
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and biological evaluation of 2-quinolyl-1,3-tropolone derivatives as new anti-cancer agents†
Evgeniy A. Gusakov‡
a,
Iuliia A. Topchu‡bc,
Aleksandra M. Mazitova‡bd,
Igor V. Dorogan a,
Emil R. Bulatovb,
Ilya G. Serebriiskiibe,
Zinaida I. Abramovab,
Inna O. Tupaevaa,
Oleg P. Demidovf,
Duong Ngoc Toang,
Tran Dai Lamh,
Duong Nghia Bangi,
Yanis A. Boumber*bc,
Yurii A. Sayapin*j and
Vladimir I. Minkina
a,
Emil R. Bulatovb,
Ilya G. Serebriiskiibe,
Zinaida I. Abramovab,
Inna O. Tupaevaa,
Oleg P. Demidovf,
Duong Ngoc Toang,
Tran Dai Lamh,
Duong Nghia Bangi,
Yanis A. Boumber*bc,
Yurii A. Sayapin*j and
Vladimir I. Minkina
aInstitute of Physical and Organic Chemistry, Southern Federal University, Rostov-on-Don, 344090, Russia
bKazan Federal University, Kazan, 420008, Russia
cRobert H Lurie Comprehensive Cancer Center, Division of Hematology/Oncology at the Department of Medicine, Feinberg School of Medicine, Northwestern University, 303 E. Superior Street, Chicago, IL 60611, USA. E-mail: yanis.boumber@northwestern.edu
dCedars-Sinai Medical Center, Department of Medicine, Los Angeles, CA 90048, USA
eFox Chase Cancer Center, Philadelphia, PA 19111, USA
fNorth Caucasus Federal University, Stavropol, 355009, Russia
gThai Nguyen University of Education, 20 Luong Ngoc Quyen, Thai Nguyen 24000, Vietnam
hInstitute for Tropical Technology, Vietnam Academy of Science and Technology, Hanoi, 10000, Vietnam
iThai Nguyen University of Sciences, Tan Thinh Ward, Thai Nguyen 24000, Vietnam
jFederal Research Centre the Southern Scientific Centre of the Russian Academy of Sciences, Chekhov Ave., 41, Rostov-on-Don, 344006, Russia. E-mail: sayapin@ipoc.sfedu.ru
First published on 22nd January 2021
Abstract
Tropolones are promising organic compounds that can have important biologic effects. We developed a series of new 2-quinolyl-1,3-tropolones derivatives that were prepared by the acid-catalyzed reaction of 4,7-dichloro-2-methylquinolines with 1,2-benzoquinones. 2-Quinolyl-1,3-tropolones have been synthesized and tested for their anti-proliferative activity against several human cancer cell lines. Two compounds (3d and mixture B of 3i–k) showed excellent activity against six cancer cell lines of different tissue of origin. The promising compounds 3d and mixture B of 3i–k also demonstrated induction of apoptotic cell death of ovarian cancer (OVCAR-3, OVCAR-8) and colon cancer (HCT 116) cell lines and affected ERK signaling. In summary, 2-quinolyl-1,3-tropolones are promising compounds for development of effective anticancer agents.
1. Introduction
Today, cancer remains one of the leading causes of death in most parts of the world and is a serious obstacle to increasing life expectancy in most countries.1 Ovarian cancer is the seventh most common type of cancer in women worldwide2 and one of the most common type so gynecological malignant tumors.3 Colon cancer is both the third most common cancer and the third most common cause of death among cancer patients.1 The standard systemic therapy for ovarian cancer is platinum chemotherapy doublet (cisplatin and carboplatin)4 in combination with taxanes (paclitaxel or docetaxel) and sometime doxorubicin,5 while for colon cancer, the most effective chemotherapy strategy is chemotherapy doublet with oxaliplatin, combined with either 5-fluorouracil or irinotecan.6However, platinum compounds have serious side effects such as nausea and vomiting, nephrotoxicity, ototoxicity, neurotoxicity, cytopenia and muscle wasting.7 In addition, in metastatic settings, very often, recurrent ovarian or colon cancers commonly acquire resistance to platinum drugs.8,9 All these factors negatively affect both the quality of life of cancer patients and their survival rate. Therefore, the search and development of new medicinal compounds with antitumor activity remains an urgent task.
Derivatives of tropolones are a class of promising organic compounds that can act as new pharmaceutical anti-inflammatory and antitumor agents. Currently, there is an increased interest in tropolone alkaloids, since many of them have a wide range of pharmacological activity. For example, some of the best-known representatives are colchicine, colchamine and β-thujaplicin (hinokitiol), which have antitumor, antibacterial, antiviral, antifungal, anti-inflammatory and antioxidant properties.10–12 In particular, high anti-bacterial and anti-inflammatory effects of natural alkaloids cordytropolone and stipitalide against Propionibacterium acnes have been recently shown.13 3-Isopropenyl-tropolone has high activity against antibiotic-resistant bacteria Staphylococcus aureus,14 while hinokitiol was effective against MRSA, Aggregatibacter actinomycetemcomitans, Streptococcus mutans, and Candida albicans.15 The antiviral properties of natural hydroxylated tropolones (β-thujaplicinol, manicol) against HIV-1.16 have also been established.16
There are a number of studies demonstrating in more detail the antitumor activity of hinokitiol. Hinokitiol blocks DNA homology recombination and repair process in breast cancer and osteosarcoma cells and is able to radiosensitize these cells and increase apoptosis in combination with PARP inhibitor.17 Hinokitiol also exhibits antitumor effect in malignant lymphoma and myeloma cells by inducing caspase-dependent apoptosis.18,19 Moreover, it can inhibit migration of melanoma cells.20 An equally interesting fact exists about the relationship between hinokitiol and DNA methylation in colon cancer cells. Hinokitiol can block DNA methylation via DNMT1 inhibition that may open up the prospects for its use as a new DNMT1 inhibitor.21
Thus, tropolone derivatives have multi-target biologic and anti-tumor activity in various types of cancer cells and are therefore of interest for further studies.
Synthetic methods for the preparation of analogs of natural tropoids with high biological activity have been described in detail earlier.22 However, related 1,3-tropolone systems with heterocyclic fragments have hardly been studied.
We have previously reported on the cytotoxic activity of halogenated derivatives of [benzo[b][1,4]oxazepino[7,6,5-de]quinolyl]-1,3-tropolones. The most significant result in the investigated series was shown by 2-[7-acetyl-9,11-di(tert-butyl)-4-methyl-5-chlorobenzo[b][1,4]oxazepino[7,6,5-de]-quinolyl]-5,6,7-trichloro-1,3-tropolone, inhibited the growth of breast cancer cells MCF-7 at IC50 concentration of 12.96 μg ml−1, as well as of epidermoid cells KB, lung cancer cells Lu and liver cancer cells Hep-G2 at IC50 concentrations >128 μg ml−1.23 In the present work, we report on the synthesis, molecular structure and cytotoxic properties of the novel 2-quinolin-2-yl-1,3-tropolones.
2. Results and discussion
2.1 Chemistry
Previously, it was established that acid-catalyzed reactions methylene-active heterocycles (substituted benzazoles or 3,3-dimethylindolines,24 quinolines,25 quinoxalines,26 benzoxazinones,27 quinazolinone28) with 1,2-benzoquinone derivatives proceed with the expansion of the o-quinone ring and can lead to the formation of 2-hetaryl-1,3-tropolones. The yields of the final 2-hetaryl-1,3-tropolones can vary in the particular series of methylene active heterocyclic compound or 1,2-benzoquinone depending on the substituents. The most optimal condition for the synthesis of 2-hetaryl-substituted 1,3-tropolones is to carry out the reaction at moderate temperatures (60–80 °C) in acetic acid solution.In this work, we used 2,8-dimethyl-4,7-dichloroquinoline (1a) and its 5-nitro derivative (1b) as the methylene-active heterocycle, and 3,5-di(tert-butyl)-1,2-benzoquinone (2a), 4,6-di(tert-butyl)-3-nitro-1,2-benzoquinone (2b), 4,6-diisopropyl-3-nitro-1,2-benzoquinone (2c),29 3,4,5,6-tetrachloro-1,2-benzoquinone (2d) as the initial 1,2-benzoquinones. The preparation of 2-quinolyl-1,3-tropolone derivatives 3 is presented in Scheme 1.
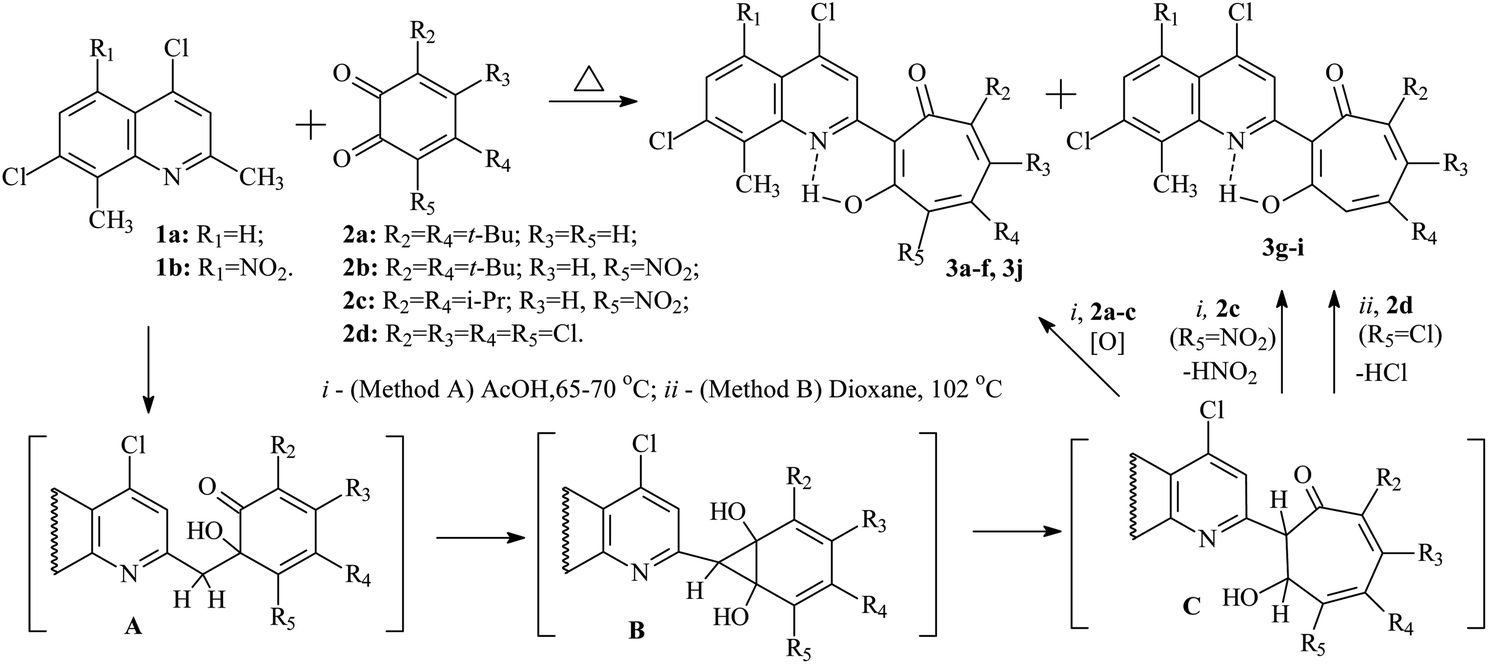 | ||
| Scheme 1 Synthesis of 2-quinolyl-1,3-tropolone derivatives 3 by an alternative pathway of the reaction. | ||
It has been established that the interaction of quinolines 1a, b with a twofold excess of quinones 2a–c leads to the formation of the corresponding 2-quinolyl-1,3-tropolones 3a–f (method A). However, in the reaction of 1a with quinone 2c, tropolone 3g is formed along with tropolone 3f in small amounts (10%). Reaction products 3f and 3g were isolated from the reaction mixture by column chromatography. On the other hand, reaction 1b with quinone 2c (method A) proceeded with the formation of only tropolone 3e.
