
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA formal [3 + 3] cycloaddition of allenyl imide and activated ketones for the synthesis of tetrasubstituted 2-pyrones†
Yu-Hao Wang‡
,
De-Hua Zhang‡,
Ze-Hun Cao,
Wang-Lai Li and
Yi-Yong Huang *
*
Department of Chemistry, School of Chemistry, Chemical Engineering and Life Science, Wuhan University of Technology, Wuhan 430070, China. E-mail: huangyy@whut.edu.cn
First published on 26th February 2021
Abstract
CsOH·H2O-catalyzed formal [3 + 3] cycloadditions of allenyl imide with β-ketoesters, 1,3-diketones or β-ketonitriles for the synthesis of tetrasubstituted 2-pyrone derivatives have been demonstrated. The allenyl imide was utilized as a C3-synthon, and a ketenyl intermediate was proposed via the process of 1,4-addition of carbon anion to allene followed by elimination of the 2-oxazolidinyl group.
The 2-pyrone (or α-pyrone) moiety1 is widely found in natural products,2 and bioactive compounds exhibiting anti-HIV, anti-bacterial, anti-infective, and anti-cancer activities (Fig. 1).3
 | ||
| Fig. 1 2-Pyrone-derived bioactive molecules. | ||
In addition, 2-pyrones have found broad application as synthetic handles in cross-coupling reactions,4 Diels–Alder reactions5 and conjugate additions6 by virtue of their aromatic, diene and enone structural characteristics. Significantly, 2-pyrones have been utilized as diene components in [4 + 2] cycloadditions for the total syntheses of natural products.7 Therefore, the development of efficient approaches to synthesize 2-pyrones has drawn much attention.8 Thus far, organometallic catalysts, base or acid-enabled the generation of 2-pyrone structures in an intermolecular or intramolecular manner have been established. Whilst partially substituted 2-pyrones can be readily synthesized, the synthetic methods to prepare tetrasubstituted 2-pyrones remains scarce.9 For instance, in 2007, Ryu and coworkers achieved tetrasubstituted 2-pyrones through the [3 + 2 + 1] cycloaddition using silylacetylenes, α,β-unsaturated ketones and CO as starting materials.10 In 2019, Yasuda and coworkers installed tetrasubstituted metalated 2-pyrones via the oxyindation of carbonyl-ene-yne compounds with indium trihalides, which could be applied into the synthesis of tetrasubstituted 2-pyrones through cross-coupling and halogenation reactions.11 In the same year, Mei and coworkers communicated the iridium-catalyst enabling C–H/O–H functionalization for alkyne annulation to install tetrasubstituted 2-pyrones.12 Furthermore, as exemplified in Fig. 1, some natural products possess tetrasubstituents in the 2-pyrone skeleton. In this context, the discovery of novel strategy to build tetrasubstituted 2-pyrones under mild conditions should be highly demanding and rewarding. To the purpose, we launched this project and documented the preliminary results.
In our previous work, we discovered that allenyl imides could be transformed into 1,4-(bis)electrophilic α,β-unsaturated ketenyl phosphonium species under nucleophilic catalysis condition, which was further utilized as C4-synthons in the [4 + 1] cycloaddition of methyl ketimines, enamines, and a primary amine (Scheme 1a).13 The 2-oxazolidinyl imide group acts as a good leaving group. Encouraged by these results, we envisaged that allenyl imide 1 should be applied into the cycloaddition of other (bis)nucleophilic partners, such as activated ketones, and thus novel types of heterocycles would be assembled; if [3 + 3] cycloaddition reaction occurs, 2-pyrone derivatives will be available (Scheme 1b).
 | ||
| Scheme 1 Cycloaddition with allenyl imide. | ||
β-Ketoester is one type of activated ketones, and known as 1C,3O-bisnucleophile in cycloaddition reactions. Ethyl benzoylacetate 2a and allenyl imide 1 were used as two substrates in the model reaction (Table 1). Under the previous [4 + 1] cycloaddition conditions by using PBu3 as a nucleophilic catalyst,13 trace amount of [3 + 3] cycloadduct 2-pyrone 3a instead of [4 + 1] cycloadduct was detected (entry 1). Then screening other base catalysts including DABCO, NEt3 and Cs2CO3 in CH2Cl2 solvent (1.0 mL) at 30 °C revealed that only Cs2CO3 could trigger an effective [3 + 3] cycloaddition reaction;14 compound 3a was delivered in 77% yield within 6 h (entry 4), where allenyl imide 1 displays dual electrophilic reactivity at βC and amide carbonyl positions. The structural assignment of 3a was spectroscopically determined and later confirmed by analogy to the X-ray crystallography of product 3e (see Table 2 below).15 The solvent effect further showed that CH2Cl2 was more suitable than other solvents. When we changed the base from Cs2CO3 to CsOH·H2O, the yield was improved to 85% (entry 11). Since CsOH·H2O is a strong base, which may better facilitate the deprotonation of β-ketoester than other bases. After investigating the lower loading of CsOH·H2O catalyst in less amount of CH2Cl2 solvent, the optimal reaction conditions for the access to product 3a (90%) were found: 0.3 equiv. of CsOH·H2O and 0.5 mL of CH2Cl2 at 30 °C (entry 14).
|
||||
|---|---|---|---|---|
| Entry | Base/equiv. | Solvent | Time/h | Yieldb (%) |
| a Reaction conditions: 1 (0.1 mmol), 2a (0.12 mmol), and solvent (1.0 mL) were stirred at 30 °C.b Isolated yield.c 0.5 mL of solvent was used. | ||||
| 1 | PBu3/0.5 | CH2Cl2 | 48 | Trace |
| 2 | DABCO/0.5 | CH2Cl2 | 48 | Trace |
| 3 | Et3N/0.5 | CH2Cl2 | 24 | Trace |
| 4 | Cs2CO3/0.5 | CH2Cl2 | 6 | 77 |
| 5 | Cs2CO3/0.5 | Et2O | 6 | 61 |
| 6 | Cs2CO3/0.5 | ClCH2CH2Cl | 20 | 63 |
| 7 | Cs2CO3/0.5 | Toluene | 20 | 72 |
| 8 | Cs2CO3/0.5 | MeCN | 6 | 73 |
| 9 | Cs2CO3/0.5 | 1,4-Dioxane | 6 | 64 |
| 10 | Cs2CO3/0.5 | EtOAc | 4 | 76 |
| 11 | CsOH·H2O/0.5 | CH2Cl2 | 6 | 85 |
| 12c | CsOH·H2O/0.1 | CH2Cl2 | 24 | 40 |
| 13c | CsOH·H2O/0.2 | CH2Cl2 | 12 | 75 |
| 14c | CsOH·H2O/0.3 | CH2Cl2 | 6 | 90 |

|
|||||
|---|---|---|---|---|---|
| Entry | R | R′ | Product | t/h | Yieldb/% |
| a Reaction condition: 1 (0.1 mmol), 2 (0.12 mmol), CsOH·H2O (30 mol%), and CH2Cl2 (0.5 mL) were stirred at 30 °C.b Isolated yield based on 1.c The data in brackets was obtained by using 4.0 mmol of 1. | |||||
| 1 | C6H5 | Et | 3a | 6 | 90 (87)c |
| 2 | C6H5 | Me | 3b | 6 | 87 |
| 3 | 4-Me–C6H4 | Et | 3c | 16 | 85 |
| 4 | 4-OMe–C6H4 | Me | 3d | 6 | 82 |
| 5 | 4-Br–C6H4 | Et | 3e | 6 | 95 |
 |
(ORTEP of 3e, CCDC 2031826) | ||||
| 6 | 4-I–C6H4 | Et | 3f | 24 | 85 |
| 7 | 4-CN–C6H4 | Et | 3g | 6 | 87 |
| 8 | 4-CF3–C6H4 | Et | 3h | 36 | 60 |
| 9 | 3,4-(CH3)2–C6H3 | Et | 3i | 24 | 85 |
| 10 | 3-CH3–C6H4 | Me | 3j | 24 | 84 |
| 11 | 3-OMe–C6H4 | Et | 3k | 6 | 95 |
| 12 | 3-F–C6H4 | Et | 3l | 24 | 95 |
| 13 | 3-Cl–C6H4 | Et | 3m | 6 | 95 |
| 14 | 3-CF3–C6H4 | Et | 3n | 12 | 90 |
| 15 | 2-OMe–C6H4 | Et | 3o | 20 | 80 |
| 16 | 2-F–C6H4 | Et | 3p | 24 | 87 |
| 17 | 2-Cl–C6H4 | Me | 3q | 12 | 80 |
| 18 | 2-Br–C6H4 | Me | 3r | 24 | 93 |
| 19 | 1-Naphthyl | Et | 3s | 5 | 94 |
| 20 | 2-Naphthyl | Et | 3t | 12 | 85 |
| 21 | 2-Thienyl | Et | 3u | 8 | 92 |
| 22 | 2-Furyl | Et | 3v | 6 | 94 |
| 23 | Cyclohexyl | Et | 3w | 20 | 80 |
| 24 | Me | Et | 3x | 12 | 85 |

After establishing the optimized conditions, the scope of β-ketoesters was carried out to synthesize various tetrasubstituted 2-pyrone derivatives (3) bearing an ester group at the 5-position, and the results were summarized in Table 2. Initially, the scale-up (4.0 mmol of 1) synthesis was carried out to provide an identical level of yield (87%, entry 1). Then the effects of steric hindrance and electronic structure properties of substituents on the phenyl ring were checked. The incorporation of electron-donating groups (Me– and MeO–) at the para-position provided slightly lower level of yields (82–85%, entries 3 and 4). 4-Br substituent provided a higher yield than the 4-I variant (95% vs. 85%, entries 5 and 6). Based on the single crystal X-ray analysis of compound 3e, the pyrone structure was determined.15 Similar reactivity and yield were observed by comparing CN-substituent (entry 7) with MeO- and Me-substituents (entries 3 and 4), however, the strong electron-withdrawing group (–CF3) was not beneficial to the yield (60%, entry 8). The use of meta,para-disubstituted β-arylketoester was found to be compatible with the reaction conditions, and 84% yield of product 3i was observed. In the case of meta-substituted substrates, good to excellent yields were obtained (entries 10–14). The incorporation of –OMe and –CF3 groups at meta-position exhibited much higher yields than those at para-position. When –OMe or halide (F, Cl and Br) groups were introduced at the ortho-position of phenyl group in β-ketoesters, up to 93% yield was obtained. Both 1-naphthyl and 2-naphthyl substitutions were tolerated, albeit much higher yield was received for the former case (94% vs. 85%). Likewise, heterocyclic β-ketoesters including 2-thienyl and 2-furyl groups underwent the [3 + 3] cycloaddition, leading to products 3u–v in excellent yields (entries 21 and 22). Except aromatic β-ketoesters, aliphatic β-ketoesters having cyclohexyl or methyl groups were also applied in the [3 + 3] cycloaddition, providing compounds 3w and 3x in 80% and 85% yields, respectively.
In order to alter the substituent at the 5-position of 2-pyrone product, β-ketonitriles and 1,3-diketones were utilized as 1C,3O-bisnucleophiles in the [3 + 3] cycloaddition of allene 1 (Table 3). Under the above-mentioned standard conditions, 9 examples of β-ketonitriles with different substituents at the phenyl group were studied. As expected, 2-pyrone 4a was successfully synthesized in 90% yield (entry 1). The use of para-Br-substituted arylketonitrile gave the corresponding product 4b in 80% yield (entry 2). More than 90% yield was obtained for three meta-substituted substrates (Me–, MeO– and F–; entries 3–5), albeit 84–88% yields for other two meta-substituted cases (Cl– and CF3–, entries 6 and 7). Similarly, ortho-substituted products 4h (–OMe) and 4i (–I) were obtained in 85% and 80% yields, respectively (entries 8 and 9). Furthermore, 2-benzoylacetophenone reacted with 1 to give the corresponding product 4j in 83% yield (entry 10). Another three symmetric 1,3-diketones also performed well in this [3 + 3] cycloaddition (entries 11–13).
|
|||||
|---|---|---|---|---|---|
| Entry | R | R′ | Product | Time/h | Yieldb/% |
| a Reaction conditions: 1 (0.1 mmol), 2 (0.12 mmol), CsOH·H2O (30 mol%), and CH2Cl2 (0.5 mL) were stirred at 30 °C.b Isolated yield based on 1. | |||||
| 1 | C6H5 | CN | 4a | 24 | 90 |
| 2 | 4-Br–C6H4 | CN | 4b | 12 | 80 |
| 3 | 3-Me–C6H4 | CN | 4c | 12 | 92 |
| 4 | 3-OMe–C6H4 | CN | 4d | 12 | 90 |
| 5 | 3-F–C6H4 | CN | 4e | 12 | 95 |
| 6 | 3-Cl–C6H4 | CN | 4f | 12 | 88 |
| 7 | 3-CF3–C6H4 | CN | 4g | 12 | 84 |
| 8 | 2-OMe–C6H4 | CN | 4h | 12 | 85 |
| 9 | 2-I–C6H4 | CN | 4i | 12 | 80 |
| 10 | C6H5 | C(O)–C6H5 | 4j | 6 | 83 |
| 11 | 3-Me–C6H4 | C(O)–(3-Me–C6H4) | 4k | 6 | 88 |
| 12 | 3-OMe–C6H4 | C(O)–(3-OMe–C6H4) | 4l | 6 | 80 |
| 13 | 3-CF3–C6H4 | C(O)–(3-CF3–C6H4) | 4m | 16 | 78 |

As shown in Scheme 2, based on our previous work,13 the proposed mechanism of this [3 + 3] annulation is presented. Firstly, β-ketoester 2a is deprotonated by the CsOH base to form nucleophilic species I, which undergoes a Michael-type addition to the Csp atom of allene 1 to give intermediate II. After eliminating the 2-oxazolidinyl anion, intermediate III containing an electrophilic ketene group is formed, which is again deprotonated at the tertiary carbon by the CsOH base to provide intermediate IV. Next, the O-containing six-member ring (V) is generated through the nucleophilic addition of oxygen anion into the ketene group. Finally, the isomerization and interception of a proton from new substrate 2a occur to produce 3a and intermediate I to initiate another catalytic cycle.
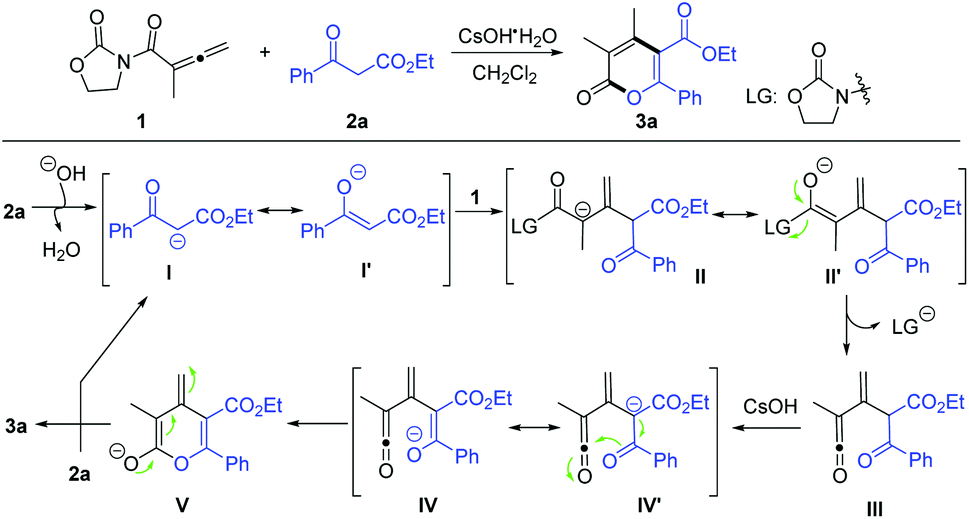 | ||
| Scheme 2 Proposed reaction pathway. | ||
In summary, we have established a novel CsOH·H2O-catalyzed [3 + 3] cycloaddition to access various tetrasubstituted 2-pyrones (37 examples, up to 95% yield), which used activated ketones as 1C,3O-bisnucleophiles and expanded the synthetic potential of allenyl imide as a C3-synthon in cycloadditions. A wide arrange of β-ketoesters, β-ketonitriles and 1,3-diketones were applied, and good to excellent yields were observed. The proposed reaction pathway including Michael addition/elimination and intramolecular nucleophilic cyclization was demonstrated. Further study of allenyl imides in other types of annulations are underway in our lab.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y.-Y. H. gratefully acknowledges the financial support for this investigation from the National Natural Science Foundation of China (21772151, 22072111), and Natural Science Foundation of Hubei Province (2018CFA084).Notes and references
- For a comprehensive review, see: A. Goel and V. J. Ram, Tetrahedron, 2009, 65, 7865–7913 CrossRef CAS.
- For reviews of 2-pyrones in nature, see: (a) J. M. Dickinson, Nat. Prod. Rep., 1993, 10, 71–98 RSC; (b) G. P. McGlacken and I. J. S. Fairlamb, Nat. Prod. Rep., 2005, 22, 369–385 RSC; (c) Z.-L. Mao, W.-B. Sun, L.-Y. Fu, H.-Y. Luo, D.-W. Lai and L.-G. Zhou, Molecules, 2014, 19, 5088–5108 CrossRef; (d) K. S. Singh, Curr. Org. Chem., 2020, 24, 354–401 CrossRef CAS; (e) A. Furstner, Angew. Chem., Int. Ed., 2017, 57, 4215–4233 CrossRef.
- For reviews, see: (a) Z. S. Bhat, M. A. Rather, M. Maqbool, H. Ul Lah, S. K. Yousuf and Z. Ahmad, Biomed. Pharmacother., 2017, 91, 265–277 CrossRef CAS; (b) M. G. Nair, A. Chandra and D. L. Thorogood, J. Antibiot., 1993, 46, 1762–1763 CrossRef CAS; (c) Y. Li, D. Ye, X. Chen, X. Lu, Z. Shao, H. Zhang and Y. Che, J. Nat. Prod., 2009, 72, 912–916 CrossRef CAS; (d) A. P. G. Macabeo, A. J. C. Cruz, A. Narmani, M. Arzanlou, A. Babai-Ahari, L. A. E. Pilapil, K. Y. M. Garcia, V. M. Huchd and M. Stadlerb, Phytochem. Lett., 2020, 35, 147–151 CrossRef CAS; (e) A. C. Giddens, L. Nielsen, H. I. Boshoff, D. Tasdemir, R. Perozzo, M. Kaiser, F. Wang, J. C. Sacchettini and B. R. Copp, Tetrahedron, 2008, 64, 1242–1249 CrossRef CAS; (f) D. Liu, X.-M. Li, L. Meng, C.-S. Li, S.-S. Gao, Z. Shang, P. Proksch, C.-G. Huang and B.-G. Wang, J. Nat. Prod., 2011, 74, 1787–1791 CrossRef CAS; (g) S. Shimizu, K. Hagiwara, H. Itoh and M. Inoue, Org. Lett., 2020, 22, 8652–8657 CrossRef CAS; (h) X.-P. Chu, Q.-F. Zhou, S. Zhao, F.-F. Ge, M. Fu, J.-P. Chen and T. Lu, Chin. Chem. Lett., 2013, 24, 120–122 CrossRef CAS.
- (a) For site-selective Sonogashira coupling of 3,5-dibromo-2-pyrone, see the following: J. H. Lee, J. S. Park and C. G. Cho, Org. Lett., 2002, 4, 1171–1173 CrossRef CAS; (b) For site-selective Stille coupling of 3,5-dibromo-2-pyrone see: W. S. Kim, H. J. Kim and C. G. Cho, J. Am. Chem. Soc., 2003, 125, 14288–14289 CrossRef CAS; (c) For site-selective Suzuki coupling of 3,5-dibromo-2-pyrone see: K. M. Ryu, A. K. Gupta, J.-W. Han, C. H. Oh and C. G. Cho, Synlett, 2004, 12, 2197–2199 Search PubMed; (d) A. M. Prendergast and G. P. McGlacken, Eur. J. Org. Chem., 2017, 32, 4827–4835 CrossRef.
- For a review, see: (a) V. J. Ram and P. Srivastava, Curr. Org. Chem., 2001, 5, 571–599 CrossRef CAS; (b) X.-W. Liang, Y. Zhao, X.-G. Si, M.-M. Xu, J.-H. Tan, Z.-M. Zhang, C.-G. Zheng, C. Zheng and Q. Cai, Angew. Chem., Int. Ed., 2019, 58, 14562–14567 CrossRef CAS; (c) V. Miskov-Pajic, F. Willig, D. M. Wanner, W. Frey and R. Peters, Angew. Chem., Int. Ed., 2020, 59, 19873–19877 CrossRef CAS; (d) X. Zhang and C. M. Beaudry, Org. Lett., 2020, 22, 6086–6090 CrossRef CAS; (e) C. J. F. Cole, L. Fuentes and S. A. Snyder, Chem. Sci., 2020, 11, 2175–2180 RSC; (f) R. P. Singh, K. Bartelson, Y. Wang, H. Su, X. Lu and L. Deng, J. Am. Chem. Soc., 2008, 130, 2422–2423 CrossRef CAS; (g) Y. Wang, H. Li, Y.-Q. Wang, Y. Liu, B. M. Foxman and L. Deng, J. Am. Chem. Soc., 2007, 129, 6364–6365 CrossRef CAS; (h) R. Shaw, I. Althagafi, A. Elagamy, R. Rai, C. Shah, V. Nemaysh, H. Singh and R. Pratap, Org. Biomol. Chem., 2020, 18, 6276–6286 RSC.
