
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrophase separation/crosslinking competition-based ternary microstructure evolution of poly(ether-b-amide)†
Yu Wangab,
Zefan Wangb,
Ping Zhub,
Xinran Liubc,
Lei Wang *a,
Xia Dong*bc and
Dujin Wangbc
*a,
Xia Dong*bc and
Dujin Wangbc
aShenzhen Key Laboratory of Polymer Science and Technology, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, P. R. China. E-mail: wl@szu.edu.cn
bCAS Key Laboratory of Engineering Plastics, Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: xiadong@iccas.ac.cn
cUniversity of Chinese Academy of Science, Beijing, 100049, P. R. China
First published on 10th February 2021
Abstract
The temperature dependence of the rheological properties of poly(ether-b-amide) (PEBA) segmented copolymer under oscillatory shear flow has been investigated. The magnitude of the dynamic storage modulus is affected by the physical microphase separation and irreversible crosslinking network, with the latter spontaneously forming between the polyamide segments and becoming the dominant factor in determining the microstructural evolution at temperatures well above the melting point of PEBA. From the rheological results, the initial temperature of the rheological properties dominated by the microphase separation  and crosslinking (Tcross) structures were determined, respectively. Based on the two obtained temperatures, the microstructure evolution upon the heating can be separated into the ternary microstructure domains: homogenous (temperature below
and crosslinking (Tcross) structures were determined, respectively. Based on the two obtained temperatures, the microstructure evolution upon the heating can be separated into the ternary microstructure domains: homogenous (temperature below  ), microphase separation dominating (between
), microphase separation dominating (between  and Tcross), and crosslinking dominating domains (above Tcross). When the PEBA is heated to above Tcross, the content of crosslinking network increases with time and temperature, leading to an irreversible and non-negligible influence on the rheological, crystallization, and mechanical properties. A more pronounced strain-hardening phenomenon during the uniaxial stretching is observed for the sample with a higher content of crosslinking network.
and Tcross), and crosslinking dominating domains (above Tcross). When the PEBA is heated to above Tcross, the content of crosslinking network increases with time and temperature, leading to an irreversible and non-negligible influence on the rheological, crystallization, and mechanical properties. A more pronounced strain-hardening phenomenon during the uniaxial stretching is observed for the sample with a higher content of crosslinking network.
Introduction
Block copolymers are composed of two or more different segments within the molecular chain, and have attracted great attention in the past few decades because of their excellent mechanical properties and processability. Depending on the architectures, block copolymers can be classified into several categories, e.g. (AB) di-block, (ABA) tri-block, and (AB)n multi-block (segmented) copolymers, etc. Due to the thermodynamic immiscibility between the segments, several long-range order structures can be formed within a sub-micrometer scale (known as microphase separation).1–5 With extensive research, the morphology of different phases is greatly affected by the Flory interaction parameters between the two segments, composition, and the overall degree of polymerization.6–14 Furthermore, the material properties such as modulus,15–17 conductivity,18,19 and rheological properties20,21 can be enhanced at the order states. In recent decades, the order-to-disorder transition temperature and its morphology evolution of di- and tri-block copolymers have been well studied by various means such as small-angle X-ray scattering (SAXS)2,10,21–23 and rheological technique.2,10,20–27 Bates et al. have demonstrated that the viscoelastic properties of di-block copolymers are sensitive to the microstructure changes, and it can be used to characterize the morphology of ordered microstructures.21,23–26 Hashimoto and Han et al. have studied the order-to-disorder transition of the polystyrene-block-polyisoprene-block-polystyrene tri-block copolymer,2,22 and trace its microstructure evolution by the SAXS and transmission electron microscope (TEM) techniques, the results are in good agreement with that determined by rheological studies. Phase diagrams of di- and tri-block copolymers can be interpreted and predicted quite well on the thermodynamic model based on Leibler's works.6,8For the segmented copolymers such as thermoplastic polyurethane (TPU),12,20 poly(ether-b-amide) (PEBA),28 poly(ester-ether) (PEE),14 olefin block copolymers (OBC),29 and transparent polyamide random copolymers,30 the structure–property relationship has been widely explored in the past three decades.7,10,30–32 Velankar and Cooper carried out a series of work to study the influence of chain architectures (including block length and block species) on the microphase separation of TPU,12,13,33 which provides an in-depth understanding on the phase behavior of segmented copolymers. The influence of microphase separation on the crystallization of segmented copolymers at quiescent or flow states also has been investigated by various techniques.28,30,34–36 It was found that the microphase separation at molten state has an effect on the morphology and size of the hard domain's crystals. Due to the co-existence nature of multi-components and multi-phase, the microstructure evolution of semi-crystalline segmented copolymer during the deformation is complicated. As the applied strain increased, the segmental chains' orientation, the crystal–crystal transition, and strain-induced crystallization can occur.37–40 Studying the microstructural evolution under the external fields is important for a better understanding of the macroscopic properties of the segmented copolymers. To date, the phase diagram of multi-block copolymers cannot be predicated well due to the more complex segmental interactions. In other words, the phase transition temperatures and the corresponding morphologies can only be determined experimentally.
PEBA segmented copolymer, which contains the polyamide hard-segment and polyether soft-segment, has been widely used in various fields such as sports equipment, polar clothing, and medical supplies. However, the studies focused on the temperature effect on the rheological properties,41 and crystallization morphology are limited.35 The systematic explore the microstructure evolution upon temperature is inadequate. It was reported that the polyamide crosslinking reaction could occur at the molten state in various polyamide (PA) systems, including PA6,42,43 PA6,6,42–45 PA6,10,42,43 PA11,46 PA12,47 and PA1012,48 especially after prolonged heating. The polyamide crosslinking is caused by the formation of the secondary amine groups,49 which reacts further with carboxyl groups to form the branched structures at high temperature.50 Therefore, the influence of the formed crosslinking structure on the microstructure evolution should not be ignored for the PEBA segmented copolymers.
The present investigation aims to probe the temperature dependence on the microstructure from apparent homogenous to crosslinked states via the small amplitude oscillatory shear (SAOS) measurement for the commercial PEBA copolymer with the trade name of Pebax®, for which contains the crystalline hard segment of PA12, while poly(tetramethylene oxide) (PTMO) as the soft segment.28,51 Based on the experimental results, there different microstructure domains of apparent homogenous, microphase separation, and crosslinking structure can be quantitatively determined. The influences of the microstructure on the rheological and mechanical properties is also studied by the thermal history under different temperature domains.