Refluxing 1a with an equivalent amount of 2d in dioxane (method B) leads to the formation of 2-quinolyl-5,6,7-trichloro-1,3-tropolone 3h. However, the reaction of 5-nitro derivative 1b with 2d gives rise the mixture of three polychloro-substituted tropolones: 5,6,7-trichloro-1,3-tropolone 3i, 4,5,6,7-tetrachloro-1,3-tropolone 3j and 4,5,6-trichloro-1,3-tropolone 3k (Scheme 2) in a ratio of 55, 30, and 15%, respectively (mixture B of 3i–k). Since it was impossible to separate a mixture of tropolones 3i–k by thin layer or column chromatography, the ratio of the mixture has been determined from the integrated signal intensity of OH-group protons.
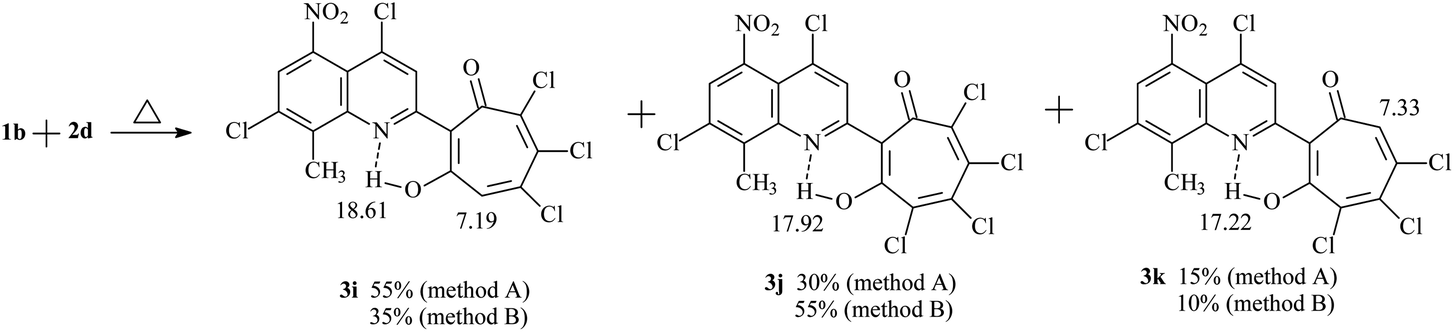 | ||
| Scheme 2 Formation of the mixture of three polychloro-substituted 1,3-tropolones 3i–k. | ||
Carrying out the reaction 1b (1 eq.) with 2d (2 eq.) under milder conditions in acetic acid (method A) leads to a change in the ratio of the mixture of polychlorotropolones (35% (3i), 55% (3j) 10% (3k)). Since the mixture B of tropolones 3i–k can easily be obtained synthetically, mixture of B was investigated further for the anticancer activity.
The mechanism of formation of the tropolone system 3 was studied earlier by theoretical methods. The multistep reaction includes aldol condensation with the formation of intermediate A, cyclization of A into the norcaradiene derivative B, and rearrangement of B into the dihydrotropolone C. Oxidation of the latter with an excess of the initial 1.2-benzoquinone 2 leads to 1,3-tropolones 3a–f (method A).
Interaction of 4,7-dichloro-2-methylquinolines 1a with 4,6-diisopropyl-3-nitro-1,2-benzoquinone 2c gives rise two competing processes: the formation of tropolone 3f and the reaction of nitrous acid elimination in C, accompanied by 1,3-sigmatropic shift of the hydrogen atom, which ultimately leads to the formation of 1,3-tropolone 3g.
o-Quinone ring expansion upon refluxing 1a,b with 3,4,5,6-tetrachloro-1,2-benzoquinone 2d (method B) proceeds according to a similar mechanism, but in this case intermediate C under goes dehydrochlorination, and as a result tropolones 3h and the mixture of tropolones 3i, k were obtained. The formation of the isomeric 4,5,6-trichloro-1,3-tropolone 3k was observed only in the case of the reaction of 2d with 1b. Tropolone 3j is formed by both method A and method B due to the competing oxidation reaction of intermediate C with 1,2-benzoquinone 2d.
The structure of the compounds 3a–k that we obtained was confirmed by 1H NMR and IR spectroscopy and mass spectrometry data. In the 1H NMR spectra of compounds 3a–j, the signal of the proton of the hydroxyl group, which forms a strong hydrogen bond with the quinoline nitrogen atom in the first position, closing the six-membered chelate ring, is observed in the downfield region δH 17–19 ppm in the form of a narrow singlet peak. 1H NMR spectrum of polychlorinated tropolones 3i–k showed in a weak field the presence of three signals of OH protons – groups 18.61 ppm (3i), 17.92 ppm (3j), 17.22 ppm (3k). Signals of protons of the tropolone cycle appear at 7.19 ppm (3i) and 7.33 ppm (3k). The HRMS (ESI) (m/z [M–H]) mass spectrum of tropolones 3i–k confirms the presence of compounds with molecular weights of 476.8777, which corresponds to 3i,k and 510.8380, which corresponds to 3j. The synthesis conditions and some physicochemical data of the obtained compounds 3a–j are shown in Table 1.
| Compd | Structure | Reaction conditions | Yield (%) | Melting point (°C) | 1H δOH(NH) (ppm) |
|---|---|---|---|---|---|
| a Method A (mixture in a ratio of 3i (35%), 3j (55%), 3k (10%)).b Method B(mixture in a ratio of 3i (55%), 3j (30%), 3k (15%)). | |||||
| 3a | 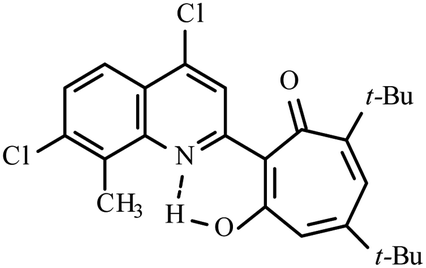 |
Method A: AcOH, 65–70 °C, 1a (1 eq.)/2a (2 eq.) | 63 | 189–190 | 18.77 |
| 3b | 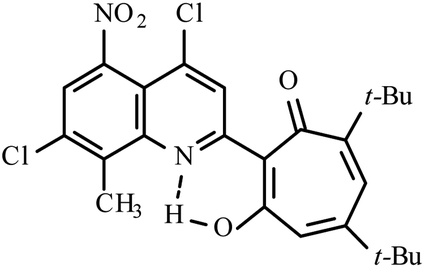 |
Method A: AcOH, 65–70 °C, 1b (1 eq.)/2a (2 eq.) | 47 | 264–265 | 17.57 |
| 3c | 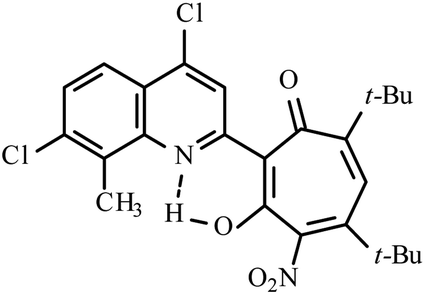 |
Method A: AcOH, 65–70 °C, 1a (1 eq.)/2b (2 eq.) | 30 | 236–237 | 18.17 |
| 3d | 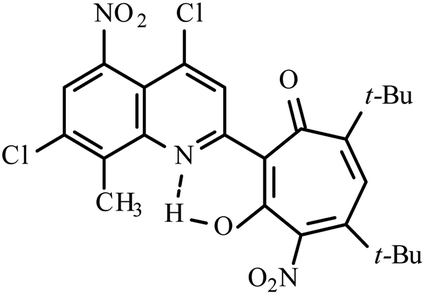 |
Method A: AcOH, 65–70 °C, 1b (1 eq.)/2b (2 eq.) | 77 | 254–255 | 18.35 |
| 3e | 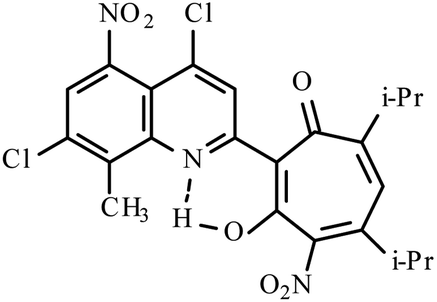 |
Method A: AcOH, 65–70 °C, 1b (1 eq.)/2c (2 eq) | 28 | 255–256 | 19.19 |
| 3f | 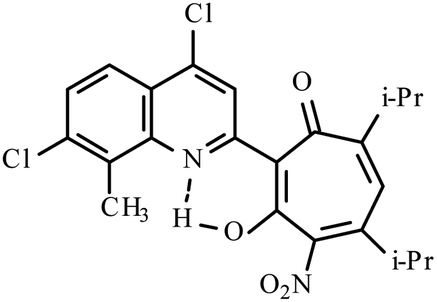 |
Method A: AcOH, 65–70 °C, 1a (1 eq.)/2c (2 eq.) | 51 | 189–190 | 19.18 |
| 3g | 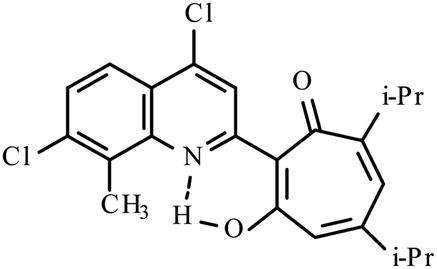 |
10 | 147–148 | 19.01 | |
| 3h | 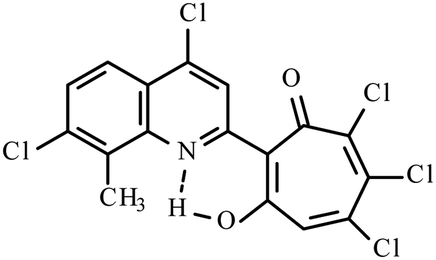 |
Method B: dioxane, 102 °C, 1a (1 eq.)/2d (1.1 eq.) | 72 | 275–276 | 19.02 |
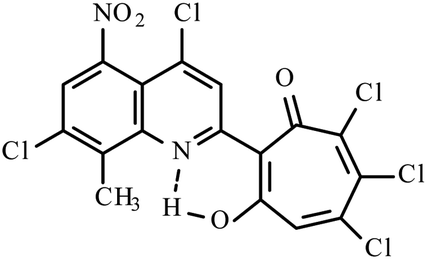 |
Method A: AcOH, 65–70 °C, 1b (1 eq.)/2d (2 eq.) | 55(A)a | 197–200A | 18.613i | |
| Mixture 3i, 3j, 3k | 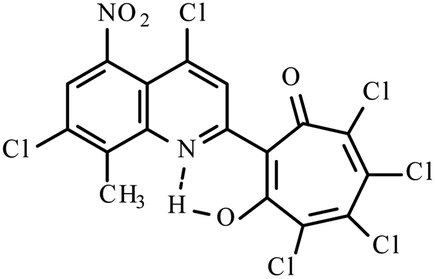 |
Method B: dioxane, 102 °C, 1b (1 eq.)/2d (1.1 eq.) | 46(B)b | 192–193B | 17.923j |
 |
17.223k | ||||
The structure of 3c was established by X-ray diffraction analysis and is shown in Fig. 1. As a result of the prototropic rearrangement, the hydrogen atom is confidently localized at the nitrogen atom N(1) at a distance of 0.98(2) Å and participates in the formation of intramolecular hydrogen bond with the oxygen atom of the carbonyl group of tropolone with the following parameters: O(2)–C(3) = 1.2785 (15) Å, H(1)⋯O(2) −1.567 Å, the angle N(1)–H(1)–O(2) in the cycle formed by the intramolecular hydrogen bond is 149.78. The bond lengths of the aminoenone system N(1)–C(8) = 1.3464(16) Å and C(8)–C(2) = 1.4454(16) Å are close in their absolute values to those for the fixed diketone form in 2-substituted N-methylquinoline30 (1.353 Å and 1.443 Å, respectively. Deposition number 1864613). This indicates the realization in the crystalline phase the aminoenone form (NH) of tropolone 3c (Fig. 1a). It should be noted that the tropolone cycle in the crystal takes on the “bath” conformation (Fig. 1b).