- B. Mao, M. Fananás-Mastral and B. L. Feringa, Org. Lett., 2013, 15, 286–289 CrossRef CAS.
- For a review, see: (a) Q. Cai, Chin. J. Chem., 2019, 37, 946–976 CrossRef CAS; (b) P. Gan, M. W. Smith, N. R. Braffman and S. A. Snyder, Angew. Chem., Int. Ed., 2016, 55, 3625–3630 CrossRef CAS; (c) J. H. Lee and C. G. Cho, Org. Lett., 2018, 20, 7312–7316 CrossRef CAS; (d) V. Palani, C. L. Hugelshofer and R. Sarpong, J. Am. Chem. Soc., 2019, 141, 14421–14432 CrossRef CAS; (e) C.-X. Zhuo and A. Fürstner, Angew. Chem., Int. Ed., 2016, 55, 6051–6056 CrossRef CAS; (f) C.-X. Zhuo and A. Fürstner, J. Am. Chem. Soc., 2018, 140, 10514–10523 CrossRef CAS.
- (a) T. Sunazuka and S. Omura, Chem. Rev., 2005, 105, 4559–4580 CrossRef CAS; (b) J. S. Lee, Mar. Drugs, 2015, 13, 1581–1620 CrossRef CAS; (c) Q.-F. Zhou, S. Zhao, X. Wang and T. Lu, Chin. J. Org. Chem., 2010, 30, 1652–1663 CAS; (d) T. F. Schaeberle, Beilstein J. Org. Chem., 2016, 12, 571–588 CrossRef CAS.
- (a) S.-M. Ma, S.-H. Yin, L.-T. Li and F.-G. Tao, Org. Lett., 2002, 4, 505–507 CrossRef CAS; (b) R. C. Larock, M. J. Doty and X. Han, J. Org. Chem., 1999, 64, 8770–8779 CrossRef CAS; (c) S. Mochida, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 6295–6298 CrossRef CAS; (d) T. Yao and R. C. Larock, J. Org. Chem., 2003, 68, 5936–5942 CrossRef CAS.
- T. Fukuyama, Y. Higashibeppu, R. Yamaura and I. Ryu, Org. Lett., 2007, 9, 587–589 CrossRef CAS.
- T. Yata, Y. Kita, Y. Nishimoto and M. Yasuda, J. Org. Chem., 2019, 84, 14330–14341 CrossRef CAS.
- Q.-L. Yang, Y.-K. Xing, X.-Y. Wang, H.-X. Ma, X.-J. Weng, X. Yang, H.-M. Guo and T.-S. Mei, J. Am. Chem. Soc., 2019, 141, 18970–18976 CrossRef CAS.
- Z.-H. Cao, Y.-H. Wang, S. J. Kalita, U. Schneider and Y.-Y. Huang, Angew. Chem., Int. Ed., 2020, 59, 1884–1890 CrossRef CAS.
- For a recent review, see: (a) Y.-N. Zhu and Y. Huang, Synthesis, 2020, 52, 1181–1202 CrossRef CASFor selected examples, see: (b) R. Na, C. Jing, Q. Xu, H. Jiang, X. Wu, J. Shi, J. Zhong, M. Wang, D. Benitez, E. Tkatchouk, W. A. Goddard III, H. Guo and O. Kwon, J. Am. Chem. Soc., 2011, 133, 13337–13348 CrossRef CAS; (c) Y.-N. Zhu and Y. Huang, Org. Lett., 2020, 22, 6750–6755 CrossRef CAS; (d) J. Hu, Y.-B. Wei and X.-F. Tong, Org. Lett., 2011, 13, 3068–3071 CrossRef CAS.
- CCDC 2031826 (compound 3e) contains the ESI crystallographic data for this paper.†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2031826. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra10686k |
| ‡ These authors are equal contribution to this work. |
| This journal is © The Royal Society of Chemistry 2021 |