Experimental
Materials and sample preparation
The commercial PEBA elastomers with different compositions employed here are Pebax® 2533 SD02, 3533 SP01, and 4033 SP01 (named as P25D, P35D, and P40D in this work), supplied by Arkema Co., Ltd., used as received. The sub-grade of SD02 and SP01 has been developed to be heat and UV resistant, and the SD02 contains mould release additive. Its repeating unit is composed of the hard-segments of PA12, soft-segments of PTMO, the chain extender of adipic acid (shown in Fig. 1).28,51 It was reported that the molecular weights of PA12 segment of P25D, P35D and P40D are 530, 674, and 800 g mol−1, while the molecular weights of PTMO segment are 2000, 1870, and 1060 g mol−1, respectively.52 The average molecular weight of the PEBAX elastomers is ∼5 × 104 g mol−1.52 By the elemental analysis, Rezac and John53 calculated the repeat unit numbers of PA12 (‘x’ in Fig. 1) and PTMO segments (‘y’ in Fig. 1) are 2.68 and 27.80 for P25D, also 3.42 and 26.00 for P35D, 4.06 and 14.72 for P40D. The weight fraction of PA12 segments (WPA) of the studied elastomers determined by 1H NMR are list in Table 1.28,40 | ||
| Fig. 1 General chemical structure of the studied Pebax® elastomers. The repeating numbers of PA12 HS and PTMO SS are denoted as “x” and “y”, respectively. | ||
| Sample | WPA | Tc,SS (°C) | Tm,SS (°C) | Tc,HS (°C) | ΔHc,HS (J g−1) | Tm,HS (°C) | ΔHm,HS (J g−1) | Xc | |
|---|---|---|---|---|---|---|---|---|---|
| a The raw pellet of the PEBA elastomers were used as received.b The films were quenched from 270 °C after the 2 hours isothermal process. | |||||||||
| P25D | Raw pelleta | 0.27 | −10.2 | 12.0 | 53.8 | 9.3 | 135.2 | 10.0 | 0.15 |
| Treated filmb | −10.4 | 12.1 | 50.2 | 6.0 | 135.4 | 2.4 | 0.04 | ||
| P35D | Raw pelleta | 0.29 | −11.9 | 9.0 | 68.9 | 14.5 | 145.0 | 14.7 | 0.21 |
| Treated filmb | −10.7 | 9.2 | 68.5 | 8.9 | 145.2 | 5.9 | 0.08 | ||
| P40D | Raw pelleta | 0.46 | −13.0 | 6.8 | 90.0 | 26.2 | 160.5 | 25.6 | 0.23 |
| Treated filmb | −12.0 | 6.0 | 89.0 | 25.7 | 158.4 | 19.5 | 0.17 | ||
The thermal properties of the three studied copolymers were determined by differential scanning calorimetry (DSC Q2000, TA instruments). The instrument was calibrated with indium before measurements. Temperature scans were performed in the range of −50 to 200 °C with a heating/cooling rate of 10 °C min−1 under the nitrogen atmosphere. The melting temperature (Tm), crystallization temperature (Tc), the corresponding melting and crystallization enthalpies (ΔHm and ΔHc) for the PA12 and PTMO crystals are denoted by the subscript “HS” and “SS”, respectively. The crystallinity of PA12 crystals (Xc) was calculated by the normalized ΔHm,HS with the fraction of PA12 HS dividing by the fusion enthalpy of perfect PA12 crystals (246 J g−1).40 The results are listed in Table 1.
To remove the absorbed moisture, the raw pellets were dried at 100 °C under vacuum for 12 hours. Both the thin films with a thickness of 0.5 mm and the rheological specimens with 25 mm diameter and a thickness of 1 mm were prepared by melt-pressed under 180 °C and 50 MPa for 3 min by mold, then quickly cooled to room temperature by cold compression for 3 min.
Rheological measurement
Linear viscoelastic properties of the PEBA elastomer at molten state with various temperatures were measured by a stress-controlled rheometer (DHR2, TA Instruments) with a 25 mm parallel plates feature, the gap-value was 1 mm. All the experiments were carried out under the nitrogen atmosphere to protect the samples from oxygen degradation.The samples were heated to different isothermal temperatures (Tiso = 170, 190, 200, 210, 230, 250, and 270 °C), kept for 3 min to ensure all the crystals were melted, then the dynamic time sweep was performed for obtaining the time dependence of the dynamic storage modulus (G′) and loss modulus (G′′) within 2 hours isothermal at each temperature. To understand the time effect on the phase state, the G′ and G′′ over a frequency (ω) range of 0.1–628 rad s−1 were measured by the SAOS mode for each sample before and after the 2 hours isothermal processes, respectively.
In order to determine the microphase separation transition temperature of P35D melt, the dynamic temperature ramp measurement at a given ω of 0.5 rad s−1 with various heating rates of 0.5, 1.0, 1.5, and 2.0 °C min−1 from 160 to 270 °C were carried out by hot nitrogen gas to control the temperature in the accuracy of ±0.1 °C. With the obtained G′ and G′′, the binodal (Tb) and spinodal temperature (Ts) can be determined based on the Ajji and Chopin's model.54–56
Fourier transform infrared spectroscopy (FTIR) measurement
The FTIR spectra of P35D films with a thickness of ca. 30 μm was obtained by using a Nicolet 6700 FTIR spectrophotometer (Thermo Scientific) operated in transmission mode at room temperature and equipped with an MCT detector. The spectra were collected by accumulating 32 scans with a resolution of 4 cm−1.Tensile testing
The tensile testing was conducted by using an Instron model 3365. The dumbbell shaped tensile bars (3 mm wide, 25 mm long, and ca. 0.35 mm thick) were cut from the melt-pressed films quenched from various Tiso (180, 230 and 270 °C) by liquid nitrogen after the 2 hours isothermal treatment. The crystallinity was as low as 6–8% for the quenched films (ESI, Table S1†). The specimens were stretched to break with jaws moving symmetrically at a constant speed of 100 mm min−1 at ambient temperature (ca. 28 °C). Under the assumption that the sample volume kept a constant during the uniaxial elongation, the true strain was used as a measurement for the deformation, according to Strobl and co-workers.57,58 With the thickness, width, and marked distance between the specific points of the initial (Wo, To, bo) and stretched (W, T, b) specimens, the true stress (σ), draw ratio (λ), and true stress (εH) can be defined as follows (where F is the detected force):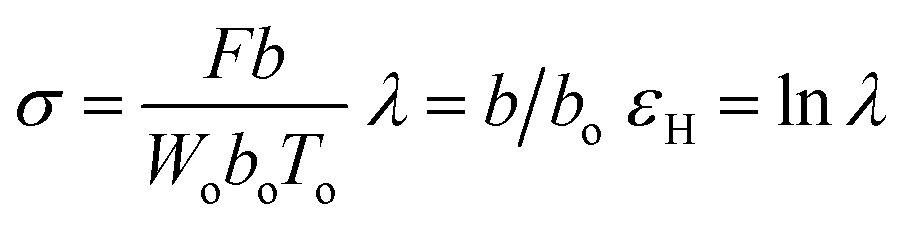
The measurement was repeated 3 times for each condition.