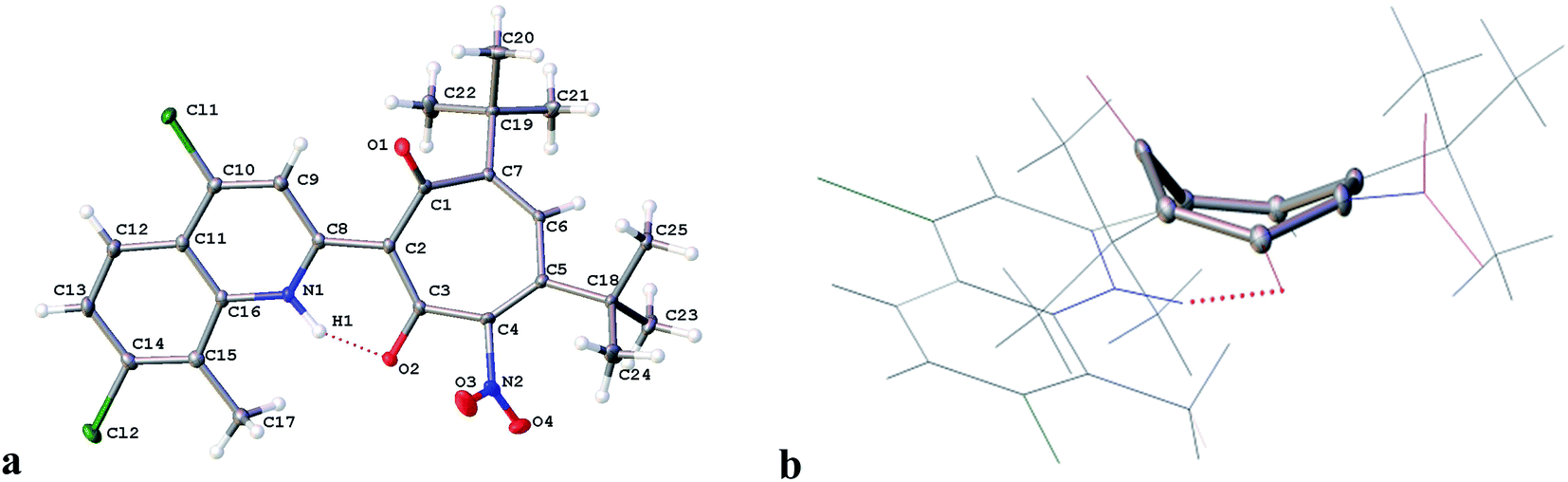 | ||
| Fig. 1 Molecular structure of 5,7-di(tert-butyl)-2-(4,7-dichloro-8-methyl-2-quinolyl)-4-nitro-1,3-tropolone 3c with representation of the atoms by thermal vibration ellipsoids of 50% probability (а); a tropolone cycle in a crystal in the “bath” conformation (b). | ||
Compounds 3 can exist in solution in two tautomeric (OH) and (NH) forms (Scheme 3). The thermodynamic equilibrium between them in the gas phase and polar solvent (DMSO) has been studied by theoretical methods (DFT) (Table 2) (Fig. S1–S3, Tables S1–S22, ESI†).
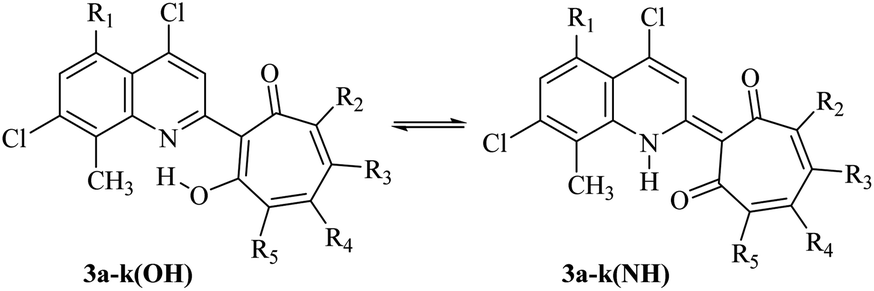 | ||
| Scheme 3 Intramolecular proton transfer in compounds 3a–k. | ||
| Structure | Etot + ZPE (g) | ΔE (g) | Etot + ZPE (s) | ΔE (s) |
|---|---|---|---|---|
| 3a (NH) | −2092.705668 | 0.0 | −2092.715974 | 0.0 |
| 3a (OH) | −2092.707205 | −0.96 | −2092.716625 | −0.41 |
| 3b (NH) | −2297.039006 | 0.0 | −2297.053321 | 0.0 |
| 3b (OH) | −2297.041444 | −1.53 | −2297.055267 | −1.22 |
| 3c (NH) | −2297.041242 | 0.0 | −2297.056554 | 0.0 |
| 3c (OH) | −2297.040005 | 0.78 | −2297.053547 | 1.89 |
| 3d (NH) | −2501.373627 | 0.0 | −2501.392772 | 0.0 |
| 3d (OH) | −2501.373616 | 0.01 | −2501.390877 | 1.19 |
| 3e (NH) | −2422.890683 | 0.0 | −2422.909876 | 0.0 |
| 3e (OH) | −2422.890567 | 0.07 | −2422.908419 | 0.91 |
| 3f (NH) | −2218.557968 | 0.0 | −2218.573183 | 0.0 |
| 3f (OH) | −2218.557252 | 0.45 | −2218.57104 | 1.34 |
| 3g (NH) | −2014.218773 | 0.0 | −2014.229307 | 0.0 |
| 3g (OH) | −2014.220275 | −0.94 | −2014.230029 | −0.45 |
| 3h (NH) | −3157.133771 | 0.0 | −3157.144041 | 0.0 |
| 3h (OH) | −3157.133633 | 0.09 | −3157.142118 | 1.21 |
| 3i (NH) | −3361.465409 | 0.0 | −3361.479782 | 0.0 |
| 3i (OH) | −3361.466531 | −0.70 | −3361.479323 | 0.29 |
| 3j (NH) | −3820.926859 | 0.00 | −3820.941783 | 0.00 |
| 3j (OH) | −3820.925688 | 0.73 | −3820.938795 | 1.87 |
| 3k (NH) | −3361.463126 | 0.00 | −3361.478029 | 0.00 |
| 3k (OH) | −3361.462296 | 0.52 | −3361.475645 | 1.50 |
According to theoretical estimates, the main influence on the equilibrium of (NH) and (OH) tautomeric forms of tropolone derivatives is exerted, on the one hand, by the donor–acceptor properties of the quinoline and tropolone fragments, and on the other, by the polarity of the environment. Variations of these factors make it possible to change the equilibrium of these isomers over a wide range (Table 2).
An acceptor substituent (nitro group) in the quinoline moiety of 3 stabilizes the (OH) isomer. On the contrary, increase of the medium polarity stabilizes the (NH) form to a greater extent. Replacement of tert-butyl substituents in the tropolone fragment with isopropyl substituents does not significantly affect the results.
The introduction of the nitro group into the tropolone fragment stabilizes (NH) isomer. In the polar solvent (DMSO), this effect is further enhanced. The introduction of the nitro group into both tropolone and quinoline fragments of molecules (compounds 3d and 3e) also stabilizes the (NH) isomer, but to a lesser extent than one nitro group in the tropolone part of the molecule. This indicates that acceptor substituents in the quinoline and tropolone fragments partially compensate for each other's influence and, on the other hand, demonstrates a higher effectiveness of acceptor substituents in the tropolone moiety. Stabilization of the (NH) isomer is also observed upon replacement of tert-butyl substituents in the tropolone fragment with chlorine atoms (compounds 3h, 3i), which is consistent with the general properties of the tautomeric equilibrium in these systems mentioned above.
2.2. Biology
This reagent contains non-toxic resazurin-based solution that is blue in color and non-fluorescent, upon entering living cells, resazurin is reduced to resorufin, a compound that is red in color and highly fluorescent. Conversion of resazurin to resorufin is used as a means to measure relative numbers of living cells. The effect of the test compounds was evaluated at concentrations up to 5 μM. This is due to the low solubility of compounds 3a–h and mixture B of 3i–k in DMSO, and due to the fact that an increase in DMSO amount can lead to the toxic effect of the solvent on cell lines under study, that can result in distortion of experimental findings. For additional controls, we used well-known analogue compound of the tropolone series, hinokitiol, and a widely used chemotherapy drug cisplatin. The IC50 values are summarized in Table 3, with the values shown representing the average of at least three independent experiments. A dash means that the compounds did not show antiproliferative activity in cell lines tested in the concentration range up to 5 μM. Representative IC50 curves for 3d and mixture B (of 3i–k) the cell lines in three cell lines are shown in Fig. S4 (see ESI†).
| Cell line | 3a, 3b, 3g, 3f, 3e | 3h | 3c | 3d | Mixture B of 3i–k | Cisplatine | Hinokitiole |
|---|---|---|---|---|---|---|---|
| a Human ovarian cancer.b Human lung cancer.c Human colon cancer.d Human pancreatic cancer.e Comparison control drugs were cisplatin and hinokitiol. | |||||||
| OVCAR-8a | >5 | 2.62 | >5 | 1.33 | 1.98 | 3.75 | >5 |
| OVCAR-3a | >5 | 4.73 | 3.93 | 3.93 | 0.63 | 5.10 | >5 |
| H441b | >5 | 1.35 | 2.62 | 0.97 | 0.70 | 3.74 | 1.97 |
| A549b | — | — | — | 4.05 | 1.61 | 4.16 | >5 |
| HCT 116c | — | — | — | 2.15 | 1.15 | 4.22 | >5 |
| Panc-1d | — | — | — | 0.95 | 0.51 | 3.25 | >5 |
The IC50 values of compounds 3a, 3b, 3e, 3g, 3f did not fall within the concentration range up to 5 μM. In contrast, the compounds 3h and 3c showed antiproliferative activity against the OVCAR-8, OVCAR-3 and H441 cell lines, and the IC50 values of these compounds were lower than the values for cisplatin and hinokitiol. Compounds 3d and mixture B of 3i–k demonstrated broad inhibitory activity against all six cell lines. The IC50 values ranged from 0.95 to 3.93 μM and from 0.51 to 1.98 μM for 3d and mixture B of 3i–k respectively. In addition, the antiproliferative activities of these compounds against OVCAR-3, OVCAR-8 and HCT 116 were generally better than against other cell lines (Table 3).
Since 3d and mixture B of 3i–k compounds showed the broadest range of biologic activity across all 6 cancer cell lines, we next focused our biologic studies on these tropolones. As a proof of concept, we decided to focus on HCT 116 colon and OVCAR-3 and OVCAR-8 ovarian cancer cell lines. Nowadays, the standard first-line therapy for both of these two types of cancer includes platinum-based doublet drug combinations, but the majority of patients ultimately experience resistance to platinum drugs or dose-limiting toxicities. Furthermore, unlike in lung cancer, immunotherapy is generally ineffective for the majority of ovarian and colon cancers, therefore only a few advances occurred in these disease in these disease in the last decades. This latter fact led us to continue looking for ways to improve treatments for these two common, yet difficult to treat types of cancers.
For the next set of biological experiments, we used compounds at IC50 and IC25 concentration values. The values IC50 of 3d compound for OVCAR-3, OVCAR-8 and HCT 116 were 3.93 μM, 1.33 μM and 2.15 μM, respectively. At the same time, IC50 of mixture B 3i–k compound for these three cell lines were 0.63 μM, 1.98 μM and 1.15 μM, respectively.
