental parameters and can readily be calculated.22 Translational diffusion coefficient, D, was calculated from the negative slope (equal to Dζ) of the linear fit of ln(I/I0) plotted against g2.
2.6 Relaxivity measurements
Relaxivity of a solute molecule is defined as the concentration (expressed in mM in most situations) normalized change in either longitudinal (R1) or transverse (R2) relaxation rate constants of water in presence of the solute. Inversion recovery method was used with 12 variable delays between 10 ms and 6 s to measure R1. R2 was measured using Carr–Purcell–Meiboom–Gill spin-echo sequence with 14 delays between 80 ms and 1.44 s. Sufficiently long recycle delay of 10 s was used for accurate measurement of the relaxation rate constants. Relaxivities were calculated from the measured R1 and R2 values.
3. Results and discussion
3.1 Role of inter-molecular hydrogen bonding in producing CEST contrast
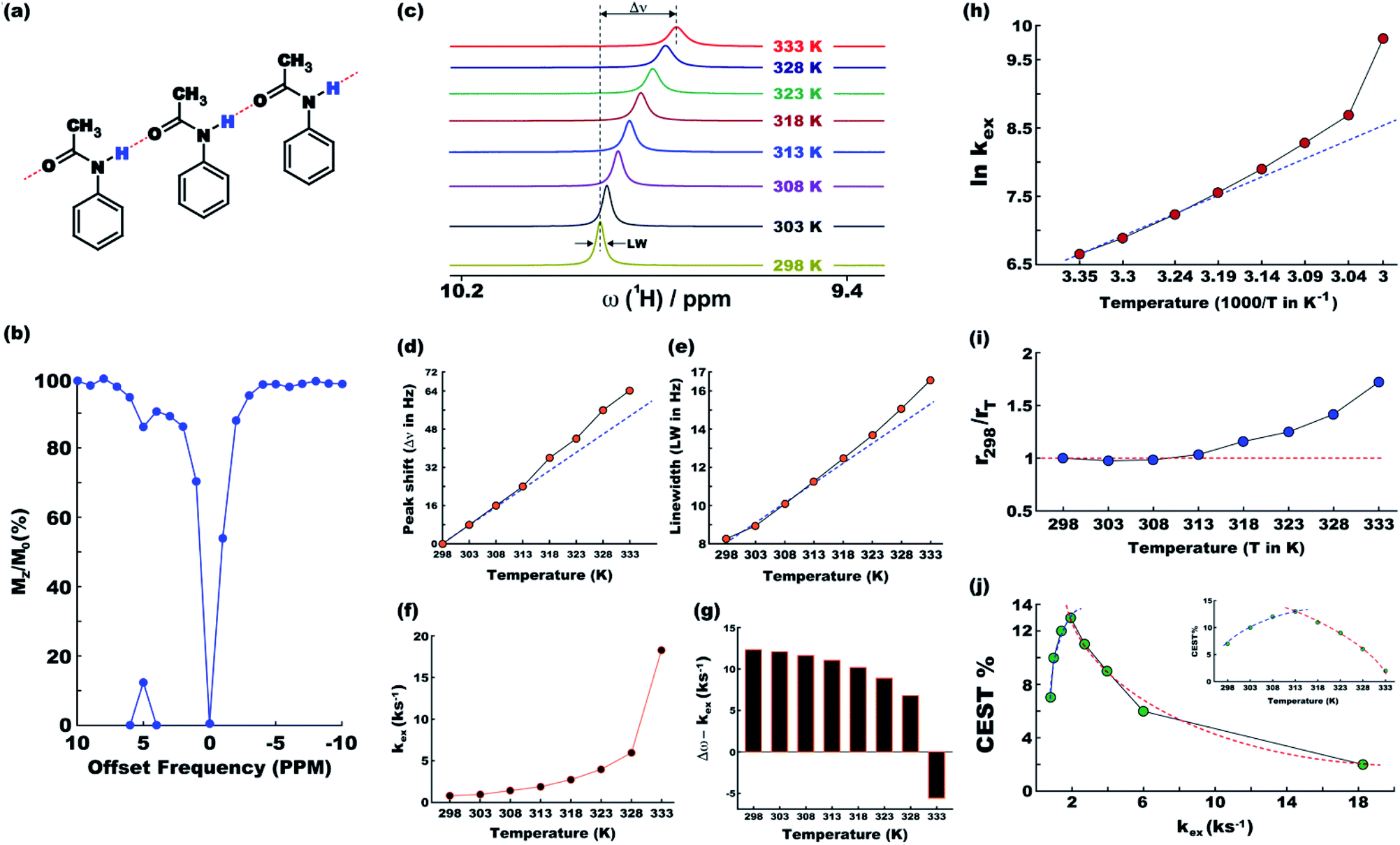
At ambient temperatures, acetanilide (1) is known18 to form molecular chains through interMHB as shown schematically in Fig. 1a. The hydrogen bond removes the partial double bond character of the C–N bond thereby facilitating a stable configuration that relieves the steric interaction between the ortho and the amine hydrogen atoms.18 We hypothesize that it is this chain formation that makes acetanilide a diaCEST agent as the interMHB lowers the exchange rate to the slow to medium exchange window. At physiological serum condition of pH 7.4 at 37 °C, acetanilide shows a significant 12.4% contrast with 15 mM concentration (Fig. 1b). To confirm our hypothesis that without the interMHB mediated molecular chains an individual acetanilide would not show appreciable CEST, we performed a series of variable temperature experiments. Assuming our hypothesis true, we anticipated that at some temperature when the interMHB chain would break, the CEST contrast would simply disappear. Fig. 1c shows the stack-plot of the labile amine proton of acetanilide acquired in DMSO-d6 as a function of temperature. The gradual upfield shift of the NH proton with temperature supports the slow reappearance of the electron density that was originally withdrawn due to the formation of the interMHB. When we plotted the peak shift (Δν) from the position at 298 K, we found an interesting behavior (Fig. 1d). Up to 313 K (40 °C) the shift followed the expected linear relationship with temperature as generally is observed for many solutes. However, beyond 313 K, the shift deviated from the linear relationship which strongly indicates the onset of an event, possibly the breaking up of the interMHB chain. The observed change in linewidth (LW) with temperature was found to be even more interesting (Fig. 1e). For small molecules, the transverse relaxation rate constant generally drops with a decrease in rotational correlation time (with an increase in temperature). As a consequence, the linewidth should also drop as the sample heats up. Contrary to this expected behaviour the linewidth was found to be monotonically increasing with temperature. Moreover, the linewidth also deviated from the expected linear behaviour (R2 changes nearly linearly with temperature for small molecules) beyond 313 K. This could only be explained if we assume that the acetanilide molecules jump between the free and the chain configurations causing exchange broadening. With an increase in temperature, the exchange broadening should naturally increase. The amine proton of acetanilide in water shows expected non-linear increase in solute–solvent exchange rate with temperature (Fig. 1f and S1–S9, ESI† for corresponding z-spectra). At 333 K the increase in exchange rate shows an avalanche which takes the exchange rate beyond the slow to medium exchange regime as can be seen from the plot of (Δω − kex) as a function of temperature (Fig. 1g). When we tried to check if the non-linear variation followed an Arrhenius type exponential equation and plotted ln(kex) as a function of 1/T, we found that the plot deviated from the expected linear behaviour beyond 313 K just as happened for the peak shift and the peak linewidth (Fig. 1h). The departure from the linear behaviour clearly indicates that the rise of the exchange rate was not entirely due to the rise of the solvent temperature but also due to the