Results and discussion
Temperature effect on the microstructure of PEBAs
To reveal the time effect on the microstructure of PEBA, the G′ of P35D melt as a function of ω obtained before and after the isothermal treatment at various Tiso are shown in Fig. 2a and b, respectively. The typical terminal flow region can be seen at 170 and 190 °C since the scaling laws of G′ ∼ ω2.0 is validated, indicating that P35D has reached the thermodynamically homogenous in the temperature range of 170–190 °C.22,24,59,60 However, G′ has deviated the scaling law from 200 °C (the deviation behavior was also seen for Pebax® 5533).41 Furthermore, a short-range plateau is formed at low frequencies after the 2 hours isothermal process since 210 °C (Fig. 2b), suggesting that a certain network structure has been developed in P35D melt. When Tiso ≥ 210 °C, the magnitude of G′ and G′′ increase rapidly with time during the isothermal process and G′ eventually surpass G′′ at low frequency (see ESI, Fig. S1†), whereas G′ and G′′ slightly decrease in the homogenous states (for example, in the temperature range of 170–190 °C). Fig. 2b shows that the magnitude of G′ plateau was enhanced with the increasing Tiso, indicating the network structure becomes stronger and contributes a greater elastic property at higher temperatures. For the three studied PEBA copolymers with different PA12 content, the time effects on G′ and G′′ at high temperatures are similar, i.e., a pronounced solid-like behavior (G′ ≫ G′′) over the detected frequency range (ESI, Fig. S2†) can be observed after the isothermal treatment at 270 °C for all the three cases. It is probably ascribed by the chemical crosslinking network. The existence of the crosslinking network in these PEBA copolymers after the isothermal at 270 °C was confirmed by the swelling experiment with the N,N-dimethylformamide (DMF) solvent (see ESI, Fig. S3†), some swollen and insoluble matters were seen by the naked eyes for the isothermal-treated films, whereas the raw pellets were all apparent dissolved in DMF. The increased intrinsic viscosity with Tiso also indicates the occurrence of crosslinking reaction (ESI, Table S2†). Based on the results, it is supposed that the polyamide crosslinking reaction can be ignored within the 2 hours rheological measurement because of the extremely low reaction rate (the time has a very weak effect on the G′ increase) when Tiso ≤ 210 °C. However, due to the high reaction rate at 270 °C, the content of crosslinking network is high enough that it cannot be ignored after the 2 hours isothermal treatment and leads to an evident G′ plateau.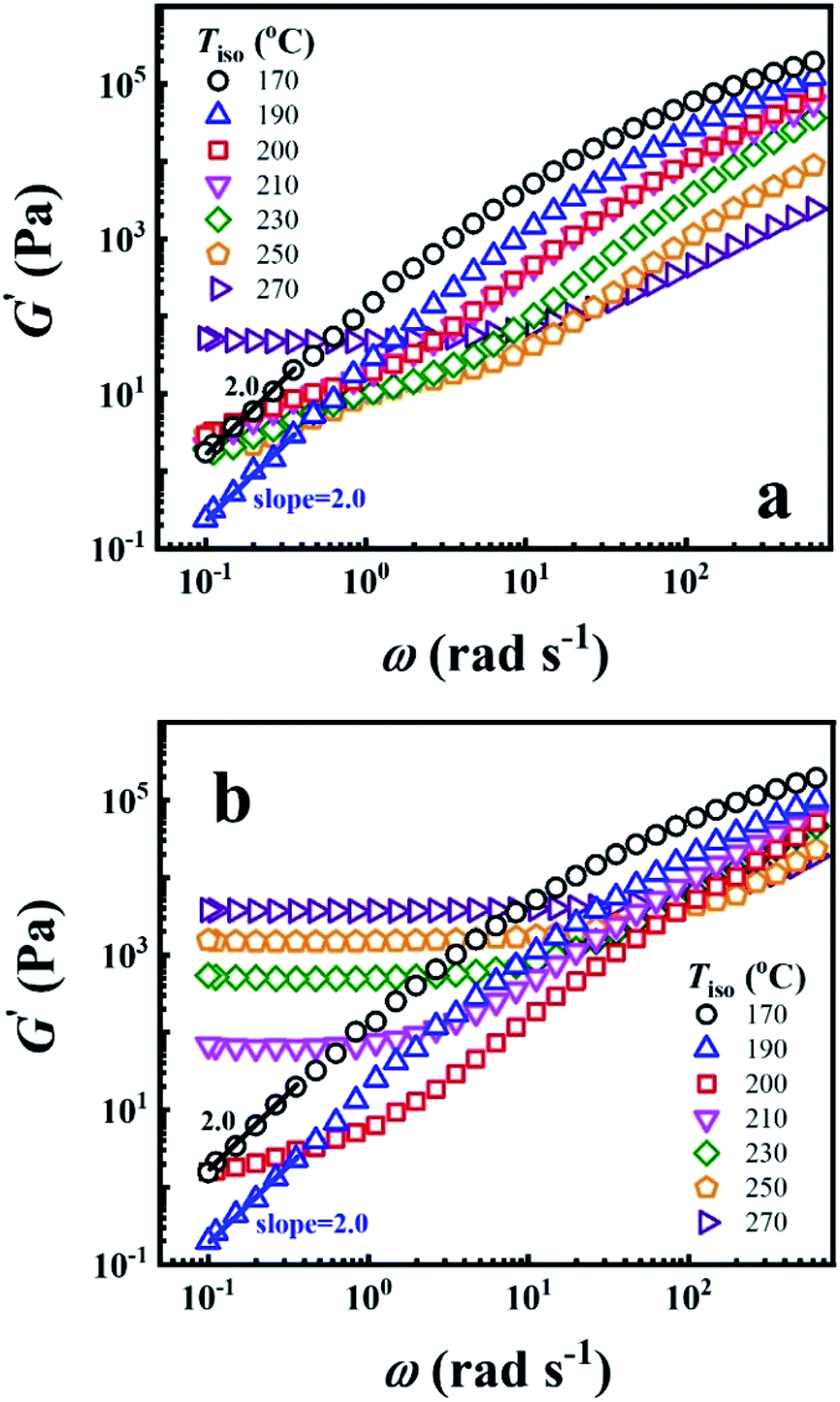 | ||
| Fig. 2 Double Logarithmic plots of G′ against ω (a) before and (b) after the 2 hours isothermal treatment at different Tiso. | ||
FTIR result provides another piece of evidence for the existence of crosslinking structure. Fig. 3 shows the two FTIR spectra of the P35D hot-pressed films with and without the 2 hours isothermal treatment at 270 °C. It was found that the band position and relative height of the bands related to amide group between the two spectra are different, i.e., the crosslinking network is closely associated with the amide groups. With the crosslinking network, the amide A band (stretching band of N–H group) at 3295 cm−1 shifts toward higher wavenumbers, indicating weakening strength of the hydrogen bonding. Moreover, some differences at 3100, 1483, 1223, 941, and 680 (amide V) cm−1 were also observed, which is consistent with our previous result in PA1012 homopolymer.48 Different from the PA1012 homopolymer, the amide II band (stretching band of C–N group and deformation vibration band of N–H group) at 1560 cm−1 shifts to lower wavenumber with the crosslinking network. For the film with crosslinking network (blue curve in Fig. 3), the absorbances of the bands at 2918 and 2859 cm−1 (corresponding to the –CH2– asymmetrical and symmetrical stretching modes, respectively) are saturated due to the thicker film-thickness, lead to the not sharp bands.
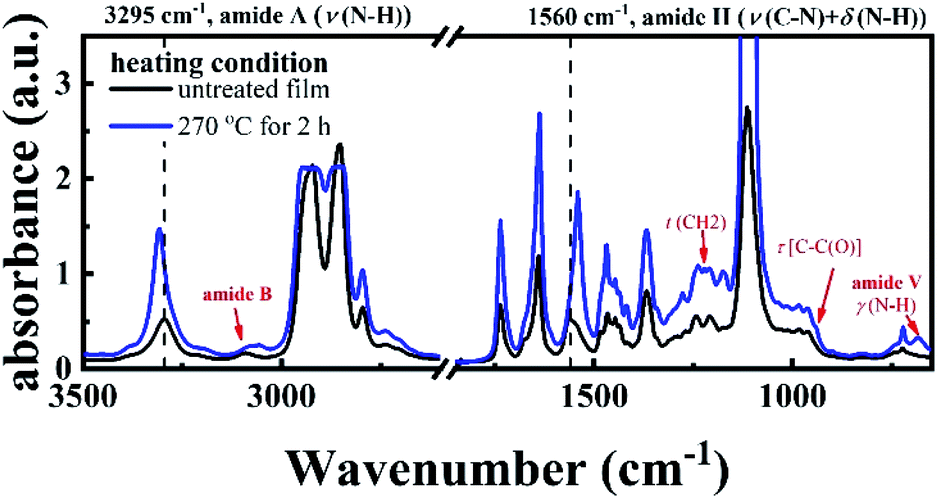 | ||
| Fig. 3 The FTIR spectra of the P35D films, including the untreated film without any isothermal treatment (black line) and the film quenched from 270 °C after the 2 hours isothermal treatment in a vacuum chamber (blue line). The differences between curves are indicated by the dash lines and red arrows. | ||
It was reported that the secondary amine groups (R1–NH–R2) are obtained by the reaction between the end groups of various polyamide homopolymers (known as diamine coupling) and act as branch points that react with carboxyl groups to form the branched structures (ESI, Scheme S1†).49,50 For polyamide melts at high temperature, the depolymerization of polyamide chains can also occur due to the ammonolysis reactions, and the depolymerized segments with –CONH2 end-group can further reacts with the secondary amine group to form the crosslinking structures.61 In other words, the chain scission and crosslinking are probably occurring simultaneously. As a result, both the end groups and the amide group in the repeating unit of polyamide chains can act as a crosslinking point. Based on the above reaction mechanisms, we propose a possible mechanism for the crosslinking reaction of PEBA elastomers as Scheme 1. The crosslinking structure established by the amide group of the repeating units of PA12 HS provides an additional elasticity for the studied PEBA elastomers. Although the proposed mechanism seems reasonable according to the literature, more research is needed to clarify it.
 | ||
| Scheme 1 Illustration of the possible crosslinking mechanism for the studied PEBA elastomers. The amide group in the repeating unit of PA12 HS first undergoes ammonolysis reactions with the ammonia molecules obtained by the reaction between end groups (known as diamine coupling), and then reacts with secondary amine group which acts as a crosslinking point to form a branched structure. | ||
It should be noted that the scaling law of G′ ∼ ω2.0 is invalidated at 200 °C (Fig. 2a), and the crosslinking reaction can be ignored at Tiso < 210 °C due to the extremely low reaction rate. According to the AFM images of Pebax® 5533 films quenched from the melt state,41 the microphase separation can be developed within several minutes. Therefore, it's reasonable the P35D melt is not homogenous one-phase but microphase separated at 200 °C, the physical microphase structure is strong enough to lead to the deviation of G′ ∼ ω.2.0 However, the microstructure of P35D melt may be dominated by the crosslinking network after the isothermal at 200 °C for an infinite period of time, even if the reaction rate is extremely low.
In block copolymers, it's well known that the microphase separation can lead to the deviation of the scaling law of G′ ∼ G′′2.0 in the Han-plot, for which is an important means of detecting the existence of microphase separation.2,21–23,27,60 Fig. 4 shows the Han-plots obtained from P25D, P35D, and P40D after the 2 hours isothermal treatment at various Tiso. It was found that the homogenous criterion of G′ ∼ G′′2.0 is only valid in 170–190 °C for P35D. Considering that no G′ plateau is detected in the Han-plots and the magnitude of G′ does not increase significantly with time (see ESI, Fig. S1†), P25D and P40D are considered to be microphase separated at 170 °C. At higher temperatures, such as 210–270 °C, the evident crosslinking network has been developed in all the three copolymers.
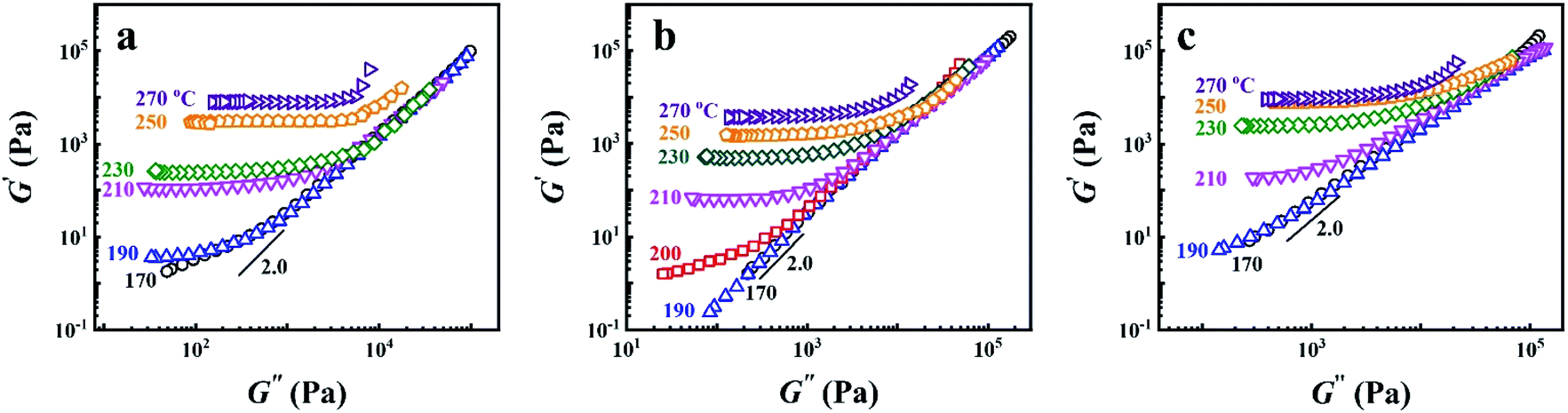 | ||
| Fig. 4 Han-plots obtained from (a) P25D, (b) P35D, and (c) P40D melts after the 2 hours isothermal treatment at various Tiso from 170–270 °C. | ||
Ternary microstructure domains of PEBA upon heating
From the rheological results, there should be two transition temperatures in PEBA copolymers: the microphase separation temperature (transition from homogenous to microphase-separated state) and the initial temperature at which the crosslinking network begins to dominate (denoted as Tcross). The microphase separation is a reversible thermodynamic behavior, the state is a function of temperature, while the crosslinking network is an irreversible evolution. Determination of these two transition temperatures is of great significance for controlling the microstructure and also the rheological properties.Due to the existence of crosslinking network, the microphase separation transition temperature cannot be determined precisely by the typical means such as Han-plot and SAXS in our system. The microphase separation originates from the interaction between the hard- and soft-segments, the apparent homogenous morphology and the possible short-range order structures may exist in the disorder state of block copolymers.2 In contrast, the long-range order structures are formed in the microphase separation region in the phase diagram. In other words, the microphase separation temperature can be defined by the relative phase domain size. Herein, the transition temperature can be analog with the binodal temperature in polymer blends, which are induced by the concentration fluctuation. Therefore, the Tb of P35D can be estimated by using the Ajji and Chopin's method.54–56 Fig. 5a shows the temperature dependence of G′ and G′′ with various heating rates. These curves merged well at low temperatures, where the homogenous criterion was satisfied. For each heating traces, the Tb was determined by the G′ minimum62 at where G′ starts to upturn because of the strong enough concentration fluctuation. Meanwhile, Ts was determined from the extrapolation of the linear regression on the x-axis of the plots of (G′′2/G′T)2/3 versus the reciprocal of temperature 1/T at a given heating-rate (shown in ESI, Fig. S4†) according to the Ajji and Chopin's model. Noted that Ts was obtained with the experimental data at the apparent homogenous region (T < Tb) for ensuring accuracy. Because the microphase separation is a time-dependent behavior, the heating rate effect on Tb and Ts should be considered. The good linear relationships for Tb and Ts versus heating rate are seen in Fig. 5b, the transition temperatures at equilibrium state (denoted as  and
and  ) are determined to be 200.8 and 229.6 °C, respectively, by the extrapolation on the y-axis. Therefore, the PEBA copolymers exhibit an LCST-type phase diagram, also called lower disorder-to-order transition behavior. It's reminded that the applied SAOS may shift the binodal line in the phase diagram and induce a temperature-gap with the quiescent state, the gap is smaller at the lower frequency63,64 In this work, all the temperature sweep measurements were performed at ω of 0.5 rad s−1, thus, the
) are determined to be 200.8 and 229.6 °C, respectively, by the extrapolation on the y-axis. Therefore, the PEBA copolymers exhibit an LCST-type phase diagram, also called lower disorder-to-order transition behavior. It's reminded that the applied SAOS may shift the binodal line in the phase diagram and induce a temperature-gap with the quiescent state, the gap is smaller at the lower frequency63,64 In this work, all the temperature sweep measurements were performed at ω of 0.5 rad s−1, thus, the 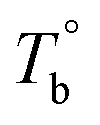 is considered to be reliable and close to that obtained at the quiescent state. The obtained
is considered to be reliable and close to that obtained at the quiescent state. The obtained 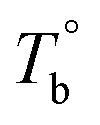 is consistent with the Han-plot result that the microphase separation has been developed in P35D at 200 °C. Although the phase diagram of Pebax® elastomers has not yet been constructed, it may be a dynamic asymmetric system because there is a considerable gap between Tc,HS and Tc,SS (Table 1).
is consistent with the Han-plot result that the microphase separation has been developed in P35D at 200 °C. Although the phase diagram of Pebax® elastomers has not yet been constructed, it may be a dynamic asymmetric system because there is a considerable gap between Tc,HS and Tc,SS (Table 1).
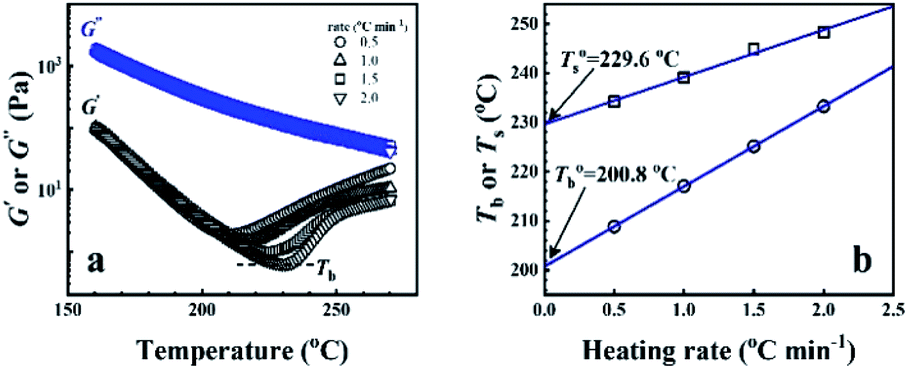 | ||
Fig. 5 (a) Temperature dependence of dynamic moduli obtained from P35D melt with various heating rates. Tb is determined by the temperature of G′ minimum for each curve. (b) Effect of heating rate on the determined Tb and Ts. The extrapolation values on the y-axis represent the temperatures at equilibrium state, denoted as 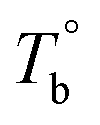 and and  . . | ||
In our system, once the microphase separation behavior occurs, it's difficult to distinguish the contribution from the crosslinking process on dynamic moduli in the initial stage. Moreover, the crosslinking reaction is time-dependent with a slow rate at lower temperatures. Therefore, the Tcross can only be approximately determined by the following experiments: one fresh sample was firstly heated to 170 °C and hold isothermal for 2 hours, then quickly jumped to various Tiso and hold isothermal for 2 hours, finally cooled back to 170 °C to check whether the microstructure is recoverable. Fig. 6a clearly shows that G′ of P35D returned to the equilibrium value (black color) at 170 °C within 2 hours in the case of Tiso = 190 °C (below  ), the reversible behavior indicates it's the homogenous state at 190 °C which is in good agreement with the Han-plots (Fig. 4b). In
), the reversible behavior indicates it's the homogenous state at 190 °C which is in good agreement with the Han-plots (Fig. 4b). In 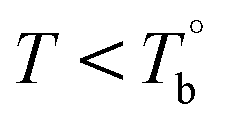 , the rheological properties are only affected by the chain entanglement. Although G′ shows an upward trend at Tiso of 203 °C within two hours, G′ eventually returns to the equilibrium value at 170 °C, implying that the content of crosslinking structure is so low that can be ignored within the measurement at 203 °C. On the other hand, the G′ cooled down from Tiso of 207 °C remained almost constants and three-folds higher than the equilibrium value at 170 °C, the considerable gap indicates the microstructure has been dominant by the crosslinking structure. It can be concluded that the exactly Tcross is in the range of 203–207 °C. When Tiso > Tcross, the crosslinking network develops rapidly, and becomes the dominant factor in determining the microstructure and rheological properties within 2 hours.