Analyzing the structure and biological activity of compounds 3, we decided that the most active compounds in the series of 2-quinoline-1,3-tropolones against various cancer cell lines (H441, A549, OVCAR-3, OVCAR-8, HCT 116, Panc-1) are tropolones containing acceptor groups (NO2, Cl) in the periphery of the tropolone ring – 5,7-di(tert-butyl)-2-(4,7-dichloro-8-methyl-2-quinolyl)-4-nitro-1,3-tropolone 3c, 5,7-di(tert-butyl)-2-(4,7-dichloro-8-methyl-5-nitro-2-quinolyl)-4-nitro-1,3-tropolone 3d and polychlorinated derivatives – 2-(4,7-dichloro-8-methyl-2-quinolyl)-5,6,7-thrichloro-1,3-tropolone 3h and mixture B of 3i–k. These tropolones, according to quantum chemical calculations, are in the tautomeric aminoenone form 3 (NH). It is likely that the tautomeric 3 (NH)-form is involved in the mechanisms of inhibition of the growth of cancer cells. Despite the fact that compounds 3e,f (5,7-di-isopropyl) are also in the aminoenone form 3 (NH), like their structural analogs 3c,3d (5,7-di-(tert-butyl)) the IC50 values of compounds 3e,f turned out to be higher than 5 μM. And tropolones 3c, 3d with bulkier tert-butyl substituents at positions 5 and 7 of the tropolone ring showed the best results in inhibiting cancer cells in this series of compounds under study.
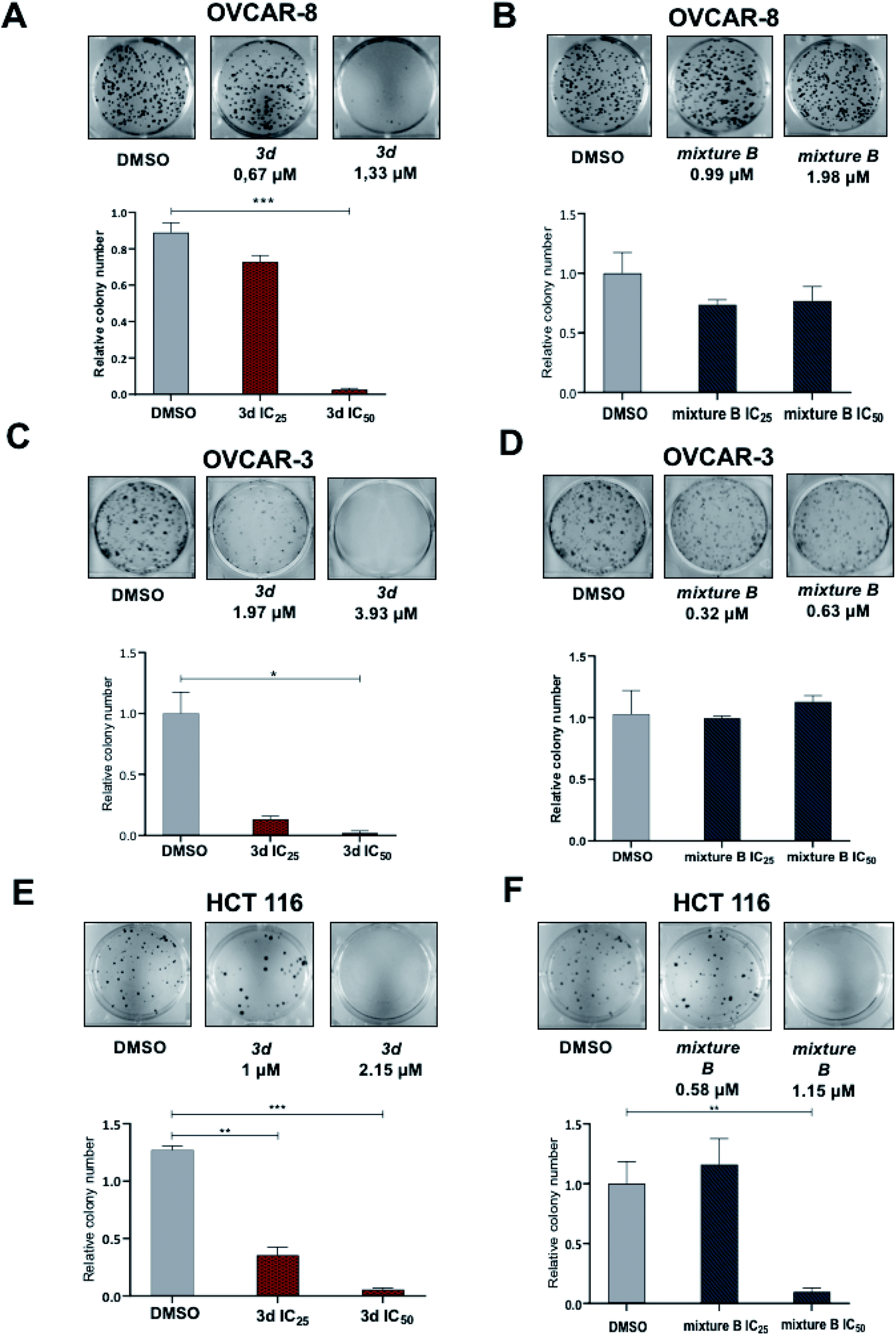 | ||
| Fig. 2 Effect of 3d and mixture B of 3i–k tropolones at IC25 and IC50 concentrations on colony formation ability of ovarian and colon cancer cells. (A) OVCAR-8 under 3d treatment for 7 days; (B) OVCAR-8 under mixture B (3i–k treatment for 7 days); (C) OVCAR-3 under 3d treatment for 7 days; (D) OVCAR-3 under mixture B of treatment of 3i–k for 7 days; (E) HCT 116 under 3d treatment for 7 days; (F) HCT 116 under mixture B of treatment of 3i–k for 7 days. | ||
 | ||
| Fig. 3 Effect of 3d on apoptosis induction in ovarian and colon cancer cell lines. Cells were treated 3d at indicated concentration, or with DMSO. Flow cytometry results after at 0 h, 24 h, 48 h using Annexin V and propidium iodide are shown for OVCAR-8 (A), OVCAR-3 (B) and HCT 116 (C) cell line. | ||
Under the treatment with mixture B (3i–k), only OVCAR-8 cells had significantly more (p-value < 0.0001) apoptotic cells relative to DMSO. The total numbers of late (Annexin-V+/PI+) apoptotic cells after the 48 hours treatment increased to 32.2 ± 1.7%. However, in OVCAR-3 and HCT 116 cells, apoptosis was no different and similar to control DMSO treatment (Fig. 4). These results confirmed that compound mixture B induces cell apoptosis in OVCAR-8 cells in a dose-dependent manner.
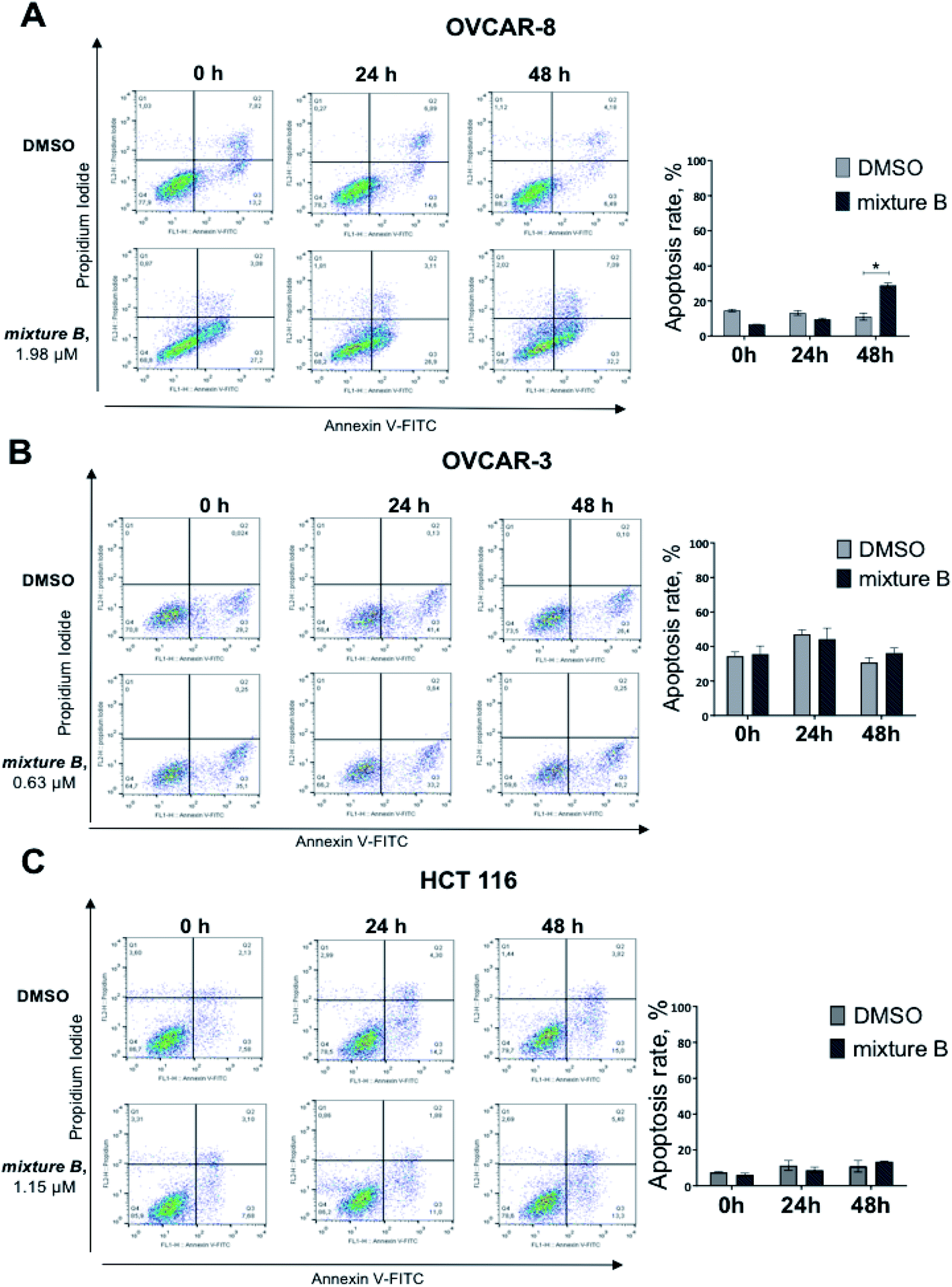 | ||
| Fig. 4 Effect of mixture B of 3i–k on apoptosis induction in ovarian and colon cancer cell lines. Flow cytometry results after at 0 h, 24 h, 48 h treatment using Annexin V and propidium iodide are shown for OVCAR-8 (A), OVCAR-3 (B) and HCT 116 (C) cell line. | ||
Here we show that the compound 3d used at IC50 concentration, during incubation at different time points induces PARP cleavage in OVCAR-8, OVCAR-3 and HCT 116 cell lines, supporting our Annexin V data (Section 2.2.3) that cells are undergoing apoptosis (Fig. 5). All three cell lines were treated for 0, 12, 24 and 48 hours with 3d, and DMSO was used as a control. In all cell lines, a statistically significant apoptosis effects appeared after 12 hours effect of 3d. In ovarian cancer cell lines, this effect extends up to 24 hours of incubation (OVCAR-3) and up to 48 in the case of OVCAR-8 (Fig. 5).