gradual breaking of the interMHB chain. Without the partial locking imposed by interMHB, the labile proton exchanged too fast. The sudden increase of kex at 333 K is possibly due to the complete disruption of the interMHB mediated molecular chain network. The discussion so far has picked up on a number of occasions the temperature 313 K as the onset temperature of an event. To further confirm that the event indeed was the breaking up of the molecular chain, we estimated at different temperatures the effective hydrodynamic radius (rT) of the molecules in the solution from the measured translational diffusion coefficient as per the following relationship,23| |
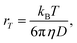 | (2) |
where, kB, T, η and D denote, respectively, the Boltzmann constant, absolute solvent temperature, solvent viscosity at temperature T and translational diffusion coefficient of acetanilide. In absence of any structural changes of the molecules, the effective hydrodynamic radius should remain unchanged and hence the ratio rT/rT′ for any two temperatures T and T′ should be equal to 1. Fig. 1i shows for different temperatures the ratio r298/rT, which deviates from the expected value of 1 beyond 313 K as has been seen previously with other experimental parameters. The fact that beyond 313 K, rT becomes smaller than r298 confirms the breaking of the longer molecular chains into smaller fragments. Finally, Fig. 1j shows CEST contrast efficiency of acetanilide as a function of the exchange constant and the temperature (Fig. 1j inset). Fig. 1j shows a smooth initial increase in the CEST contrast as a function of increasing kex (and also temperature) but beyond 313 K, the CEST efficiency starts abruptly dropping. A clear change in the slope of the curves (the blue and red dotted guidelines) indicate once again the onset of the event of breaking of interMHB chains. The population of the free acetanilide molecules in the dynamic mixture cannot produce CEST contrast due to very fast exchange and as with increasing temperature the fractional population of free molecules increases, the CEST efficiency gradually drops. At 333 K when the free molecule population dominates heavily, the CEST effect nearly disappears. The observation that the onset temperature (313 K) for all the events is so close to the physiological temperature of 310 K made us believe that without the interMHB, acetanilide would have had an exchange rate at the physiological temperature too high for any meaningful CEST contrast.
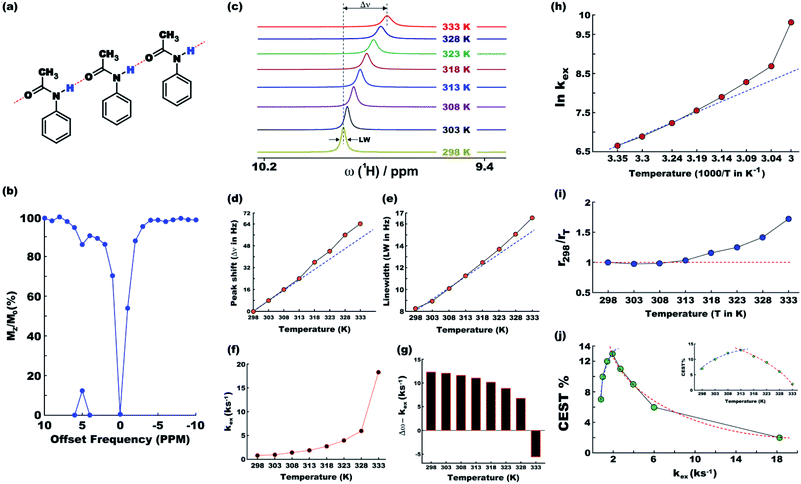 |
| | Fig. 1 (a) Schematic representation of molecular chain formation through inter-molecular hydrogen bonding by acetanilide in solution. (b) The CEST z-spectrum of acetanilide at the physiological condition (pH 7.4, 37 °C). MTRassym peak on the x-axis shows 12.4% CEST contrast. (c) Stack 1D plot of the labile amine peak of acetanilide in DMSO-d6 as a function of temperature. Temperature dependent peak-shift with respect to the position at 298 K (d) and peak line-width (e) of the acetanilide amine proton. (f) Exchange rate of the amine proton in water as a function of temperature. The exchange rate crosses the important limit of Δω (offset with respect to water peak position) at 333 K (g). (h) Natural logarithm of the exchange constant against the inverse of temperature. (i) The ratio of effective hydrodynamic radii at temperature T and at 298 K. (j) CEST contrast percentage as a function of exchange rate constant and temperature (inset). None of the dotted lines are mathematical fitting of data. They simply help to visualize the pattern of the data points. Please refer to the main text for analysis of the data towards confirming the gradual breaking up of the chain formation with increasing temperature. | |
3.2 Variation of exchange rate and CEST efficiency in different acetanilide derivatives
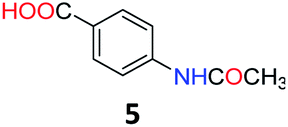
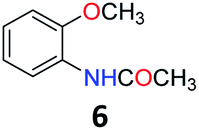
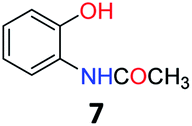
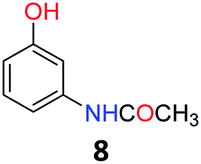
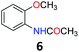
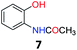
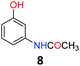
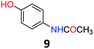
Upon being convinced that the acetanilide molecule as a single entity is not effective and needs to form a colony through interMHB to show contrast, we synthesized a number of water-soluble derivatives of acetanilide to explore the system while keeping the interMHB chain formation intact (Table 1 and Fig. S10–S25, ESI†). The steric and electronic effects brought in by different substitutions generally plays an important role in determining the frequency offset, exchange rate and CEST efficiency of a molecule. In the present scenario, the additional influence of interMHB was expected to play a pivotal role. In presence of electron withdrawing groups like fluorine or carboxylic acid at the para position, the electron density around the NH proton decreases resulting in a downfield shift of the amide proton with respect to acetanilide. Contrary to this, electron donating group at the para position increases the electron density and causes an upfield shift of the amide proton. In case of ortho substitutions, the field effect produces further upfield shift. For para hydroxy acetanilide the combination of inductive as well as the mesomeric effect of the hydroxyl group is instrumental in producing maximum upfield shift for the NH proton among all the derivatives studied. We found that for all the para-substitutions, a negative correlation exists between pKa of the molecule and the kex (Fig. S26–S32, ESI† for corresponding z-spectra). In the presence of electron-withdrawing substituents like fluorine (2), the electron density decreases at the nitrogen centre because of −I effect, which in turn increases the lability and hence kex of the amine proton. However, we did not find the CEST contrast increasing in accordance in comparison to 1. For para methyl (3) and para methoxy (4) substituents both kex and the CEST efficiency dropped from 1. In the case of carboxylic acid substitution at the para-position (5) the electron density increases at the site of hydrogen bond donation due to the presence of carboxylate anion at the basic pH. Thus the exchange rate as well as the CEST percentage quite expectedly decreased considerably. Among the ortho substituents, o-methoxy (6) and o-hydroxy (7) acetanilide showed a drastic increase in kex most likely due to the solvation of the molecules with the bulk water. Interestingly, even with such a steep increase in kex, the CEST efficiency was found to be very moderate for both 6 and 7. In the case of hydroxy substitutions, both meta (8) and the para (9) substituted acetanilide derivatives produced a higher CEST effect than o-hydroxy substitution, with 9 being the top of the lot in spite of the fact that 9 does not show the highest exchange rate among the hydroxy-substituted acetanilide compounds. As CEST contrast depends on several other factors including relaxation rate constants of the labile proton of the solute, and in the presence of interMHB network these parameters can widely vary, we could not see any direct correlation of the exchange rate and/or offset frequency with the CEST contrast efficiency of one acetanilide derivative with respect to the other. Paracetamol (9) shows better CEST efficiency than most other derivatives in spite of the fact that it has one of the smallest frequency offsets.
3.3 Study of CEST properties of paracetamol
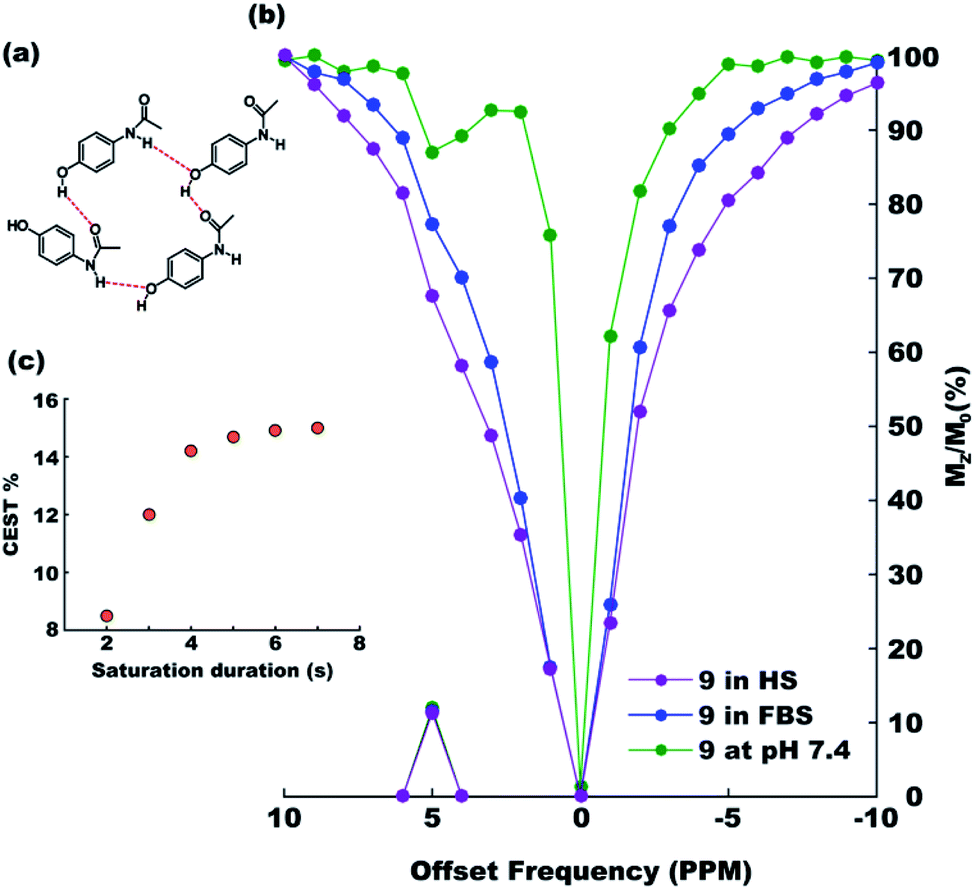
After exploring the electronic and steric influences of different substitutions on the CEST properties of acetanilide, we propose para-hydroxyl acetanilide (also known popularly as paracetamol) as a potential CEST contrast agent. Paracetamol is a widely used analgesic having a proven safety track record for human consumption.24 It is easily synthesizable in pure form from 4-aminophenol using the procedure discussed earlier. At ambient temperatures, paracetamol makes two-dimensional sheet through interMHB as shown in Fig. 2a.25 Both the hydroxyl and the carbonyl oxygen atoms take part in an alternative arrangement to form the sheet. Fig. 2b shows the overlaid CEST z-spectra of paracetamol at physiological temperature of 37 °C and pH 7.4 in the buffer and two biological media, Fetal Bovine Serum (FBS) and Horse Serum (HS). At 15 mM concentration, paracetamol produces 12% contrast in the buffer as well as in biological media with 3 s saturation at 5 μT irradiation power. The CEST contrast increases with saturation duration (observed in buffer) and saturates at 15% above 6 s saturation duration (Fig. 2c).
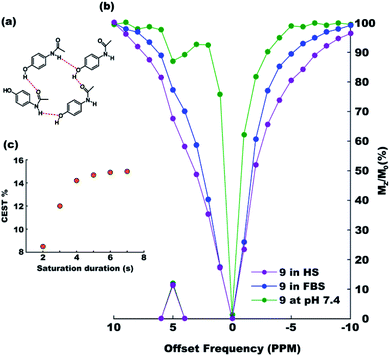 |
| | Fig. 2 (a) The intermolecular hydrogen bond mediated molecular sheet formation by paracetamol.23 (b) Overlaid z-spectra of 15 mM solutions of paracetamol in 10 mM (1×) PBS buffer (green), Fetal Bovine Serum (FBS, blue) and Horse Serum (HS, purple) at the physiological condition acquired with 3 s saturation with 5 μT power. (c) CEST efficiency (MTRassym) as a function of saturation duration at 5 μT r.f. power. | |
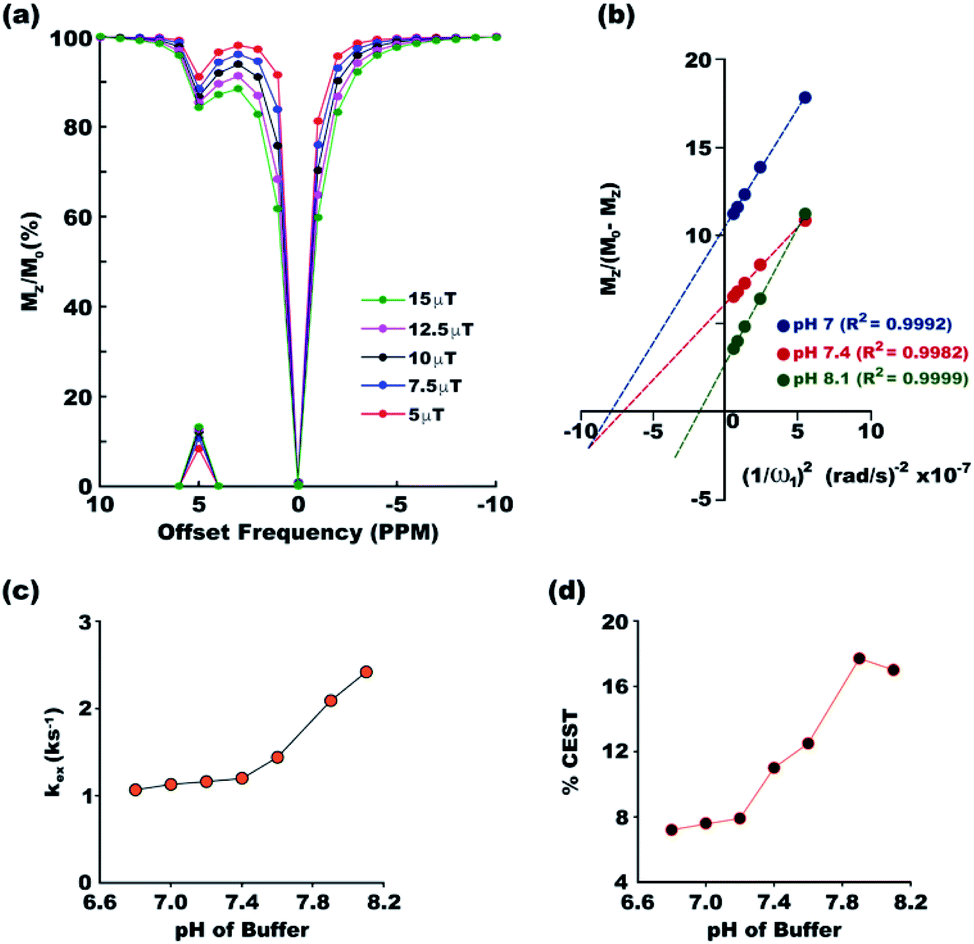
Exchange rates between the two pools of protons, one constituted by the solute and the other by the solvent, dictates the efficiency of a CEST contrast agent. To ensure accurate measurement of exchange rates, all the experimental conditions required for exchange rate measurement using the method proposed by Dixon et al. were satisfied. Long 6 s saturation was used for complete saturation (as indicated by Fig. 2c), the maximum power used was 639 Hz (corresponding to 15 μT field strength) which is well within Δω = 3276 Hz at 16.4 T (the highest magnetic field available in the laboratory) and DS was measured to be less than 3% even with the maximum applied r.f power. Fig. 3a shows the overlaid z-spectra of paracetamol at different saturation fields at the physiological condition. The corresponding linear relationship between MZ/(M0 − MZ) and 1/ω12 is shown in Fig. 3b for the physiological pH 7.4 and also for the two endpoints of the pH range (pH 7 and pH 8.1) for the current study. The goodness of linear fit is evident in all the plots. At the physiological condition, paracetamol shows an exchange rate of 1.26 ± 0.04 ks−1 which is well within the desired slow to medium exchange window. Paracetamol shows monotonically increasing exchange rate as the buffer pH moves from acidic to basic (Fig. 3c). Such a pattern is well observed in most CEST contrast agents. Following the thumb rule of CEST agents, the CEST efficiency also increases along with the exchange rate and reaches a maximum of 17.7% at pH 7.9 (Fig. 3d). However, the CEST efficiency drops marginally at a very basic pH of 8.1 due to exchange broadening.
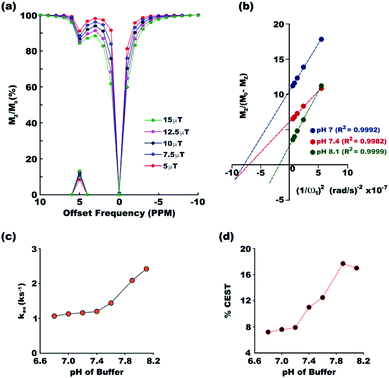 |
| | Fig. 3 (a) Overlaid z-spectra of paracetamol at different irradiation powers with 3 s saturation in the physiological condition. The peaks on the x-axis show corresponding MTRassym. (b) The linear fit for MZ/(M0 − MZ) plotted against 1/ω12 for three pH values, 7, 7.4 and 8.1. The goodness of fitting is given by the R2 values. Plots at other pH values are given in Fig. S33–S39 of ESI.† Exchange rate constant (c) and CEST efficiency (d) at different pH of the medium show gradual increase of both at basic pH. | |
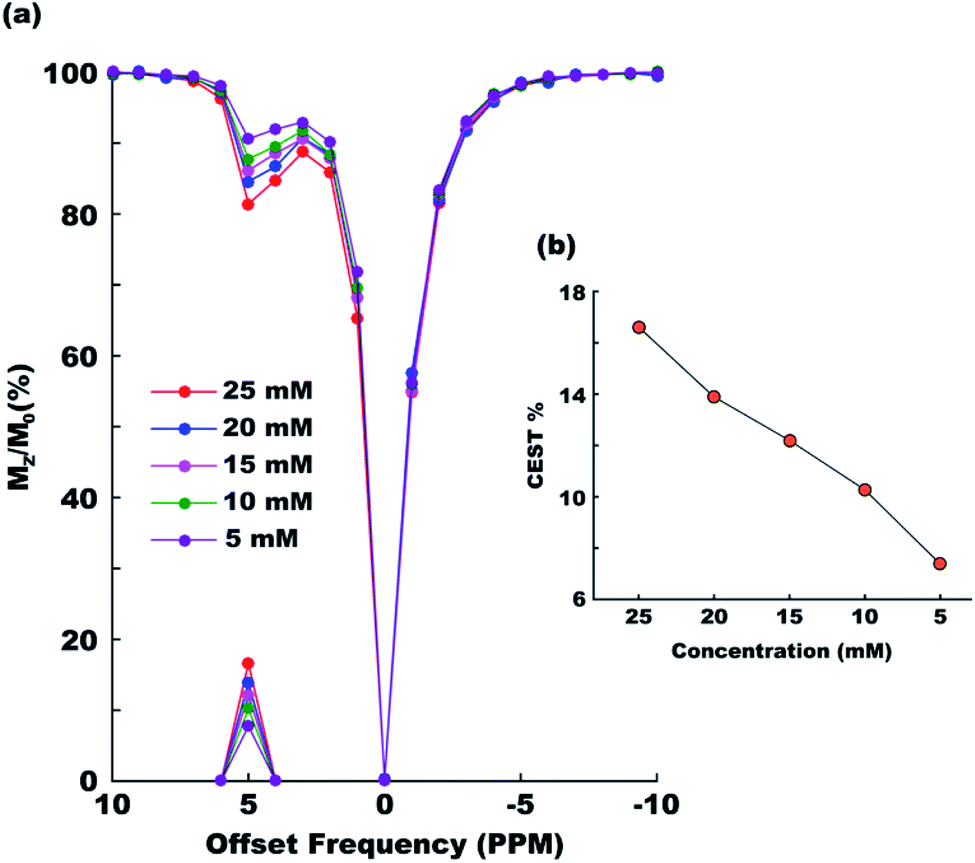
Paracetamol has a proven safety record for human consumption. But even then it is always desired that any foreign object be administered in the human body at a minimal possible concentration. To check the efficiency of paracetamol as a CEST agent in the range of concentrations generally considered safe we recorded CEST spectra at six different concentrations ranging between 25 mM and 5 mM (Fig. 4a). The CEST contrast was found dropping nearly linearly from approximately 17% (25 mM) to 7% (5 mM) in the said range (Fig. 4b).
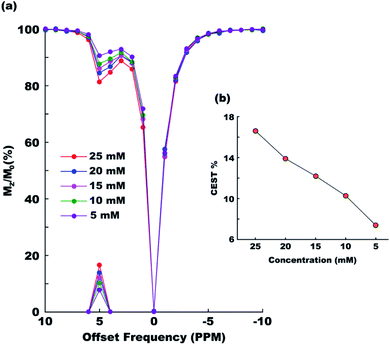 |
| | Fig. 4 CEST contrast efficiency of paracetamol at different concentrations between 5 mM and 25 mM. (a) Overlaid z-spectra and (b) MTRassym as a function of concentration. All experiments were performed with 3 s saturation duration at 5 μT irradiation power. | |
Finally, we calculated the relaxivities of paracetamol by measuring concentration normalized changes in longitudinal (for R1 relaxivity) and transverse (for R2 relaxivity) relaxation times of water in presence of paracetamol. The R1 and R2 relaxivities of paracetamol were found, respectively, 0.02 s−1 mM−1 and 0.01 s−1 mM−1. The calculated value for R2 relaxivity matched a previous report where it has been discussed that the high relaxivity is due to the phenolic proton of paracetamol.26 The phenolic proton executes a much faster exchange in comparison to the CEST producing amine proton and hence shows R2ex contrast instead. This makes paracetamol a very special molecule in which two different labile protons produce MRI contrast following two completely different mechanisms.
4. Conclusions
In summary, we have demonstrated that the formation of molecular chains or sheets through inter-molecular hydrogen bonding can transform a collection of otherwise not suitable acetanilide derivatives into diamagnetic CEST MRI contrast agents. The hydrogen bonding network shifts the contrast producing labile protons downfield and lowers their exchange constants so optimally that the performance of the contrast agents peaks just about at the physiological condition. A notable member of the group is the well-known analgesic drug ‘paracetamol’ which shows 12% contrast with 15 mM concentration at the physiological temperature and pH. Paracetamol is regularly administered both orally and through intravenous route in humans and hence paracetamol has the potential of being used as either oral contrast agent (OCA)27 or as extracellular fluid contrast agent (ECF).27 Nearly 95% of paracetamol metabolism occurs in the liver and is generally considered safe for human consumption.24 Although one can expect marginal drop in CEST efficiency at the lower magnetic field of clinical MRI scanners, we believe our findings make it a good candidate nonetheless for in vivo applications as a diaCEST agent.
5. Spectral characterization
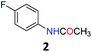
5.1 N-(4-Fluorophenyl)acetamide (2)28
1H NMR (400 MHz, DMSO) δ 9.97 (s, 1H), 7.60–7.56 (m, 2H), 7.12 (t, J = 8.9 Hz, 1H), 2.03 (s, 3H). 13C NMR (176 MHz, DMSO) δ 168.60, 159.00, 157.64, 136.19, 121.24, 121.20, 115.66, 115.54, 24.28.
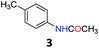
5.2 N-(4-Tolyl)acetamide (3)29
1H NMR (700 MHz, DMSO) δ 9.78 (s, 1H), 7.45 (d, J = 8.2 Hz, 2H), 7.08 (d, J = 8.2 Hz, 2H), 2.24 (s, 3H), 2.02 (s, 3H). 13C NMR (176 MHz, DMSO) δ 168.50, 137.28, 129.45, 119.56, 24.35, 20.85.
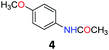
5.3 N-(4-Methoxyphenyl)acetamide (4)29
1H NMR (400 MHz, DMSO) δ 9.76 (s, 1H), 7.47 (d, J = 9.1 Hz, 1H), 6.86 (d, J = 9.0 Hz, 1H), 3.71 (s, 3H), 2.00 (s, 3H). 13C NMR (101 MHz, DMSO) δ 168.18, 155.48, 132.99, 121.00, 114.25, 55.60, 24.25.
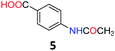
5.4 4-Acetamidobenzoic acid (5)30
1H NMR (400 MHz, DMSO) δ 12.64 (s, 1H), 10.23 (s, 1H), 7.88 (d, J = 8.7 Hz, 2H), 7.69 (d, J = 8.8 Hz, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO) δ 169.33, 167.40, 143.80, 130.84, 125.36, 118.64, 24.62.
5.5 N-(2-Methoxyphenyl)acetamide (6)31
1H NMR (700 MHz, DMSO) δ 9.11 (s, 1H), 7.92–7.91 (m, 1H), 7.09–7.04 (m, 1H), 7.03 (dd, J = 8.2, 1.6 Hz, 1H), 6.89 (td, J = 7.7, 1.6 Hz, 1H), 3.83 (s, 3H), 2.08 (s, 3H). 13C NMR (176 MHz, DMSO) δ 168.88, 150.00, 127.88, 124.65, 122.49, 120.61, 111.53, 56.05, 24.31.
5.6 N-(2-Hydroxyphenyl)acetamide (7)32
1H NMR (700 MHz, DMSO) δ 9.77 (s, 1H), 9.31 (s, 1H), 7.66 (dd, J = 8.0, 1.7 Hz, 1H), 7.01–6.91 (m, 1H), 6.86 (dd, J = 8.0, 1.5 Hz, 1H), 6.76 (t, J = 7.7 Hz, 1H), 2.09 (s, 3H). 13C NMR (176 MHz, DMSO) δ 169.54, 148.36, 126.82, 125.16, 122.88, 119.44, 116.37, 24.03.
5.7 N-(3-Hydroxyphenyl)acetamide (8)33
1H NMR (700 MHz, DMSO) δ 9.78 (s, 1H), 9.33 (s, 1H), 7.18 (d, J = 2.1 Hz, 1H), 7.04 (t, J = 8.0 Hz, 1H), 6.91 (dd, J = 8.0, 2.0 Hz, 1H), 6.42 (dd, J = 8.0, 2.4 Hz, 1H), 2.01 (s, 3H). 13C NMR (176 MHz, DMSO) δ 168.61, 158.01, 140.82, 129.73, 110.55, 110.18, 106.59, 24.52.
5.8 N-(4-Hydroxyphenyl)acetamide (9)34
1H NMR (700 MHz, DMSO) δ 9.64 (s, 1H), 9.13 (s, 1H), 7.33 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.7 Hz, 1H), 1.98 (s, 3H). 13C NMR (176 MHz, DMSO) δ 167.96, 153.57, 131.50, 121.25, 115.4.
Author contributions
SC conceptualized the project. SP designed and synthesized the compounds. SC collected and analysed data using in-house MATLAB packages written by AG. Overall supervision of the project was done by AG. Original draft was written by AG while review and editing on final draft was done by all.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The infrastructure provided by NISER and DAE, India is duly acknowledged. SC thanks NISER, DAE for financial support.
References
- V. P. B. Grover, J. M. Tognarelli, M. M. E. Crossey, I. J. Cox, S. D. Taylor-Robinson and M. J. W. McPhail, J. Clin. Exp. Hepatol., 2015, 5, 246–255 CrossRef.
- E. Seeram, Radiol. Technol., 2018, 89, 279CT–302CT Search PubMed.
-
(a) Y.-D. Xiao, R. Paudel, J. Liu, C. Ma, Z.-S. Zhang and S.-K. Zhou, Int. J. Mol. Med., 2016, 38, 1319–1326 CrossRef CAS;
(b) C. F. G. C. Geraldes and S. Laurent, Contrast Media Mol. Imaging, 2009, 4, 1–23 CrossRef CAS.
- I. R. Young, G. J. Clarke, D. R. Baffles, J. M. Pennock, F. H. Doyle and G. M. Bydder, J. Comput. Tomogr., 1981, 5, 543–547 CrossRef CAS.
-
(a) D. V. Hingorani, A. S. Bernstein and M. D. Pagel, Contrast Media Mol. Imaging, 2015, 10, 245–265 CrossRef CAS;
(b) F. Ye, S. Laurent, A. Fornara, L. Astolfi, J. Qin, A. Roch, A. Martini, M. S. Toprak, R. N. Muller and M. Muhammed, Contrast Media Mol. Imaging, 2012, 7, 460–468 CrossRef CAS.
- K. M. Ward, A. H. Aletras and R. S. Balaban, J. Magn. Reson., 2000, 143, 79–87 CrossRef CAS.
-
(a) A. D. Sherry and M. Woods, Annu. Rev. Biomed. Eng., 2008, 10, 391–411 CrossRef CAS;
(b) P. C. M. van Zijl and N. N. Yadav, Magn. Reson. Med., 2011, 65, 927–948 CrossRef CAS;
(c) G. Liu, X. Song, K. W. Y. Chan and M. T. McMahon, NMR Biomed., 2013, 26, 810–828 CrossRef CAS.
-
(a) S. Aime, M. Botta, M. Fasano and E. Terreno, Chem. Soc. Rev., 1998, 27, 19–29 RSC;
(b) S. Aime, A. Barge, D. Delli Castelli, F. Fedeli, A. Mortillaro, F. U. Nielsen and E. Terreno, Magn. Reson. Med., 2002, 47, 639–648 CrossRef CAS;
(c) S. Aime, D. Delli Castelli and E. Terreno, Angew. Chem., Int. Ed., 2002, 41, 4334–4336 CrossRef CAS;
(d) S. Zhang, M. Merritt, D. E. Woessner, R. E. Lenkinski and A. D. Sherry, Acc. Chem. Res., 2003, 36, 783–790 CrossRef CAS;
(e) M. Woods, D. E. Woessner and A. D. Sherry, Chem. Soc. Rev., 2006, 35, 500–511 RSC;
(f) S. Aime, D. D. Castelli, S. G. Crich, E. Gianolio and E. Terreno, Acc. Chem. Res., 2009, 42, 822–831 CrossRef CAS;
(g) S. Viswanathan, Z. Kovacs, K. N. Green, S. J. Ratnakar and A. D. Sherry, Chem. Rev., 2010, 110, 2960–3018 CrossRef CAS;
(h) I. Hancu, W. T. Dixon, M. Woods, E. Vinogradov, A. D. Sherry and R. E. Lenkinski, Acta Radiol., 2010, 51, 910–923 CrossRef;
(i) P. B. Tsitovich, J. M. Cox, J. A. Spernyak and J. R. Morrow, Inorg. Chem., 2016, 55, 12001–12010 CrossRef CAS;
(j) P. J. Burns, J. M. Cox and J. R. Morrow, Inorg. Chem., 2017, 56, 4545–4554 CrossRef;
(k) R. N. Pradhan, S. Chakraborty, P. Bharti, J. Kumar, A. Ghosh and A. K. Singh, Dalton Trans., 2019, 48, 8899–8910 RSC.
- C. Rydahl, H. S. Thomsen and P. Marckmann, Invest. Radiol., 2008, 43, 141–144 CrossRef CAS.
-
(a) P. C. van Zijl, C. K. Jones, J. Ren, C. R. Malloy and A. D. Sherry, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4359–4364 CrossRef CAS;
(b) W. Ling, R. R. Regatte, G. Navon and A. Jerschow, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2266 CrossRef CAS;
(c) M. Haris, K. Cai, A. Singh, H. Hariharan and R. Reddy, NeuroImage, 2011, 54, 2079–2085 CrossRef CAS;
(d) K. Cai, M. Haris, A. Singh, F. Kogan, J. H. Greenberg, H. Hariharan, J. A. Detre and R. Reddy, Nat. Med., 2012, 18, 302–306 CrossRef CAS;
(e) G. Liu, M. Moake, Y.-e. Har-el, C. M. Long, K. W. Y. Chan, A. Cardona, M. Jamil, P. Walczak, A. A. Gilad, G. Sgouros, P. C. M. van Zijl, J. W. M. Bulte and M. T. McMahon, Magn. Reson. Med., 2012, 67, 1106–1113 CrossRef CAS;
(f) K. W. Y. Chan, M. T. McMahon, Y. Kato, G. Liu, J. W. M. Bulte, Z. M. Bhujwalla, D. Artemov and P. C. M. van Zijl, Magn. Reson. Med., 2012, 68, 1764–1773 CrossRef CAS;
(g) A. Bar-Shir, G. Liu, M. M. Greenberg, J. W. M. Bulte and A. A. Gilad, Nat. Protoc., 2013, 8, 2380–2391 CrossRef CAS;
(h) A. Bar-Shir, G. Liu, Y. Liang, N. N. Yadav, M. T. McMahon, P. Walczak, S. Nimmagadda, M. G. Pomper, K. A. Tallman, M. M. Greenberg, P. C. M. van Zijl, J. W. M. Bulte and A. A. Gilad, J. Am. Chem. Soc., 2013, 135, 1617–1624 CrossRef CAS;
(i) K. W. Y. Chan, G. Liu, X. Song, H. Kim, T. Yu, D. R. Arifin, A. A. Gilad, J. Hanes, P. Walczak, P. C. M. van Zijl, J. W. M. Bulte and M. T. McMahon, Nat. Mater., 2013, 12, 268–275 CrossRef CAS;
(j) S. Walker-Samuel, R. Ramasawmy, F. Torrealdea, M. Rega, V. Rajkumar, S. P. Johnson, S. Richardson, M. Gonçalves, H. G. Parkes, E. Arstad, D. L. Thomas, R. B. Pedley, M. F. Lythgoe and X. Golay, Nat. Med., 2013, 19, 1067–1072 CrossRef CAS;
(k) M. Haris, A. Singh, K. Cai, K. Nath, R. Crescenzi, F. Kogan, H. Hariharan and R. Reddy, J. Neurosci. Methods, 2013, 212, 87–93 CrossRef CAS;
(l) A. Bar-Shir, G. Liu, K. W. Y. Chan, N. Oskolkov, X. Song, N. N. Yadav, P. Walczak, M. T. McMahon, P. C. M. van Zijl, J. W. M. Bulte and A. A. Gilad, ACS Chem. Biol., 2014, 9, 134–138 CrossRef CAS;
(m) K. W. Chan, T. Yu, Y. Qiao, Q. Liu, M. Yang, H. Patel, G. Liu, K. W. Kinzler, B. Vogelstein, J.
W. Bulte, P. C. van Zijl, J. Hanes, S. Zhou and M. T. McMahon, J. Controlled Release, 2014, 180, 51–59 CrossRef CAS;
(n) K. W. Y. Chan, J. W. M. Bulte and M. T. McMahon, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 111–124 CAS;
(o) S. Maruyama, J. Ueda, A. Kimura and K. Murase, Magn. Reson. Med. Sci., 2016, 15, 324–334 CrossRef CAS.
-
(a) X. Yang, X. Song, Y. Li, G. Liu, S. Ray Banerjee, M. G. Pomper and M. T. McMahon, Angew. Chem., Int. Ed., 2013, 52, 8116–8119 CrossRef CAS;
(b) W. G. Lesniak, N. Oskolkov, X. Song, B. Lal, X. Yang, M. Pomper, J. Laterra, S. Nimmagadda and M. T. McMahon, Nano Lett., 2016, 16, 2248–2253 CrossRef CAS.
- X. Song, X. Yang, S. Ray Banerjee, M. G. Pomper and M. T. McMahon, Contrast Media Mol. Imaging, 2015, 10, 74–80 CrossRef CAS.
- X. Yang, N. N. Yadav, X. Song, S. Ray Banerjee, H. Edelman, I. Minn, P. C. M. van Zijl, M. G. Pomper and M. T. McMahon, Chem.–Eur. J., 2014, 20, 15824–15832 CrossRef CAS.
- X. Yang, X. Song, S. Ray Banerjee, Y. Li, Y. Byun, G. Liu, Z. M. Bhujwalla, M. G. Pomper and M. T. McMahon, Contrast Media Mol. Imaging, 2016, 11, 304–312 CrossRef CAS.
-
(a) X. Zhang, Y. Yuan, S. Li, Q. Zeng, Q. Guo, N. Liu, M. Yang, Y. Yang, M. Liu, M. T. McMahon and X. Zhou, Magn. Reson. Med., 2019, 82, 577–585 CrossRef CAS;
(b) S. Chakraborty, M. Das, A. Srinivasan and A. Ghosh, New J. Chem., 2021, 45, 1262–1268 RSC.
-
(a) S. Aime, L. Calabi, L. Biondi, M. De Miranda, S. Ghelli, L. Paleari, C. Rebaudengo and E. Terreno, Magn. Reson. Med., 2005, 53, 830–834 CrossRef CAS;
(b) D. L. Longo, W. Dastrù, G. Digilio, J. Keupp, S. Langereis, S. Lanzardo, S. Prestigio, O. Steinbach, E. Terreno, F. Uggeri and S. Aime, Magn. Reson. Med., 2011, 65, 202–211 CrossRef CAS;
(c) D. L. Longo, P. Z. Sun, L. Consolino, F. C. Michelotti, F. Uggeri and S. Aime, J. Am. Chem. Soc., 2014, 136, 14333–14336 CrossRef CAS;
(d) S. Sinharay, E. A. Randtke, C. M. Howison, N. A. Ignatenko and M. D. Pagel, Mol. Imaging Biol., 2018, 20, 240–248 CrossRef CAS;
(e) X. Cai, J. Zhang, J. Lu, L. Yi, Z. Han, S. Zhang, X. Yang and G. Liu, Chem.–Eur. J., 2020, 26, 11705–11709 CrossRef CAS.
-
(a) A. Greenberg, C. M. Breneman and J. F. Liebman, The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, Wiley, 2000 Search PubMed;
(b) J. Pitzer and K. Steiner, J. Biotechnol., 2016, 235, 32–46 CrossRef CAS;
(c) A. K. Agrahari, A. K. Singh, A. S. Singh, M. Singh, P. Maji, S. Yadav, S. Rajkhowa, P. Prakash and V. K. Tiwari, New J. Chem., 2020, 44, 19300–19313 RSC;
(d) C. W. Evans, S. Edwards, J. A. Kretzmann, G. L. Nealon, R. Singh, T. D. Clemons, M. Norret, C. A. Boyer and K. S. Iyer, New J. Chem., 2020, 44, 20013–20020 RSC;
(e) S. Zhang, J. Li, J. Li, N. Du, D. Li, F. Li and J. Man, RSC Adv., 2020, 10, 34308–34322 RSC;
(f) L. Posada, D. Davyt and G. Serra, RSC Adv., 2020, 10, 43653–43659 RSC.
- J. Binoy, N. B. Prathima, C. Murali Krishna, C. Santhosh, I. Hubert Joe and V. S. Jayakumar, Laser Phys., 2006, 16, 1253–1263 CrossRef CAS.
- B. J. Anderson, Pediatr. Anesth., 2008, 18, 915–921 CrossRef.
- B. S. Furniss, A. J. Hannaford, P. W. G. Smith and A. R. Tatchell, Vogel's textbook of practical organic chemistry, Longman Scientific & Technical, 5th edn, 1989, p. 985 Search PubMed.
- W. T. Dixon, J. Ren, A. J. M. Lubag, J. Ratnakar, E. Vinogradov, I. Hancu, R. E. Lenkinski and A. D. Sherry, Magn. Reson. Med., 2010, 63, 625–632 CrossRef CAS.
- D. H. Wu, A. D. Chen and C. S. Johnson, J. Magn. Reson., Ser. A, 1995, 115, 260–264 CrossRef CAS.
- C. C. Miller and J. Walker, Proc. R. Soc. London, Ser. A, 1924, 106, 724–749 CAS.
-
(a) L. L. Mazaleuskaya, K. Sangkuhl, C. F. Thorn, G. A. FitzGerald, R. B. Altman and T. E. Klein, Pharmacogenet. Genomics, 2015, 25, 416–426 CrossRef CAS;
(b) J. A. Forrest, J. A. Clements and L. F. Prescott, Clin. Pharmacokinet., 1982, 7, 93–107 CrossRef CAS.
- G.-W. An, H. Zhang, X.-L. Cheng, Q.-L. Zhuo and Y.-C. Lv, Struct. Chem., 2008, 19, 613 CrossRef CAS.
- J. Zhang, Y. Li, S. Slania, N. N. Yadav, J. Liu, R. Wang, J. Zhang, M. G. Pomper, P. C. van Zijl, X. Yang and G. Liu, Chem.–Eur. J., 2018, 24, 1259–1263 CrossRef CAS.
- Y.-D. Xiao, R. Paudel, J. Liu, C. Ma, Z.-S. Zhang and S.-K. Zhou, Int. J. Mol. Med., 2016, 38, 1319–1326 CrossRef CAS.
- G. K. S. Prakash, M. D. Moran, T. Mathew and G. A. Olah, J. Fluorine Chem., 2009, 130, 806–809 CrossRef CAS.
- S. R. Thopate, S. R. Kote, S. V. Rohokale and N. M. Thorat, J. Chem. Res., 2011, 35, 124–125 CrossRef CAS.
- S. Mönch, M. Netzel, G. Netzel and M. Rychlik, Anal. Biochem., 2010, 398, 150–160 CrossRef.
- B. P. Fors, K. Dooleweerdt, Q. Zeng and S. L. Buchwald, Tetrahedron, 2009, 65, 6576–6583 CrossRef CAS.
- R. Moreno-Corral, H. Höpfl, L. Machi-Lara and K. O. Lara, Eur. J. Org. Chem., 2011, 2148–2162, DOI:10.1002/ejoc.201001312.
- S. S. van Berkel, B. van der Lee, F. L. van Delft and F. P. J. T. Rutjes, Chem. Commun., 2009, 4272–4274, 10.1039/B906762K.
- A. S. El-Shahawy, S. M. Ahmed and N. K. Sayed, Spectrochim. Acta, Part A, 2007, 66, 143–152 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C spectra of all the compounds. Additional CEST z-spectra. Additional omega plots for exchange rate calculation. See DOI: 10.1039/d0ra10410h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
S. Peruncheralathan
,
S. Peruncheralathan