
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and evaluation of novel indole-2-carboxamides for growth inhibition of Mycobacterium tuberculosis and paediatric brain tumour cells†
Shahinda S. R. Alsayed a,
Shichun Lunb,
Anders W. Baileyc,
Amreena Suric,
Chiang-Ching Huangd,
Mauro Mocerinoe,
Alan Paynee,
Simone Treiger Sredni*cf,
William R. Bishai*bg and
Hendra Gunosewoyo*a
a,
Shichun Lunb,
Anders W. Baileyc,
Amreena Suric,
Chiang-Ching Huangd,
Mauro Mocerinoe,
Alan Paynee,
Simone Treiger Sredni*cf,
William R. Bishai*bg and
Hendra Gunosewoyo*a
aCurtin Medical School, Faculty of Health Sciences, Curtin University, Bentley, Perth, WA 6102, Australia. E-mail: Hendra.Gunosewoyo@curtin.edu.au
bCenter for Tuberculosis Research, Department of Medicine, Division of Infectious Disease, Johns Hopkins School of Medicine, 1550, Orleans Street, Baltimore, Maryland 21231-1044, USA. E-mail: wbishai1@jhmi.edu
cDivision of Pediatric Neurosurgery, Ann and Robert H. Lurie Children's Hospital of Chicago, Chicago, IL 60611, USA
dDepartment of Biostatistics, Zilber School of Public Health, University of Wisconsin, Milwaukee, WI 53205, USA
eSchool of Molecular and Life Sciences, Curtin University, Perth, WA 6102, Australia
fDepartment of Surgery, Northwestern University, Feinberg School of Medicine, Chicago, IL 60611, USA. E-mail: ssredni@northwestern.edu
gHoward Hughes Medical Institute, 4000 Jones Bridge Road, Chevy Chase, Maryland 20815-6789, USA
First published on 26th April 2021
Abstract
The omnipresent threat of tuberculosis (TB) and the scant treatment options thereof necessitate the development of new antitubercular agents, preferably working via a novel mechanism of action distinct from the current drugs. Various studies identified the mycobacterial membrane protein large 3 transporter (MmpL3) as the target of several classes of compounds, including the indole-2-caboxamides. Herein, several indoleamide analogues were rationally designed, synthesised, and evaluated for their antitubercular and antitumour activities. Compound 8g displayed the highest activity (MIC = 0.32 μM) against the drug-sensitive (DS) Mycobacterium tuberculosis (M. tb) H37Rv strain. This compound also exhibited high selective activity towards M. tb over mammalian cells [IC50 (Vero cells) = 40.9 μM, SI = 128], suggesting its minimal cytotoxicity. In addition, when docked into the MmpL3 active site, 8g adopted a binding profile similar to the indoleamide ligand ICA38. A related compound 8f showed dual antitubercular (MIC = 0.62 μM) and cytotoxic activities against paediatric glioblastoma multiforme (GBM) cell line KNS42 [IC50 (viability) = 0.84 μM]. Compound 8f also showed poor cytotoxic activity against healthy Vero cells (IC50 = 39.9 μM). Compounds 9a and 15, which were inactive against M. tb, showed potent cytotoxic (IC50 = 8.25 and 5.04 μM, respectively) and antiproliferative activities (IC50 = 9.85 and 6.62 μM, respectively) against KNS42 cells. Transcriptional analysis of KNS42 cells treated with compound 15 revealed a significant downregulation in the expression of the carbonic anhydrase 9 (CA9) and the spleen tyrosine kinase (SYK) genes. The expression levels of these genes in GBM tumours were previously shown to contribute to tumour progression, suggesting their involvement in our observed antitumour activities. Compounds 9a and 15 were selected for further evaluations against three different paediatric brain tumour cell lines (BT12, BT16 and DAOY) and non-neoplastic human fibroblast cells HFF1. Compound 9a showed remarkable cytotoxic (IC50 = 0.89 and 1.81 μM, respectively) and antiproliferative activities (IC50 = 7.44 and 6.06 μM, respectively) against the two tested atypical teratoid/rhabdoid tumour (AT/RT) cells BT12 and BT16. Interestingly, compound 9a was not cytotoxic when tested against non-neoplastic HFF1 cells [IC50 (viability) = 119 μM]. This suggests that an indoleamide scaffold can be fine-tuned to confer a set of derivatives with selective antitubercular and/or antitumour activities.
1. Introduction
Mycobacterium tuberculosis (M. tb), the causative agent of tuberculosis (TB), is a highly contagious airborne pathogen infecting around 10 million people and claiming more than one million lives worldwide every year.1 In 2019 before the COVID-19 pandemic, the inexorable TB burden together with the currently estimated one-quarter latent infections of global population ranked TB as the number one cause of mortality/morbidity from a single infectious agent (surpassing HIV/AIDS).1 Due to the poor patient compliance to the onerous multi-drug first-line regimen, administered for at least 6 months, and the co-epidemic with HIV, vicious drug-resistant TB strains have emerged, perpetuating TB as a global health threat.2,3 The insufficiency and limited efficacy of the drug options of multi- and extensively drug resistant TB (MDR- and XDR-TB, respectively) accompanied with their high cost, toxicity and drawn-out duration of therapy (up to 2 years) further exacerbate the TB resistance crisis.4 Accordingly, it is imperative to revitalise the anti-TB drug discovery efforts and identify novel lead compounds inhibiting cellular targets different from the ones targeted by the current anti-TB drugs. This will presumably synergise their efficacy in combination with current agents in combating drug-sensitive (DS) and drug-resistant (DR) M. tb.The mycobacterial membrane protein large 3 (MmpL3) is a vital transporter currently considered as one of the most druggable TB targets.5 It is responsible for the translocation of mycolic acids (MAs) precursor, trehalose monomycolate (TMM), across the plasma membrane.6–11 MAs are the major lipid component of the mycobacterial outer membrane and the main driving force behind its hydrophobicity and impermeability.12–14 We previously illustrated the detailed biosynthetic machinery of MAs and their assembly into the mycobacterial cell envelope.15 After the flipping and release of TMM from cytoplasm to periplasm via MmpL3, the cell wall core components serve as acceptors anchoring MAs and forming an intricate protective lipid bilayer. This coating insulates M. tb against exogenous substances, including antibiotics, and the host immune system.15 The indispensable role of MmpL3 in M. tb growth and survival through shuttling TMM across the cytoplasmic membrane was verified when the downregulation of mmpl3 expression led to an abrogation of cell division and rapid cell death. This was preceded by an intracellular accumulation of TMM and significant reduction of cell wall mycolation.16,17
A conspicuous trend of “high lipophilicity = high activity” was entrenched in many of the MmpL3 inhibitors reported to date. Recent literature revealed that five structurally distinct MmpL3 inhibitors indeed directly interact with MmpL3, while three of them additionally impact the proton motive force (PMF) in M. tb.18 Of particular interest are the indoleamides in which NITD-304 (1, Fig. 1) and its analogous 4,6-difluoro derivative NITD-349 were identified as preclinical agents for treating MDR-TB.7 We also identified the N-(1-adamantyl)-4,6-dimethylindole-2-carboxamide (2) as potent anti-TB agent (MIC = 0.012 μM) with no cytotoxicity to healthy cells (IC50 > 200 μM).19 Subsequent efforts, mainly directed towards non-tuberculous mycobacteria (NTM), identified the N-(1-adamantyl)-indole-2-carboxamide (3) and the N-rimantadine-4,6-dimethylindole-2-carboxamide (4) as highly potent anti-TB compounds with minimum inhibitory concentration (MIC) values of 0.68 and 0.88 μM, respectively (Fig. 1).20
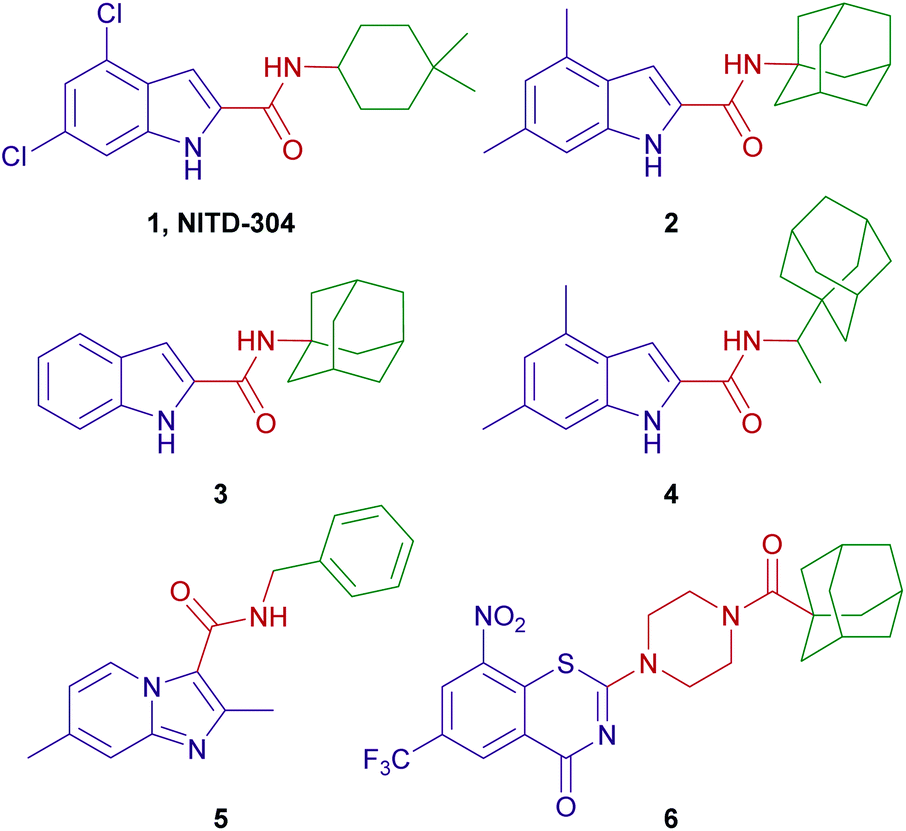 | ||
| Fig. 1 Potent anti-TB compounds reported in the literature. | ||
To date, several groups have reported the dual anti-TB and antineoplastic activities for a variety of scaffolds, including indole-2-carboxamides.21–25 Our group has also demonstrated the selective cannabinoid receptor 2 (CB2) agonistic activity of many indole-2-carboxamide derivatives, for instance compound 3 (EC50 = 0.98 μM).26 Interestingly, in the same year, Franz et al. reported the potent anti-TB activity of compound 3 (MIC = 0.68 μM).20 Similarly, rimonabant, a cannabinoid receptor 1 (CB1) inverse agonist, was found to inhibit the growth of M. tb via targeting MmpL3.9 The aforementioned two examples demonstrate the polypharmacological profile of various small molecules, a phenomenon that has been harnessed for drug repurposing, whereby new indications of existing agents/drugs can be identified.27,28 This is well exemplified by the current repositioning of drugs in the COVID-19 era.29 Towards this end, we were interested in exploring the antitumour activity that might be harboured in compound 3. We were particularly drawn into the paediatric brain tumours that are categorised as the highest-grade (grade IV) tumours according to the World Health Organisation (WHO). Paediatric gliomas represent the most common type of brain tumour and the leading cause of cancer-related death in children.30,31 Glioblastoma multiforme (GBM) is a grade IV glioma that originate from the supportive cells of the brain and the spinal cord, known as the glial cells.32–34 GBMs are among the most malignant, invasive and aggressive primary brain tumours which are usually refractory to current treatment, wherefore they remain largely incurable and are nearly universally fatal.35,36 The mainstays of GBM treatment include gross total resection when feasible followed by focal radiotherapy combined with chemotherapy.37 Many chemotherapeutic agents have been tested including temozolomide which is used in the treatment of adult GBM, but no clear benefit or remarkable improvement on survival has been achieved yet in paediatric GBM, likely due to its non-selective alkylating actions.38 The dismal prognosis of GBM necessitates the discovery and development of new, more effective therapies for these tumours.
In the present study, we describe the design, synthesis, and in vitro biological evaluation of novel indole-2-carboxamide analogues as antitubercular agents. Upon assessing the drug-like properties in silico using ACD Labs/Percepta, the designed compounds were predicted to cross the blood brain barrier (BBB). Accordingly, all synthesised compounds as well as compound 3 were also screened for their cytotoxic and antiproliferative activities against the KNS42 tumor cell line (paediatric GBM). We subsequently performed a transcriptional analysis on the KNS42 cells treated with one of the most potent compounds in order to determine the potential mechanism of action of our indoleamide analogues. The top two potent compounds were then subjected to further evaluations against a panel of non-GBM high-grade paediatric brain tumor cell lines as well as non-tumor cells as control.
2. Results and discussion
2.1. Chemistry
The design of final compounds 8a–h, 9a,b, 11a,b, 15 and 20 is depicted in Fig. 2. The indoleamides 8a–h and 9a,b were prepared in a one-pot amide coupling reaction as outlined in Scheme 1. First, since the N-rimantadine-indoleamide skeleton proved to have potent anti-TB activity,20 we were interested in exploring the effect of different substitutions at various positions on the indole ring on the anti-TB activity. Towards this, commercially available indole-2-carboxylic acids 7a–h were coupled with rimantadine hydrochloride. The amide coupling was carried out in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl), hydroxybenzotriazole hydrate (HOBt) and N,N-diisopropylethylamine (DIPEA) to deliver the requisite rimantadine-derived indoles 8a–h. Compound 8a was previously prepared using N,N′-dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) coupling reagents in a poor yield (31%).20 The other N-rimantadine analogue 4 prepared in the same report was also obtained in a similar low yield (29%).20 However, upon employing EDC·HCl and HOBt coupling conditions in the current study, the N-rimantadine indoleamide derivatives were generated in significantly higher yields (66–99%).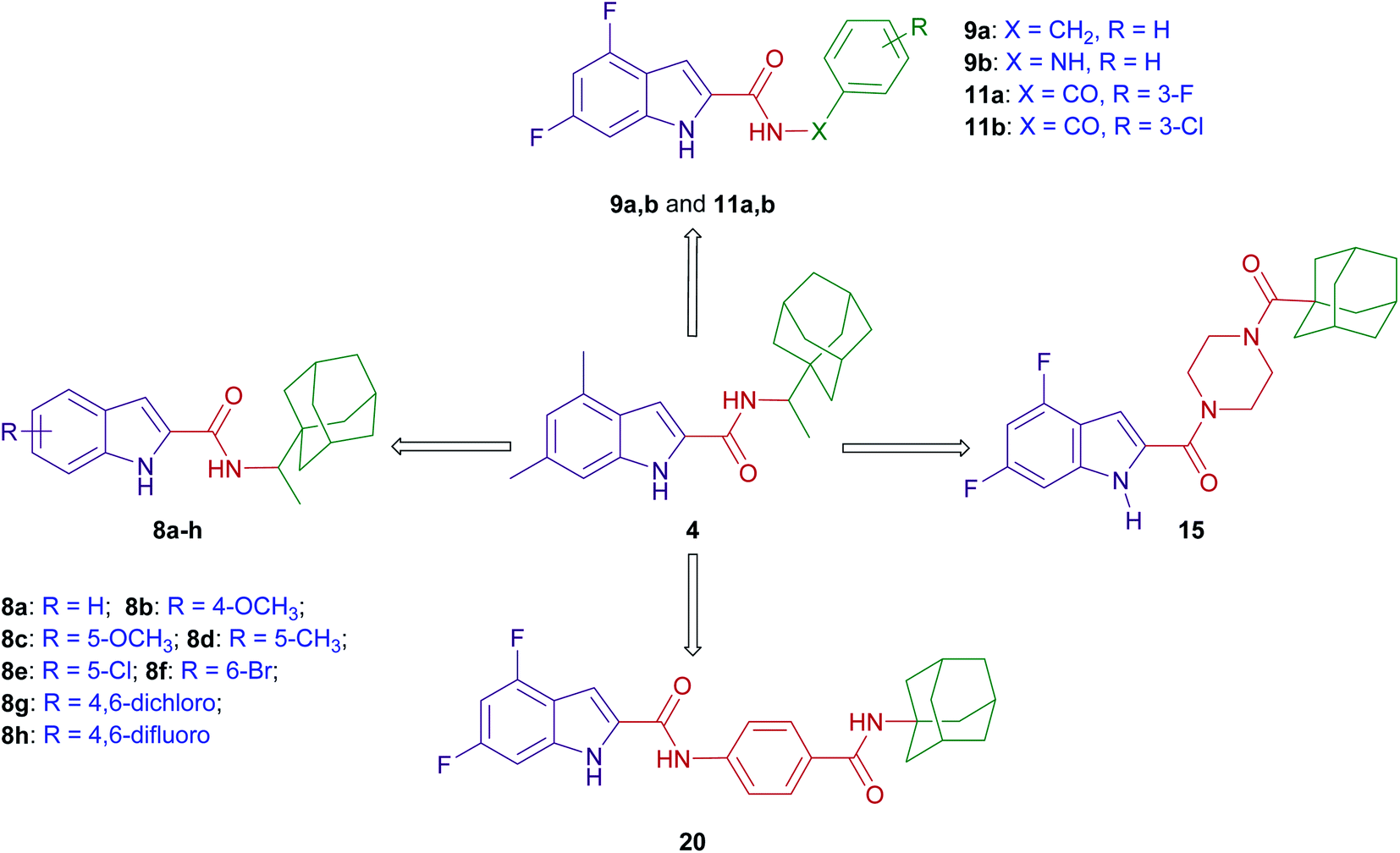 | ||
| Fig. 2 A diagram summarising the design strategy of the target indoleamides. | ||
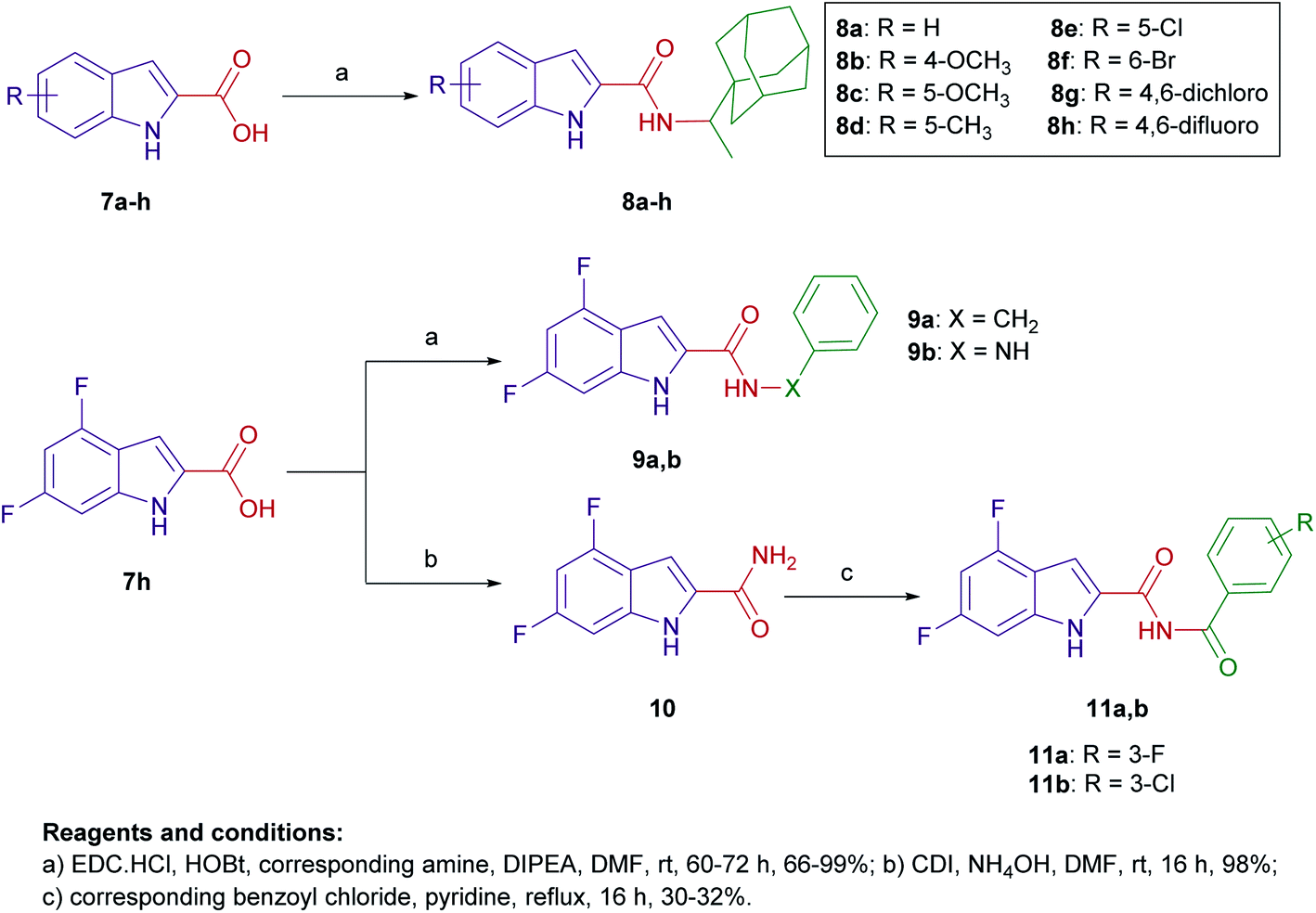 | ||
| Scheme 1 General synthetic routes for compounds 8a–h, 9a,b, and 11a,b. | ||
Likewise, amide coupling of 4,6-difluoroindole-2-carboxylic acid 7h with benzylamine or phenylhydrazine rendered amides 9a,b in yields 98% and 93%, respectively. We used fragment-based drug design in compound 9a in which we incorporated the benzyl group from the previously reported anti-TB compound 5 (ref. 39) into the indoleamide scaffold (Fig. 1 and 2). In Scheme 1, we also reacted the 4,6-difluoroindole 7h with 1,1′-carbonyldiimidazole (CDI) and the resulting N-acylimidazole intermediate was treated in situ with ammonia (nucleophilic substitution reaction) to yield the amide 10. The final imide derivatives 11a,b were then formed via reacting 10 with either 3-fluorobenzoyl chloride or 3-chlorobenzoyl chloride, respectively, in pyridine.
Similar to 9a,b and 11a,b, we further probed the effect of extending the linker tethering the indole scaffold and the adamantane ring by introducing a carbonyl piperazine fragment as seen in the reported anti-TB derivative 6 (Fig. 1 and 2).40 The diamides 15 and 20 were synthesised following the protocol delineated in Scheme 2. The synthesis of compound 15 was accomplished in three steps starting from coupling N-(tert-butoxycarbonyl) piperazine (N-Boc piperazine, 12) with the carboxylic acid 7h to give the amide analogue 13. Subsequent N-Boc deprotection using trifluoroacetic acid (TFA) yielded the key indoleamide analogue 14 which was then converted to the final diamide 15 through a standard EDC-mediated coupling with 1-adamantanecarboxylic acid. On the other hand, compound 20, entailing a phenyl group as a linker in place of piperazine, was obtained in a four-step pathway. The amino group of the aniline 16 was initially protected using di-tert-butyl dicarbonate (Boc)2O to form the N-Boc derivative 17. An amide linkage was formed thereafter connecting both 17 with the adamantane moiety, providing the crude N-Boc intermediate 18. Next, cleaving the Boc group under acidic conditions (TFA) furnished the penultimate amidoadamantyl compound 19. Finally, amide coupling of 19 with the carboxylic acid 7h generated the desired dicarboxamide derivative 20.
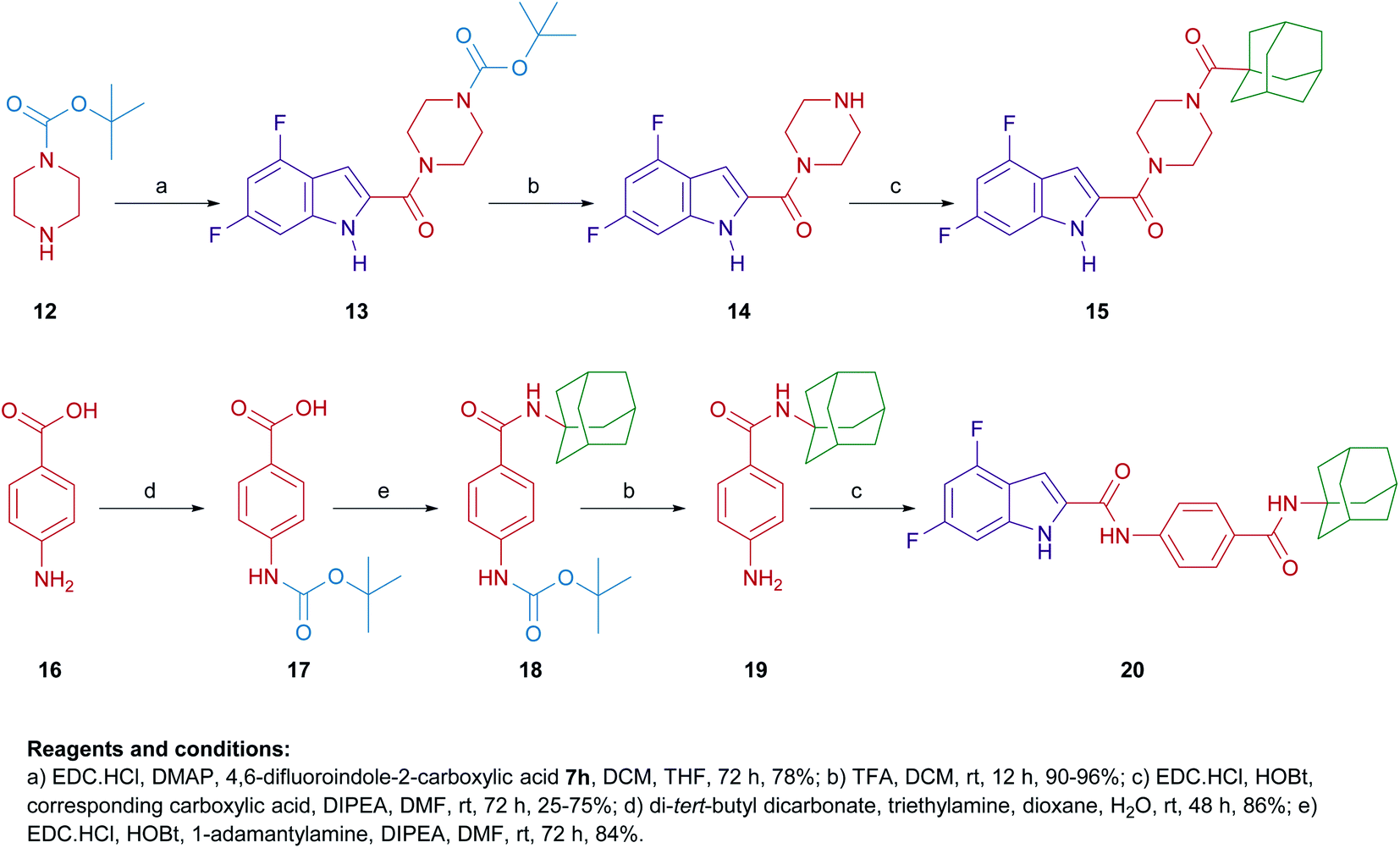 | ||
| Scheme 2 General synthetic routes for compounds 15 and 20. | ||
2.2. Antitubercular activity
Target compounds 8a–h, 9a,b, 11a,b, 14, 15 and 20 were screened in vitro against the wild-type M. tb strain (H37Rv) to obtain their respective MIC values (Table 1). Compounds 3 and 4 together with the two front-line anti-TB drugs, isoniazid (INH) and ethambutol (EMB) were used as positive controls. Franz et al. assessed the activity of both N-(1-adamantyl)-indole-2-carboxamide (3) and the N-rimantadine-4,6-dimethylindole-2-carboxamide analogue (4) against several mycobacterial strains, including H37Rv M. tb strain (MIC = 0.68 and 0.88 μM, respectively).20 Within this context, we previously conducted a thorough antimycobacterial screening for several indole-2-carboxamide derivatives.6,19,41–43 We found that the methyl groups located at the indole moiety in compound 2 present a potential metabolic liability (susceptible to metabolic oxidation). Accordingly, in order to explore the effects of various substituents on the indole ring, we synthesised several indoleamide derivatives 8a–h bearing a rimantadine substituent in place of the adamantane ring. All of the tested N-rimantadine indoleamides had single digit micro- and submicro-molar MIC values, with compounds 8f–h exhibiting potencies (MIC = 0.32–0.70 μM) higher than compound 4.
|
|||||||
|---|---|---|---|---|---|---|---|
| Cpd | R | X | H37Rv, MICa (μg mL−1) | H37Rv, MICb (μM) | KNS42 viability, IC50c (μM) | KNS42 prolif., IC50d (μM) | Clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Pe Pe |
| a The lowest concentration of drug causing at least 90% reduction of bacterial growth by the microplate alamarBlue assay (MABA). The reported MIC values are an average of three individual measurements, in μg mL−1.b The reported H37Rv MIC values converted to μM.c Compound dose required to achieve 50% inhibition of KNS42 cell viability, reflecting cytotoxicity.d Compound dose required to achieve 50% inhibition of KNS42 cell proliferation.e Calculated using ChemDraw 16.0.f Not determined. | |||||||
| 8a | H | — | 2 | 6.20 | >10 | >10 | 6.07 |
| 8b | 4-OCH3 | — | 1 | 2.84 | 4.17 | >10 | 6.09 |
| 8c | 5-OCH3 | — | 2 | 5.67 | 4.94 | >10 | 6.09 |
| 8d | 5-CH3 | — | 1 | 2.97 | >10 | >10 | 6.57 |
| 8e | 5-Cl | — | 1 | 2.80 | 3.13 | >10 | 6.89 |
| 8f | 6-Br | — | 0.25 | 0.62 | 0.84 | >10 | 7.04 |
| 8g | 4,6-Dichloro | — | 0.125 | 0.32 | >10 | >10 | 7.64 |
| 8h | 4,6-Difluoro | — | 0.25 | 0.70 | 5.66 | >10 | 6.50 |
| 9a | H | CH2 | >32 | >112 | 8.25 | 9.85 | 3.84 |
| 9b | H | NH | >32 | >111 | 4.45 | >10 | 2.42 |
| 11a | 3-F | CO | >32 | >101 | 2.16 | >10 | 4.49 |
| 11b | 3-Cl | CO | >32 | >96 | 6.65 | >10 | 5.06 |
| 14 | H | — | >32 | >121 | 1.34 | >10 | 1.54 |
| 15 | Adamantane-1-carbonyl | — | >32 | >75 | 5.04 | 6.62 | 3.80 |
| 20 | — | — | >32 | >71 | >10 | >10 | 5.29 |
| INH | — | 0.04 (ref. 6) | 0.29 | NDf | NDf | −0.67 | |
| EMB | — | 1 (ref. 6) | 4.89 | NDf | NDf | 0.12 | |
| 3 | — | 0.2 (ref. 20) | 0.68 (ref. 20) | 0.33 | >10 | 4.11 | |
| 4 | — | 0.31 (ref. 20) | 0.88 (ref. 20) | NDf | NDf | 7.07 | |
| 6 | — | — | — | 0.031 (ref. 40) | NDf | NDf | 4.25 |
| ICA38 | — | — | — | 0.003 (ref. 42) | NDf | NDf | 5.90 |
Upon re-evaluating the activity of the unsubstituted indole derivative 8a, it displayed an MIC value of 6.20 μM, which was lower than the published one (MIC > 15.5 μM (ref. 20)). The 4-methoxyindole analogue 8b showed a two-fold enhancement of activity compared to the 5-methoxy counterpart 8c (MIC = 2.84 and 5.67 μM, respectively). To further probe the effect of monosubstitution at position 5, more lipophilic methyl and chloro groups were installed. The observed activities of compounds 8d,e (MIC = 2.97 and 2.80 μM, respectively) were 2-fold higher than the respective 5-methoxy analogue 8c. This could be attributed to the higher lipophilicity of 8d,e (ClogP = 6.57 and 6.89, respectively) compared to 8c (ClogP = 6.09). These results suggest the lipophilicity-driven bioactivity of this series. Nevertheless, the fact that the 5-substituted indoles 8d,e are equipotent to the 4-substituted analogue 8b, despite the lower lipophilicity of the latter compared to the former derivatives, reflects the substitution preference for certain positions on the indole ring. Interestingly, the presence of a single bromo group at position 6 of the indole ring (8f: MIC = 0.62 μM) led to an approximately 10- and 4.5-fold increase in the activity in comparison to the unsubstituted and 5-halosubstituted indoles 8a,e, respectively.
Next, we replaced the two methyl groups in compound 4 with metabolically stable halogens 8g,h. It was pleasing to find that compound 8g possessing 4,6-dichloro substituents on the indole core exhibited nearly a three-fold rise in activity (MIC = 0.32 μM) relative to compound 4 and a comparable potency to INH (MIC = 0.88 and 0.29 μM, respectively). Of note is the fact that compound 8g is the most lipophilic compound in our study (ClogP = 7.64) that was translated into the highest activity as well which once again signifies the apparent influence of lipophilicity on the anti-TB activity. The 4,6-difluoro analogue 8h (MIC = 0.70 μM) was more active than 4 and 2-fold less potent than the 4,6-dichloroindole counterpart 8g. The consequential impact of the substitution pattern in the indole ring on activity, depicted earlier in compounds 8d,e versus 8b, was recapitulated in compound 8h versus 8f in which both compounds exhibited similar activities, although 8f is more lipophilic than 8h (ClogP = 7.04, 6.50, respectively). Importantly, the potency rendered by all the rimantadine analogues was comparable to or significantly higher than the first-line anti-TB drug EMB (MIC = 4.89 μM).
Similar to the other MmpL3 inhibitors, the inherent high lipophilicity of the foregoing series (ClogP = 6.07–7.64) likely endowed them with facilitated diffusion through the lipid-rich bilayer of M. tb where they presumably interacted with MmpL3 and elicited potent anti-TB activity. Nonetheless, it is worth noting that the anti-TB activities of the N-(rimantadine)-indole-2-carboxamides in our study are generally lower than the previously evaluated N-(1-adamantyl)-indole-2-carboxamide counterparts. Indeed, this was counterintuitive due to the high lipophilicity of the rimantadine derivatives compared to the corresponding 1-adamantane analogues.43 The SAR hallmarks of these analogues, in which substitutions at the 4- and/or 6-positions of the indole ring was optimal for activity, together with the apparent impact of lipophilicity on the anti-TB activity, were consistent with our previous findings.19,42,43 Therefore, in our subsequent evaluations in which we used the fragment-based drug design technique, 4,6-difluoroindole 7h was the scaffold of choice, to which we attached different fragments from previously reported anti-TB agents with diverse mechanism of actions 5 and 6 (Fig. 1).
First, Moraski et al. called attention to the remarkable anti-TB potency of the N-benzyl imidazo[1,2-a]pyridine-3-carboxamide 5 (MIC = 0.37–1.9 μM in three different media).39 This compound also maintained its excellent potency when tested against a panel of single DR strains in addition to several MDR and XDR clinical isolates.39 Imidazo[1,2-a]pyridines were previously found to exert their anti-TB activity via inhibiting M. tb cytochrome b subunit of the cytochrome bc1 complex (QcrB).44,45 We, thereupon, appended a benzyl moiety to the 4,6-difluoroindoleamide architecture. Disappointingly, the tested benzylamide analogue 9a was bereft of anti-TB activity (MIC > 112 μM). We also presumed that incorporating an NH or a carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group, instead of CH2, between the carboxamide moiety and the phenyl group would result in the formation of more hydrogen bonds with the hydrophilic residues in the S4 hydrophilic subsite of MmpL3,9 leading to an improved binding and better activity. As reported previously,46 when we introduced a CO group between the 4,6-difluoroindoleamide and the adamantane moiety, the obtained imide derivative displayed moderate anti-TB activity (MIC = 22 μM). Unfortunately, this NH or CO extension in compounds 9b and 11a,b, did not show any improved anti-TB activity (MIC > 96 μM).
O) group, instead of CH2, between the carboxamide moiety and the phenyl group would result in the formation of more hydrogen bonds with the hydrophilic residues in the S4 hydrophilic subsite of MmpL3,9 leading to an improved binding and better activity. As reported previously,46 when we introduced a CO group between the 4,6-difluoroindoleamide and the adamantane moiety, the obtained imide derivative displayed moderate anti-TB activity (MIC = 22 μM). Unfortunately, this NH or CO extension in compounds 9b and 11a,b, did not show any improved anti-TB activity (MIC > 96 μM).
It is noteworthy that the estimated lipophilicities of compounds 11a,b (ClogP = 4.49 and 5.06, respectively) are higher than that of 9a,b (ClogP = 3.84 and 2.42, respectively) which did not seemingly impact the anti-TB activity. Of interest is the fact that two N-phenyl indole-2-carboxamide analogues exhibited good anti-TB activities in our previous work (MIC = 1.7 and 3.8 μM).19 This in turn suggests that introducing an extra spacer to the amide linker tethering the indole ring and the phenyl moiety is unfavourable. In the same report, on the contrary, we found that adding a methylene spacer between the amide nitrogen and a cyclohexyl ring elicited almost the same high potency as the desmethylene analogue (MIC = 0.88 and 0.93 μM, respectively).19 Evidently, the potent anti-TB activity manifested by the N-rimantadine indoleamide analogues 8a–h in the present study corroborated the notion that incorporating an extra spacer next to the cycloaliphatic motifs is tolerated.
On the other hand, the nitrobenzothiazinone derivative 6, bearing a carbonyl piperazine moiety (Fig. 1), was reported to have a potent anti-TB activity (MIC = 0.031 μM (ref. 40)). The 1,3-benzothiazin-4-ones (BTZs) are a potent class of antimycobacterial agents that kill M. tb in vitro, ex vivo and in TB mouse models via targeting decaprenylphosphoryl-β-D-ribose 2′-epimerase (DprE1) enzyme. Inhibition of the DprE1 enzymatic activity compromises the formation of a key precursor in the synthesis of cell wall arabinans, namely decaprenylphosphoryl arabinose, resulting in cell lysis and bacterial death.47 Introducing a 4-carbonyl piperazine segment into the BTZ framework was well tolerated and led to compounds with enhanced aqueous solubility compared to other BTZ analogues in earlier reports.40 This prompted us to integrate the carbonyl piperazine fragment into the N-(adamantyl)indoleamide structure core to verify its tolerability within this template. Initially, we examined the activity of the piperazinylindole intermediate 14 which displayed an MIC value > 121 μM. This dramatic loss in activity could be ascribed to its diminished lipophilicity (ClogP = 1.54) and the absence of the amide NH which is a crucial element for the hydrogen bonding with Asp645 in MmpL3.9 The NH necessity was further emphasised when we increased the lipophilicity of 14 via attaching an adamantane ring, furnishing the 1,4-dicarbonyl piperazine 15 which was devoid of activity (MIC > 75 μM) despite having a ClogP value of 3.80 that is higher than that of 14.
A similar trend was observed in the analogous dicarboxamide 20, in which a phenyl motif, in lieu of piperazine, was introduced in between the indole and adamantane rings. Despite its higher lipophilicity (ClogP = 5.29) and that the amide NH was preserved, compound 20 was inactive (MIC > 71 μM). Hence, it could be surmised that stretching the linker between the indole scaffold and the adamantane moiety via ring insertion is detrimental to the anti-TB activity, suggesting some steric requirements of the S4 subsite of MmpL3. The abolition of anti-TB activity pertaining to overextending the middle linker resonates with our previous findings.46 Overall, whilst a positive correlation between lipophilicity and anti-TB activity was discernible in compounds 8a–h, the rest of the tested analogues did not conform with this trend.
Motivated by the potent in vitro anti-TB activities of the rimantadine-derived indole derivatives 8a–h, two compounds 8f and 8g (MIC = 0.62, 0.32 μM, respectively) were selected for further cytotoxicity assessment and their selectivity index (SI) were subsequently calculated (Table 2). We were pleased to find that both compounds displayed high IC50 values against Vero cells (IC50 = 39.9 and 40.9 μM, respectively). These values correspond to SI values of 64 and 128, respectively, indicating minimal primary cytotoxicity of these derivatives against healthy mammalian cells.
| Cpd | MIC (μM) | IC50a (μM) | SIb | Docking scorec (kcal mol−1) | No. of H-bonds | Distance | Amino acids | Ligand atoms |
|---|---|---|---|---|---|---|---|---|
| a Cytotoxicity against Vero cells expressed as the half maximal inhibitory concentration of the drug by MABA.b Selectivity index (SI) = IC50(Vero)/MIC(H37Rv).c The lowest binding energy corresponding to the top-ranked pose.d Not determined. | ||||||||
| 8f | 0.62 | 39.9 | 64 | −13.9 | 2 | 2.51 | Asp645 | Amide NH |
| 2.66 | Asp645 | Indole NH | ||||||
| 8g | 0.32 | 40.9 | 128 | −14.4 | 2 | 2.55 | Asp645 | Amide NH |
| 2.62 | Asp645 | Indole NH | ||||||
| ICA38 | 0.003 (ref. 42) | NDd | NDd | −22.7 | 1 | 2.54 | Asp645 | Amide NH |
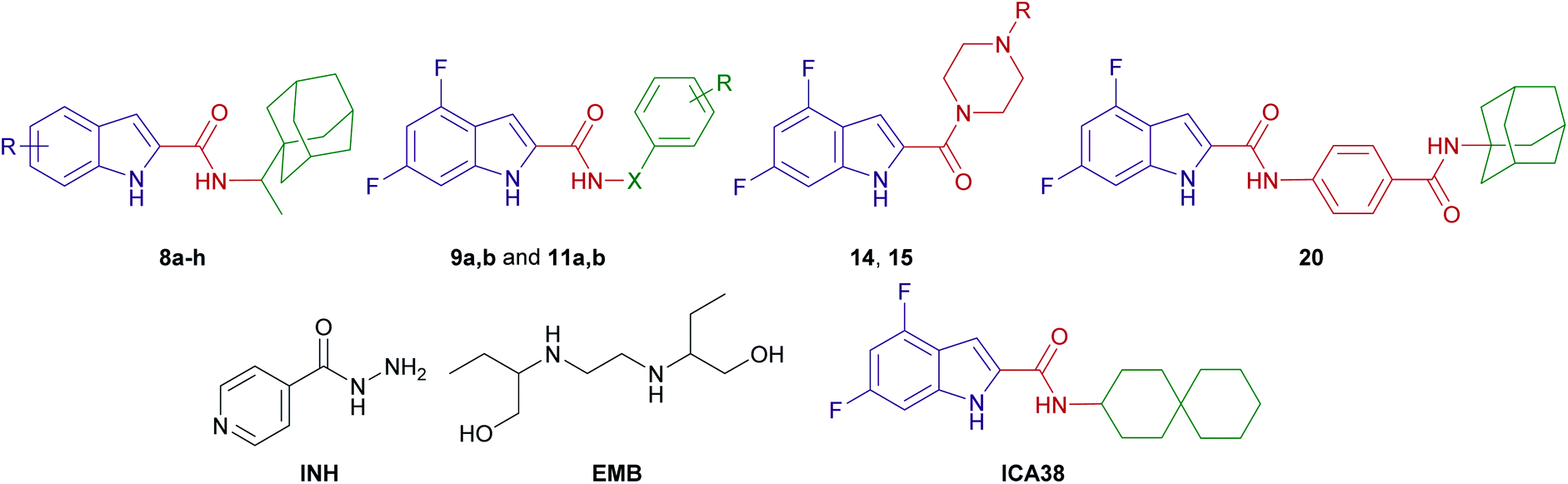
Taking into consideration that MmpL3 is the most relevant potential enzymatic target of our analogues, molecular docking simulations were carried out to evaluate the possible binding mechanism of the rimantadine-based indoles within the MmpL3 active site (Table 2; Fig. 3 and 4). First, the co-crystallised ligand ICA38 was re-docked into the MmpL3 binding site to validate the docking protocol as we reported before (Fig. 3).46 In consonance with the previous findings,9 the indole moiety and the spirocarbocyclic group of ICA38 were both lodged in the bulky S3 and S5 hydrophobic subsites, respectively, forming hydrophobic contacts with the surrounding residues. The amide linker was positioned in the S4 hydrophilic subsite, wherein the amide NH of ICA38 formed a hydrogen bond with Asp645. This Asp645 is a crucial part of the two Asp–Tyr pairs implicated in proton relay. Upon binding, ICA38 inhibits the MmpL3 by occupying S3–S5 subsites in the proton transportation channel, disrupting the key components of the S4 subsite, namely the two Asp–Tyr pairs (Fig. 3), thereby blocking the PMF for substrate translocation.9
 | ||
| Fig. 3 Re-docking of ICA38 (Cyan) in the MmpL3 active site (A), showing the S3–S5 subsites. The ICA38 top pose was oriented almost at the same position as the original ICA38 co-crystallised ligand (purple). The two key Asp–Tyr pairs, implicated in proton relay, located in the S4 subsite are marked in orange hashed ovals. The putative binding interactions of ICA38 with the MmpL3 binding pocked is represented in 2D on the right panel (B). | ||
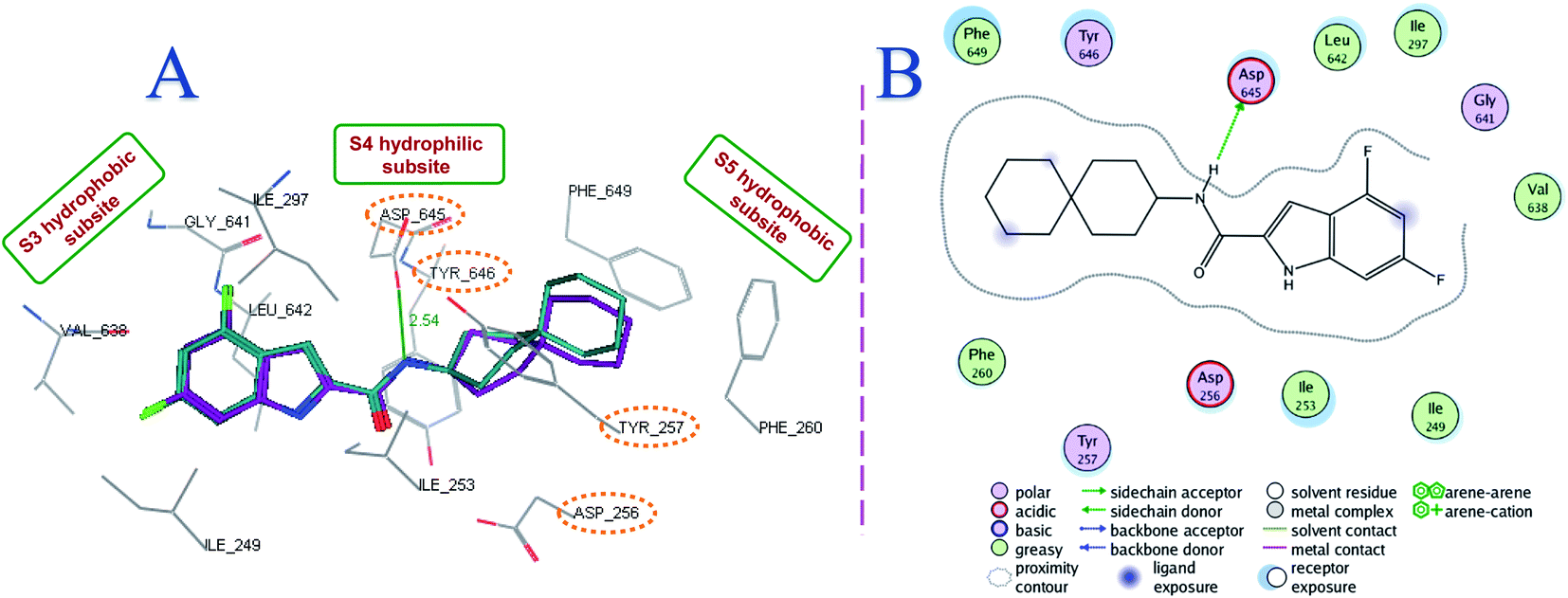
 | ||
| Fig. 4 Superposition of the top ranked docking pose of 8f (A) and 8g (B) (brown) and the co-crystallised ligand ICA38 (purple), showing the putative binding mode of both compounds in the MmpL3 active site. | ||
Both compounds 8f and 8g, typifying the rimantadine class, were oriented inside the MmpL3 binding pocket in a manner resembling ICA38 (Fig. 4). The most favourable docking pose (i.e. lowest binding energy) retained the key interactions observed in ICA38. The S3 hydrophobic subsite accommodated the indole nucleus, while the rimantadine moiety was embedded in the bulky S5 hydrophobic subsite, overlapping with the spirocyclic group of ICA38. Meanwhile, the amide NH in 8f and 8g was inserted in the S4 hydrophilic subsite, forming a hydrogen bond with Asp645 (distance = 2.51 and 2.55 Å, respectively). An additional hydrogen bond was formed between the indole NH of 8f and 8g with Asp645 (distance = 2.66 and 2.62 Å, respectively). They also showed high binding affinity to the MmpL3 active site (docking score = −13.9 and −14.4 kcal mol−1, respectively). The similarities between the binding modes of 8f and 8g and the MmpL3 inhibitor ICA38 suggest that this potent class of N-rimantadine indoleamides likely inhibit the same target via disrupting the two Asp–Tyr pairs that play a pivotal role in the proton translocation. It is worth mentioning that a recent study used a combination of in vitro and whole-cell-based approaches to determine whether a structurally distinct panel of MmpL3 inhibitors directly target the MmpL3 or dissipate the PMF from which this transporter derives its energy (indirect mechanism). They provided evidence that all compounds, including the indole-2-carboxamides, directly inhibit MmpL3, whether or not they exert an additional impact on the PMF that potentiate their activity.18
2.3. Cytotoxic and antiproliferative activity against paediatric GBM cells
All synthesised final compounds in addition to reference compound 3 were initially evaluated for their cytotoxicity and antiproliferative activities against paediatric KNS42 GBM cell line (Table 1). Compound 3, which previously showed potent antitubercular activity against M. tb H37Rv strain (MIC = 0.68 μM (ref. 20)), displayed potent cytotoxicity against KNS42 cells with sub-micromolar inhibitory activity on cell viability (IC50 = 0.33 μM). The cytotoxicity of compound 3 was previously evaluated against human THP-1 cells in which it was non-cytotoxic up to 50 μM.20 Similarly, compound 8f demonstrated excellent dual antitubercular and antitumour activity [MIC (H37Rv) = 0.62 μM, IC50 (KNS42 viability) = 0.84 μM]. Importantly, compound 8f also exhibited minimal cytotoxicity against healthy mammalian Vero cells with an IC50 value of 39.9 μM (Table 2). Nonetheless, both 8f and reference compound 3 showed weak antiproliferative activities against KNS42 (IC50 > 10 μM). Compounds 8b, 8c, 8e and 8h showed potent antitubercular activity and reduction of KNS42 cell viability [MIC (M. tb) and IC50 (KNS42) < 6 μM].On the other hand, compound 9a, which was bereft of antitubercular activity, displayed good inhibitory activities against KNS42 cells in the viability and proliferation assays with IC50 values of 8.25 and 9.85 μM, respectively. Similarly, compound 15 exhibited potent cytotoxic and antiproliferative activity against KNS42 cells (IC50 = 5.04 and 6.62 μM, respectively). Additionally, compounds 9b, 11a, 11b and 14 displayed high cytotoxic activities (IC50 = 1.34–6.65 μM) and poor antiproliferative activity (IC50 > 10 μM) against KNS42 cells.
2.4. Transcriptional analysis of KNS42 cells and compound 15-induced alterations to gene expression
To shed light on the potential mechanism of action of our indole-2-carboxamides as antitumour agents, we characterised the gene transcriptional response of the KNS42 cells upon exposure to compound 15, using DNBSEQ Eukaryotic Stranded Transcriptome Resequencing. Of note, compound 15 was chosen in this respect due to its potent activity against KNS42 cells in both viability and proliferation assays (IC50 = 5.04 and 6.62 μM, respectively). When we analysed the differential expression of the genes, in comparison to the control cells (untreated), we found that compound 15 downregulated the expression of 9 genes (fold change ≥ 8) with statistical significance (P < 0.05). Two genes, namely the carbonic anhydrase 9 (CA9) and the spleen tyrosine kinase (SYK) stood out as potential targets of this compound, whereby it exerts its antitumour activity. Several reports previously showed that the expression levels of CA9 and SYK in GBM tumours are positively correlated to tumour growth/progression and negatively correlated to survival rates.48–50 Therefore, downregulating the expression of these two genes could be conducive to the antitumour effects observed in compound 15.First, we found that the expression level of CA9 was downregulated by 15-fold (p < 0.01) upon treating the KNS42 cells with compound 15. CA9 is a hypoxia-responsive gene which is used as a hypoxia marker and a regulator of the pH of tumour cells.51 Hypoxia is a common feature of the majority of malignant tumours, in which extensively expanding and proliferating tumour tissues outgrow their blood supply resulting in hampered oxygen diffusion and hypoxic milieus.52 CA9 expression is strongly induced by hypoxia, therefore it can serve as a marker that signifies aggressive rapidly proliferating tumours.50 Hence, CA9 expression was found to be correlated to tumour progression in various human cancers.53 Importantly, strong CA9 immunoreactivity was previously detected in HGGs, including GBM, whereas the normal brain cells showed no expression of CA9.53 This tumour-exclusive expression pattern establishes CA9 as a feasible antitumour target. Indeed, Proescholdt et al. found that CA9 is expressed in the GBM tumours in all of the patients they investigated, whereby the expression levels were generally correlated with the overall prognosis.50 Their survival analysis revealed that high expression levels of CA9 was associated with poor survival in GBM patients (15 months) compared to GBM patients with low CA9 expression (34 months). Therefore, high CA9 expression is considered a prognostic marker for poor survival in GBM patients.48,50 Selective gene silencing of CA9 in GBM cells resulted in a profound reduction of cell attachment and invasion as well as a strong enhanced susceptibility to adjuvant chemotherapy and radiation treatment.50 This was substantiated by Boyd et al.'s recent findings, in which concurrently using a CA9 inhibitor with temozolomide inhibited the growth of GBM cells and induced cell cycle arrest in vitro.51 This combination was also efficacious in extending the survival of GBM-bearing mice. Taken together, the significant downregulation of CA9 gene in KNS42 cells induced by compound 15 suggest the involvement of CA9 in the cytotoxic and antiproliferative activities observed in this compound.
On the other hand, compound 15 also repressed the expression of the SYK by 9-fold with statistical significance (p < 0.05). SYK is a non-receptor protein tyrosine kinase which is an essential component of the signalling machinery in the immune system.54 It is mainly expressed in hematopoietic cells, including macrophages, B cells, neutrophils, monocytes, and natural killer cells.55 SYK plays a key role in the oncogenesis and tumour promotion of various cancers, including gliomas.55 Indeed, Moncayo and colleagues have recently found that SYK is overexpressed in the malignant gliomas, including GBM.49 Their in vitro experiments showed that inhibiting or knocking down SYK activity resulted in blocking the proliferation and migration of GBM cell lines. Importantly, these findings were mirrored in vivo in nude mice injected with human GBM cells overexpressing SYK.49 In this respect, SYK inhibition reduced GBM cell proliferation and invasiveness in vivo. In addition, blocking the activity of SYK genetically or by inhibitors in tumour cells prolonged the life of treated mice and increased their survival.49 Of note, our indoleamide derivative 15, which significantly downregulated the expression of SYK protein, displayed the most potent antiproliferative activity in our set of analogues. This is in fact consistent with the Moncayo et al. findings. Overall, the evident downregulation of CA9 and SYK expression induced by compound 15 suggest that our observed antitumour effects could be correlated to modulating the activity of these two targets.
2.5. Cytotoxic and antiproliferative activities of the top potent compounds against different non-GBM paediatric brain tumour cells and non-neoplastic fibroblasts
Both compounds 9a and 15 were selected for further in vitro viability and proliferation inhibition studies against different grade IV paediatric brain tumour cell lines and non-neoplastic mammalian cells (Table 3). Both compounds retained their potent cytotoxic activities against two atypical teratoid/rhabdoid tumour (AT/RT) cell lines BT12 and BT16. Remarkably, compound 9a showed approximately 9- and 5-fold higher cytotoxic activity, respectively, when tested against the two forenamed cell lines compared to KNS42 [IC50 = 0.89 (BT12), 1.81 (BT16) and 8.25 (KNS42) μM]. Compound 9a also exhibited high antiproliferative activities against BT12 and BT16 cells [IC50 = 7.44 and 6.06 μM, respectively]. Despite the potent antiproliferative activity of compound 15 against KNS42, this activity was not maintained against the two tested AT/RT cell lines (IC50 > 10 μM). On the other hand, both compounds demonstrated a drop in both cytotoxic and antiproliferative activities when tested against the medulloblastoma cell line DAOY (IC50 > 10 μM). To our delight, compound 9a displayed negligible cytotoxicity when tested against the non-neoplastic human fibroblast cell line HFF1 (IC50 = 119 μM), suggesting its selective cytotoxicity with an SI value up to 134 against tumour cells. Compound 15 displayed moderate cytotoxicity against HFF1 cells (IC50 = 19.35 μM), giving low SI values.| Cpd | BT12 viability, IC50 (μM) | BT12 prolif., IC50 (μM) | BT16 viability, IC50 (μM) | BT16 prolif., IC50 (μM) | DAOY viability, IC50 (μM) | DAOY prolif., IC50 (μM) | HFF1 viability, IC50 (μM) | HFF1 prolif., IC50 (μM) |
|---|---|---|---|---|---|---|---|---|
| 9a | 0.89 | 7.44 | 1.81 | 6.06 | >10 | >10 | 119 | 65 |
| 15 | 3.16 | >10 | 5.89 | >10 | >10 | >10 | 19.35 | 17.41 |
The physicochemical properties of compound 9a were predicted using ACD/Labs Percepta 2016 Build 2911 (13 Jul 2016). We evaluated the compliance of this compound with Lipinski's rule of five (RO5) in order to evaluate its drug-likeness.56 Compound 9a showed zero violations to the RO5, indicating the drug-like attributes of this compound, including its prospective in vivo bioavailability. Importantly, this compound is also expected to cross the BBB, whereupon it can elicit its antitumour activity. Overall, the aforementioned results establish compound 9a as a promising antitumour agent against various aggressive paediatric brain cancer cells, whilst being safe towards healthy cells.
3. Conclusions and future directions
In summary, we established a body of preliminary SAR for the N-rimantadine indoleamides 8a–h as anti-TB agents, based on compound 4 which was obtained from a previous screening20 against several mycobacterial strains. The SAR outcomes of this class were in harmony with our published findings,19,42,43 highlighting the significance of both the substitution pattern of the indole moiety and the lipophilic nature of the substituents. In this study, a subsequent massive drop in the anti-TB activity ensued from the structural modifications implemented in compounds 9a,b, 11a,b, 14, 15 and 20. Although lipophilicity is a key driver for the anti-TB efficacy in 8a–h, it is not the sole parameter influencing the potency in our study. The most active anti-TB compound identified in our study 8g exhibited limited cytotoxic activity against Vero cells. A docking analysis was performed on compounds 8g and 8f, exemplifying the binding mode of the N-rimantadine indoleamide series. The similarity between the binding fashion of both compounds and ICA38 in the same MmpL3 binding pocket support the high in vitro anti-TB potency of the amidorimantadine class. Since the indoleamide architecture was previously reported to have antitubercular and antitumour activities,22,23 all compounds in the present study were tested for their antitumour activity against the paediatric GBM cell line KNS42. Some of the tested compounds demonstrated potent anti-TB activity and cytotoxicity towards KNS42 cells. Compound 8f displayed the most potent dual activity while showing minimal cytotoxicity against non-neoplastic mammalian cells. Additionally, several compounds, which were devoid of anti-TB activity, showed potent cytotoxicity and/or antiproliferative activities against the KNS42 cell line.Differential expression analysis of the genes in the KNS42 cells in response to compound 15 versus untreated cells revealed that this compound repressed the expression of CA9 (p < 0.01) and SYK (p < 0.05) genes by 15-fold and 9-fold, respectively. The expression levels of these genes in GBM tumours were previously shown to be directly correlated to the tumour growth and progression and inversely correlated to survival rates.48–50 Indeed, knocking down the activity of each of these two genes was also found to inhibit the growth of GBM cells in vitro.49,51 Accordingly, the cytotoxic and antiproliferative effects of compound 15 could be attributed to downregulating the activity of these two genes. Two compounds 9a and 15, which potently reduced cell viability and proliferation of KNS42 cells, were further evaluated against three paediatric brain tumour cell lines and non-neoplastic human fibroblasts. Compound 9a stood out as the most promising antitumour agent, displaying excellent cytotoxic and antiproliferative activities against two AT/RT cell lines (BT12 and BT16) in addition to the GBM KNS42 cell line. The cytotoxicity of 9a was nullified against non-neoplastic human cells, suggesting its selective activity towards tumour cells. Overall, the high activity against brain tumour cells, negligible cytotoxicity towards non-neoplastic cells and in silico drug-like profile of the N-benzyl indoleamide 9a establish this compound as a potential therapeutic agent that merits further investigation.
These findings foreground the indoleamides as a new class of compounds that could be fine-tuned to modulate their dual anti-TB and antitumour activities. In addition, our gene expression results lay the foundation for future studies to identify more indoleamide analogues as antitumour agents that can modulate the expression of CA9 and SYK genes. Unlike the conventional target identification approaches which employ determining the activity of a small molecule at a receptor or enzyme level, the transcriptional approach has the advantage of identifying the compound's impact on the absolute concentration of a target protein.57 Future efforts will focus on confirming if the observed antitumour activity of the indoleamide scaffold in pediatric brain cancer cells is the direct consequence of inhibiting CA9 and/or SYK.
4. Experimental
4.1. Chemistry
N-(1-(Adamantan-1-yl)ethyl)-1H-indole-2-carboxamide (8a). The title compound was obtained from indole-2-carboxylic acid (7a) and rimantadine hydrochloride employing method A and its 1H NMR data matched the one reported in the literature.20 White solid, yield: 91%. 1H NMR (DMSO-d6) δ 11.50 (s, 1H), 7.86 (d, J = 9.5 Hz, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.42 (dd, J = 8.2, 0.6 Hz, 1H), 7.23 (d, J = 1.4 Hz, 1H), 7.19–7.13 (m, 1H), 7.05–6.99 (m, 1H), 3.83 (overlapping dq, J = 7.0 Hz, 1H), 1.94 (s, 3H), 1.72–1.44 (m, 12H), 1.08 (d, J = 7.1 Hz, 3H).
N-(1-(Adamantan-1-yl)ethyl)-4-methoxy-1H-indole-2-carboxamide (8b). The title compound was prepared from 4-methoxyindole-2-carboxylic acid (7b) and rimantadine hydrochloride employing method A. Yellow solid, yield: 95%. 1H NMR (DMSO-d6) δ 11.49 (s, 1H), 7.83 (d, J = 9.6 Hz, 1H), 7.33 (d, J = 1.6 Hz, 1H), 7.11–6.97 (m, 2H), 6.50 (d, J = 7.5 Hz, 1H), 3.88 (s, 3H), 3.81 (overlapping dq, J = 7.0 Hz, 1H), 1.94 (s, 3H), 1.69–1.47 (m, 12H), 1.06 (d, J = 6.9 Hz, 3H); 13C NMR (DMSO-d6) δ 160.9, 154.0, 138.2, 131.0, 124.6, 118.6, 105.8, 100.8, 99.6, 55.4, 52.6, 38.6, 37.2, 36.7, 28.3, 14.5; HRMS (ESI) m/z calcd for C22H28N2O2 ([M + H]+) m/z 353.2224; found 353.2217.
N-(1-(Adamantan-1-yl)ethyl)-5-methoxy-1H-indole-2-carboxamide (8c). 5-Methoxyindole-2-carboxylic acid (7c) and rimantadine hydrochloride were used to synthesise the title compound employing method A. White solid, yield: 99%. 1H NMR (DMSO-d6) δ 11.34 (s, 1H), 7.81 (d, J = 9.5 Hz, 1H), 7.31 (d, J = 8.9 Hz, 1H), 7.13 (d, J = 1.5 Hz, 1H), 7.06 (d, J = 2.3 Hz, 1H), 6.83 (dd, J = 8.9, 2.4 Hz, 1H), 3.83 (overlapping dq, J = 7.0 Hz, 1H), 3.76 (s, 3H), 1.94 (s, 3H), 1.72–1.48 (m, 12H), 1.08 (d, J = 7.0 Hz, 3H); 13C NMR (DMSO-d6) δ 161.0, 154.1, 132.8, 132.1, 127.9, 114.6, 113.5, 103.0, 102.4, 55.7, 52.6, 38.6, 37.1, 36.7, 28.3, 14.5; HRMS (ESI) m/z calcd for C22H28N2O2 ([M + H]+) m/z 353.2224; found 353.2223.
N-(1-(Adamantan-1-yl)ethyl)-5-methyl-1H-indole-2-carboxamide (8d). This compound was obtained from 5-methylindole-2-carboxylic acid (7d) and rimantadine hydrochloride according to method A. White solid, yield: 66%. 1H NMR (DMF-d7) δ 11.40 (s, 1H), 7.86 (d, J = 9.6 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.40 (d, J = 0.6 Hz, 1H), 7.27 (dd, J = 2.0, 0.6 Hz, 1H), 7.04 (dd, J = 8.4, 1.4 Hz, 1H), 3.94 (overlapping dq, J = 7.0 Hz, 1H), 2.38 (s, 3H), 1.93 (s, 3H), 1.75–1.53 (m, 12H), 1.12 (d, J = 7.0 Hz, 3H); 13C NMR (DMF-d7) δ 161.6, 136.0, 133.2, 129.2, 128.7, 125.7, 121.5, 112.6, 102.7, 53.2, 39.1, 37.6, 37.2, 29.1, 21.4, 14.4; HRMS (ESI) m/z calcd for C22H28N2O ([M + H]+) m/z 337.2274; found 337.2269.
N-(1-(Adamantan-1-yl)ethyl)-5-chloro-1H-indole-2-carboxamide (8e). The title compound was synthesised from 5-chloroindole-2-carboxylic acid (7e) and rimantadine hydrochloride employing method A. White solid, yield: 98%. 1H NMR (DMSO-d6) δ 11.68 (s, 1H), 7.97 (d, J = 9.5 Hz, 1H), 7.67 (d, J = 2.0 Hz, 1H), 7.43 (d, J = 8.7 Hz, 1H), 7.21 (d, J = 0.8 Hz, 1H), 7.16 (dd, J = 8.7, 2.1 Hz, 1H), 3.81 (overlapping dq, J = 7.0 Hz, 1H), 1.93 (s, 3H), 1.67–1.46 (m, 12H), 1.06 (d, J = 7.0 Hz, 3H); 13C NMR (DMSO-d6) δ 160.7, 135.2, 133.9, 128.6, 124.6, 123.7, 120.9, 114.3, 102.8, 52.8, 38.6, 37.1, 36.7, 28.3, 14.4; HRMS (ESI) m/z calcd for C21H25ClN2O ([M + H]+) m/z 357.1728; found 357.1732.
N-(1-(Adamantan-1-yl)ethyl)-6-bromo-1H-indole-2-carboxamide (8f). This compound was obtained from 6-bromoindole-2-carboxylic acid (7f) and rimantadine hydrochloride according to method A. White solid, yield: 91%. 1H NMR (DMSO-d6) δ 11.66 (s, 1H), 7.97 (d, J = 9.5 Hz, 1H), 7.63–7.52 (m, 2H), 7.28 (d, J = 1.4 Hz, 1H), 7.16 (dd, J = 8.6, 1.7 Hz, 1H), 3.83 (overlapping dq, J = 6.9 Hz, 1H), 1.94 (s, 3H), 1.75–1.38 (m, 12H), 1.08 (d, J = 7.0 Hz, 3H); 13C NMR (DMSO-d6) δ 160.7, 137.5, 133.3, 126.6, 123.8, 123.1, 116.3, 115.1, 103.3, 52.8, 38.6, 37.1, 36.7, 28.3, 14.4; HRMS (ESI) m/z calcd for C21H25BrN2O ([M + H]+) m/z 401.1223; found 401.1224.
N-(1-(Adamantan-1-yl)ethyl)-4,6-dichloro-1H-indole-2-carboxamide (8g). This compound was prepared from 4,6-dichloroindole-2-carboxylic acid (7g) and rimantadine hydrochloride employing method A. White solid, yield: 87%. 1H NMR (DMSO-d6) δ 12.01 (d, J = 1.7 Hz, 1H), 8.13 (d, J = 9.5 Hz, 1H), 7.45–7.39 (m, 2H), 7.19 (d, J = 1.7 Hz, 1H), 3.82 (overlapping dq, J = 6.9 Hz, 1H), 1.91 (s, 3H), 1.66–1.45 (m, 12H), 1.06 (d, J = 7.0 Hz, 3H); 13C NMR (DMSO-d6) δ 160.2, 137.2, 134.1, 128.0, 126.7, 125.3, 119.8, 111.5, 101.2, 52.9, 38.6, 37.1, 36.7, 28.3, 14.4; HRMS (ESI) m/z calcd for C21H24Cl2N2O ([M + H]+) m/z 391.1338; found 391.1339.
N-(1-(Adamantan-1-yl)ethyl)-4,6-difluoro-1H-indole-2-carboxamide (8h). 4,6-Difluoroindole-2-carboxylic acid (7h) and rimantadine hydrochloride were used to afford the title compound following method A. White solid, yield: 85%. 1H NMR (DMSO-d6) δ 11.93 (s, 1H), 7.96 (d, J = 9.5 Hz, 1H), 7.38 (s, 1H), 7.02 (dd, J = 9.4, 1.7 Hz, 1H), 6.86 (td, J = 10.4, 2.0 Hz, 1H), 3.82 (overlapping dq, J = 6.9 Hz, 1H), 1.94 (s, 3H), 1.76–1.45 (m, 12H), 1.07 (d, J = 7.0 Hz, 3H); 13C NMR (DMSO-d6) δ 160.3, 159.6 (dd, J = 238.1, 12.1 Hz), 156.2 (dd, J = 248.5, 15.5 Hz), 138.0 (dd, J = 15.2, 13.3 Hz), 133.4 (d, J = 3.2 Hz), 113.6 (d, J = 21.6 Hz), 98.9, 95.4 (dd, J = 29.6, 23.3 Hz), 95.0 (dd, J = 25.9, 4.3 Hz), 52.8, 38.6, 37.1, 36.7, 28.3, 14.4; HRMS (ESI) m/z calcd for C21H24F2N2O ([M + H]+) m/z 359.1929; found 359.1924.
N-Benzyl-4,6-difluoro-1H-indole-2-carboxamide (9a). This compound was obtained from 4,6-difluoroindole-2-carboxylic acid (7h) and benzylamine employing method A. White solid, yield: 98%. 1H NMR (DMSO-d6) δ 12.05 (s, 1H), 9.13 (t, J = 5.9 Hz, 1H), 7.38–7.28 (m, 5H), 7.28–7.20 (m, 1H), 7.04 (dd, J = 9.4, 1.3 Hz, 1H), 6.87 (td, J = 10.4, 1.9 Hz, 1H), 4.52 (d, J = 6.0 Hz, 2H); 13C NMR (DMSO-d6) δ 160.8, 159.7 (dd, J = 237.0, 12.0 Hz), 156.2 (dd, J = 248.7, 15.6 Hz), 139.8, 138.1 (dd, J = 15.2, 13.2 Hz), 133.1 (d, J = 3.3 Hz), 128.8, 127.7, 127.3, 113.6 (d, J = 21.7 Hz), 98.9, 95.7 (dd, J = 29.6, 23.3 Hz), 95.1 (dd, J = 25.9, 4.4 Hz), 42.7; HRMS (ESI) m/z calcd for C16H12F2N2O ([M + H]+) m/z 287.0987; found 287.0990.
4,6-Difluoro-N′-phenyl-1H-indole-2-carbohydrazide (9b). Compound 7h and phenylhydrazine were reacted to deliver the title compound following method A. White solid, yield: 93%. 1H NMR (DMSO-d6) δ 12.11 (s, 1H), 10.45 (d, J = 2.0 Hz, 1H), 7.99 (d, J = 1.7 Hz, 1H), 7.38 (s, 1H), 7.16 (dd, J = 8.4, 7.4 Hz, 2H), 7.04 (dd, J = 9.4, 1.4 Hz, 1H), 6.92 (td, J = 10.4, 2.0 Hz, 1H), 6.78 (d, J = 7.7 Hz, 2H), 6.73 (t, J = 7.3 Hz, 1H); 13C NMR (DMSO-d6) δ 161.1, 159.8 (dd, J = 237.4, 12.1 Hz), 156.2 (dd, J = 248.9, 15.6 Hz), 149.8, 138.3 (dd, J = 15.3, 13.0 Hz), 131.4 (d, J = 3.3 Hz), 129.3, 119.2, 113.6 (d, J = 21.7 Hz), 112.7, 99.2, 95.9 (dd, J = 29.6, 23.3 Hz), 95.1 (dd, J = 25.9, 4.3 Hz); HRMS (ESI) m/z calcd for C15H11F2N3O ([M + H]+) m/z 288.0943; found 288.0939.
4,6-Difluoro-1H-indole-2-carboxamide (10). The title compound was prepared following the procedure published in our previous report and the 1H NMR data matched the one therein.46 Buff solid, yield: 98%. 1H NMR (DMSO-d6) δ 11.97 (s, 1H), 8.03 (s, 1H), 7.44 (s, 1H), 7.22 (s, 1H), 7.01 (dd, J = 9.4, 1.9 Hz, 1H), 6.86 (td, J = 10.5, 2.0 Hz, 1H).
4,6-Difluoro-N-(3-fluorobenzoyl)-1H-indole-2-carboxamide (11a). The title compound was prepared from 7h and 3-fluorobenzoyl chloride employing method B. White solid, yield: 32%. 1H NMR (DMSO-d6) δ 12.31 (s, 1H), 11.31 (s, 1H), 7.79–7.69 (m, 3H), 7.64–7.55 (m, 1H), 7.54–7.46 (m, 1H), 7.07 (dd, J = 9.3, 1.3 Hz, 1H), 6.95 (td, J = 10.4, 2.0 Hz, 1H); 13C NMR (DMSO-d6) δ 166.6 (d, J = 2.6 Hz), 162.2 (d, J = 244.7 Hz), 160.6 (dd, J = 240.6, 12.1 Hz), 160.0, 156.7 (dd, J = 250.4, 15.7 Hz), 139.1 (dd, J = 15.4, 12.6 Hz), 136.8 (d, J = 7.1 Hz), 131.5 (d, J = 3.2 Hz), 131.0 (d, J = 8.0 Hz), 125.3 (d, J = 2.8 Hz), 119.8 (d, J = 21.2 Hz), 115.9 (d, J = 23.1 Hz), 113.5 (dd, J = 22.0, 0.5 Hz), 103.8, 96.3 (dd, J = 30.0, 23.1 Hz), 95.2 (dd, J = 26.0, 4.6 Hz); HRMS (ESI) m/z calcd for C16H9F3N2O2 ([M + H]+) m/z 319.0689; found 319.0686.
N-(3-Chlorobenzoyl)-4,6-difluoro-1H-indole-2-carboxamide (11b). This compound was synthesised from 7h and 3-chlorobenzoyl chloride following method B. White solid, yield: 30%. 1H NMR (DMSO-d6) δ 12.30 (s, 1H), 11.35 (s, 1H), 7.95 (s, 1H), 7.85 (d, J = 7.8 Hz, 1H), 7.72 (s, 1H), 7.70 (d, J = 1.0 Hz, 1H), 7.57 (t, J = 7.9 Hz, 1H), 7.07 (dd, J = 9.3, 1.2 Hz, 1H), 6.95 (td, J = 10.4, 1.8 Hz, 1H); 13C NMR (DMSO-d6) δ 166.7, 160.6 (dd, J = 240.5, 12.2 Hz), 160.1, 156.7 (dd, J = 250.4, 15.6 Hz), 139.1 (dd, J = 15.4, 12.6 Hz), 136.6, 133.5, 132.7, 131.5 (d, J = 2.5 Hz), 130.8, 128.8, 127.8, 113.5 (d, J = 21.9 Hz), 103.8, 96.3 (dd, J = 29.9, 23.1 Hz), 95.2 (dd, J = 25.9, 4.6 Hz); HRMS (ESI) m/z calcd for C16H9ClF2N2O2 ([M + H]+) m/z 335.0393; found 335.0393.
tert-Butyl 4-(4,6-difluoro-1H-indole-2-carbonyl)piperazine-1-carboxylate (13). A mixture of 4,6-difluoroindole 7h (1.5 mmol), N-Boc-piperazine 12 (1.8 mmol), EDC·HCl (1.8 mmol), and DMAP (1.8 mmol) in a 1
:1 mixture of tetrahydrofuran (THF) and DCM (10 mL each) was stirred at rt for 72 h. The reaction mixture was then quenched with saturated aqueous solution of ammonium chloride (50 mL) and extracted with DCM (3 × 25 mL) and ethyl acetate (3 × 25 mL). The combined organic layers were washed with brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The obtained residue was used for the next reaction without further purification; buff solid, yield: 78%.
(4,6-Difluoro-1H-indol-2-yl)(piperazin-1-yl)methanone (14). The crude product 13 was N-Boc deprotected employing method C to afford the title compound which was >95% pure after flash chromatography. Buff solid, yield: 90%. 1H NMR (DMSO-d6) δ 7.03 (dd, J = 9.4, 1.4 Hz, 1H), 6.77 (d, J = 0.6 Hz, 1H), 6.71 (td, J = 10.3, 2.0 Hz, 1H), 3.64 (s, 4H), 2.73 (s, 4H); 13C NMR (DMSO-d6) δ 161.5, 159.5 (dd, J = 238.3, 12.1 Hz), 156.0 (dd, J = 248.8, 15.5 Hz), 137.6 (dd, J = 15.2, 13.2 Hz), 131.5 (d, J = 3.3 Hz), 113.2 (d, J = 21.7 Hz), 99.9, 95.6 (dd, J = 29.6, 23.3 Hz), 94.8 (dd, J = 25.9, 4.4 Hz), 46.1 (br, 4C).
(Adamantan-1-yl)(4-(4,6-difluoro-1H-indole-2-carbonyl)piperazin-1-yl)methanone (15). This compound was prepared following method A entailing compound 14 and 1-adamantanecarboxylic acid. White solid, yield: 75%. 1H NMR (DMSO-d6) δ 12.02 (s, 1H), 7.04 (dd, J = 9.3, 1.3 Hz, 1H), 6.92 (d, J = 1.3 Hz, 1H), 6.88 (dd, J = 10.4, 1.6 Hz, 1H), 3.70 (s, 8H), 1.98 (s, 3H), 1.92 (s, 6H), 1.69 (q, J = 12.1 Hz, 6H); 13C NMR (DMSO-d6) δ 175.1, 161.6, 159.7 (dd, J = 238.5, 12.0 Hz), 156.1 (dd, J = 248.8, 15.4 Hz), 137.7 (dd, J = 15.2, 13.2 Hz), 131.2 (d, J = 3.3 Hz), 113.3 (d, J = 21.6 Hz), 100.5, 95.7 (dd, J = 29.6, 23.3 Hz), 94.9 (dd, J = 25.9, 4.3 Hz), 45.1 (br, 4C), 41.4, 38.8, 36.5, 28.4; HRMS (ESI) m/z calcd for C24H27F2N3O2 ([M + H]+) m/z 428.2131; found 428.2130.
4-((tert-Butoxycarbonyl)amino)benzoic acid (17). To a solution of 4-aminobenzoic acid (16, 3 mmol) in a 1
:2 mixture of water (10 mL) and dioxane (20 mL), di-tert-butyl dicarbonate (Boc2O, 6 mmol) and triethylamine (Et3N, 6 mmol) were added and the reaction mixture was stirred at rt for 48 h. Three quarters of the solvent were then evaporated in vacuo and the residue was acidified with 3 M aqueous HCL solution. The formed precipitate was filtered off, washed with water, and dried. The 1H NMR data was in agreement with the reported one.58 White solid, yield: 86%. 1H NMR (DMSO-d6) δ 9.71 (s, 1H), 7.83 (d, J = 8.5 Hz, 2H), 7.55 (d, J = 8.4 Hz, 2H), 1.48 (s, 9H).
tert-Butyl (4-((adamantan-1-yl)carbamoyl)phenyl)carbamate (18). Compound 17 and 1-adamantylamine were reacted according to method A and the residue, obtained after evaporating the EtOAc extract, was used in the next step without further purification; white solid, yield: 84%.
N-(1-Adamantyl)-4-aminobenzamide (19). The crude product 18 was N-Boc deprotected following method C to deliver the title compound. Brown solid, yield: 96%. 1H NMR (DMSO-d6) δ 7.51 (dd, J = 8.5, 1.8 Hz, 2H), 7.02 (s, 1H), 6.50 (dd, J = 8.5, 1.9 Hz, 2H), 5.50 (s, 2H), 2.03 (s, 9H), 1.64 (s, 6H); 13C NMR (DMSO-d6) δ 166.4, 151.7, 129.2, 123.0, 112.8, 51.5, 41.6, 36.6, 29.4.
N-(4-((Adamantan-1-yl)carbamoyl)phenyl)-4,6-difluoro-1H-indole-2-carboxamide (20). The indole 7h was coupled with 19 following method A to afford the title compound. White solid, yield: 25%. 1H NMR (DMSO-d6) δ 12.21 (s, 1H), 10.42 (s, 1H), 7.89–7.80 (m, 4H), 7.58 (d, J = 1.5 Hz, 1H), 7.49 (s, 1H), 7.08 (dd, J = 9.3, 1.4 Hz, 1H), 6.94 (td, J = 10.4, 2.0 Hz, 1H), 2.08 (s, 6H), 2.06 (s, 3H), 1.67 (s, 6H); 13C NMR (DMSO-d6) δ 165.9, 160.1 (dd, J = 239.4, 12.1 Hz), 159.5, 156.4 (dd, J = 249.2, 15.6 Hz), 141.4, 138.5 (dd, J = 15.3, 12.9 Hz), 132.7 (d, J = 3.3 Hz), 131.4, 128.6, 119.5, 113.6 (d, J = 21.8 Hz), 100.5, 96.0 (dd, J = 29.7, 23.2 Hz), 95.2 (dd, J = 25.9, 4.4 Hz), 51.9, 41.4, 36.6, 29.4; HRMS (ESI) m/z calcd for C26H25F2N3O2 ([M + H]+) m/z 450.1988; found 450.1987.
4.2. Biology
4.2.2.1. Cell culture. The four well-established paediatric brain tumour cell lines were all derived from humans and were used to assess the effects on proliferation and viability when treated with the indole-2-carboxamides. KNS42 (glioblastoma multiforme – GBM), BT-12 and BT-16 (atypical teratoid rhabdoid tumour – AT/RT) cell lines were gifts from Dr Hashizume, Northwestern University, whereas DAOY cells (medulloblastoma – MB) were obtained from ATCC. The human fibroblasts HFF1 (obtained from ATCC) were used as non-neoplastic controls. The KNS42, BT-12 and BT-16 cell lines were maintained in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum (FBS). The DAOY cell line was maintained in Eagle's minimal essential medium (EMEM) supplemented with 10% FBS. The HFF1 were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 15% FBS. All media contained 1% penicillin/streptomycin. The cells were incubated at 37 °C in 5% CO2.
4.2.2.2. Screening. After extensive optimization at various concentrations and time-points, the compounds were screened in the GBM cell line (KNS42) to assess the impact on proliferation and viability (described below). The cells were treated with 0.5, 1, 2.5, 5, 7.5 and 10 μM of each compound and assessed after 72 hours of treatment, whereupon their IC50 values were calculated. The top potent compounds with IC50 values <10 μM were further screened in multiple paediatric brain cancer cell lines, BT-12, BT-16 and DAOY in addition to the non-neoplastic cell line HFF1 to assess their impact on proliferation and viability (described below). The cells were treated with 0.5, 1, 2.5, 5, 7.5 and 10 μM of each compound and assessed after 72 h of treatment.
4.2.2.3. Cell proliferation assay. All cell lines were seeded in 96-well plates, at 2000 cells per well, except KNS42 which was seeded at 4000 cell per well. After incubating overnight in normal growth media, the media was aspirated off and 100 μl of treatment media was added. Media containing 0.1% DMSO was used as a control. After 72 h of incubation, the MTT proliferation assay reagents were added as previously described.61 The absorbance of the assay (570 nm/600 nm) was measured using the CLARIOstar (BMG Lab Tech, USA) and exported into excel, where the data was normalized by removing the background absorbance. The data was analysed and IC50 values were calculated using PRISM 8 (GraphPad, USA).
4.2.2.4. Cell viability assay. All cell lines were seeded at 2000 cells per well, except KNS42 which was seeded at 4000 cell per well, in 96-well plates. After an overnight incubation in normal growth media, the media was aspirated off and 100 μl of treatment media was added. 0.1% DMSO was used in the control media. After 72 h of incubation, the PrestoBlue Viability (ThermoFisher Scientific, USA) was added, per the manufacturer's instructions. The fluorescence of the assay (560 nm/590 nm) was measured using the CLARIOstar (BMG Lab Tech, USA) and exported into Excel, where the data was normalized by removing the background fluorescence. The data was analysed and IC50 values were calculated using PRISM 8 (GraphPad, USA).
4.2.2.5. Transcriptional analysis of KNS42 cells. The KNS42 cell line was maintained in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin and incubated at 37 °C in 5% CO2. The cells were treated with 10 μM of compound 15. The cells were washed with 1× PBS, scraped with a cell scraper and centrifuged to collect cell pellets after 72 hours of treatment. RNA was extracted from the cell pellets using TRIzol reagent (Thermo Fisher, USA) and following the protocol from Cold Spring Harbor Laboratory Press.62 The RNA pellet was resuspended in 50 μl of sterile diethylpyrocarbonate (DEPC) treated water. Concentration of the RNA was measured using the standard Qubit RNA Broad-Range Assay kit (Thermo Fisher, USA) according to the manufacturer's instructions. The RNA samples were submitted to BGI Americas for DNBSEQ Eukaryotic Stranded Transcriptome Resequencing.
4.3. Molecular modelling
In silico molecular docking analysis was undertaken using the Molecular Operating Environment MOE software version 2008.10 (Chemical Computing Group, Montreal, Canada). The crystal structure of MmpL3 in complex with ICA38 (6AJJ) was retrieved from the protein data bank (PDB) and the docking protocol was similar to our previous report.46Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
SSRA acknowledges Curtin University for the support through Curtin International Postgraduate Research Scholarship (CIPRS). WRB is grateful for the support of NIH grants AI 37856 and HL 133190. HG is thankful for the ARC Discovery Early Career Researcher Award DE160100482.References
- World Health Organization, Global Tuberculosis Report, Geneva, 2020, retrieved from https://apps.who.int/iris/bitstream/handle/10665/336069/9789240013131-eng.pdf Search PubMed.
- K. Dheda, C. E. Barry III and G. Maartens, Lancet, 2016, 387, 1211–1226 CrossRef.
- S. Keshavjee and P. E. Farmer, N. Engl. J. Med., 2012, 367, 931–936 CrossRef CAS PubMed.
- J. A. Caminero, G. Sotgiu, A. Zumla and G. B. Migliori, Lancet Infect. Dis., 2010, 10, 621–629 CrossRef CAS PubMed.
- N. Chim, R. Torres, Y. Liu, J. Capri, G. Batot, J. P. Whitelegge and C. W. Goulding, Chem. Biol., 2015, 22, 1098–1107 CrossRef CAS PubMed.
- S. Lun, H. Guo, O. K. Onajole, M. Pieroni, H. Gunosewoyo, G. Chen, S. K. Tipparaju, N. C. Ammerman, A. P. Kozikowski and W. R. Bishai, Nat. Commun., 2013, 4, 2907 CrossRef PubMed.
- S. P. Rao, S. B. Lakshminarayana, R. R. Kondreddi, M. Herve, L. R. Camacho, P. Bifani, S. K. Kalapala, J. Jiricek, N. L. Ma, B. H. Tan, S. H. Ng, M. Nanjundappa, S. Ravindran, P. G. Seah, P. Thayalan, S. H. Lim, B. H. Lee, A. Goh, W. S. Barnes, Z. Chen, K. Gagaring, A. K. Chatterjee, K. Pethe, K. Kuhen, J. Walker, G. Feng, S. Babu, L. Zhang, F. Blasco, D. Beer, M. Weaver, V. Dartois, R. Glynne, T. Dick, P. W. Smith, T. T. Diagana and U. H. Manjunatha, Sci. Transl. Med., 2013, 5, 214ra168 CrossRef PubMed.
- K. Tahlan, R. Wilson, D. B. Kastrinsky, K. Arora, V. Nair, E. Fischer, S. W. Barnes, J. R. Walker, D. Alland, C. E. Barry III and H. I. Boshoff, Antimicrob. Agents Chemother., 2012, 56, 1797–1809 CrossRef CAS PubMed.
- B. Zhang, J. Li, X. Yang, L. Wu, J. Zhang, Y. Yang, Y. Zhao, L. Zhang, X. Yang, X. Yang, X. Cheng, Z. Liu, B. Jiang, H. Jiang, L. W. Guddat, H. Yang and Z. Rao, Cell, 2019, 176, 636–648 CrossRef CAS PubMed.
- Z. Xu, V. A. Meshcheryakov, G. Poce and S. S. Chng, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7993–7998 CrossRef CAS PubMed.
- A. E. Grzegorzewicz, H. Pham, V. A. Gundi, M. S. Scherman, E. J. North, T. Hess, V. Jones, V. Gruppo, S. E. Born, J. Kordulakova, S. S. Chavadi, C. Morisseau, A. J. Lenaerts, R. E. Lee, M. R. McNeil and M. Jackson, Nat. Chem. Biol., 2012, 8, 334–341 CrossRef CAS PubMed.
- P. J. Brennan, Tuberculosis, 2003, 83, 91–97 CrossRef CAS PubMed.
- C. Hoffmann, A. Leis, M. Niederweis, J. M. Plitzko and H. Engelhardt, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3963–3967 CrossRef CAS.
- J. Liu, C. E. Barry III, G. S. Besra and H. Nikaido, J. Biol. Chem., 1996, 271, 29545–29551 CrossRef CAS PubMed.
- S. S. R. Alsayed, C. C. Beh, N. R. Foster, A. D. Payne, Y. Yu and H. Gunosewoyo, Curr. Mol. Pharmacol., 2019, 12, 27–49 CrossRef CAS PubMed.
- G. Degiacomi, A. Benjak, J. Madacki, F. Boldrin, R. Provvedi, G. Palu, J. Kordulakova, S. T. Cole and R. Manganelli, Sci. Rep., 2017, 7, 43495 CrossRef PubMed.
- W. Li, A. Obregon-Henao, J. B. Wallach, E. J. North, R. E. Lee, M. Gonzalez-Juarrero, D. Schnappinger and M. Jackson, Antimicrob. Agents Chemother., 2016, 60, 5198–5207 CrossRef CAS PubMed.
- W. Li, C. M. Stevens, A. N. Pandya, Z. Darzynkiewicz, P. Bhattarai, W. Tong, M. Gonzalez-Juarrero, E. J. North, H. I. Zgurskaya and M. Jackson, ACS Infect. Dis., 2019, 5, 1001–1012 CrossRef CAS PubMed.
- O. K. Onajole, M. Pieroni, S. K. Tipparaju, S. Lun, J. Stec, G. Chen, H. Gunosewoyo, H. Guo, N. C. Ammerman, W. R. Bishai and A. P. Kozikowski, J. Med. Chem., 2013, 56, 4093–4103 CrossRef CAS PubMed.
- N. D. Franz, J. M. Belardinelli, M. A. Kaminski, L. C. Dunn, V. Calado Nogueira de Moura, M. A. Blaha, D. D. Truong, W. Li, M. Jackson and E. J. North, Bioorg. Med. Chem., 2017, 25, 3746–3755 CrossRef CAS PubMed.
- L. F. Castano, V. Cuartas, A. Bernal, A. Insuasty, J. Guzman, O. Vidal, V. Rubio, G. Puerto, P. Lukac, V. Vimberg, G. Balikova-Novtona, L. Vannucci, J. Janata, J. Quiroga, R. Abonia, M. Nogueras, J. Cobo and B. Insuasty, Eur. J. Med. Chem., 2019, 176, 50–60 CrossRef CAS PubMed.
- G. Cihan-Ustundag and G. Capan, Mol. Diversity, 2012, 16, 525–539 CrossRef PubMed.
- G. Cihan-Ustundag, D. Satana, G. Ozhan and G. Capan, J. Enzyme Inhib. Med. Chem., 2016, 31, 369–380 CAS.
- I. Correia, P. Adao, S. Roy, M. Wahba, C. Matos, M. R. Maurya, F. Marques, F. R. Pavan, C. Q. F. Leite, F. Avecilla and J. Costa Pessoa, J. Inorg. Biochem., 2014, 141, 83–93 CrossRef CAS PubMed.
- T. Taj, R. R. Kamble, T. M. Gireesh, R. K. Hunnur and S. B. Margankop, Eur. J. Med. Chem., 2011, 46, 4366–4373 CrossRef CAS PubMed.
- Y. Shi, Y. H. Duan, Y. Y. Ji, Z. L. Wang, Y. R. Wu, H. Gunosewoyo, X. Y. Xie, J. Z. Chen, F. Yang, J. Li, J. Tang, X. Xie and L. F. Yu, J. Med. Chem., 2017, 60, 7067–7083 CrossRef CAS PubMed.
- A. Moya-Garcia, T. Adeyelu, F. A. Kruger, N. L. Dawson, J. G. Lees, J. P. Overington, C. Orengo and J. A. G. Ranea, Sci. Rep., 2017, 7, 10102 CrossRef PubMed.
- A. S. Reddy and S. Zhang, Expert Rev. Clin. Pharmacol., 2013, 6, 41–47 CrossRef CAS PubMed.
- A. A. Elfiky, Life Sci., 2020, 248, 117477 CrossRef CAS PubMed.
- Q. T. Ostrom, H. Gittleman, P. Liao, C. Rouse, Y. Chen, J. Dowling, Y. Wolinsky, C. Kruchko and J. Barnholtz-Sloan, Neuro-Oncology, 2014, 16(Suppl 4), iv1–iv63 CrossRef PubMed.
- A. T. Reddy, Curr. Neurol. Neurosci. Rep., 2001, 1, 137–143 CrossRef CAS PubMed.
- F. He and Y. E. Sun, Int. J. Biochem. Cell Biol., 2007, 39, 661–665 CrossRef CAS PubMed.
- K. R. Jessen and R. Mirsky, Nature, 1980, 286, 736–737 CrossRef CAS PubMed.
- D. N. Louis, A. Perry, G. Reifenberger, A. von Deimling, D. Figarella-Branger, W. K. Cavenee, H. Ohgaki, O. D. Wiestler, P. Kleihues and D. W. Ellison, Acta Neuropathol., 2016, 131, 803–820 CrossRef PubMed.
- K. Aldape, K. M. Brindle, L. Chesler, R. Chopra, A. Gajjar, M. R. Gilbert, N. Gottardo, D. H. Gutmann, D. Hargrave, E. C. Holland, D. T. W. Jones, J. A. Joyce, P. Kearns, M. W. Kieran, I. K. Mellinghoff, M. Merchant, S. M. Pfister, S. M. Pollard, V. Ramaswamy, J. N. Rich, G. W. Robinson, D. H. Rowitch, J. H. Sampson, M. D. Taylor, P. Workman and R. J. Gilbertson, Nat. Rev. Clin. Oncol., 2019, 16, 509–520 CrossRef CAS PubMed.
- F. Hanif, K. Muzaffar, K. Perveen, S. M. Malhi and S. U. Simjee, Asian Pac. J. Cancer Prev., 2017, 18, 3–9 Search PubMed.
- J. Fangusaro, Front. Oncol., 2012, 2, 105 Search PubMed.
- Z. Miklja, A. Pasternak, S. Stallard, T. Nicolaides, C. Kline-Nunnally, B. Cole, R. Beroukhim, P. Bandopadhayay, S. Chi, S. H. Ramkissoon, B. Mullan, A. K. Bruzek, A. Gauthier, T. Garcia, C. Atchison, B. Marini, M. Fouladi, D. W. Parsons, S. Leary, S. Mueller, K. L. Ligon and C. Koschmann, Neuro-Oncology, 2019, 21(8), 968–980 CrossRef CAS.
- G. C. Moraski, L. D. Markley, P. A. Hipskind, H. Boshoff, S. Cho, S. G. Franzblau and M. J. Miller, ACS Med. Chem. Lett., 2011, 2, 466–470 CrossRef CAS PubMed.
- C. T. Peng, C. Gao, N. Y. Wang, X. Y. You, L. D. Zhang, Y. X. Zhu, Y. Xv, W. Q. Zuo, K. Ran, H. X. Deng, Q. Lei, K. J. Xiao and L. T. Yu, Bioorg. Med. Chem. Lett., 2015, 25, 1373–1376 CrossRef CAS PubMed.
- A. P. Kozikowski, O. K. Onajole, J. Stec, C. Dupont, A. Viljoen, M. Richard, T. Chaira, S. Lun, W. Bishai, V. S. Raj, D. Ordway and L. Kremer, J. Med. Chem., 2017, 60, 5876–5888 CrossRef CAS PubMed.
- J. Stec, O. K. Onajole, S. Lun, H. Guo, B. Merenbloom, G. Vistoli, W. R. Bishai and A. P. Kozikowski, J. Med. Chem., 2016, 59, 6232–6247 CrossRef CAS PubMed.
- S. S. R. Alsayed, S. Lun, A. Payne, W. R. Bishai and H. Gunosewoyo, Bioorg. Chem., 2021, 106, 104486 CrossRef CAS.
- K. A. Abrahams, J. A. Cox, V. L. Spivey, N. J. Loman, M. J. Pallen, C. Constantinidou, R. Fernandez, C. Alemparte, M. J. Remuinan, D. Barros, L. Ballell and G. S. Besra, PLoS One, 2012, 7, e52951 CrossRef CAS PubMed.
- G. C. Moraski, P. A. Miller, M. A. Bailey, J. Ollinger, T. Parish, H. I. Boshoff, S. Cho, J. R. Anderson, S. Mulugeta, S. G. Franzblau and M. J. Miller, ACS Infect. Dis., 2015, 1, 85–90 CrossRef CAS PubMed.
- S. S. R. Alsayed, S. Lun, G. Luna, C. C. Beh, A. D. Payne, N. Foster, W. R. Bishai and H. Gunosewoyo, RSC Adv., 2020, 10, 7523–7540 RSC.
- V. Makarov, G. Manina, K. Mikusova, U. Mollmann, O. Ryabova, B. Saint-Joanis, N. Dhar, M. R. Pasca, S. Buroni, A. P. Lucarelli, A. Milano, E. De Rossi, M. Belanova, A. Bobovska, P. Dianiskova, J. Kordulakova, C. Sala, E. Fullam, P. Schneider, J. D. McKinney, P. Brodin, T. Christophe, S. Waddell, P. Butcher, J. Albrethsen, I. Rosenkrands, R. Brosch, V. Nandi, S. Bharath, S. Gaonkar, R. K. Shandil, V. Balasubramanian, T. Balganesh, S. Tyagi, J. Grosset, G. Riccardi and S. T. Cole, Science, 2009, 324, 801–804 CrossRef CAS PubMed.
- B. Cetin, I. I. Gonul, O. Gumusay, I. Bilgetekin, E. Algin, A. Ozet and A. Uner, Neuropathology, 2018, 38, 457–462 CrossRef CAS PubMed.
- G. Moncayo, M. Grzmil, T. Smirnova, P. Zmarz, R. M. Huber, D. Hynx, H. Kohler, Y. Wang, H. R. Hotz, N. E. Hynes, G. Keller, S. Frank, A. Merlo and B. A. Hemmings, Neuro-Oncology, 2018, 20, 621–631 CrossRef CAS PubMed.
- M. A. Proescholdt, M. J. Merrill, E. M. Stoerr, A. Lohmeier, F. Pohl and A. Brawanski, Neuro-Oncology, 2012, 14, 1357–1366 CrossRef CAS.
- N. H. Boyd, K. Walker, J. Fried, J. R. Hackney, P. C. McDonald, G. A. Benavides, R. Spina, A. Audia, S. E. Scott, C. J. Libby, A. N. Tran, M. O. Bevensee, C. Griguer, S. Nozell, G. Y. Gillespie, B. Nabors, K. P. Bhat, E. E. Bar, V. Darley-Usmar, B. Xu, E. Gordon, S. J. Cooper, S. Dedhar and A. B. Hjelmeland, JCI Insight, 2017, 2(24), e92928 CrossRef PubMed.
- B. Muz, P. de la Puente, F. Azab and A. K. Azab, Hypoxia, 2015, 3, 83–92 CrossRef PubMed.
- S. Ivanov, S. Y. Liao, A. Ivanova, A. Danilkovitch-Miagkova, N. Tarasova, G. Weirich, M. J. Merrill, M. A. Proescholdt, E. H. Oldfield, J. Lee, J. Zavada, A. Waheed, W. Sly, M. I. Lerman and E. J. Stanbridge, Am. J. Pathol., 2001, 158, 905–919 CrossRef CAS.
- M. O. Krisenko and R. L. Geahlen, Biochim. Biophys. Acta, 2015, 1853, 254–263 CrossRef CAS PubMed.
- J. Fueyo, M. M. Alonso, B. C. Parker Kerrigan and C. Gomez-Manzano, Neuro-Oncology, 2018, 20, 582–583 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- A. Heguy, A. A. Stewart, J. D. Haley, D. E. Smith and J. G. Foulkes, Gene Expression, 1995, 4, 337–344 CAS.
- M. W. Jones, G. Mantovani, C. A. Blindauer, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 7406–7413 CrossRef CAS PubMed.
- L. Collins and S. G. Franzblau, Antimicrob. Agents Chemother., 1997, 41, 1004–1009 CrossRef CAS PubMed.
- M. Pieroni, S. K. Tipparaju, S. Lun, Y. Song, A. W. Sturm, W. R. Bishai and A. P. Kozikowski, ChemMedChem, 2011, 6, 334–342 CrossRef CAS PubMed.
- S. T. Sredni, M. Suzuki, J. P. Yang, J. Topczewski, A. W. Bailey, T. Gokirmak, J. N. Gross, A. de Andrade, A. Kondo, D. R. Piper and T. Tomita, Pediatr. Blood Cancer, 2017, 64(11), e26551 CrossRef PubMed.
- D. C. Rio, M. Ares Jr, G. J. Hannon and T. W. Nilsen, Cold Spring Harb. Protoc., 2010, 2010, pdb prot5439 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10728j |
| This journal is © The Royal Society of Chemistry 2021 |