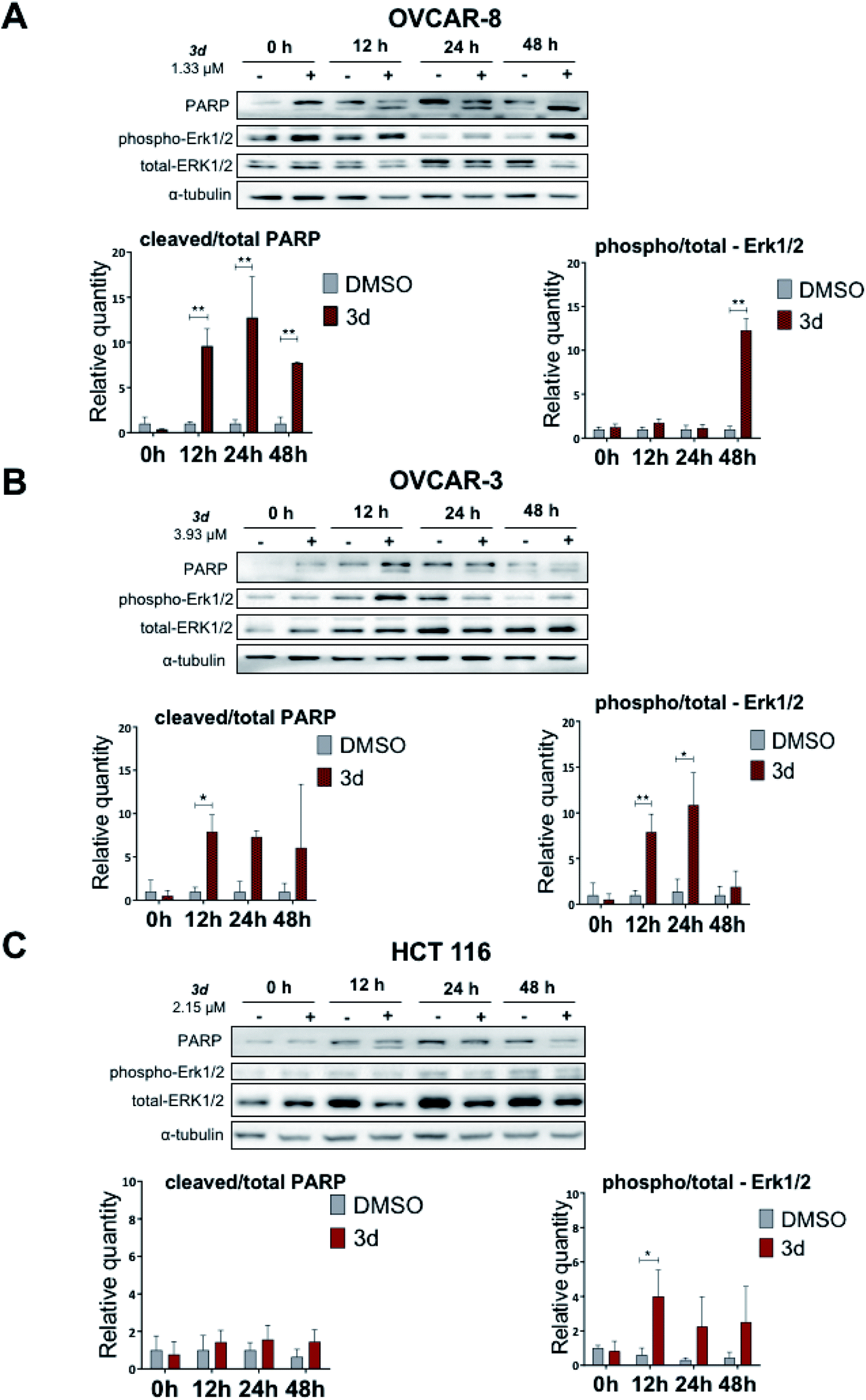 | ||
| Fig. 5 Effect of the 3d compound on apoptosis, as measured by PARP cleavage, and ERK-signaling pathway in OVCAR-8, OVCAR-3 and HCT 116 cell lines. Cells were treated with 3d compound for 0, 12, 24 and 48 hours, and western blot analysis was done. (A) OVCAR-8 cell line; (B) OVCAR-3 cell line; (C) HCT 116 cell line. | ||
Protein-serine/threonine kinases ERK 1/2 are involved in cell survival, proliferation, as well as adhesion and cell migration. Similar to PARP cleavage kinetics, phospho-ERK1/2 kinases phosphorylation occurred with 3d treatment for 12 hours. In OVCAR-3 and OVCAR-3 cell lines, high level of phospho-ERK1/2 is maintained for up to 48 hours. In the HCT 116 cell line, there was no statistically significant activation of ERK1/2-kinase, but there is a trend to increase with 48 hours of incubation (Fig. 5).
Finally, we investigated apoptosis and signaling effects of tropolone mixture B (3i–k) treatment, as described in Fig. 6. Cells were treated with IC50 concentration of mixture B for 0, 12, 24 and 48 hours. There was a significant induction of cleaved PARP between the two conditions under DMSO and mixture B in OVCAR-8 cells on 12 and 24 hours (Fig. 6A). Meanwhile, in OVCAR-3 and HCT 116 cell lines, slight non-significant increase in the cleaved/total PARP 24 and 48 hours can be clearly seen. Turning now to the experimental evidence on protein-serine/threonine kinases ERK1/2, we did not find significant ERK1/2 phosphorylation in across all three cell lines (Fig. 6A–C).
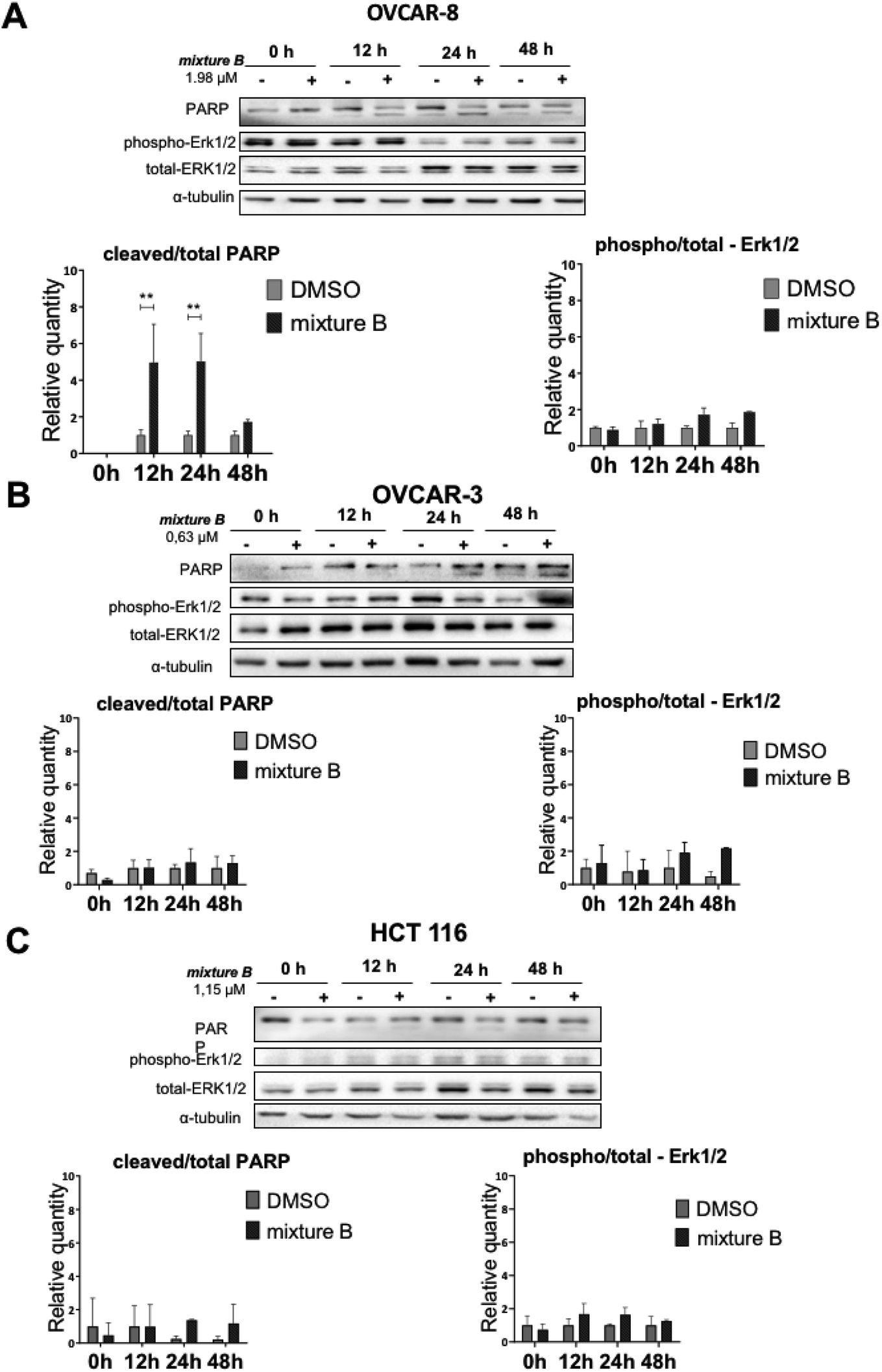 | ||
| Fig. 6 Effect of the mixture B of 3i–k compound on apoptosis, as measured by PARP cleavage and ERK-signaling pathway in OVCAR-8, OVCAR-3 and HCT 116 cell lines. Cells were treated with mixture B of 3i–k compound for 0, 12, 24 and 48 hours. Western blot analysis: (A) OVCAR-8 cell line; (B) OVCAR-3 cell line; (C) HCT 116 cell line. | ||
3. Conclusions
In this study, a series of novel 2-quinolyl-substituted 1,3-tropolones 3 was obtained. The structure of compounds 3 was confirmed by physicochemical methods of analysis (NMR 1H, 13C, IR and Mass spectrometry). The structure of 5,7-di(tert-butyl)-2-(4,7-dichloro-8-methyl-2-quinolyl)-4-nitro-1,3-tropolone established by X-ray analysis. The relative thermodynamic stability of (OH) and (NH) tautomeric forms of 2-quinolyl-substituted 1,3-tropolones 3 has been studied. In subsequent testing for biological activity, compound 3d and tropolone mixture B of 3i–k exhibited the broadest and potent activities against six various human cancer cell lines with IC50 values ranging from 0.63 to 3.93 μM. Furthermore, compound 3d reduced colony formation induced apoptosis in ovarian cancer (OVCAR-3, OVCAR-8) and colon cancer (HCT 116) cell lines. However, it should be noted that when testing the effect of compounds on the ability to colony formation, tropolone mixture B of 3i–k did not show activity, although the proliferation assay nominated it as a strong active compound. Also, cell death was induced by tropolone mixture B of 3i–k treatment appeared only in OVCAR-8 cell line, but not in OVCAR-3 and HCT 116 as no significant changes of percent apoptotic cells and accumulation of cleaved PARP this cell line under compound treatment were detected. In summary, tropolone mixture B of 3i–k can lead to apoptosis activation but had a narrower range of activity in this study and has less potent anti-tumor activity than 3d. Finally, the ERK pathway was broadly induced by 3d compound, but not by tropolone mixture B of 3i–k treatment. Taken together, our study suggests that 2-quinolyl-1,3-tropolones are a promising new class of heterocyclic compounds that should be used for the future development of effective anticancer agents.4. Materials and methods
4.1. General
The 1H, 13C NMR spectra were recorded on a Bruker Avance 600 spectrometer. The chemical shifts are given with respect to the signal of SiMe4 as the internal standard. Attenuated total internal reflectance IR (ATR-IR) spectra were measured on a Varian 3100 FT-IR Excalibur Series spectrometer. HRMS were registered on a Bruker UHR-TOF MaxisTM Impact instrument. Chromatography was performed on columns packed with Al2O3 (Brockmann activity II–III). The melting point was measured on a Fisher–Johns apparatus. The IR and NMR spectra were recorded using equipment of the Shared Use Center “Molecular spectroscopy” of the Southern Federal University.4.2. Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) collecting the first bright yellow fraction (Rf ∼ 0.9). After removal of the solvent in vacuo, the residue was recrystallized from propane-2-ol. Yellow crystals of 3a. Yield 2.8 g (63%), mp 189–190 °C. IR (ν): 3117, 1644, 1623, 1586, 1494, 1462, 1455, 1409, 1378, 1364, 1330, 1271, 1240, 1188, 1149, 1087, 1017, 942, 879, 853, 822, 808, 767, 728, 691 cm−1. 1