, the rheological properties are only affected by the chain entanglement. Although G′ shows an upward trend at Tiso of 203 °C within two hours, G′ eventually returns to the equilibrium value at 170 °C, implying that the content of crosslinking structure is so low that can be ignored within the measurement at 203 °C. On the other hand, the G′ cooled down from Tiso of 207 °C remained almost constants and three-folds higher than the equilibrium value at 170 °C, the considerable gap indicates the microstructure has been dominant by the crosslinking structure. It can be concluded that the exactly Tcross is in the range of 203–207 °C. When Tiso > Tcross, the crosslinking network develops rapidly, and becomes the dominant factor in determining the microstructure and rheological properties within 2 hours.
 | ||
| Fig. 6 (a) The effect of thermal history (isothermal treatment at various Tiso) on the magnitude of G′ of P35D with respect to time at 170 °C. The applied ω is 0.1 rad s−1. (b) Schematic representation of the microstructure domains in PEBA copolymers at the respective temperature regions. The critical temperatures of P35D are indicated on the temperature axis. | ||
Based on our experiments, the microstructure evolution of PEBAs upon heating can be divided into ternary domains: the apparent homogenous, microphase separation, and crosslinking domains, by the determined  and Tcross. Taking P35D as an example, it is apparent homogenous state (the hard and soft segments are compatible at the molecular level) in the temperature range of
and Tcross. Taking P35D as an example, it is apparent homogenous state (the hard and soft segments are compatible at the molecular level) in the temperature range of  . Meanwhile, the rheological properties and microstructure are reversible and depending on the temperature. In the range of
. Meanwhile, the rheological properties and microstructure are reversible and depending on the temperature. In the range of 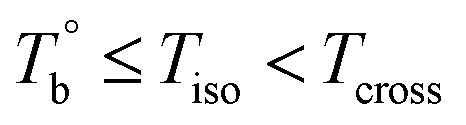 , a microphase-separated P35D melt was obtained after the 2 hours isothermal treatment, the content of crosslinking network is negligible due to the slow reaction rate. The microstructure is mainly affected by the microphase separation, the equilibrium state at a given temperature can be reached over a period of time. However, once the P35D melt was heated to temperatures above Tcross, the content of crosslinking network (or crosslinking density) increases significantly, and becomes the dominant factor within a short period, resulting in the irreversible evolutions in microstructure. As presented, the rheological properties are also irreversible with temperature/time in this temperature domain. Not only PEBA copolymers, the reported irreversible effect of thermal history on the rheological properties in the TPU elastomers20 can also be explained by this mechanism.
, a microphase-separated P35D melt was obtained after the 2 hours isothermal treatment, the content of crosslinking network is negligible due to the slow reaction rate. The microstructure is mainly affected by the microphase separation, the equilibrium state at a given temperature can be reached over a period of time. However, once the P35D melt was heated to temperatures above Tcross, the content of crosslinking network (or crosslinking density) increases significantly, and becomes the dominant factor within a short period, resulting in the irreversible evolutions in microstructure. As presented, the rheological properties are also irreversible with temperature/time in this temperature domain. Not only PEBA copolymers, the reported irreversible effect of thermal history on the rheological properties in the TPU elastomers20 can also be explained by this mechanism.
Unfortunately, no intensity peak associated with the long-range ordered structures was detected after the isothermal-treatment in the P35D's SAXS experiments (ESI, Fig. S5†), even in the microphase-separated domains. It may be caused by the poor contrast of electron cloud density between the hard- and soft-segments. In addition, no evident long-range order structure was observed by the TEM technique for the ultra-thin sections of P35D films quenched from Tiso above  after the 2 hours isothermal process (shown in ESI, Fig. S6†). There are two possibilities: the first one is that interdomain distance is too short to be clearly seen. The interdomain distance can be as small as serval nanometers to tens of nanometers for Pebax® elastomers,41,65 which is much smaller than the specimen thickness (ca. 90 nm). Therefore, the obtained ultra-thin section may contain tens of layers of this kind of structure, resulting in the homogenous-like morphology. The other possibility is that the crosslinking network inhibits the formation of the long-range order structures at T > Tcross. For the three studied elastomers, the lower ΔHm,HS and ΔHc,HS of the treated films after the isothermal process at 270 °C compared to the respective raw pellets implies that the presence of crosslinking network can reduce the regularity of the polymer chains. In other words, the chain mobility of P35D melt is probably restricted by the formed crosslinking structures.
after the 2 hours isothermal process (shown in ESI, Fig. S6†). There are two possibilities: the first one is that interdomain distance is too short to be clearly seen. The interdomain distance can be as small as serval nanometers to tens of nanometers for Pebax® elastomers,41,65 which is much smaller than the specimen thickness (ca. 90 nm). Therefore, the obtained ultra-thin section may contain tens of layers of this kind of structure, resulting in the homogenous-like morphology. The other possibility is that the crosslinking network inhibits the formation of the long-range order structures at T > Tcross. For the three studied elastomers, the lower ΔHm,HS and ΔHc,HS of the treated films after the isothermal process at 270 °C compared to the respective raw pellets implies that the presence of crosslinking network can reduce the regularity of the polymer chains. In other words, the chain mobility of P35D melt is probably restricted by the formed crosslinking structures.
Effect of the crosslinking network on mechanical properties
In the last section, the effect of the crosslinking network on the mechanical properties of P35D at room temperature is investigated by comparing the quenched samples that have been isothermally treated at different Tiso. The true stress–strain curves of P35D after the isothermal-treatment at various Tiso are shown in Fig. 7, the typical elastomer behavior was seen for the untreated sample without any isothermal treatment (black circle). In the case of Tiso of 180 °C (lower than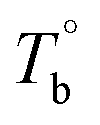 ), the curve (blue triangle) was almost identified with the untreated one because of the reversible entanglement network, whereas the steeper increase trend of stress versus strain was detected in the case of Tiso of 230 and 270 °C (both of them are higher than Tcross). From Fig. 2b, the magnitude of the G′ plateau obtained from Tiso of 270 °C is ca. 7-fold of that from 230 °C, suggesting that the crosslinking density at 270 °C is several times higher than that of 230 °C. The stronger network provides an additional elasticity. Compared with the untreated sample, the stress-at-break was enhanced to 326.3 ± 60.2 MPa at Tiso of 270 °C, as 1.47-fold of that of the untreated sample. It should be noted that the strain-hardening phenomenon becomes more pronounced as the crosslinking density increases. Our previous work has demonstrated that the strain-hardening phenomena in PEBA copolymers is related to the strain-induced crystallization of PTMO segments and the orientations of molecular segments.39 Our true stress–strain curves implied that the existence of the crosslinking network has a considerable influence on the strain-induced crystallization of the PTMO segment at the large deformation for PEBA elastomers, although the mechanical properties do not show a significant difference with the increasing Tiso (Table 2).