:1) collecting the first bright yellow fraction (Rf ∼ 0.9). After removal of the solvent in vacuo, the residue was recrystallized from propane-2-ol. Yellow crystals of 3a. Yield 2.8 g (63%), mp 189–190 °C. IR (ν): 3117, 1644, 1623, 1586, 1494, 1462, 1455, 1409, 1378, 1364, 1330, 1271, 1240, 1188, 1149, 1087, 1017, 942, 879, 853, 822, 808, 767, 728, 691 cm−1. 1![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) NMR (600 MHz, CDCl3) δ, ppm: 1.25 (9H, s, C(CH3)3), 1.38 (9H, s, C(CH3)3), 2.76 (3H, s, CH3), 6.67 (1H, d, CHtrop, J = 1.8 Hz), 6.77 (1H, d, CHtrop, J = 1.8 Hz), 7.53 (1H, d, CHAr, J = 8.9 Hz), 7.94 (1H, d, CHAr, J = 8.9 Hz), 8.23 (1H, s, CHAr), 18.77 (1H, s, OH). 13
NMR (600 MHz, CDCl3) δ, ppm: 1.25 (9H, s, C(CH3)3), 1.38 (9H, s, C(CH3)3), 2.76 (3H, s, CH3), 6.67 (1H, d, CHtrop, J = 1.8 Hz), 6.77 (1H, d, CHtrop, J = 1.8 Hz), 7.53 (1H, d, CHAr, J = 8.9 Hz), 7.94 (1H, d, CHAr, J = 8.9 Hz), 8.23 (1H, s, CHAr), 18.77 (1H, s, OH). 13![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) NMR (151 MHz, CDCl3) δ, ppm: 14.97, 29.95, 31.12, 37.19, 38.43, 114.37, 119.78, 122.74, 122.86, 123.18, 124.47, 127.94, 129.86, 137.41, 141.96, 144.12, 154.70, 155.59, 155.80, 173.84, 194.71. HRMS (ESI): m/z [M–H]− calcd for C25H26Cl2NO2: 442.1346; found: 442.1345. Calcd for C25H27Cl2NO2 (%): C, 67.57; H, 6.12; N, 3.15. Found (%): C, 67.38; H, 6.00; N, 3.02 (Fig. S5 and S6, ESI†).:1) collecting the first bright yellow fraction (Rf ∼ 0.4). After removal of the solvent in vacuo, the residue was recrystallized from propane-2-ol. Yellow crystals of 3b. Yield 2.3 g (47%), mp 264–265 °C. IR (ν): 3115, 1658, 1595, 1577, 1539, 1486, 1463, 1455, 1392, 1367, 1317, 1287, 1245, 1192, 1100, 1025, 1003, 942, 893, 872, 864, 836, 813, 763, 727, 704 cm−1. 1 NMR (600 MHz, CDCl3) δ, ppm: 1.26 (9H, s, C(CH3)3), 1.38 (9H, s, C(CH3)3), 2.78 (3H, s, CH3), 6.69 (1H, d, CHtrop, J = 1.8 Hz), 6.85 (1H, d, CHtrop, J = 1.8 Hz), 7.67 (1H, s, CHAr), 8.28 (1H, s, CHAr), 17.57 (1H, s, OH). 13 NMR (151 MHz, CDCl3) δ, ppm: 15.88, 29.95, 31.10, 37.46, 38.57, 114.41, 114.53, 123.11, 123.17, 123.81, 123.89, 135.20, 135.50, 139.96, 144.17, 145.01, 156.20, 156.43, 157.25, 171.36, 194.08. HRMS (ESI): m/z [M–H]− calcd for C25H25Cl2N2O4: 487.1197; found: 487.1190. Calcd for C25H26Cl2N2O4 (%): C, 61.36; H, 5.35; N, 5.72. Found (%): C, 61.12; H, 5.08; N, 5.56 (Fig. S7 and S8, ESI†).:1) collecting the second bright yellow fraction (Rf ∼ 0.6). After removal of the solvent in vacuo, the residue was recrystallized from propane-2-ol. Yellow crystals of 3c. Yield 1.45 g (30%), mp 236–237 °C. IR (ν): 3107, 1659, 1626, 1597, 1589, 1561, 1539, 1485, 1466, 1393, 1377, 1280, 1245, 1197, 1151, 1086, 1045, 1013, 966, 941, 892, 873, 851, 825, 814, 784, 767, 731, 670 cm−1. 1 NMR (600 MHz, CDCl3) δ, ppm: 1.28 (9H, s, C(CH3)3), 1.30 (9H, s, C(CH3)3), 2.72 (3H, s, CH3), 6.38 (1H, s, CHtrop), 7.56 (1H, d, CHAr, J = 9.0 Hz), 7.93 (1H, d, CHAr, J = 9.0 Hz), 8.28 (1H, s, CHAr), 18.17 (1H, s, OH). 13 NMR (151 MHz, CDCl3) δ, ppm: 14.67, 29.07, 30.40, 37.40, 38.01, 114.16, 118.69, 119.33, 122.11, 123.13, 126.92, 128.15, 137.73, 139.09, 143.21, 146.06, 149.96, 152.02, 154.20, 174.54, 194.09. HRMS (ESI): m/z [M–H]− calcd for C25H25Cl2N2O4: 487.1197; found: 487.1193. Calcd for C25H26Cl2N2O4 (%): C, 61.36; H, 5.35; N, 5.72. Found (%): C, 61.18; H, 5.10; N, 5.58 (Fig. S9 and S10, ESI†).:1) collecting the second bright yellow fraction (Rf ∼ 0.3). After removal of the solvent in vacuo, the residue was recrystallized from propane-2-ol. Yellow crystals of 3d. Yield 4.1 g (77%), mp 254–255 °C. IR (ν): 3111, 1664, 1602, 1547, 1490, 1464, 1366, 1329, 1281, 1244, 1199, 1096, 1053, 1020, 965, 944, 889, 869, 828, 813, 764, 735, 695 cm−1. 1 NMR (600 MHz, CDCl3) δ, ppm: 1.29 (9H, s, C(CH3)3), 1.31 (9H, s, C(CH3)3), 2.77 (3H, s, CH3), 6.48 (1H, s, CHtrop), 7.68 (1H, s, CHAr) 8.31 (1H, s, CHAr), 18.35 (1H, s, OH). 13 NMR (151 MHz, CDCl3) δ, ppm: 15.60, 29.06. 30.38, 37.78, 38.16, 114.49, 120.36, 122.83, 123.09, 132.25, 137.03, 140.06, 141.52, 144.94, 145.02, 148.76, 152.77, 154.59, 172.38, 193.48. HRMS (ESI): m/z [M–H]− calcd for C25H24Cl2N3O6: 532.1048; found: 532.1042. Calcd for C25H25Cl2N3O6 (%): C, 56.19; H, 4.72; N, 7.86. Found (%): C, 56.00; H, 4.54; N, 7.68 (Fig. S11 and S12, ESI†).:4) collecting the second yellow fraction (Rf ∼ 0.45). After removal of the solvent in vacuo, the residue was recrystallized from propane-2-ol. Yellow crystals of 3e. Yield 0.35 g (28%), mp 255–256 °C. IR (ν): 3117, 1609, 1592, 1547, 1506, 1463, 1409, 1368, 1310, 1287, 1246, 1200, 1105, 1071, 1042, 947, 880, 841, 803, 760, 723, 692 cm−1. 1 NMR (600 MHz, CDCl3) δ, ppm: 1.23 (6H, d, CH(C3)2, J = 6.9 Hz), 1.24 (6H, d, CH(C3)2, J = 6.9 Hz), 2.76 (1H, sep, C(CH3)2, J = 6.9 Hz), 2.80 (3H, s, CH3), 3.39 (1H, sep, C(CH3)2, J = 6.9 Hz), 6.49 (1H, s, CHtrop), 7.72 (1H, s, CHAr), 8.35 (1H, s, CHAr), 19.19 (1H, s, OH). 13 NMR (151 MHz, CDCl3) δ, ppm: 15.76, 21.51, 22.66, 32.97, 33.49, 113.94, 114.41, 119.15, 123.66, 124.03, 133.24, 136.81, 140.45, 141.64, 143.53, 144.96, 149.53, 155.92, 158.70, 169.39, 190.02. HRMS (ESI): m/z [M–H]− calcd for C23H20Cl2N3O6: 504.0735; found: 504.0725. Calcd for C23H21Cl2N3O6 (%): C, 54.56; H, 4.18; N, 8.30. Found (%): C, 54.38; H, 4.02; N, 8.16 (Fig. S13 and S14, ESI†).:1) collecting the first and the second yellow fractions. The first fraction is compound 3g (Rf ∼ 0.65), the second fraction is compound 3f (Rf ∼ 0.35). After removal of the solvent in vacuo, the residues were recrystallized from propane-2-ol.
NMR (151 MHz, CDCl3) δ, ppm: 14.97, 29.95, 31.12, 37.19, 38.43, 114.37, 119.78, 122.74, 122.86, 123.18, 124.47, 127.94, 129.86, 137.41, 141.96, 144.12, 154.70, 155.59, 155.80, 173.84, 194.71. HRMS (ESI): m/z [M–H]− calcd for C25H26Cl2NO2: 442.1346; found: 442.1345. Calcd for C25H27Cl2NO2 (%): C, 67.57; H, 6.12; N, 3.15. Found (%): C, 67.38; H, 6.00; N, 3.02 (Fig. S5 and S6, ESI†).:1) collecting the first bright yellow fraction (Rf ∼ 0.4). After removal of the solvent in vacuo, the residue was recrystallized from propane-2-ol. Yellow crystals of 3b. Yield 2.3 g (47%), mp 264–265 °C. IR (ν): 3115, 1658, 1595, 1577, 1539, 1486, 1463, 1455, 1392, 1367, 1317, 1287, 1245, 1192, 1100, 1025, 1003, 942, 893, 872, 864, 836, 813, 763, 727, 704 cm−1. 1 NMR (600 MHz, CDCl3) δ, ppm: 1.26 (9H, s, C(CH3)3), 1.38 (9H, s, C(CH3)3), 2.78 (3H, s, CH3), 6.69 (1H, d, CHtrop, J = 1.8 Hz), 6.85 (1H, d, CHtrop, J = 1.8 Hz), 7.67 (1H, s, CHAr), 8.28 (1H, s, CHAr), 17.57 (1H, s, OH). 13 NMR (151 MHz, CDCl3) δ, ppm: 15.88, 29.95, 31.10, 37.46, 38.57, 114.41, 114.53, 123.11, 123.17, 123.81, 123.89, 135.20, 135.50, 139.96, 144.17, 145.01, 156.20, 156.43, 157.25, 171.36, 194.08. HRMS (ESI): m/z [M–H]− calcd for C25H25Cl2N2O4: 487.1197; found: 487.1190. Calcd for C25H26Cl2N2O4 (%): C, 61.36; H, 5.35; N, 5.72. Found (%): C, 61.12; H, 5.08; N, 5.56 (Fig. S7 and S8, ESI†).:1) collecting the second bright yellow fraction (Rf ∼ 0.6). After removal of the solvent in vacuo, the residue was recrystallized from propane-2-ol. Yellow crystals of 3c. Yield 1.45 g (30%), mp 236–237 °C. IR (ν): 3107, 1659, 1626, 1597, 1589, 1561, 1539, 1485, 1466, 1393, 1377, 1280, 1245, 1197, 1151, 1086, 1045, 1013, 966, 941, 892, 873, 851, 825, 814, 784, 767, 731, 670 cm−1. 1 NMR (600 MHz, CDCl3) δ, ppm: 1.28 (9H, s, C(CH3)3), 1.30 (9H, s, C(CH3)3), 2.72 (3H, s, CH3), 6.38 (1H, s, CHtrop), 7.56 (1H, d, CHAr, J = 9.0 Hz), 7.93 (1H, d, CHAr, J = 9.0 Hz), 8.28 (1H, s, CHAr), 18.17 (1H, s, OH). 13 NMR (151 MHz, CDCl3) δ, ppm: 14.67, 29.07, 30.40, 37.40, 38.01, 114.16, 118.69, 119.33, 122.11, 123.13, 126.92, 128.15, 137.73, 139.09, 143.21, 146.06, 149.96, 152.02, 154.20, 174.54, 194.09. HRMS (ESI): m/z [M–H]− calcd for C25H25Cl2N2O4: 487.1197; found: 487.1193. Calcd for C25H26Cl2N2O4 (%): C, 61.36; H, 5.35; N, 5.72. Found (%): C, 61.18; H, 5.10; N, 5.58 (Fig. S9 and S10, ESI†).:1) collecting the second bright yellow fraction (Rf ∼ 0.3). After removal of the solvent in vacuo, the residue was recrystallized from propane-2-ol. Yellow crystals of 3d. Yield 4.1 g (77%), mp 254–255 °C. IR (ν): 3111, 1664, 1602, 1547, 1490, 1464, 1366, 1329, 1281, 1244, 1199, 1096, 1053, 1020, 965, 944, 889, 869, 828, 813, 764, 735, 695 cm−1. 1 NMR (600 MHz, CDCl3) δ, ppm: 1.29 (9H, s, C(CH3)3), 1.31 (9H, s, C(CH3)3), 2.77 (3H, s, CH3), 6.48 (1H, s, CHtrop), 7.68 (1H, s, CHAr) 8.31 (1H, s, CHAr), 18.35 (1H, s, OH). 13 NMR (151 MHz, CDCl3) δ, ppm: 15.60, 29.06. 30.38, 37.78, 38.16, 114.49, 120.36, 122.83, 123.09, 132.25, 137.03, 140.06, 141.52, 144.94, 145.02, 148.76, 152.77, 154.59, 172.38, 193.48. HRMS (ESI): m/z [M–H]− calcd for C25H24Cl2N3O6: 532.1048; found: 532.1042. Calcd for C25H25Cl2N3O6 (%): C, 56.19; H, 4.72; N, 7.86. Found (%): C, 56.00; H, 4.54; N, 7.68 (Fig. S11 and S12, ESI†).:4) collecting the second yellow fraction (Rf ∼ 0.45). After removal of the solvent in vacuo, the residue was recrystallized from propane-2-ol. Yellow crystals of 3e. Yield 0.35 g (28%), mp 255–256 °C. IR (ν): 3117, 1609, 1592, 1547, 1506, 1463, 1409, 1368, 1310, 1287, 1246, 1200, 1105, 1071, 1042, 947, 880, 841, 803, 760, 723, 692 cm−1. 1 NMR (600 MHz, CDCl3) δ, ppm: 1.23 (6H, d, CH(C3)2, J = 6.9 Hz), 1.24 (6H, d, CH(C3)2, J = 6.9 Hz), 2.76 (1H, sep, C(CH3)2, J = 6.9 Hz), 2.80 (3H, s, CH3), 3.39 (1H, sep, C(CH3)2, J = 6.9 Hz), 6.49 (1H, s, CHtrop), 7.72 (1H, s, CHAr), 8.35 (1H, s, CHAr), 19.19 (1H, s, OH). 13 NMR (151 MHz, CDCl3) δ, ppm: 15.76, 21.51, 22.66, 32.97, 33.49, 113.94, 114.41, 119.15, 123.66, 124.03, 133.24, 136.81, 140.45, 141.64, 143.53, 144.96, 149.53, 155.92, 158.70, 169.39, 190.02. HRMS (ESI): m/z [M–H]− calcd for C23H20Cl2N3O6: 504.0735; found: 504.0725. Calcd for C23H21Cl2N3O6 (%): C, 54.56; H, 4.18; N, 8.30. Found (%): C, 54.38; H, 4.02; N, 8.16 (Fig. S13 and S14, ESI†).:1) collecting the first and the second yellow fractions. The first fraction is compound 3g (Rf ∼ 0.65), the second fraction is compound 3f (Rf ∼ 0.35). After removal of the solvent in vacuo, the residues were recrystallized from propane-2-ol.Yellow crystals of 3f. Yield 0.7 g (51%), mp 189–190 °C. IR (ν): 3120, 3087, 1620, 1599, 1567, 1537, 1483, 1462, 1434, 1392, 1374, 1325, 1287, 1246, 1234, 1192, 1152, 1091, 1063, 1036, 1013, 988, 942, 889, 876, 851, 838, 825, 805, 789, 766, 722, 681 cm−1. 1 NMR (600 MHz, CDCl3) δ, ppm: 1.21 (6H, d, CH(C3)2, J = 6.9 Hz), 1.22 (6H, d, CH(C3)2, J = 6.9 Hz), 2.73 (1H, sep, C(CH3)2, J = 6.9 Hz), 2.76 (3H, s, CH3), 3.41 (1H, sep, C(CH3)2, J = 6.9 Hz), 6.40 (1H, s, CHtrop), 7.60 (1H, d, CHAr, J = 8.9 Hz), 7.97 (1H, d, CHAr, J = 8.9 Hz), 8.40 (1H, s, CHAr), 19.18 (1H, s, OH). 13 NMR (151 MHz, CDCl3) δ, ppm: 14.8, 21.45, 22.64, 32.66, 33.19, 113.77, 118.11, 120.08, 122.76, 123.06, 127.71, 128.67, 138.02, 138.86, 141.50, 146.15, 151.12, 154.92, 157.75, 172.23, 190.33. HRMS (ESI): m/z [M–H]− calcd for C23H21Cl2N2O4: 459.0884; found: 459.0880. Calcd for C23H22Cl2N2O4 (%): C, 59.88; H, 4.81; N, 6.07. Found (%): C, 59.64; H, 4.60; N, 5.90 (Fig. S15 and S16, ESI†).
Yellow crystals of 3g. Yield 0.12 g (10%), mp 147–148 °C. IR (ν): 3115, 3086, 1605, 1525, 1488, 1463, 1400, 1378, 1339, 1318, 1260, 1188, 1150, 1112, 1091, 1049, 1007, 990, 953, 931, 878, 865, 831, 812, 780, 769, 723, 682 cm−1. 1H NMR (600 MHz, CDCl3) δ, ppm: 1.21 (6H, d, CH(C3)2, J = 6.8 Hz), 1.24 (6H, d, CH(C3)2, J = 6.8 Hz), 2.68 (1H, sep, C(CH3)2, J = 6.8 Hz), 2.78 (3H, s, CH3), 3.46 (1H, sep, C(CH3)2, J = 6.8 Hz), 6.54 (1H, s, CHtrop), 6.65 (1H, s, CHtrop), 7.55 (1H, d, CHAr, J = 8.9 Hz), 7.96 (1H, d, CHAr, J = 8.9 Hz), 8.31 (1H, s, CHAr), 19.01 (1H, s, OH). 13 NMR (151 MHz, CDCl3) δ, ppm: 15.02, 22.48, 23.06, 32.64, 37.63, 115.21, 121.00, 122.72, 123.25, 125.94, 128.28, 130.28, 137.33, 141.88, 144.12, 153.08, 156.64, 157.48. HRMS (ESI): m/z [M–H]− calcd for C23H22Cl2NO4: 414.1033; found: 414.1034. Calcd for C23H23Cl2NO2 (%): C, 66.35; H, 5.57; N, 3.36. Found (%): C, 66.14; H, 5.28; N, 3.18 (Fig. S17 and S18, ESI†).
NMR (600 MHz, CDCl3) δ, ppm: 2.81 (3H, s, CH3), 7.09 (1H, s, CHtrop), 7.64 (1H, d, CHAr, J = 9 Hz), 8.02 (1H, d, CHAr, J = 9 Hz), 8.44 (1H, s, CHAr), 19.02 (1H, s, OH). HRMS (ESI): m/z [M–H]−calcd for C17H7Cl5NO2: 431.8925; found: 431.8925. Calcd for C17H8Cl5NO2 (%): C, 46.88; H, 1.85; N, 3.22. Found (%): C, 46.62; H, 1.60; N, 3.02 (Fig. S19, ESI†).Method A. A solution of 4,7-dichloro-2,8-dimethyl-5-nitroquinoline 1b (1.35 g, 5 mmol) and of o-chloranil 2d (2.46 g, 10 mmol) in glacial acetic acid (15 ml) was stirred for 36 h at 50 °C. After cooled to the room temperature, the reaction mixture was diluted with water and extracted with dichloromethane (2 × 100 ml). The combined organic layers were washed with water (2 × 100 ml) and then dried with anhydrous Na2SO4 and the solvent was removed in vacuo. The residue was purified by silica gel column chromatography with dichloromethane collecting the bright yellow fraction (Rf ∼ 0.9). After removal of the solvent in vacuo, the residues was recrystallized from benzene to give inseparable mixture of compounds 3i–k (35% – 3i, 55% – 3j and 10% – 3k), mp 197–200 °C (benzene) (1.4 g, yield 55%).
Method B. To the boiling solution of 4,7-dichloro-2,8-dimethyl-5-nitroquinoline 1b (1.35 g, 5 mmol) in dioxane (20 ml) was added portions out o-chloranil 2d (1.23 g, 5 mmol). The reaction mixture was refluxed for 30 min. After cooled to the room temperature and the solvent was removed in vacuo. The residue was purified by silica gel column chromatography with dichloromethane collecting the second bright yellow fraction (Rf ∼ 0.9). After removal of the solvent in vacuo, the residues was recrystallized from benzene to give inseparable mixture of compounds 3i–k (55% – 3i, 30% – 3j and 15% – 3k), mp 192–193 °C (benzene) (1.1 g, yield 46%).
Yellow crystals of mixture of 3i–k (method B). IR (ν): 3113, 1646, 1594, 1539, 1463, 1376, 1286, 1224, 1197, 1159, 1124, 1104, 1046, 962, 902, 885, 767, 718, 682 cm−1. 1 NMR (600 MHz, CDCl3) δ, ppm: 2.833i (3H, s, CH3), 2.883j (3H, s, CH3), 2.913k (3H, s, CH3), 7.193i (1H, s, CHtrop), 7.333k (1H, s, CHtrop), 7.73–7.763i–k (1H, m, CHAr), 8.34–8.463i–k (1H, m, CHAr), 17.723k (1H, s, OH), 17.923j (1H, s, OH), 18.613i (1H, s, OH). HRMS (ESI): m/z[M–H]− calcd for C17H6Cl5N2O4 (3i, 3k): 476.8774; found: 476.8777. HRMS (ESI): m/z[M–H]−calcd for C17H5Cl6N2O4 (3j): 510.8458; found: 510.8380 (Fig. S20, ESI†).
4.3. X-ray analysis
The single crystals of 3c have been grown in dimethyl sulfoxide solution. The data collection have been performed on an Agilent SuperNova diffractometer using microfocus X-ray source with copper anode (λ = 1.54184) and Atlas S2 CCD detector. The diffraction data of 3c were obtained at room temperature (293 K). Single crystals of C25H26Cl2N2O4 are triclinic: a = 7.6690(2) Å, b = 9.8456(4) Å, c = 15.9190(6) Å, α = 88.165(3)°, β = 87.926(3)°, γ = 73.153(3)°,V = 1149.35(7) Å3, Z = 2, T = 100 K, μ(Cu Kα) = 2.838 mm−1, Dcalc = 1.414 g cm−3, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2). 21532 reflections measured (9.388° ≤ 2Θ ≤ 148.056°), 4600 unique (Rint = 0.0282, Rsigma = 0.0187) which were used in all calculations. The final R1 was 0.0324 (I > 2σ(I)) and wR2 was 0.0851 (all data). The collection of reflexes, determination and refinement of unit cell parameters were performed by using the specialized CrysAlisPro 1.171.38.41 software suite.32 The structures were solved by using ShelXT program,33 structure refinement was also performed with ShelXL program.34 Molecular graphics were rendered and prepared for publication with the Olex2 version 1.2.10 software suite.35 The complete X-ray diffraction datasets were deposited at the Cambridge Crystallographic Data Center (deposit CCDC 2040512) (Tables S23 and S24, ESI†).