), the curve (blue triangle) was almost identified with the untreated one because of the reversible entanglement network, whereas the steeper increase trend of stress versus strain was detected in the case of Tiso of 230 and 270 °C (both of them are higher than Tcross). From Fig. 2b, the magnitude of the G′ plateau obtained from Tiso of 270 °C is ca. 7-fold of that from 230 °C, suggesting that the crosslinking density at 270 °C is several times higher than that of 230 °C. The stronger network provides an additional elasticity. Compared with the untreated sample, the stress-at-break was enhanced to 326.3 ± 60.2 MPa at Tiso of 270 °C, as 1.47-fold of that of the untreated sample. It should be noted that the strain-hardening phenomenon becomes more pronounced as the crosslinking density increases. Our previous work has demonstrated that the strain-hardening phenomena in PEBA copolymers is related to the strain-induced crystallization of PTMO segments and the orientations of molecular segments.39 Our true stress–strain curves implied that the existence of the crosslinking network has a considerable influence on the strain-induced crystallization of the PTMO segment at the large deformation for PEBA elastomers, although the mechanical properties do not show a significant difference with the increasing Tiso (Table 2).
 | ||
| Fig. 7 True stress–strain curves of P35D, including the untreated sample without any thermal-treatment and the samples after the 2 hours isothermal at various Tiso: 180, 210, and 270 °C. The applied crosshead speed is 100 mm min−1. The solid lines at large strain are used to guide the eyes. | ||
| Tiso (°C) | Young's modulus (MPa) | Strain-at-break (%) | Stress-at-break (MPa) |
|---|---|---|---|
| Untreated | 5.0 ± 0.3 | 228.7 ± 1.4 | 222.8 ± 41.7 |
| 180 | 4.8 ± 0.1 | 219.0 ± 11.5 | 270.6 ± 48.5 |
| 230 | 5.5 ± 0.2 | 235.1 ± 0.7 | 316.4 ± 57.7 |
| 270 | 5.2 ± 0.2 | 211.7 ± 10.4 | 326.3 ± 60.2 |
Conclusions
In this work, the microstructure evolution of the poly(ether-b-amide) segmental copolymer upon heating was investigated via rheological measurement. Based on our experiments, the crosslinking network by the inherent crosslinking reaction in the polyamide segments is confirmed to be formed, and dominates the microstructure of the three studied PEBA copolymers at temperatures higher than Tcross. With the obtained and Tcross, the microstructure evolution of PEBA upon the heating process can be classified into ternary domains, they are the homogenous
and Tcross, the microstructure evolution of PEBA upon the heating process can be classified into ternary domains, they are the homogenous  , microphase separated
, microphase separated 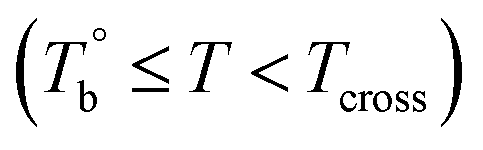 , and the crosslinking (Tcross < T) domains. Our experiments have demonstrated that when P35D melt is once heated above Tcross (ca. 58–62 °C higher than Tm,HS in P35D), the content of the crosslinking network increases rapidly with time and temperature, leading to an irreversible and non-negligible influence on the rheological, crystallization, and mechanical properties. To the best of our knowledge, the present work is the first to systematically explore the microstructure evolution from the apparent homogenous to crosslinked state in PEBA copolymers.
, and the crosslinking (Tcross < T) domains. Our experiments have demonstrated that when P35D melt is once heated above Tcross (ca. 58–62 °C higher than Tm,HS in P35D), the content of the crosslinking network increases rapidly with time and temperature, leading to an irreversible and non-negligible influence on the rheological, crystallization, and mechanical properties. To the best of our knowledge, the present work is the first to systematically explore the microstructure evolution from the apparent homogenous to crosslinked state in PEBA copolymers.
Determination of the critical temperatures (Tm,HS,  and Tcross) is important for regulating the microstructure and studying PEBA's structure–property relationship. It should be noted that these critical temperatures are depending on the content of PA component. For example, the Tm,HS of PEBA increases with the content of polyamide segment, it is possible to even higher than
and Tcross) is important for regulating the microstructure and studying PEBA's structure–property relationship. It should be noted that these critical temperatures are depending on the content of PA component. For example, the Tm,HS of PEBA increases with the content of polyamide segment, it is possible to even higher than  . In that case, no homogenous state can be obtained. Although this work has demonstrated the competition between the microphase separation and crosslinking structures for the microstructure of PEBA, the mechanism is still not well understood from the molecular level viewpoint.
. In that case, no homogenous state can be obtained. Although this work has demonstrated the competition between the microphase separation and crosslinking structures for the microstructure of PEBA, the mechanism is still not well understood from the molecular level viewpoint.
These results provide a deep understanding of the microstructure evolution of the PEBA elastomers. Further investigations of the effects of microstructure evolution and chemical crosslinking on the crystallization behaviors of PEBA are undertaking in our lab, and the results will be reported in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51820105005) and the STS Program of Chinese Academy of Sciences (KFJ-STS-QYZX-113).Notes and references
- G. H. Fredrickson and E. Helfand, J. Chem. Phys., 1987, 87, 697–705 CrossRef CAS.
- N. Sakamoto, T. Hashimoto, C. D. Han, D. Kim and N. Y. Vaidya, Macromolecules, 1997, 30, 1621–1632 CrossRef CAS.
- F. Kayser, G. Fleury, S. Thongkham, C. Navarro, B. Martin-Vaca and D. Bourissou, Macromolecules, 2018, 51, 6534–6541 CrossRef CAS.
- M. Qiu, X. Z. Zhao, D. P. Liu and C. J. He, RSC Adv., 2015, 5, 17851–17861 RSC.
- K. Takano, T. Nyu, T. Maekawa, T. Seki, R. Nakatani, T. Komamura, T. Hayakawa and T. Hayashi, RSC Adv., 2020, 10, 70–75 RSC.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- P. He, W. Shen, W. Yu and C. Zhou, Macromolecules, 2014, 47, 807–820 CrossRef CAS.
- M. W. Matsen and M. Schick, Phys. Rev. Lett., 1994, 72, 2660–2663 CrossRef CAS.
- R. H. Colby, Curr. Opin. Colloid Interface Sci., 1996, 1, 454–465 CrossRef CAS.
- H. E. Park, J. M. Dealy, G. R. Marchand, J. Wang, S. Li and R. A. Register, Macromolecules, 2010, 43, 6789–6799 CrossRef CAS.
- L. Zhu, Y. Chen, A. Zhang, B. H. Calhoun, M. Chun, R. P. Quirk, S. Z. D. Cheng, B. S. Hsiao, F. Yeh and T. Hashimoto, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 10022–10031 CrossRef CAS.
- S. Velankar and S. L. Cooper, Macromolecules, 1998, 31, 9181–9192 CrossRef CAS.
- S. Velankar and S. L. Cooper, Macromolecules, 2000, 33, 382–394 CrossRef CAS.
- W. Gabriëlse, M. Soliman and K. Dijkstra, Macromolecules, 2001, 34, 1685–1693 CrossRef.
- P. Ping, W. Wang, X. Chen and X. Jing, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 557–570 CrossRef CAS.
- L. L. Wang, X. Dong, P. Zhu, X. Q. Zhang, X. R. Liu and D. J. Wang, Eur. Polym. J., 2017, 90, 171–182 CrossRef CAS.
- L. L. Wang, X. Dong, P. Zhu, X. Q. Zhang and D. J. Wang, Acta Polym. Sin., 2017, 5, 752–760 Search PubMed.
- R. Y. Wang, X. S. Guo, B. Fan, S. F. Zou, X. H. Cao, Z. Z. Tong, J. T. Xu, B. Y. Du and Z. Q. Fan, Macromolecules, 2018, 51, 2302–2311 CrossRef CAS.
- R. Y. Wang, J. Huang, X. S. Guo, X. H. Cao, S. F. Zou, Z. Z. Tong, J. T. Xu, B. Y. Du and Z. Q. Fan, Macromolecules, 2018, 51, 4727–4734 CrossRef CAS.
- P. J. Yoon and C. D. Han, Macromolecules, 2000, 33, 2171–2183 CrossRef CAS.
- M. B. Kossuth, D. C. Morse and F. S. Bates, J. Rheol., 1999, 43, 167–196 CrossRef CAS.
- S. Choi, N. Y. Vaidya, C. D. Han, N. Sota and T. Hashimoto, Macromolecules, 2003, 36, 7707–7720 CrossRef CAS.
- K. Almdal, F. S. Bates and K. Mortensen, J. Chem. Phys., 1992, 96, 9122–9132 CrossRef CAS.
- J. H. Rosedale and F. S. Bates, Macromolecules, 1990, 23, 2329–2338 CrossRef CAS.
- F. S. Bates, Science, 1991, 251, 898–905 CrossRef CAS.
- F. S. Bates, Macromolecules, 1984, 17, 2607–2613 CrossRef CAS.
- C. D. Han, J. Kim and J. K. Kim, Macromolecules, 1989, 22, 383–394 CrossRef CAS.
- J. P. Sheth, J. Xu and G. L. Wilkes, Polymer, 2003, 44, 743–756 CrossRef CAS.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714–719 CrossRef CAS.
- S. Y. Dong, P. Zhu, J. G. Liu, D. J. Wang and X. Dong, Acta Polym. Sin., 2019, 50, 189–198 Search PubMed.
- S. Fakirov, Handbook of Condensation Thermoplastic Elastomer, WiLEY-VCH, Weinheim, 2005 Search PubMed.
- F. Zuo, C. G. Alfonzo and F. S. Bates, Macromolecules, 2011, 44, 8143–8153 CrossRef CAS.
- S. Velankar and S. L. Cooper, Macromolecules, 2000, 33, 395–403 CrossRef CAS.
- B. Tavernier, J. Mewis, P. Van Puyvelde, M. Takenaka, B. Ernst and T. Hashimoto, Polym. Eng. Sci., 2008, 48, 2418–2425 CrossRef CAS.
- Y. Y. Cao, P. Zhu, Z. F. Wang, Y. Zhou, H. M. Chen, A. J. Müller, D. J. Wang and X. Dong, Polym. Cryst., 2018, 1, e100121 Search PubMed.
- P. Zhu, X. Dong, Y. Y. Cao, L. L. Wang, X. R. Liu, Z. F. Wang and D. J. Wang, Eur. Polym. J., 2017, 93, 334–346 CrossRef CAS.
- H. S. Lee, H. D. Park and C. K. Cho, J. Appl. Polym. Sci., 2000, 77, 699–709 CrossRef CAS.
- B. B. Sauer, R. S. McLean, D. J. Brill and D. J. Londono, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 1727–1740 CrossRef CAS.
- P. Zhu, X. Dong and D. J. Wang, Macromolecules, 2017, 50, 3911–3921 CrossRef CAS.
- P. Zhu, X. Dong, M. M. Huang, L. L. Wang, S. X. Qi and D. J. Wang, J. Polym. Sci., Part B: Polym. Phys., 2018, 56, 855–864 CrossRef CAS.
- I. K. Yang and P. H. Tsai, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 2557–2567 CrossRef CAS.
- R. Hill, Chem. Ind., 1954, 36, 1083–1089 Search PubMed.
- G. Meacock, J. Appl. Chem., 1954, 4, 172–177 CrossRef CAS.
- I. Goodman, J. Polym. Sci., 1955, 17, 587–590 CrossRef CAS.
- L. H. Peebles and M. W. Huffman, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 1807–1822 CrossRef CAS.
- G. Filippone, S. C. Carroccio, R. Mendichi, L. Gioiella, N. T. Dintcheva and C. Gambarotti, Polymer, 2015, 72, 134–141 CrossRef CAS.
- J. Zhang and A. Adams, Polym. Degrad. Stab., 2016, 134, 169–178 CrossRef CAS.
- X. R. Liu, Y. Wang, L. Y. Liu, X. Dong and D. J. Wang, Chin. J. Polym. Sci., 2020, 38, 993–998 CrossRef CAS.
- R. E. Kirk, D. F. Othmer, J. I. Kroschwitz and M. Howe-Grant, Encyclopedia of Chemical Technology, ed. K. Othmer, Wiley VCH, 1991, vol. 19, pp. 224–229 Search PubMed.
- V. Korshak and T. Frunze, Synthetic Hetero-Chain Polyamides, IPST, Israel, 1964 Search PubMed.
- D. W. Janes, V. Chandrasekar, S. E. Woolford and K. B. Ludwig, Macromolecules, 2017, 50, 6137–6148 CrossRef CAS.
- Y. Song, H. Yamamoto and N. Nemoto, Macromolecules, 2004, 37, 6219–6226 CrossRef CAS.
- M. E. Rezac and T. John, Polymer, 1998, 39, 599–603 CrossRef CAS.
- A. Ajji and L. Choplin, Macromolecules, 1991, 24, 5221–5223 CrossRef CAS.
- M. Pakravan, M. C. Heuzey and A. Ajji, Macromolecules, 2012, 45, 7621–7633 CrossRef CAS.
- J. Khademzadeh Yeganeh, F. Goharpey and R. Foudazi, Macromolecules, 2010, 43, 8670–8685 CrossRef CAS.
- R. Hiss, S. Hobeika, C. Lynn and G. Strobl, Macromolecules, 1999, 32, 4390–4403 CrossRef CAS.
- Y. Men and G. Strobl, Macromolecules, 2003, 36, 1889–1898 CrossRef CAS.
- C. D. Han and J. Kim, J. Polym. Sci., Part B: Polym. Phys., 1987, 25, 1741–1764 CrossRef CAS.
- C. D. Han, D. M. Baek and J. K. Kim, Macromolecules, 1990, 23, 561–570 CrossRef CAS.
- L. H. Peebles Jr and M. W. Huffman, J. Polym. Sci., Part A: Polym. Chem., 1971, 9, 1807–1822 CrossRef.
- W. Liu, X. Dong, F. S. Zou, J. Yang, D. J. Wang and C. C. Han, Polymer, 2014, 55, 2744–2750 CrossRef CAS.
- F. S. Zou, X. Dong, D. Lin, W. Liu, D. J. Wang and C. C. Han, Polymer, 2012, 53, 4818–4826 CrossRef CAS.
- F. S. Zou, X. Dong, W. Liu, J. Yang, D. Lin, A. Liang, W. Li and C. C. Han, Macromolecules, 2012, 45, 1692–1700 CrossRef CAS.
- R. S. McLean and B. B. Sauer, Macromolecules, 1997, 30, 8314–8317 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10627e |
| This journal is © The Royal Society of Chemistry 2021 |