(no. 2). 21532 reflections measured (9.388° ≤ 2Θ ≤ 148.056°), 4600 unique (Rint = 0.0282, Rsigma = 0.0187) which were used in all calculations. The final R1 was 0.0324 (I > 2σ(I)) and wR2 was 0.0851 (all data). The collection of reflexes, determination and refinement of unit cell parameters were performed by using the specialized CrysAlisPro 1.171.38.41 software suite.32 The structures were solved by using ShelXT program,33 structure refinement was also performed with ShelXL program.34 Molecular graphics were rendered and prepared for publication with the Olex2 version 1.2.10 software suite.35 The complete X-ray diffraction datasets were deposited at the Cambridge Crystallographic Data Center (deposit CCDC 2040512) (Tables S23 and S24, ESI†).
4.4. Computational methods
All computations were carried out with Gaussian 09 (ref. 36) program package using DFT methods, employing PBE0 exchange–correlation functional37,38 and 6-311+G(d,p) basis set. The stationary points (minima) on the potential energy surface (PES) were verified by calculating the eigenvalues of the Hessian matrix. Solvent effects were modeled by using polarizable continuum model (PCM)39 within the integral equation formalism (IEFPCM).404.5. Biological experiments
:1000. For the western analysis, phospho-p44/42 MAPK (T202/Y204) (#4370S, Cell Signaling, MA, USA), p44/42 MAPK (Erk1/2) (L34F12) (#4696S, Cell Signaling, MA, USA), PARP and cleaved PARP (#9542S, Cell Signaling, MA, USA), tubulin (3873S, Cell Signaling, MA, USA) antibodies were used. For the detection of the signal from the membranes was used The ChemiDoc XRS+ System (Bio-Rad, USA) and for quantitative analysis of western blots was used Image Lab software (Bio-Rad, USA).000 events were calculated for each experiment.4.6. Statistical analysis
All the data are mean values of at least 3 independent experiments and expressed as means (±SEM). For multiple-group comparison multiple t-tests were used by statistical software GraphPad Prism 6, p-value less than 0.05 was considered to be statistically significant.Author contributions
Evgeniy A. Gusakov: methodology, investigation, data curation, visualization, writing – original draft, funding acquisition. Iuliia A. Topchu: investigation, formal analysis, data curation, visualization, writing – original draft. Aleksandra M. Mazitova: investigation, formal analysis, data curation, visualization, writing – original draft. Igor V. Dorogan: investigation, formal analysis, visualization, writing – original draft. Emil R. Bulatov: methodology, data curation, writing – original draft, funding acquisition. Ilya G. Serebriiskii: methodology, data curation, funding acquisition. Zinaida I. Abramova: methodology, investigation, data curation. Inna O. Tupaeva: investigation, data curation, visualization, writing – review & editing. Oleg P. Demidov: investigation, visualization, data curation, writing – original draft. Duong Ngoc Toan: investigation, funding acquisition. Tran Dai Lam: investigation, data curation. Duong Nghia Bang: conceptualization, investigation, data curation, visualization, writing – original draft. Yanis A. Boumber: project administration, conceptualization, investigation, writing – original draft, writing – review & editing, funding acquisition. Yurii A. Sayapin: project administration, conceptualization, investigation, writing – original draft, writing – review & editing, resources, funding acquisition. Vladimir I. Minkin: supervision, conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Vietnam National Foundation for Science and Technology Development “NAFOSTED” [grant 104.01-2015.68]; E. G. thanks the scholarship of the President RF [grant number SP-3728.2019.4]. Y. B. and I. T. were supported in part by the NIH R21 CA223394, NIH R01 CA218802 grants and by the NCI NIH Core Grant P30 CA060553 to the Robert H Lurie Comprehensive Cancer Center at Northwestern University. I. S. was supported by the NCI NIH Core Grant P30 CA006927 to Fox Chase Cancer Center. Yu. S. worked within the framework of the State Assignment of Southern Scientific Centre of RAS [grant number 01201354239]. E. B. was supported by RSF [grant number 19-74-10022]. Finally, this study was supported by the program for Competitive Growth of the Kazan Federal University (E. B., I. T., A. M., I. S., Z. A., and Y. B.).References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2020, 70(1), 7–30 CrossRef.
- S. Muñoz-Galván, B. Felipe-Abrio, M. García-Carrasco, J. Domínguez-Piñol, E. Suarez-Martinez, E. M. Verdugo-Sivianes, A. Espinosa-Sánchez, L. E. Navas, D. Otero-Albiol, J. J. Marin, M. P. Jiménez-García, J. M. García-Heredia, A. G. Quiroga, P. Estevez-Garcia and A. Carnero, J. Exp. Clin. Cancer Res., 2019, 38, 234 CrossRef.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68(6), 394–424 CrossRef.
- L. Y. Guan and Y. Lu, Discov. Med., 2018, 26(144), 219–229 Search PubMed.
- K. Brasseur, N. Gévry and E. Asselin, Oncotarget, 2017, 8, 4008–4042 CrossRef.
- I. Mármol, J. Quero, M. J. Rodríguez-Yoldi and E. Cerrada, Cancers, 2019, 11, 780 CrossRef.
- A. Moreira-Paisa, R. Ferreira and R. G. da Costa, Life Sci., 2018, 208, 1–9 CrossRef.
- Y. K. Lee, J. Lim, S. Y. Yoon, J. C. Joo, S. J. Park and Y. J. Park, Int. J. Mol. Sci., 2019, 20, 2443 CrossRef.
- H. C. Zheng, Oncotarget, 2017, 8, 59950–59964 CrossRef.
- J. Zhao, Curr. Med. Chem., 2007, 14, 2597–2621 CrossRef CAS.
- Y. H. Shih, K. W. Chang, S. M. Hsia, C. C. Yu, L. J. Fuh, T. Y. Chi and T. M. Shieh, Microbiol. Res., 2013, 168, 254–262 CrossRef CAS.
- M. Elagawany, L. Hegazy, F. Cao, M. J. Donlin, N. Rath, J. Tavis and B. Elgendy, RSC Adv., 2018, 8(52), 29967–29975 RSC.
- W. Sonyot, S. Lamlertthon, J. J. Luangsa-ard, S. Mongkolsamrit, K. Usuwanthim, K. Ingkaninan, N. Waranuch and N. Suphrom, Antibiotics, 2020, 9(5), 274 CrossRef CAS.
- K. Nakano, T. Chigira, T. Miyafusa, S. Nagatoishi, J. M. M. Caaveiro and K. Tsumoto, Sci. Rep., 2015, 5, 15337 CrossRef CAS.
- H. Domon, T. Hiyoshi, T. Maekawa, D. Yonezawa, H. Tamura, S. Kawabata, K. Yanagihara, O. Kimura, E. Kunitomo and Y. Terao, Microbiol. Immunol., 2019, 63(6), 213–222 CrossRef CAS.
- S. R. Budihas, I. Gorshkova, S. Gaidamakov, A. Wamiru, M. K. Bona, M. A. Parniak, R. J. Crouch, J. B. McMahon, J. A. Beutler and S. F. J. Le Grice, Nucleic Acids Res., 2005, 33(4), 1249–1256 CrossRef CAS.
- L. Zhang, Y. Peng, I. P. Uray, J. Shen, L. Wang, X. Peng, P. H. Brown, W. Tu and G. Peng, DNA Repair, 2017, 60, 89–101 CrossRef CAS.
- J. Li, E. R. Falcone, S. A. Holstein, A. C. Anderson, D. L. Wright and A. J. Wiemer, Pharmacol. Res., 2016, 113(Pt A), 438–448 CrossRef CAS.
- S. L. Haney, C. Allen, M. L. Varney, K. M. Dykstra, E. R. Falcone, S. H. Colligan, Q. Hu, A. M. Aldridge, D. L. Wright, A. J. Wiemer and S. A. Holstein, Oncotarget, 2017, 8, 76085–76098 CrossRef.
- C. H. Huang, S. H. Lu, C. C. Chang, P. A. Thomas, T. Jayakumar and J. R. Sheu, Eur. J. Pharmacol., 2015, 746, 148–157 CrossRef CAS.
- J. S. Seo, Y. H. Choi, J. W. Moon, H. S. Kim and S. H. Park, BMC Cell Biol., 2017, 18, 14 CrossRef.
- N. Liu, W. Song, C. M. Schienebeck, M. Zhang and W. Tang, Tetrahedron, 2014, 70, 9281–9305 CrossRef CAS.
- D. N. Bang, Yu. A. Sayapin, H. Lam, N. D. Duc and V. N. Komissarov, Chem. Heterocycl. Compd., 2015, 51, 291–294 CrossRef CAS.
- Yu. A. Sayapin, I. O. Tupaeva, A. A. Kolodina, E. A. Gusakov, V. N. Komissarov, I. V. Dorogan, N. I. Makarova, A. V. Metelitsa, V. V. Tkachev, S. M. Aldoshin and V. I. Minkin, Beilstein J. Org. Chem., 2015, 11, 2179–2188 CrossRef CAS.
- Yu. A. Sayapin, D. N. Bang, V. N. Komissarov, I. V. Dorogan, N. I. Makarova, I. O. Bondareva, V. V. Tkachev, G. V. Shilov, S. M. Aldoshin and V. I. Minkin, Tetrahedron, 2010, 66, 8763–8771 CrossRef CAS.
- Yu. A. Sayapin, V. N. Komissarov, D. N. Bang, I. V. Dorogan, V. I. Minkin, V. V. Tkachev, G. V. Shilov, S. M. Aldoshin and V. N. Charushin, Mendeleev Commun., 2008, 18, 180–182 CrossRef CAS.
- Yu. A. Sayapin, E. A. Gusakov, I. V. Dorogan, I. O. Tupaeva, M. G. Teimurazov, N. K. Fursova, K. V. Ovchinnikov and V. I. Minkin, Russ. J. Bioorg. Chem., 2016, 42, 224–228 CrossRef CAS.
- Yu. A. Sayapin, E. A. Gusakov, A. A. Kolodina, V. N. Komissarov, I. V. Dorogan, V. V. Tkachev, G. V. Shilov, E. V. Nosova, S. M. Aldoshin, V. N. Charushin and V. I. Minkin, Russ. Chem. Bull., 2014, 63, 1364–1372 CrossRef CAS.
- Yu. A. Sayapin, D. N. Bang, E. A. Gusakov, I. V. Dorogan, V. V. Tkachev, V. S. Gorkovets, V. N. Komissarov, N. T. Duong, D. D. Nguyen, G. V. Shilov, S. M. Aldoshin and V. I. Minkin, Russ. Chem. Bull., 2016, 65, 2461–2468 CrossRef CAS.
- S. Sangher, C. Kesornpun, T. Aree, C. Mahidol, S. Ruchirawat and P. Kittakoop, Dyes Pigm., 2020, 178, 108341 CrossRef CAS.
- D. Tang, R. Kang, T. V. Berghe, P. Vandenabeele and G. Kroemer, Cell Res., 2019, 29, 347–364 CrossRef CAS.
- CrysAlisPro, version 1.171.38.41, Rigaku Oxford Diffraction, 2015 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2015, 71, 3–8 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.01, Gaussian. Inc., Wallingford CT, 2013 Search PubMed.
- M. Ernzerhof and G. E. J. Scuseria, J. Chem. Phys., 1999, 111, 911–915 CrossRef CAS.
- C. Adamo, G. E. Scuseria and V. J. Barone, J. Chem. Phys., 1999, 111, 2889–2899 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Chem. Phys., 1997, 107, 3210–3221 CrossRef CAS.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental data (Fig. S1–S20 and Tables S1–S24). CCDC 2040512. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra10610k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |