DOI:
10.1039/D0RA10755G
(Review Article)
RSC Adv., 2021,
11, 5832-5858
Synthetic efforts on the road to marine natural products bearing 4-O-2,3,4,6-tetrasubstituted THPs: an update
Received
22nd December 2020
, Accepted 26th January 2021
First published on 3rd February 2021
Abstract
Scientific literature is inundated with secondary metabolites from marine sources. In this ocean of natural products, the presence of recurring patterns has traditionally led scientists to unravel the biosynthetic mechanisms that naturally yield these products, as well as to imitate Nature to prepare them in the laboratory, especially when promising bioactivities and stimulating molecular architectures are involucrate. For instance, natural products containing multisubstituted oxygenated rings and macrocyclic lactones are recurrently selected as targets for developing total syntheses. Thus, in the last decades a noteworthy number of synthetic works regarding miyakolide, madeirolide A and representative compounds of polycavernosides, lasonolides and clavosolides have come to fruition. Up to now, these families of macrolides are the only marine natural products bearing a tetrasubstituted tetrahydropyran ring with carbon substituents at positions 2, 3 and 6, as well as an oxygen at position 4. Their splendid structures have received the attention of the synthetic community, up to the point of starring in dozens of articles, and even some reviews. This work covers all the synthetic studies towards miyakolide and madeirolide A, as well as the synthetic efforts performed after the previous specialised reviews about lasonolide A, polycavernoside A and clavosolides, published in 2006, 2007 and 2016, respectively. In total, this review summarises 22 articles in which these marine natural products with 4-O-2,3,4,6-tetrasubstituted tetrahydropyrans have the leading role.
 Marta Fariña-Ramos | Marta Fariña Ramos was born in 1990 in Tenerife (Canary Islands). She received her BSc degree in Chemistry in 2014 at the University of La Laguna (ULL). In 2015, she obtained her MSc in Chemistry at the same University. She is currently pursuing her PhD in Organic Chemistry under the supervision of Prof. Víctor S. Martín and Dra. Celina García. Her research focuses on the access to six-membered oxacycles by simultaneous C–C and C–O bond formation. |
 Celina García | Celina García received her PhD degree in Organic Chemistry at the University of La Laguna (ULL) in 2001, working on the enantioselective total synthesis of polyfunctionalized oxacyclic compounds, with Prof. Víctor S. Martín. The same year, she joined the laboratories of Prof. Patrick J. Walsh at the University of Pennsylvania as a postdoctoral researcher, working in the area of organocatalysis. In 2005 she came back as a researcher from a Ramón y Cajal Program of the Spanish Government to the ULL, where she is currently Associate Professor. |
 Víctor S. Martín | Víctor S. Martín was born in La Palma (Canary Islands), and received his PhD at the University of La Laguna (ULL) in 1978. From 1980–82 he was a postdoctoral fellow with Prof. K. Barry Sharpless in Stanford University and the Massachusetts Institute of Technology. He moved to the ULL as Associate Professor in 1984 and since 1992 he holds a Full Professor position. His research interests include asymmetric synthesis, methodology and total synthesis of bioactive natural products. He is the director of the Institute of Bio-Organic Antonio González and recipient of the 2016 Felix Serratosa Medal awarded by the Spanish Royal Society of Chemistry. |
 Sergio J. Álvarez-Méndez | Sergio J. Álvarez-Méndez was born in 1987 in La Palma (Canary Islands). He received his BSc degree in Chemistry in 2010 at the Universidad de La Laguna (ULL), with honours “Extraordinary Award to the Best Student's Academic Record”. At the same university, he obtained his MSc in Research in Organic Chemistry (2011) and his PhD in Chemistry and Chemical Engineering (2016), under the supervision of Prof. Víctor S. Martín and Dra. Celina García. After that, he has been working at the Technical College of Agricultural Engineering (ULL), with research interests in the application of organic chemistry to plant physiology and anaerobic digestion. |
1. Introduction
Mother Nature has been an endless source of inspiration for drug discovery throughout history.1 Over the past decades, natural products (NPs) from marine sources have been progressively catching the attention and interest of the scientific community due to their biological potential.2 Marine NPs show a panoply of molecular scaffolds,3 as a consequence of the biosynthetic pathways implied in their formation.4 Tetrahydropyrans (THPs) and macrocyclic lactones (macrolides) are interesting examples of this structural diversity. On the one hand, macrolides constitute an important subgroup of polyketides, formed in nature from acyl-CoA precursors by polyketide synthases.5 Artificial pathways have also been developed in laboratories worldwide to access these secondary metabolites.6 Pharmacological activities of macrolides are wide-ranging, including antimicrobial, anti-inflammatory, antitumour, antimalarial, and so on.7 On the other hand, THPs are also structural motifs recurrently found in NPs,8 likewise display numerous bioactivities.9 The synthesis of THPs in the laboratory is generally challenging, specially when they are highly substituted. Thus, reviews focusing on the building of THPs in the context of NPs syntheses are recurrently issued.8 Moreover, in last years, several methodological studies concretely targeting 2,3,4,6-tetrasubstituted THPs have also been published.10 For this work, we have directed our attention to marine NPs owning these 2,3,4,6-tetrasubstituted THPs, and besides, restricting our search to those in which the substituent at C4 is an oxygen, either ester, ether or hydroxy group. To the best of our knowledge, only 5 families of compounds come to terms with our baseline, i.e., miyakolide (1), polycavernosides (2), lasonolides (3), clavosolides (4) and madeirolide A (5a) (Fig. 1).
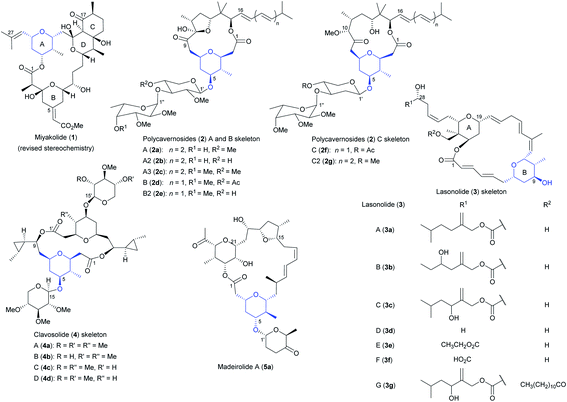 |
| | Fig. 1 NPs bearing a 4-O-2,3,4,6-tetrasubstituted THP. This figure has been adapted from ref. 15, 36, 41, 87, 132 and 162 with permission from ACS PUBLICATIONS, copyright 1999; ELSEVIER, copyright 2007; WILEY, copyright 2007; SPRINGER, copyright 2006; ACS PUBLICATIONS, copyright 2002; and ACS PUBLICATIONS, copyright 2016. | |
NPs 1–5 have in common a core formed by a macrolactone of, respectively, 16, 13 (or 14 in the case of 2f and 2g), 20, 16 and 21 members. The presence of oxygenated rings such as tetrahydrofurans (THFs) (2a–e and 5a), methylated deoxy sugars (2 and 4) and cyclohexanones (1 and 5a) is also widespread, being our target 2,3,4,6-tetrasubstituted THPs the leitmotif of all of them. These densely substituted THPs also display the C–O bond at the position 4 of the ring, being part of an ether linkage (2, 4 and 5a), the ester group of the macrolide (1) or a hydroxy group (3). Table 1 shows that the molecular weight of representative compounds of these families of macrolides ranges from 676 to 857 g mol−1. Table 1 also illustrates the noteworthy bioactivities exhibited by most of these marine NPs, highlighting the importance of the development of new synthetic pathways to access them in an efficient fashion. Indeed, the irrefutable importance of theses metabolites is laid bare by the previously published reviews focusing on syntheses of lasonolide A (3a), polycavernoside A (2a) and clavosolides (4). After their respective releases in 2006, 2007 and 2016, more approaches to these NPs have appeared in the literature, highlighting the everlasting interest of the synthetic community in them. Thus, we compile herein all the new synthetic efforts towards the obtaining of marine macrolides 2–4. Additionally, we gather for the first time all the approaches which tackle the synthesis of 1 and 5a, updating in this way the existing information about syntheses of marine NPs owning 2,3,4,6-tetrasubstituted THPs.
Table 1 Molecular formulas, molecular weights and most relevant bioactivities for characteristic macrolides 1–5
| NP |
Formula |
g mol−1 |
Relevant bioactivity |
| 1 |
C36H54O12 |
678.36 |
Antitumour: IC50 17.5 mg mL−1 (P388 mouse leukaemia), 17.1 mg mL−1 (A549 human lung carcinoma cells)11 |
| 2a |
C43H68O15 |
824.46 |
Toxin: LCLo 0.2–0.4 mg kg−1 in mice12 |
| 3a |
C41H60O9 |
696.42 |
Antitumour: IC50 2 ng mL−1 (P388 mouse leukaemia), 40 ng mL−1 (A549 human lung carcinoma cells)13 |
| 4a |
C44H72O16 |
856.48 |
No cytotoxic or antiproliferative activities described |
| 5a |
C37H56O11 |
676.38 |
Antifungal: MIC 12.5 μg mL−1 (Candida albicans)14 |
2. Miyakolide
Miyakolide (1) was isolated in 1992 from a sponge of the genus Polyfibrospongia, collected in Miyako island (Okinawa Prefecture, Japan).11 NMR and X-ray analyses allowed the characterization of the molecule. It was initially proposed as its enantiomer until absolute configuration was corrected after the total synthesis performed by Evans' group (vide infra).15 Fig. 1 shows the real miyakolide structure: a 16-membered macrolide bearing three THP rings (A, B and D) in chair conformations, being THP D trans-fused to the cyclohexanone ring C. The tetrasubstituted THP (ring A) is separated from THP D via a methylene bridge at C2, and also owns a methyl group at C3, the oxygen of the ester of the macrocyclic structure at C4 and a 2-methylprop-1-en-1-yl group at C6 (Fig. 1). Despite its structural similarity to anti-cancer bryostatins,16 miyakolide exhibited a weaker in vitro (IC50 17.5 μg mL−1 against P388 mouse leukemia, IC50 17.1 μg mL−1 against A-549 human lung carcinoma cells, and >20 μg mL−1 against HT-29 human colon adenocarcinoma cells) and in vivo (T/C 127% at 800 μg kg−1 against P388 mouse leukemia, and T/C 123% at 400 μg kg−1 against B-16 melanoma) antitumor activities.11 Curiously, the authors speculate that these differences in bioactivity may be indeed related, among other reasons, with the structure of the tetrasubstituted THP presented by miyakolide. Herein, the ethereal oxygen of this THP is not oriented to the inner of the macrocycle, as occurs with the three THPs of bryostatin-1.16 This difference may affect the ability of miyakolide to bind cations and, consequently, disrupt its bioactivity. With respect to synthetic works towards 1, the three up-to-date approaches are summarised below, maintaining the original nomenclature of the rings above described (A–D) for clarity.
2.1 Masamune's contribution (1997)17
In 1997, Masamune and co-workers presented a synthetic approach to the rings A and D of ent-miyakolide,17 as an application of the enantioselective anti-aldol tactic previously developed by the same research group.18 The synthesis of the ring A began with the preparation of the known aldehyde 6,19 which was submitted to a stereoselective aldol reaction with the enol borinate 7 to provide the anti-aldol 8 (Scheme 1). Cleavage of the chiral auxiliary with LAH provided diol 9, which was transformed into the O-benzylated alcohol 10 in excellent yield. Swern oxidation20 and subsequent hydrolysis of the acetonide in acidic conditions, followed by an in situ cyclisation, provided the hemiacetal 11 in excellent yield. The selective silylation and PCC oxidation of 11 gave the lactone 12, which was then converted into the aldol adduct 13 by treatment with LiHMDS and EtOAc. The stereoselective reduction of 13 using triethyl silane in the presence of boron trifluoride diethyl etherate (i.e., through the formation of an oxocarbenium ion), furnished the THP 14, which was finally transformed into desired THP 15 via the tandem PDC oxidation/Wittig olefination. Thus, THP A was obtained from 6 after 13 steps with an overall yield of 19%.
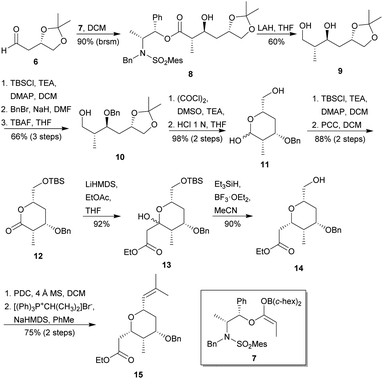 |
| | Scheme 1 Masamune's approach to ring A of miyakolide (1). This figure has been adapted from ref. 17 with permission from ACS PUBLICATIONS, copyright 1997. | |
Regarding the synthesis of the precursor of the ring D, it commenced with the stereoselective formation of the anti-aldol 18, treating the aldehyde 16 with the borinate 17 (Scheme 2).
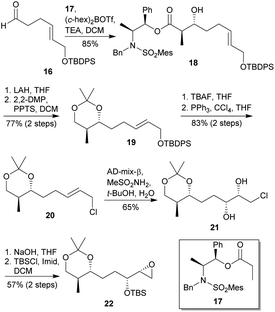 |
| | Scheme 2 Masamune's approach to ring D of miyakolide (1). This figure has been adapted from ref. 17 with permission from ACS PUBLICATIONS, copyright 1997. | |
Then, the aldol 18 was converted into acetonide 19, after removal of the chiral auxiliary and protection as acetonide of the resulting diol. Selective deprotection of 19 followed by an Appel reaction21 gave the chlorinated compound 20, which then underwent to an asymmetric Sharpless dihydroxylation to afford the chlorohydrin 21.22 Finally, the epoxidation of 2122 followed by TBS protection gave the targeted compound 22 with an overall yield of 24% (8 steps from 16).
2.2 Evans' synthesis (1999)15
The first total synthesis of the originally proposed structure of miyakolide (ent-1) was reported by Evans and co-workers in 1999.15 They envisioned the construction of the NP in a convergent fashion by means of the assembly of three different fragments (Scheme 3). Astonishingly, the three synthons (with minimal structural differences indicated as R1 and R2, see Scheme 3) were successfully assembled via the same three reactions in different order: aldol addition, [3 + 2] dipolar cycloaddition and esterification.
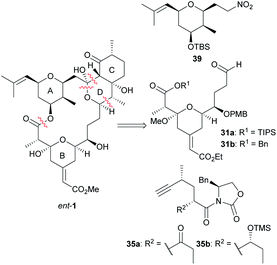 |
| | Scheme 3 Evans' retrosynthesis of miyakolide (1). This figure has been adapted from ref. 15 with permission from ACS PUBLICATIONS, copyright 1999. | |
The synthetic route towards THPs 31a and 31b (future ring B) started with the keto alcohol 24, previously obtained from the epoxide 23 in five steps via addition of allyl magnesium bromide, protection of the resulting secondary alcohol as PMB ether, trityl deprotection, Swern oxidation20 and Chan's diene addition (Scheme 4).23 Then, the stereoselective reduction of the keto group of 24 afforded a syn diol which was protected as acetonide prior to the DIBAL-H reduction of the ester group to yield aldehyde 25. Addition of chiral auxiliary 26 to aldehyde 25 via Evans aldol protocol24 yielded diastereoselectively the aldol adduct, which was subsequently oxidised20 to give the β-ketoimide 27. THP 28 was furnished from 27 via acidic deprotection, in situ cyclisation and Swern oxidation.20 Peterson–Yamamoto olefination25 of 28 conduced quantitatively to the exocyclic olefin 30, which was finally transformed to 31a (R = TIPS) through auxiliary hydrolysis, osmium-catalysed dihydroxylation of the terminal olefin, periodate cleavage and O-silylation (15% overall yield after 17 steps from 23). Alternatively, 31b (R = Bn) was synthesised from 30 via hydrolysis, benzylation, hydroxylation and treatment with periodate (24% overall yield after 17 steps from 23).
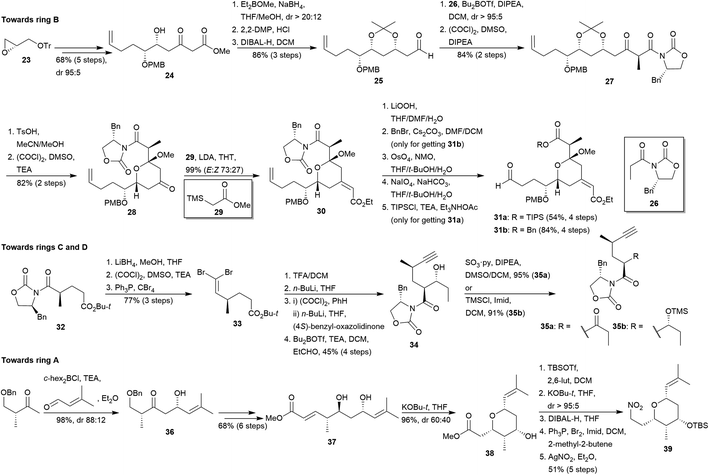 |
| | Scheme 4 Evans' preparation of miyakolide (1) fragments. This figure has been adapted from ref. 15 with permission from ACS PUBLICATIONS, copyright 1999. | |
The approach to the region of future rings C and D started with the preparation of the fragments 35a and 35b via the cleavage of the chiral auxiliary in 32 to give a primary alcohol which was oxidised to the corresponding aldehyde20 and submitted to olefination to yield dibromoalkene 33 (Scheme 4). Then, secondary alcohol 34 was obtained via successive ester cleavage, elimination of the dibromoalkene, N-acylation of the acyl chloride using lithiated (4S)-benzyl-oxazolidin-2-one, and aldol addition. Subsequent oxidation of 34 delivered compound 35a (32% after 9 steps), whereas O-silylation yielded 35b (31% after 9 steps). Synthesis of the tetrasubstituted THP 39 (future ring A) was achieved in 13 steps starting from commercially available (R)-4-(benzyloxy)-3-methylbutan-2-one and 3-methylbut-2-enal (Scheme 4). Aldol addition involving these reagents quantitatively yielded aldol 36. It was transformed into the key intermediate compound 37 after six steps: anti reduction, di-silylation of the diol, deprotection and oxidation of the primary alcohol, Horner–Wadsworth–Emmons (HWE) homologation26 and removal of the protecting silyl groups. Cyclisation under basic condition of 37 gave THP 38 in excellent yield though with low diastereoselectivity (dr 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40).
:40).
This setback was resolved protecting the epimeric mixture as silyl ethers and submitting them again to the base-mediated cyclisation (dr > 95:5). Then, desired THP 39 was obtained via reduction, bromination and nitration27 (33%, 13 steps from commercial starting materials).
Evans' group designed fragments 31, 35 and 39 to be interconnected through any of these three reaction sequences: aldol addition, [3 + 2] dipolar cycloaddition and esterification; cycloaddition, aldol addition and esterification; and aldol addition, esterification and cycloaddition. In all of the cases, compound 40 was the final key intermediate (Scheme 5). Finally, it was submitted to N–O bond reduction using Mo(CO)6 (Nitta's procedure),28 followed by the subsequent treatment with TsOH and DDQ. Thus, they achieved the first total synthesis of ent-1 after 29 linear steps and in 6.8% overall yield in the shortest and most efficient pathway.
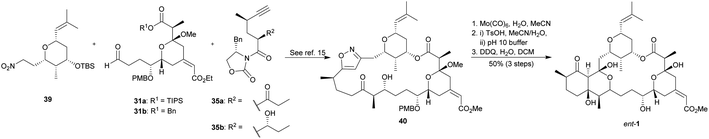 |
| | Scheme 5 Evans' fragments coupling and final steps on the way to miyakolide (1). This figure has been adapted from ref. 15 with permission from ACS PUBLICATIONS, copyright 1999. | |
2.3 Trost's contribution (2008)29
In 2008, Trost's group displayed the synthesis of the miyakolide THPs A and B (according to the original nomenclature).29 As key reactions for the rings formation, their approach involved the Pd-catalysed alkyne–alkyne coupling30 and the Ru-catalysed alkene–alkyne coupling31 previously developed by them. The route towards THP B began with the preparation of the compound 42 from bisepoxide 41 through a TMS-acetylide addition followed by TBS protection of the resulting secondary alcohol (Scheme 6). A subsequent addition of lithiated methyl propiolate to epoxide 42 yielded diyne 43. Later, the alkyne–alkyne coupling between 43 and 44 in the presence of Pd(OAc)2 afforded product 45.30 Its treatment with PdCl2(MeCN)2 followed by removal of the TMS group yielded 46, a precursor of the ring B of 1 (19% after 6 steps). The synthesis of tetrasubstituted THP A started with the preparation of 49 by Ru-catalysed alkene–alkyne coupling between 47 and 48 (Scheme 6).31 Then, treatment of epoxide 49 with Et2AlCN provided the allylic alkylation precursor 50, which was submitted to a Pd-catalysed allylic alkylation in the presence of DIPEA and S,S-L to obtain the desired cis isomer 51. Chemoselective epoxidation of 5132 followed by cleavage with HIO4 afforded the pyranone 52. It was transformed into ketone 53 after reduction with NaBH4, TBS protection and treatment with MeLi/CeCl3. Finally, cross metathesis using Grubbs II catalyst (54)33 yielded the THP A (55) with an overall yield of 20% after 9 steps.
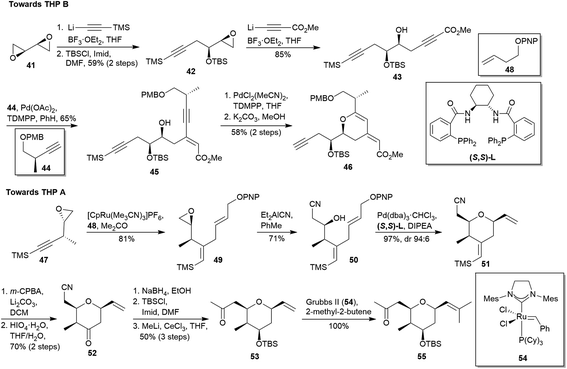 |
| | Scheme 6 Trost's approach to THPs A and B of miyakolide (1). This figure has been adapted from ref. 29 with permission from ACS PUBLICATIONS, copyright 2008. | |
3. Polycavernosides
Gracilaria edulis (also known as Polycavernosa tsudai) is an edible red alga of the Gracilaria genus, widely used in the gastronomy of the Pacific area. It was the cause of a grave human intoxication suffered in Guam by 13 people in April 1991, of which 3 died after consumption. For the purpose of isolating the involved toxins, Yasumoto's group collected 2.6 kg of the alga in June 1991, and its extract was purified by mice bioassays guided column chromatography.34 Thus, they isolated 400 μg and 200 μg of two compounds which were named polycavernoside A (2a) and B (2d), respectively (Fig. 1). It was observed that both presented high toxicity on mice, causing less intense but identical symptoms that those previously registered in sick patients (diarrhea, hypersalivation, lachrymation, muscle spasms, and cyanosis).34 The incident rose again the next year in the same season, though at lower levels. Thus, the same group published the isolation and structural determination of polycavernosides A2 (2b), A3 (2c) and B2 (2e) (Fig. 1) from P. tsudai collected in 1991 and 1992.35 Further studies about the same extract led to the description of analogues polycavernosides C (2f) and C2 (2g) (Fig. 1).36 The seasonal nature of this phenomenon was laid bare by the identification of 2a as the causative agent of new fatal food poisoning incidents (8 deaths of 36 patients) in the Philippines.37 All these polycavernosides differ mainly in the substituents carried by the disaccharide fragment (a fucosyl-xylose), as well as in the hemiacetal THF owned by 2a–e (Fig. 1). However, as part of the same family, they have in common several structural characteristics, such as a conjugated lateral chain contiguous to the macrolide ester and the presence of an all-trans tetrasubstituted THP with the following substituents: at C2 a methylene unit is joined to the carbonyl of the macrocyclic ester, a methyl group at C3, a glycosidic bond at C4 and a keto group connected to C6 via a methylene bridge. It should be now remarked that a new analogue of this family (polycavernoside D) was isolated in 2015 from the marine cyanobacterium Okeania sp.38 Besides some structural changes regarding the other polycavernosides, its THP is pentasubstituted due to an extra methyl group at C3, hence further details exceeds the scope of this review. Nevertheless, it is worth mentioning that its isolation was the first evidence that these macrocyclic toxins are actually cyanobacterial secondary metabolites, what explains the difficulty for re-isolating them from algae. Whatever the biosynthetic origin is, it is clear that the toxicity of these products deserves a careful attention. Both polycavernosides A (2a) and B (2d) exhibit a minimal lethal dose in mice of 0.2–0.4 mg kg−1, higher values than some synthetic analogues prepared.12 Pharmacological studies developed with a synthetic derivative of 2a (in which the middle double bond of the lateral triene moiety was replaced by an alkyne group), gave a hint about the mechanism of action of this neurotoxin, suggesting that polycavernosides evokes a cytosolic calcium influx and a membrane potential depolarization.39 Keeping in mind the aforementioned stimulant molecular architecture and potent toxic bioactivities, it was reasonable that synthetic strategies were developed to attain polycavernosides in the laboratory. The first total synthesis of 2a performed by Murai's group in 1998 allowed the unambiguous establishment of its absolute stereochemistry.40 After that pioneering work, other synthetic efforts were performed, as reviewed by Paquette and Yotsu-Yamashita in 2007.41 Due to the considerable time interval that has elapsed since then, our intention herein is bringing polycavernosides syntheses up to date.
3.1 Markó's contribution (2009)42
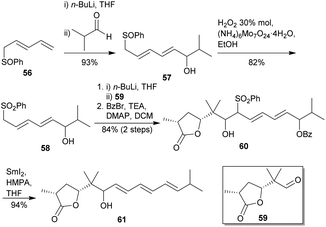
Following their previous synthesis of the THP fragment of polycavernoside A (2a) via an intramolecular Sakurai cyclisation,43 Markó and co-workers reported the synthesis of the fragment containing both the 5-membered ring and the triene side chain (Scheme 7).42 At first, starting compound 56 was lithiated and treated with isobutyraldehyde to yield sulphoxide 57 after a double sigmatropic rearrangement. Secondly, chemoselective oxidation of 57 gave sulphone 58. Then, it was treated with n-BuLi and aldehyde 59 to provide the corresponding diol-sulphone, which was selectively protected as benzoyl ester to afford compound 60. A Julia-type 1,6-reductive elimination in the presence of SmI2 and HMPA furnished the desired triene 61 with an overall yield of 60% after 5 steps.
 |
| | Scheme 7 Markó's approach to polycavernoside A (2a). This figure has been adapted from ref. 42 with permission from ELSEVIER, copyright 2009. | |
3.2 Lee's synthesis (2010)44
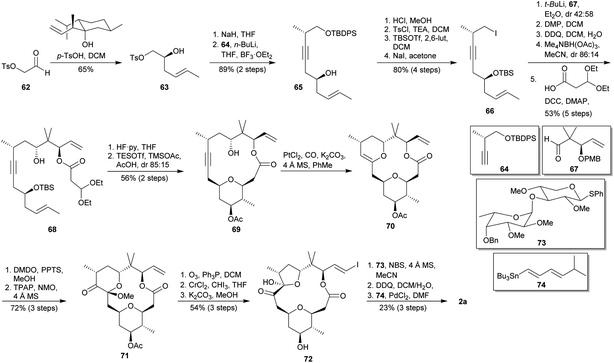
The key feature of the total synthesis of polycavernoside A (2a) published in 2010 by Lee and co-workers is the use of an intramolecular Prins bicyclisation to simultaneously construct the macrolactone and the tetrasubstituted THP.44 As illustrated in Scheme 8, the synthesis began with an enantioselective allyl-transfer using Nokami alcohol over aldehyde 6245 to yield the homoallylic alcohol 63. Basic treatment of 63 followed by opening of the epoxide with lithiated alkyne 6446 provided secondary alcohol 65, which was submitted to a sequence of desilylation, regioselective tosylation, silylation of the secondary alcohol and iodide substitution to afford the iodide 66. Lithium-iodide exchange followed by reaction with aldehyde 6747 gave a mixture of secondary alcohols, which were transformed into the corresponding ketone by treatment with Dess–Martin Periodinane (DMP).48 After PMB deprotection, the desired anti-relationship was accomplished by means of reduction mediated by Me4NBH(OAc)3. Condensation of the resulting anti-diol with appropriate activated carboxylic acid provided the compound 68. Acidic cleavage of TBS group in 68 provided the key homoallylic alcohol, which was submitted to a stereoselective intramolecular Prins cyclisation to furnish the desired 2,6-cis-THP 69. The intramolecular alkyne hydration of 69 proceed in the 6-endo mode to give 70 in the presence of PtCl2 as catalyst.49 Oxidation of 70 with DMDO followed by MeOH addition delivered the corresponding hydroxyl ketal intermediate, which was submitted to Ley oxidation conditions50 to yield 71. The 5-membered ring hemiacetal 72 was procured via ozonolysis of the terminal olefin, iodoolefination and rearrangement by prolonged exposition to Takai conditions,51 and basic hydrolysis to release the hydroxy group at C4 position of the THP. NBS-mediated glycosylation52 of 72 was carried out by using the thioglycoside 73.53 Posterior debenzylation and Stille-type coupling reaction employing dienylstannane 7454 finally provided (−)-polycavernoside A (2a) with an overall yield of 1.5% after 22 linear steps.
 |
| | Scheme 8 Lee's total synthesis of polycavernoside A (2a). This figure has been adapted from ref. 44 with permission from ACS PUBLICATIONS, copyright 2010. | |
3.3 Sasaki's synthesis (2012)55
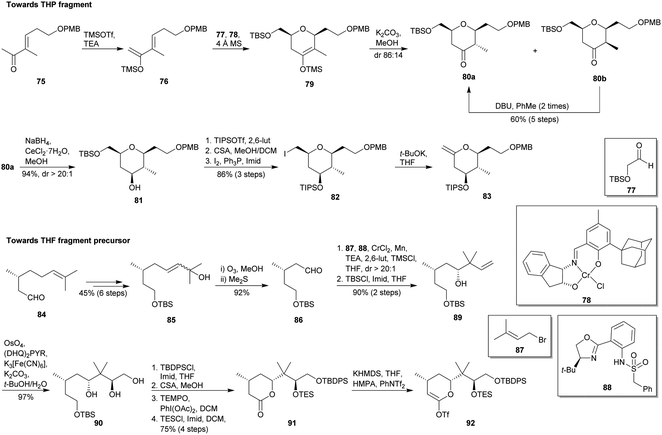
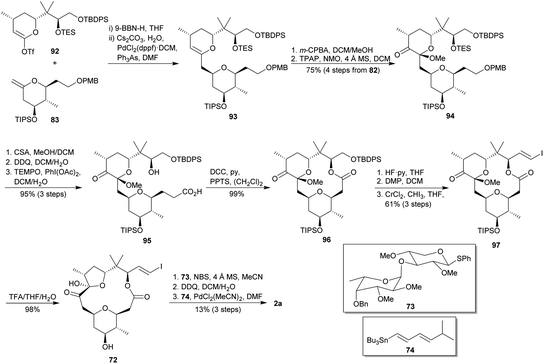
Sasaki and co-workers reported the total synthesis of polycavenroside A (2a) in 2012 (Scheme 9).55 They proposed a convergent approach starting from two fragments and employing as key reactions Suzuki–Miyaura coupling56 and Keck macrolactonization.57 As shown in Scheme 9, the construction of the THP fragment started with a catalytic asymmetric hetero Diels–Alder reaction between diene 76 (derived from 75)58 and aldehyde 77 by using Jacobsen tridentate chromium(III) catalyst (78)59 to yield dihydropyran 79. Then, treatment of 79 with K2CO3 in MeOH afforded a separable mixture of ketones 80a and 80b. Full conversion of minor 80b into desired isomer 80a was possible after two cycles of isomerization by means of DBU in toluene. Then, alcohol 81 was diastereoselectively obtained from 80a by Luche reduction.60 Subsequently, compound 81 was sequentially submitted to a silylation, deprotection of primary alcohol and iodination to deliver iodide 82, whose treatment with t-BuOK allowed the obtaining of desired fragment 83. Thereafter, they embarked on the future THF fragment synthesis (Scheme 9). (R)-(+)-citronellal (84) was transformed into enol 85,61 which was submitted to an ozonolysis to yield aldehyde 86. Then, 86 was submitted to a catalytic asymmetric allylation with bromide 87 and using sulphonamide 88 as chiral ligand,62 affording 89 (31%) together with the corresponding diol (69%), which was selectively re-silylated to give 89. Asymmetric dihydroxylation of alkene 89 using (DHQ)2PYR as chiral ligand22 provided triol 90. Later, it was transformed into lactone 91 by selective protection of the primary alcohol as TBDPS ether, cleavage of the TBS group, oxidative lactonisation63 and protection of the secondary alcohol as TES ether. Treatment of 91 with KHMDS and PhNTf2 afforded desired triflate 92. The Suzuki–Miyaura coupling of fragments 83 and 92 allowed the obtaining of desired 2,6-cis-THP 93 (Scheme 10).64 Oxidation of the enol 93 using m-CPBA provided a mixture of two epimeric alcohols which were individually oxidised to the same tetrahydropyranone 94. After that, 94 was submitted to TES and PMB deprotection followed by selective oxidation of the primary alcohol,65 providing carboxylic acid 95. Macrolactonisation under Keck conditions led to 96, which was converted into iodoolefin 97 after cleavage of the TBDPS, oxidation with DMP48 and Takai reaction.51 Afterwards, TFA-mediated removal of the TIPS and concomitant rearrangement to 5-membered hemiacetal yielded the intermediate 72. Finally, NBS-mediated glycosylation52 of 72 with the known glycosyl sulphide 73,53 followed by the oxidative cleavage of the benzyl ether group using DDQ and a Stille-type coupling54 with 74, consummated the total synthesis of 2a with an overall yield of 2.4% from 84 after 29 steps in the longest linear sequence.
 |
| | Scheme 9 Preparation of the Sasaki's fragments of polycavernoside A (2a). This figure has been adapted from ref. 55 with permission from ACS PUBLICATIONS, copyright 2012. | |
 |
| | Scheme 10 Sasaki's fragments assembly and final steps in the direction of polycavernoside A (2a). This figure has been adapted from ref. 55 with permission from ACS PUBLICATIONS, copyright 2012. | |
3.4 Fürstner's formal synthesis (2013)66
The key feature of the formal synthesis of 2a proposed by Fürstner and co-workers is the ring-closing alkyne metathesis (RCAM) to form the alkyne-macrolactone 69,66 the late-stage intermediate obtained by Lee and co-workers (vide supra).44 Scheme 11 shows the preparation of the three required fragments and the subsequent assembly. They started with an aldol addition of 98 with acrolein to yield racemic aldol 99, immediately converted into chiral acetated 100 through a lipase-catalysed kinetic resolution. Subsequently, 101 was achieved after basic hydrolysis, silylation and again basic treatment to hydrolyse undesired bis-silylated precursor. Parallelly, the Sonogashira coupling67 of propyne with 102 afforded alkyne 103, which was submitted to a Mn-salen (104a) catalysed epoxidation followed by a Co-salen (104b) catalysed hydrolytic kinetic resolution to provide chiral epoxide 105.68 Epoxide opening with MeLi and subsequent O-tosylation yielded 106. Treatment of 106 with LiBr supplied the volatile bromide derivate, which was exposed to Zn dust to provide the organozinc compound 107. In third place, asymmetric hydrogenation of 108 followed by PMB protection of the resulting secondary alcohol and DIBAL-H reduction of the ester allowed the obtaining of 109. Treatment of this aldehyde with organosilicon compound 110 yielded β,γ-unsaturated alcohol 111, consecutively submitted to O-silylation, DDQ-based deprotection, epoxidation and cross metathesis with methyl acrylate to give epoxide 113. Epoxide opening with the propynyl borane obtained in situ from lithiated propyne and BF3·OEt2 yielded 114,69 the proper precursor for a DBU/LiCl-promoted cis-selective oxy-Michael addition that led to the desired THP ring, subsequently reduced with DIBAL-H to yield 115.
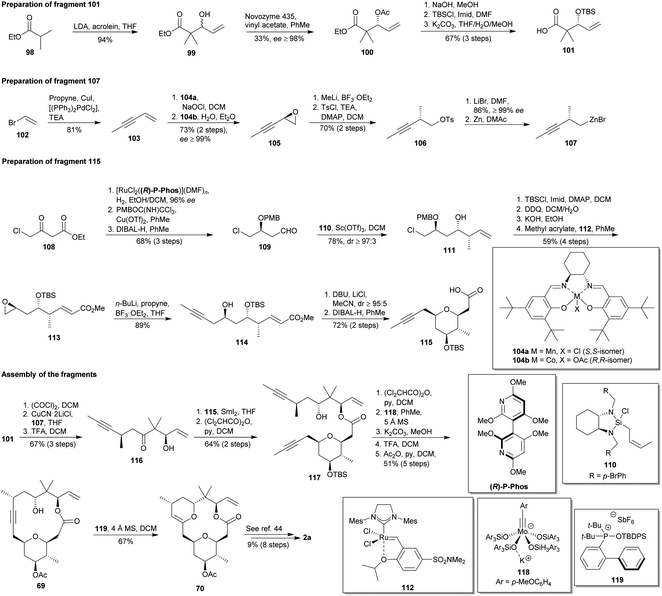 |
| | Scheme 11 Fürstner's formal synthesis of polycavernoside A (2a). This figure has been adapted from ref. 66 with permission from WILEY, copyright 2013. | |
Once synthesized the three fragments 101, 107 and 115, their assembly started with the CuCN·2LiCl-mediated70 acylation of organozinc 107 with the acid chloride of 101 to furnish ketone 116 after desilylation (Scheme 11). Then, 116 was employed in an Evans–Tischenko reaction71 with THP 115 to yield diyne 117, which was protected as dichloroacetate and submitted to a RCAM catalysed by Mo-complex 118.72 The removal of the dichloroacetate and the change of the protective group in the THP afforded Lee's cycloalkyne 69.44 Transannular hydroalkoxylation of 69 using the complex 119 yielded enol ether 70, thus ending the formal synthesis of 2a.
3.5 Sasaki's synthesis (2017)73
In 2017, Sasaki and co-workers reported an improved version of their previously reported synthesis of polycavernoside A (2a) (vide supra),55 as well as the application of this protocol to the synthesis of polycavernoside B (2d).73 Fragment 83 was again prepared as shown in Scheme 9 without any modifications, albeit synthesis of fragment 92 was modified. The new strategy towards 92 began with the Kiyooka aldol reaction74 between acetal 120 and known aldehyde 121,75 affording enantioselectively β-hydroxy ester 122 which was, in turn, converted into Weinreb amide 123 (Scheme 12). Alkylation of Weinreb amide 123 was achieved by using 2-methylallylmagnesium chloride to afford the β-hydroxy ketone 124, subsequently protected with TESCl and diastereoselectively reduced to give anti compound 125. Diene 126 was accessed by O-acylation of alcohol 125, and submitted to ring-closing metathesis (RCM) conditions76 to prepare lactone 127. Its hydrogenation allowed 91, which was transformed after 12 steps in the late intermediate 72, as reported in their previous work (see Schemes 9 and 10).55 This is the common precursor to complete both synthesis of 2a and 2d, based on the employment of sugar moieties 73 and 128 for the glycosylation52 and stannous polyene 74 and 129 for Stille-type coupling,54 respectively. Thus, Sasaki and co-workers achieved the synthesis of 2a and 2d in 0.96% and 2.0% overall yield after 23 and 22 steps, respectively.
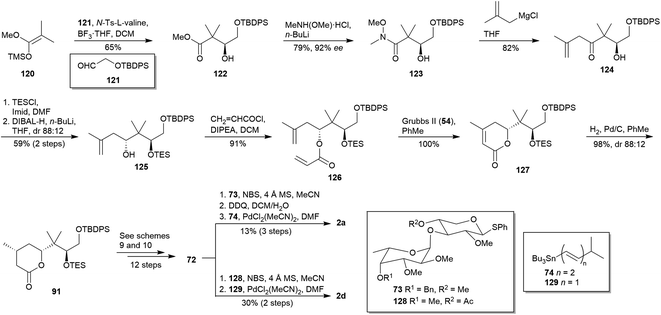 |
| | Scheme 12 Sasaki's syntheses of polycavernosides A (2a) and B (2d). This figure has been adapted from ref. 73 with permission from ACS PUBLICATIONS, copyright 2017. | |
3.6 Kadari & Yerrabelly's contribution (2018)77
In 2018, Kadari et al. reported the synthesis of the γ-butyrolactone ring of polycavernoside A (2a).77 The starting sequence implied a mono-benzylation of diol 130 followed by a Swern oxidation20 to access aldehyde 131 (Scheme 13). Its HWE homologation26 afforded (E)-α,β-unsaturated ester 132. Ensuing ester reduction and Sharpless asymmetric epoxidation78 yielded epoxide 133, which was transformed into iodide derivate 134. Treatment with Zn and NaI in MeOH allowed the obtaining of allylic alcohol 135, whose O-acylation with 136 gave the bis-olefin 137. Subsequently, 137 was submitted to RCM employing Grubbs II (54) as catalyst33 and providing thus γ-butyrolactone 138. Diastereofacial reduction of 138 with concomitant debenzylation and subsequent O-silylation finally yielded target 139.
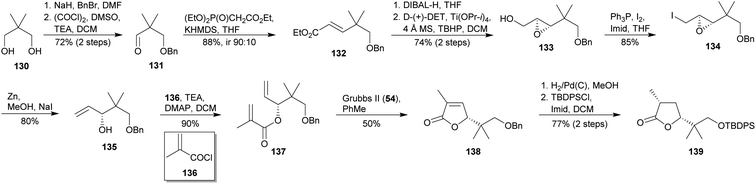 |
| | Scheme 13 Kadari & Yerrabelly's approach to the THF segment of polycavernoside A (2a). This figure has been adapted from ref. 77 with permission from WILEY, copyright 2018. | |
4. Lasonolide A
Lasonolide A (3a) was isolated from a Caribbean species of sponges of the genus Forcepia, collected close to Guana Island, British Virgin Islands. Its originally proposed structure (3a′) was published in 1994 by McConnell and co-workers (Fig. 2),13 and the same group published the initial elucidation of lasonolide B (3b′) in 1997.79 Some years later, Lee's group published a series of works regarding the first synthesis of proposed 3a′, highlighting certain inconsistencies in spectroscopic data, optical rotation and bioactivity compared to those originally reported.80 As consequence, structure of 3a was unambiguously established (Fig. 2). Lasonolides possess a 2,3,3,4,6-pentasubstituted THP (ring A) and a 2,3,4,6-tetrasubstituted THP (ring B). Their 20-membered macrolide core own a polyene linkage (4 E double bonds and a Z double bond between C12–C13). The initially proposed Z geometry of C17–C18 double bond was corrected to E, as well as the C25–C26 E geometry was re-assigned as Z after Lee's work.80 In 2004, 3a was again isolated from Forcepia sponges collected in the Gulf of Mexico, together with other new metabolites called lasonolides C–G (3c–3g, Fig. 2).81 All these lasonolides share the lasonopyran skeleton, and they differ in the nature of the R1 and R2 side chains.
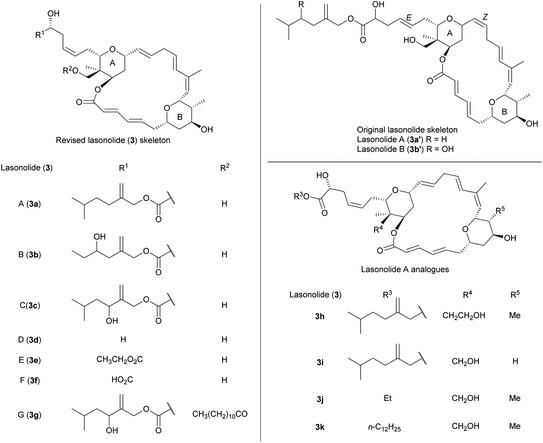 |
| | Fig. 2 Lasonolides (3). This figure has been adapted from ref. 82 and 87 with permission from ELSEVIER, copyright 2004 and SPRINGER, copyright 2006. | |
The interest in lasonolides lie in the potent in vitro antiproliferative activity exhibited by 3a against A549 human lung adenocarcinomatous epithelial cells (IC50 40 ng mL−1, i.e., 0.057 μM) and P388 murine leukaemia cell lines (IC50 2 ng mL−1, i.e., 0.003 μM).13 In addition, it was found that 3a inhibits cell adhesion in the EL-4.IL2 cell line (IC50 19 ng mL−1, i.e., 0.027 μM), that correlates with signal transduction activity, although toxicity against this line is greater than 25 μg mL−1.13 Indeed, the term lasonolide derives from the word lason, which means poison or toxin in Philippine. 3c–3g showed cytotoxicity against A549, human pancreatic carcinoma PANC-1 and the resistant human breast NCI/ADR-RES cell lines, although 3a showed the lowest values in all the cases.81 In 2004, four synthetic analogues (3h–3k, Fig. 2) were prepared and biologically evaluated against A549, human colon carcinoma HCT116 and human non-small cell lung carcinoma NCI-H460 cell lines, showing cytotoxicity but greater values than 3a.82 Once established the biopotential of 3a, further studies digging into its unique mechanism of action were developed. Thus, it was found that 3a acts as activator of protein kinase C in Panc-1 pancreatic carcinoma cells, inducing the formation of blebs and contraction of the cells shortly after the exposure and the final loss of adherence.83 3a also incites rapid, massive and reversible protein hyperphosphorylation and premature chromosome condensation at all phases of the cell-division cycle independently of cyclin-dependent kinases,84 in a process mediated by RAF1 gene.85 Very recently, it has also been reported the connection between 3a and lipid droplets, where it becomes a toxic metabolite by a lipid droplet-associated hydrolase.86
In the light of its stimulating structure, and mainly to respond to the request of more amount of product to explore the promising anticancer activity, it is evident that scientific community has directed considerable efforts towards the synthesis of 3a. Herein we discuss all the synthetic contributions published after the previous review, issued in 2006.87
4.1 Jennings' contribution (2006)88
Jenning and co-workers presented in 2006 the asymmetric synthesis of the C1–C14 fragment of lasonolide A (3a),88 using a SmI2-mediated Molander–Reformatsky intramolecular aldol addition89 as key reaction to form the oxygenated 6-membered ring. Their approach started with carbocupration under Corey conditions90 of the known ester 140,91 providing an α,β-unsaturated ester which yielded aldehyde 141 after a reduction/oxidation sequence (Scheme 14). Evans aldol condensation92 between aldehyde 141 and pre-formed (Z)-enolate of 142 diastereoselectively gave syn-aldol 143. The chiral auxiliary was then cleavage by treatment with NaBH4,93 providing a diol whose primary alcohol was protected as TBS ether (144) prior to be submitted to an esterification of its secondary alcohol to yield 145. Selective desilylation of 145 by using PPTS supplied a terminal alcohol which was then oxidised with DMP48 to give aldehyde 146. The SmI2-mediated intramolecular Reformatsky reaction of 146 under Molander conditions89 provided a samarium(III) intermediate, which underwent an intramolecular cyclisation to yield β-hydroxy lactone 147. Alkylation of 147 with allyl magnesium bromide followed by the treatment with TFA allowed the formation of an oxocarbenium ion which was reduced using TES to afford the 2,6-cis-THP 148 with the desired configuration.94 Subsequently, the terminal olefin in 148 was subjected to a chemo and diastereoselective cross-metathesis using Grubbs II catalyst (54)33 to afford an α,β-unsaturated aldehyde which was, in turn, homologated with appropriate phosphonium ylide to deliver target compound 149 in 3% overall yield after 14 steps.
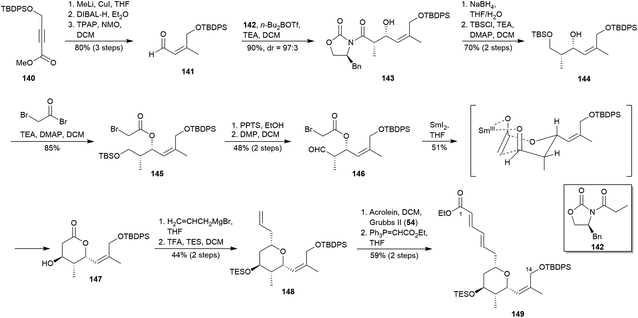 |
| | Scheme 14 Jennings' approach to the 2,3,4,6-tetrasubstituted THP ring of lasonolide A (3a). This figure has been adapted from ref. 88 with permission from ELSEVIER, copyright 2006. | |
4.2 Jennings' contribution (2007)95
Shortly after, Jenning and co-workers acceded to the C17–C25 fragment of lasonolide A (3a).95 This approach involves a Tsuchihashi–Yamamoto rearrangement,96 and again a SmI2-mediated aldol reaction89 to achieve the oxygenated 6-membered ring (Scheme 15). Thus, Wittig olefination of known aldehyde 15097 yielded ester 151, whose reduction and Sharpless asymmetric epoxidation78 led to the epoxy alcohol 152. The alcohol to aldehyde oxidation followed by nucleophilic addition of vinyl magnesium bromide and subsequent protection of the resulting alcohol as its TMS ether gave compound 153. As reported by Tsuchihashi–Yamamoto,96 the addition of TiCl4 triggered the 1,2-vinyl migration to deliver 154. Esterification of 154 with 2-bromoacetyl bromide gave 155, whose treatment with SmI2 provided the corresponding Sm(III) enolate which undergo a cyclisation via a 6-membered transition state to yield δ-lactone 156 as a single diastereoisomer.94 Nucleophilic addition of allyl magnesium bromide to 156 afforded lactol 157, whose treatment with TFA and TES provided, via formation of an oxocarbenium ion, the THP 158. Finally, simultaneous ozonolysis of both terminal alkenes followed by NaBH4-mediated reduction and subsequent treatment with PPTS yielded desired acetonide 159, completing thus the synthesis of 3a fragment C17–C25 with an overall yield of 0.51% after 14 steps from 151.
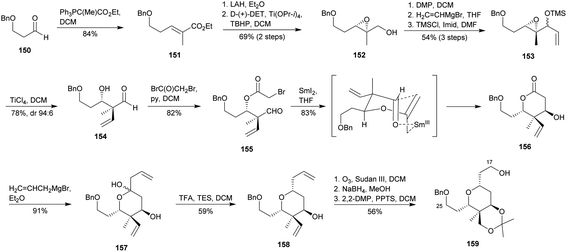 |
| | Scheme 15 Jennings' approach to the 2,3,3,4,6-pentasubstituted THP ring of lasonolide A (3a). This figure has been adapted from ref. 95 with permission from ELSEVIER, copyright 2007. | |
4.3 Ghosh's synthesis (2007)98
The total synthesis of lasonolide A (3a) published by Ghosh and co-workers employ as key steps an intramolecular [3 + 2] dipolar cycloaddition and a hetero-Diels–Alder reaction to construct the penta- and tetrasubstituted THPs, respectively (Scheme 16).98 The pathway towards the tetrasubstituted THP B fragment started from commercial alkynol 160, which was O-benzylated, lithiated and treated with n-propanal to yield alkynol 161. It underwent alkyne reduction, alcohol oxidation and treatment with TESOTf to afford desired silylated dienol 162. After that, it was submitted to a hetero-Diels–Alder reaction with TBSOCH2CHO in the presence of the chiral Schiff base chromium(III) complex 78 (Scheme 9) developed by Jacobsen,59 providing the 2,6-cis-tetrahydropyranone 163. Reduction of ketone 163 with DIBAL-H gave a 1/2 mixture of axial/equatorial alcohols, albeit an extra amount of desired axial alcohol 164 was achieved after tedious separation of equatorial alcohol, Swern oxidation and again DIBAL-H reduction. O-Silylation of secondary alcohol in 164, followed by hydrogenative removal of benzyl group provided primary alcohol 165. Then, olefin 166 was accomplished through the sequence Dess–Martin oxidation,48 Wittig reaction and acidic deprotection of the primary alcohol. Cross-metathesis between olefins 166 and 167 by means of Grubbs II catalyst (54)33 led mainly to the (E)-olefin 168. Successive oxidation of its primary alcohol followed by HWE olefination under Ando conditions99 afforded the (Z)-α,β-unsaturated ester 170. Finally, a tandem reduction/oxidation yielded aldehyde 171.
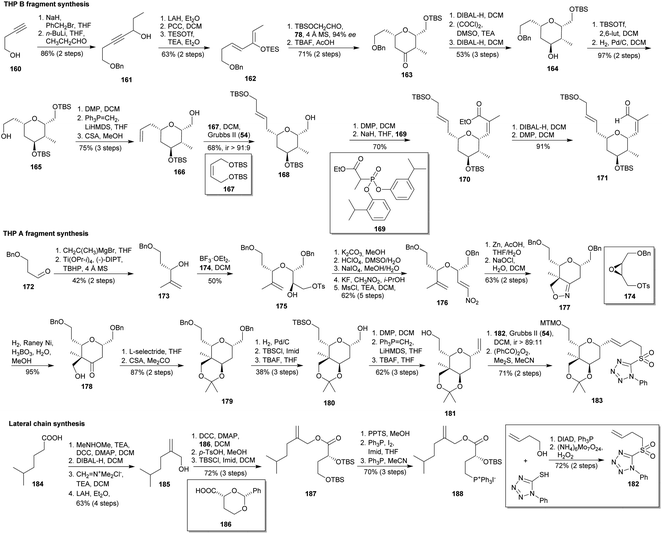 |
| | Scheme 16 Ghosh's syntheses of the fragments of lasonolide A (3a). This figure has been adapted from ref. 98b with permission from ACS PUBLICATIONS, copyright 2007. | |
The synthesis of the pentasubstituted A-ring THP began with the addition of isopropenyl magnesium bromide to aldehyde 172 and subsequent Sharpless kinetic resolution100 to access alcohol 173. This alcohol allowed the opening of the epoxide of tosylate 174 to yield 175.100 This compound underwent successive epoxidation to remove the O-tosyl moiety, acid epoxide opening to get a diol, oxidative cleavage of the diol to install an aldehyde, condensation with nitromethane and mesylation followed by an in situ elimination to finally obtain nitroalkene 176. Its reduction afforded the corresponding oxime, which was subsequently subjected to an intramolecular [3 + 2] cycloaddition to diastereoselectively provide the isoxazoline 177. Hydrogenolysis of isoxazoline ring yielded the hydroxy ketone 178, which was submitted to a L-selectride reduction followed by the diol protection to afford acetonide 179. Then, it was transformed into alcohol 180 by cleavage of benzyl groups, bis-protection as bis-TBS ether and treatment with TBAF (the also obtained diol was again bis-silylated and treated with TBAF). Alcohol 180 was transformed into olefin 181 via DMP oxidation,48 Wittig olefination and desilylation. Finally, cross-metathesis33 between 181 and the sulphone 182, followed by protection of the alcohol as MTM ether101 yielded target compound 183.
The approach to the lateral chain fragment employed commercial acid 184 as starting material. It was converted into a Weinreb amide, reduced to aldehyde, methylenated with Eschenmoser's salt,102 and reduced to allylic alcohol 185. Next it was submitted to esterification with known acid 186,103 removal of benzylidene group and bis-silylation to access 187. Later, primary alcohol was deprotected, converted into iodide and finally into the required phosphonium salt 188.
The final steps of this synthesis started with the fragment A (183) and B (171) coupling under Julia–Kocienski conditions104 to provide the tetraene 189 (Scheme 17). Acetonide and TBS protecting groups cleavage followed by bis-silylation of both primary alcohols yielded tris-TBS ether 190. Its secondary free alcohol was treated with activated phosphonoacetic acid, and the allylic TBS-ether was selectively deprotected to afford alcohol 191. After oxidation to aldehyde, the macrolactonization was carried out via an intramolecular HWE olefination,26 followed by MTM removal to yield macrolide 192. Lasonolide A (3a) was finally obtained via oxidation, Wittig olefination with phosphonium salt 188 and TBS bis-deprotection, with an overall yield of 0.12% after 32 steps in the longest sequence.
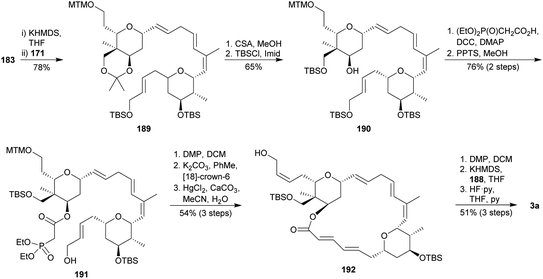 |
| | Scheme 17 Ghosh's fragments assembly and final steps on the way to lasonolide A (3a). This figure has been adapted from ref. 98b with permission from ACS PUBLICATIONS, copyright 2007. | |
4.4 Ghosh's contribution (2008)98a
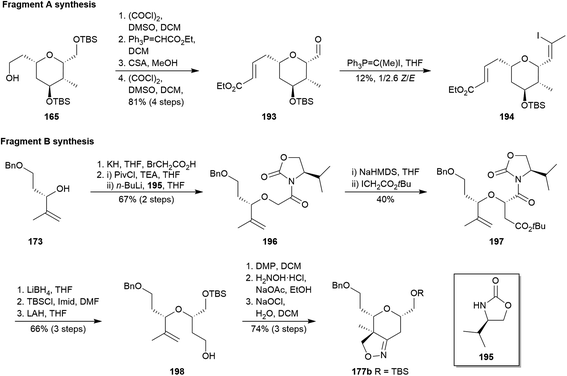
One year later, Ghosh and co-workers published a comprehensive explanation of their previous reported synthesis of 3a (vide supra).98a Additionally, in that work they related preliminary strategies through the obtaining of the THP fragments, which were finally replaced by the above described pathways. Nevertheless, within the context of this review, it is adequate to highlight the most relevant points of the original approaches. On the one hand, effort towards fragment A started from THP 165,98 which was successively submitted to Swern oxidation, Wittig reaction, selective desilylation and again to Swern oxidation to yield aldehyde 193 (Scheme 18). However, the attempts to exclusively transform 193 into desired Z-194 via a Stork–Zhao–Wittig reaction105 were infructuous. On the other hand, the pathway to the fragment B began with allylic alcohol 173 (Scheme 16),98 which was alkylated with bromoacetic acid, and the resulting acid was activated and treated with lithiated oxazolidin-2-one 195 to yield 196. Key Evans alkylation106 of N-acyl oxazolidin-2-one 196 was not as diastereoselective as expected, and undesired diastereoisomer was always the main product even when different chiral auxiliaries were employed (dr 48:52 in the shown best conditions). Even so, alkylated product 197 was isolated and transformed into 198 after reductive cleavage of the auxiliary, silylation and reduction of the ester group. After that, alcohol 198 was oxidized to aldehyde and transformed into an oxime, which underwent an intramolecular 1,3-dipolar cycloaddition via nitrile oxide to yield desired isooxazoline 177b.
 |
| | Scheme 18 Ghosh's original approaches to the THP rings of lasonolide A (3a). This figure has been adapted from ref. 98a with permission from WILEY, copyright 2008. | |
4.5 Ghosh's contribution (2012)107
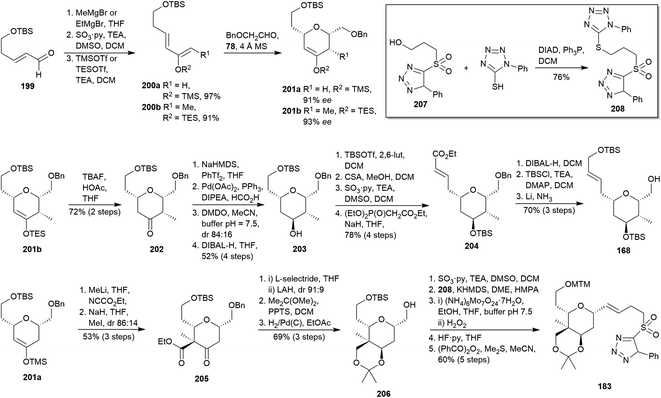
Four years later, Ghosh et al. envisioned the syntheses of 168 and 183 (the lasonolide A THP fragments previously prepared, see Scheme 16)98 from two similar precursors: dihydropyrans 201a and 201b (Scheme 19).107 At first, known aldehyde 199108 was submitted to a nucleophilic addition with both methyl and ethyl magnesium bromide, Parikh–Doering oxidation109 and silylation with both TMS and TES to yield silyl enol ethers 200a and 200b, respectively. An asymmetric hetero-Diels–Alder reaction was carried out using 2-(benzyloxy)acetaldehyde110 in the presence of Jacobsen catalyst 7859 in order to obtain 201a and 201b from 200a and 200b, respectively. On the one hand, 201b was transformed into ketone 202. Its DIBAL-H reduction to get alcohol 203 was inefficient since a diastereoselectivity standpoint, as previously occurred in the conversion of 163 to 164 (Scheme 16). Thus, it was replaced by conversion of 202 to 203 via formation of an enol triflate, Pd-catalysed reduction to alkene, diastereoselective DMDO-based epoxidation and diaxial epoxide opening by DIBAL-H. Protection of secondary alcohol in 203 as its TBS ether followed by selective primary TBS acidic deprotection, Parikh–Doering oxidation109 and HWE olefination26 yielded the α,β-unsaturated ester 204. Finally, DIBAL-H reduction, protection of the resulting alcohol as TBS ether and selective cleavage of the benzyl group afforded target 168.
 |
| | Scheme 19 Ghosh's preparation of lasonolide A (3a) THPs from a quasi-common dihydropyran precursor. This figure has been adapted from ref. 107 with permission from ACS PUBLICATIONS, copyright 2012. | |
On the other hand, 201a was lithiated and treated with ethyl cyanoformate to yield a β-keto ester, straightaway methylated to give tetrahydropyranone 205. It underwent a double reduction protocol: L-selectride provided the axial alcohol and then LAH reduced the ester moiety. The corresponding diol was protected as acetonide, and benzyl hydrogenation supplied alcohol 206. Finally, it was submitted to Parikh–Doering oxidation,109 Julia–Kocienski olefination104 with sulphone 208 (previously obtained from known alcohol 207111 via a Mitsunobu reaction), oxidation of sulphide to sulphone using ammonium heptamolybdate and hydrogen peroxide,112 desilylation and protection as MTM ether101 to yield target 183.
4.6 Trost's synthesis (2014)113
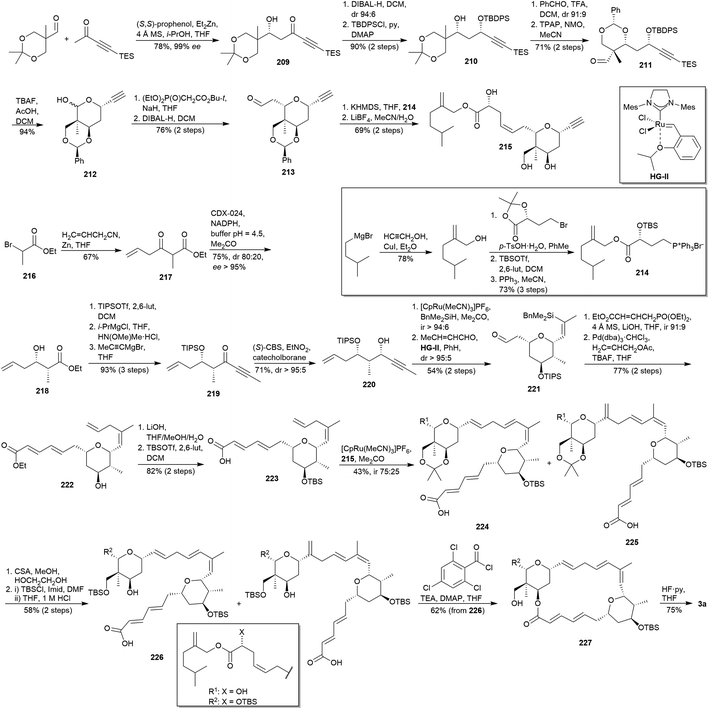
Trost and co-workers disclosed the enantioselective total synthesis of lasonolide A (3a) in 2014.113 The highlighted feature of this route is the Ru-catalysed alkene–alkyne coupling31 to assemble the two THPs moieties. Initially, β-hydroxy-ynone 209 was obtained via a Zn/prophenol-catalysed aldol addition114 between appropriate aldehyde and ynone (Scheme 20). The new stereocentre orientated the subsequent DIBAL-H reduction of 209 to afford the corresponding 1,3-syn-alcohol, which was selectively protected to yield silyl ether 210. Transacetalization of 210 and subsequent oxidation provided aldehyde 211. Then, cleavage of silyl groups in 211 led to an in situ cyclisation affording lactol 212, immediately submitted to an HWE olefination26 and DIBAL-H reduction to aldehyde 213. Finally, the alkyne 215 was achieved by Wittig olefination of 213 with the phosphonium salt 214 (obtained from isopentylmagnesium bromide via carbocupration, transesterification, silylation and nucleophilic substitution, see Scheme 20), followed by removal of the benzylidene acetal.
 |
| | Scheme 20 Trost's synthesis of lasonolide A (3a). This figure has been adapted from ref. 113 with permission from ACS PUBLICATIONS, copyright 2014. | |
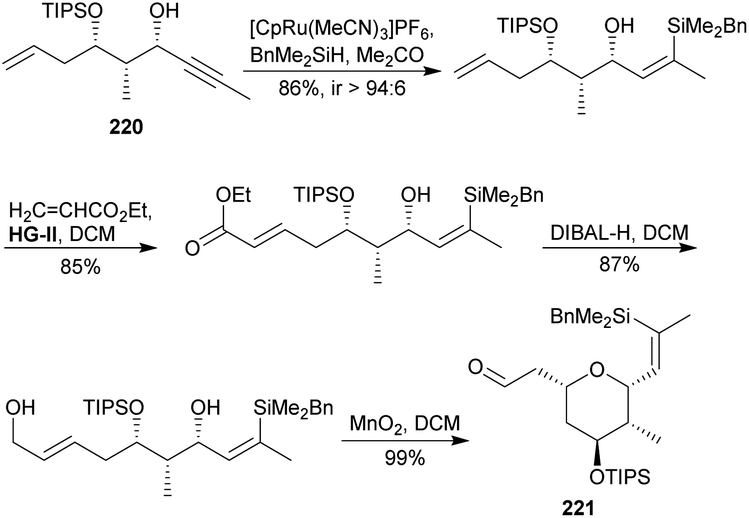
On the other hand, α-bromo keto ester 216 was subjected to a Blaise reaction115 with allyl cyanide to supply the β-ketoester 217. An enzymatic kinetic asymmetric reduction116 upon 217 by an enzyme called CDX-024 allowed the obtaining of β-hydroxyester 218. It was transformed into alkyne 219 following the sequence silylation, conversion into Weinreb amide and nucleophilic addition.117 (S)-Corey–Bakshi–Shibata (CBS) catalysed reduction diastereoselectively afforded 220.118 Afterwards, its Ru-catalysed hydrosilylation119 gave a (Z)-vinylsilane, which was underwent Hoveyda–Grubbs120 cross metathesis with crotonaldehyde and simultaneous oxa-Michael conjugated addition to provide 221.121 Terminal aldehyde of the 2,6-cis-THP 221 was submitted to an HWE olefination,26 and then the vinyl silyl group was involved in a Pd-catalysed Hiyama coupling122 with allyl acetate to furnish desilylated 222. Ester saponification followed by secondary alcohol protection as its TBS ether yielded 223.
Fragments 223 and 215 were joined via an alkene–alkyne coupling employing [CpRu(MeCN)3]PF6 as catalyst in acetone,31 providing thus a 3:1 inseparable mixture of linear:branched acetonides 224 and 225, respectively. The mixture was treated with CSA to remove the acetonides and then with TBSCl to yield required tris-silyl ether 226 together with its undesired branched isomer. Both were submitted to Yamaguchi macrolactonization to provide, after separation by chromatography, macrolide 227 (9% yield from 223 after 4 steps). Removal of silyl group finally led to lasonolide A (3a), obtained in 1.6% overall yield after 16 linear steps from 216.
4.7 Trost's contribution (2016)123
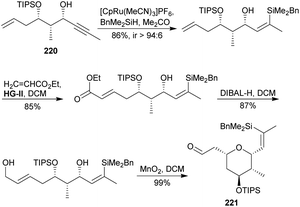
Two years later, Trost's group reported a detailed description of their previously published synthesis of 3a.113,123 In the context of this review, it is interesting to show briefly an alternative pathway that they tried on the road to the tetrasubstituted THP. As shown in Scheme 21, alkynol 220 was transformed into target THP 221 through the following sequence: Ru-catalyzed hydrosilylation,119 cross-metathesis with ethyl acrylate employing Hoveyda–Grubbs catalyst,120 reduction of the ester to alcohol, and eventually allylic oxidation of the alcohol to aldehyde with MnO2, which triggered a diastereoselective intramolecular addition to access 221 in 63% overall yield after 4 steps.
 |
| | Scheme 21 Trost's alternative approach to the tetrasubstituted THP of lasonolide A (3a). This figure has been adapted from ref. 123 with permission from ACS PUBLICATIONS, copyright 2016. | |
4.8 Hong's synthesis (2018)124
In 2018, Hong and co-workers published an enantioconvergent total synthesis of lasonolide A (3a).124 The modular approach employed an iterative Julia olefination to link the THP-containing fragments A and B, previously accessed from both enantiomers of α-allenic alcohol rac-228 after an enzymatic kinetic resolution (Scheme 22).125 The ring A synthesis began with the direct hydroboration of (R)-228 followed by allylation of the 3-(benzyloxy)propanal126 to provide the anti–anti diol 229 as a single diastereoisomer. Thereafter, nitroalkene 230 was prepared through sequential acetalisation of the diol, desilylation, oxidation of the resulting primary alcohol, Henry reaction and elimination. Treatment of 230 with PTSA triggered a cascade process involving removal of the acetal, oxa-Michael addition, Nef reaction and acetalisation to yield 2,6-cis-tetradropyran 231. Subsequent ozonolysis of the terminal alkene in 231 followed by reduction and silylation afforded 232. Compound 233 was achieved by deprotection of benzyl group (after an in situ BBN protection of secondary alcohol), TEMPO/BAIB oxidation and Wittig olefination with 214 (Scheme 20).111 Then, 233 was deacetylated and submitted to a selective Julia olefination127 with disulphone 234 to furnish sulphone 235 in good yield.
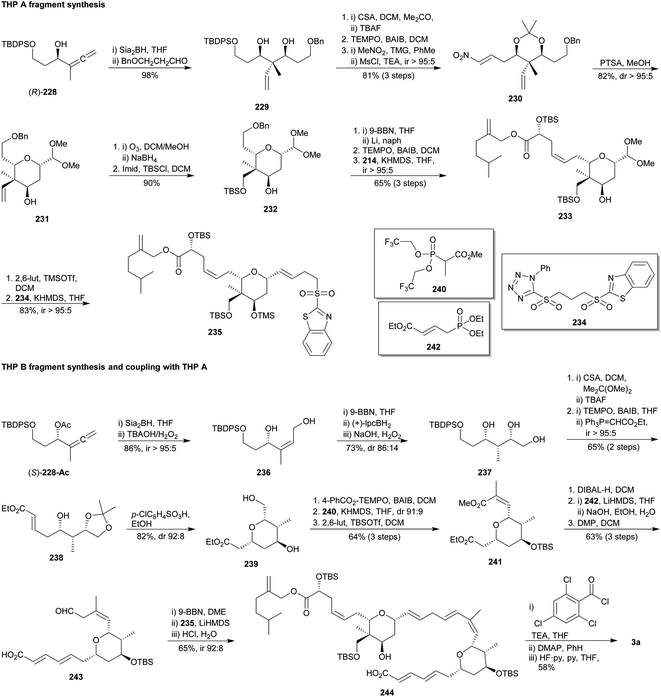 |
| | Scheme 22 Hong's synthesis of lasonolide A (3a). This figure has been adapted from ref. 124c with permission from WILEY, copyright 2018. | |
After that, Hong's group embarked on the ring B synthesis starting from the acetylated S-enantiomer of 228 ((S)-228-Ac). A novel iterative hydroboration/oxidation protocol allowed them to transform the allene into the Z-alkene 236 first, and then into the syn:syn-triol 237. Compound 237 was subjected to acetalisation, desilylation, oxidation of the primary alcohol and Wittig reaction to supply the E-α,β-unsaturated ester 238. Exposure of ester 238 to acidic conditions gave rise to a cascade acetal cleavage and 6-exo oxa-Michael addition,128 affording the corresponding 2,6-cis-THP 239. It was selectively oxidized,129 treated with the Still–Gennani phosphonate 240,130 and silylated to yield 241. DIBAL-H reduction led to an allylic alcohol but also to an aldehyde, which immediately submitted to an HWE olefination26 with phosphonate 242 and then to a saponification. Lastly, a DMP-based oxidation48 of the allylic alcohol provided aldehyde 243. In situ BBN-protection of the carboxylic acid in 243 followed by the Julia olefination with sulphone 235 yielded intermediate 244, which was transformed into the final natural product via a macrolactonisation/desilylation protocol.113 Thus, lasonolide A (3a) was synthesized in 12% overall yield after 15 steps from rac-228.
5. Clavosolide A
Clavosolides A and B (4a,b) were first isolated by Faulkner's group from the Philippine marine sponge Myriastra clavosa in 2002.131 A short while after, Gustafson group reported the isolation of clavosolides A–D (4a–d) from the same source, as well as the absence of cytotoxic or antiproliferative activities regarding these compounds.132 Clavosolides are 16-membered macrolides with a heterodimeric structure (Fig. 1). All the clavosolides have in common the monomer owning a 2,3,4,6-tetrasubstituted THP, a cyclopropane moiety and a totally methylated D-xylose. Clavosolides B (4b) and C (4c) differ from A in the presence of a free hydroxy group in one of the glycoside units, whereas clavosolide D (4d) exhibit a 2,4,6-trisubstituted THP instead the tetrasubstituted one found in A (Fig. 1). The absence of exhaustive biostudies concerning these metabolites, their unique biosynthetic origin, and specially their stimulating molecular architectures, encouraged the scientific community to dedicate enormous synthetic efforts to access these natural products. Thus, from 2005 to 2016, 16 syntheses towards clavosolides have been described.133 Herein, we summarise the two synthetic works regarding clavosolide A (4a) subsequent to that date.
5.1 Raghavan's contribution (2017)134
In 2017, Raghavan and co-workers reported a convergent approach to clavosolide A (4a),134 employing as key feature two HWE cyclopropanations26 over the starting epoxides 245 and 252 (Scheme 23). On the one hand, the HWE cyclopropanation of the epoxide 245135 was carried out in order to access ester 246. Its treatment with lithiated commercial (R)-methyl p-tolyl sulphoxide afforded the keto sulphoxide 247. The diastereoselective reduction136 of ketone in 247 yielded alcohol 248 with the required stereochemistry. Then, the sulphinyl moiety was employed as an internal nucleophile to open the Hg(II)-activated cyclopropane ring,137 providing thus mercuric bromide 249, whose subsequent demercuration138 led to diol 250. Protection of 250 as acetonide followed by sulphoxide reduction139 yielded sulphide 251. On the other hand, the HWE cyclopropanation of the epoxide 252 allowed the obtaining of an ester which was transformed into Weinreb amide 253. Addition of the corresponding alkynylmagnesium chloride followed by a stereoselective transfer hydrogenation employing (R,R)-Noyori catalyst (254)140 led to propargylic alcohol 255. Removal of the TMS and silylation yielded terminal alkyne 256. It was treated with isopropyl magnesium chloride, trans-metalated with ZnBr2 and added to previously chlorinated 251 to furnish 257 after TBS deprotection. Trost regioselective hydrosilylation141 afforded the silyl alkene 258. Finally, acetylation, RANEY® Ni desulphurisation and Tamao–Fleming oxidation142 of 258, yielded ketone 259, an advanced intermediate of clavosolide A (4a).
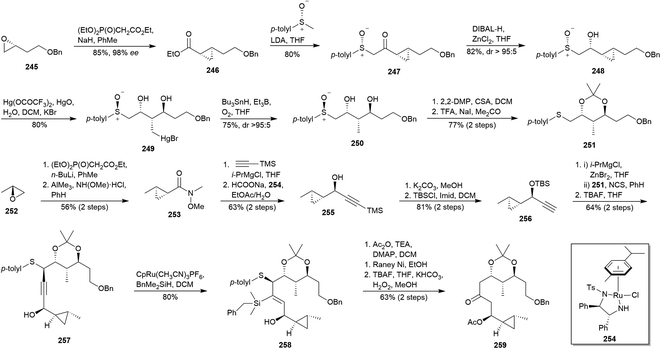 |
| | Scheme 23 Raghavan's approach to clavosolide A (4a). This figure has been adapted from ref. 134 with permission from ELSEVIER, copyright 2017. | |
5.2 Krische's synthesis (2019)143
The absence of protecting groups and chiral auxiliaries are the distinctive features of Krische's total synthesis of clavosolide A (4a).143 Firstly, a bidirectional asymmetric allylation144 employing (S)-IrBINAP (261), allyl acetate and 4-cyano-3-nitrobenzoic acid allowed the transformation of starting diol 260 into diol 262 (Scheme 24). Then, Fenton–Semmelhack alkoxypalladation/carbonylation145 of 262 provided the 2,6-cis-THP 263. A Schmidt-type glycosylation146 of 263 using trichloroacetimidate 264 and (R)-PA-II (265)147 furnished selectively the β-isomer 266. DIBAL-H reduction of ester group in 266 proceed smoothly to provide aldehyde 267. In contrast to Cram–Reetz model dictate,148 the use of (−)-sparteine149 in the carbonyl addition between 267 and Li-treated 268 allowed the access to desired 269. Finally, terminal alkene oxidation gave carboxylic acid 270, whose macrolactonisation led to clavosolide A (4a) in only 7 steps from 260 with an overall yield of 6.3%.
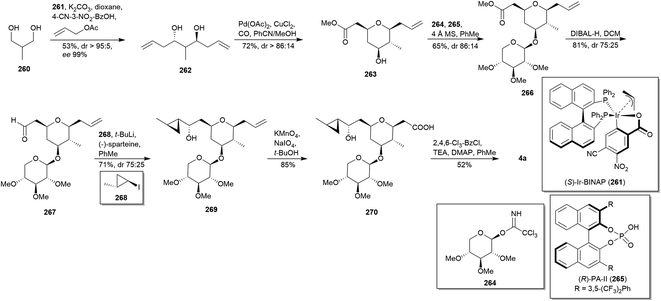 |
| | Scheme 24 Krische's total synthesis of clavosolide A (4a). This figure has been adapted from ref. 143 with permission from WILEY, copyright 2019. | |
6. Madeirolide A
Madeirolide family (5) was described in 2009 by Winder after its isolation from a species of sponge (genus Leiodermatium) harvested from the deep sea off the coast of Madeira (Portugal).14 Madeirolide A (5a) consists of a 21-membered macrolide with a 2,3,4,6-tetra and a 2,3,4,5,6-pentasubstituted THP, as well as a 2,3,5-trisubstituted THF, all of them with a cis ring closure (Fig. 1). A trans ring closure is also found in the peripheral 2-methyl-3-oxo-6-oxy-trisubstituted THP linked to the 4-oxy position of the tetrasubstituted THP. 5a displayed antifungal activity against Candida albicans (fungicidal MIC = 12.5 μg mL−1).14 However, further biostudies remain unexplored due to the low amount of isolated 5a. The synthetic efforts performed towards its obtaining are gathered herein.
6.1 Paterson's contribution (2013)150
In 2013, Paterson and co-workers, reported the C1–C11 fragment synthesis of madeirolide A (5a).150 This approach involves a hetero Michael cyclisation, a Takai olefination and glycosylation as key reactions steps (Scheme 25). The synthesis was initiated with reduction of known (S)-ester 271151 to alcohol, mesylation and iodination to access 272. Reaction of lithiated dithiane 273152 with 272, followed by treatment of the resulting dithiane adduct with aqueous iodine, provided ketone 274.153 Aldol reaction between ketone 274 and aldehyde 275,154 mediated by chiral ligand (−)-(lpc)2BOTf, led to alcohol 276 as a single diastereoisomer. 1,3-anti reduction of 276 under Evans–Tishchenko conditions71 led to an ester, immediately transformed via methanolysis into the corresponding diol which was, in turn, protected as acetonide 277. TBS-deprotection and Dess–Martin oxidation48 provided the necessary aldehyde 278. Condensation between aldehyde 278 and phosphonate 279 under Masamune–Roush conditions155 delivered unsaturated ester 280. Acidic removal of the acetal of 280 provided the corresponding diol, which immediately underwent a Michael cyclisation to yield a 2,6-cis-THP,156 then converted into compound 281 after PMBO cleavage. Selective oxidation of the primary alcohol in 281, followed by Takai olefination51 supplied vinyl iodide 282. Finally, BF3·OEt2-mediated glycosylation of 282 with 284 (previously synthesised in 3 steps from 283 via Noyori reduction,140 Achmatowicz rearrangement followed by in situ acetylation of the lactol157 and hydrogenation) provided the C1–C11 fragment (285) of 5a in 11% overall yield after 17 steps.
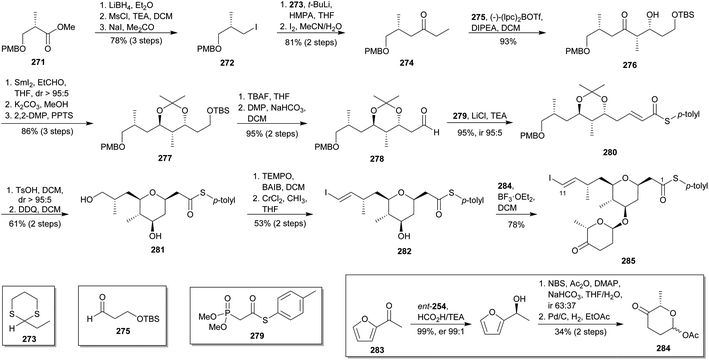 |
| | Scheme 25 Paterson's approach to madeirolide A (5a). This figure has been adapted from ref. 150 with permission from ACS PUBLICATIONS, copyright 2013. | |
6.2 Carter's contribution (2016)158
In 2016, Carter and co-workers prepared the C1–C12 fragment of madeirolide A (5a) as a showcase of their silver catalyzed cyclisations to obtain THP.158 The approach began with the Sonogashira coupling67 between both known iodide 286159 and alkyne 287160 to provide the propargylic alkene 288 (Scheme 26). Then, it was submitted to a Shi epoxidation161 to afford epoxide 290, whose opening by utilizing LiAlMe4 supplied homo-propargylic alcohol 291. Treatment of 291 with AgBF4 delivered a 58:42 mixture of the desired dihydropyran 293 together with dihydrofuran 292. Once separated, treatment of 293 with NaOMe afforded ketone 294, which was, in turn, underwent to a stereoselective reduction to yield alcohol 295. The glycosylation step was carried out in the presence of BF3·OEt2 and glycoside 284150 (see preparation in Scheme 25) to furnish compound 296. Finally, they acceded to C1–C12 fragment (297) via TIPS-deprotection, DMP-based oxidation of the resulting primary alcohol,48 and Wittig olefination.
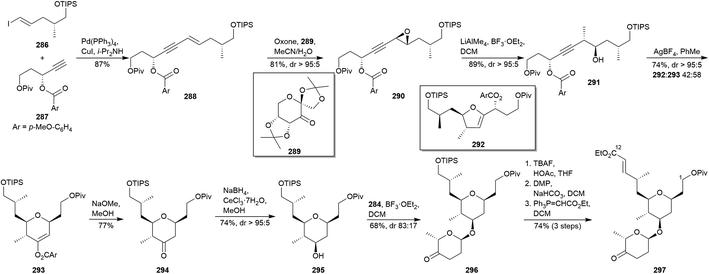 |
| | Scheme 26 Carter's approach to madeirolide A (5a). This figure has been adapted from ref. 158 with permission from ACS PUBLICATIONS, copyright 2016. | |
6.3 Lee's contribution (2016)162
Lee and co-workers disclosed their approach towards the C1–C10 fragment of madeirolide A (5a).162 Their approach involved the use of an Ir-catalysed visible light induced radical cyclisation to construct the THP ring and a Pd-catalysed glycosylation to assembly the saccharide fragment. Known anti-aldol 298 was selected as starting material (Scheme 27).163 It was submitted to a reductive removal of the chiral auxiliary and selectively silylated to yield alcohol 299. O-Acylation allowed the access to 300, whose Ireland–Claisen rearrangement yielded acid 301 with excellent stereoselectivity. Stereoselective iodolactonization, reductive deiodination of 302 using iridium catalyst 303, Hantzsch ester (304), DIPEA and visible light (λmax = 454 nm) as photocatalyst, and subsequent reduction of the lactone provided 305. This diol was then submitted to an acetalisation with dimethyl acetal 306 to furnish the diastereomeric mixture of cyclic acetal 307. Chemo and stereoselective basic elimination induced ring opening to yield acrylate 308,164 whose primary alcohol was transformed into iodide derivate 309. Once again, exposure of 309 to visible light in the presence of 303, 304 and DIPEA led to a radical mediated cyclisation165 to obtain the 2,6-cis-THP 310. After benzyl hydrogenolysis, a subsequent Pd-catalysed glycosylation of 310 with pyranone 311166 gave glycoside 312167 Eventually, hydrogenation of the alkene in 312 furnished the C1–C10 fragment (313) of madeirolide A (5a).
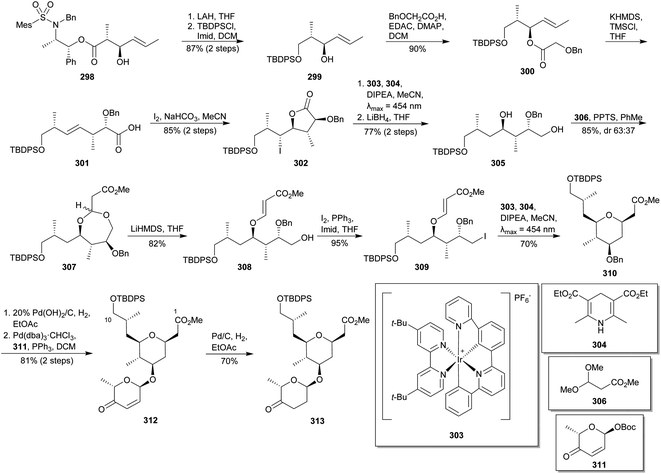 |
| | Scheme 27 Lee's approach to madeirolide A (5a). This figure has been adapted from ref. 162 with permission from ACS PUBLICATIONS, copyright 2016. | |
7. Conclusions
In this review we have provided a comprehensive study about the synthetic efforts performed towards the preparation of some marine macrolides. Due to the vast amount of these metabolites, we focused our interest in those families containing 4-O-2,3,4,6-tetrasubstituted THPs in their structures. Thus, synthesis directed at miyakolide, polycavernosides A and B, lasonolide A, clavosolide A and madeirolide A were tackled in detail, with a special emphasis on the strategies used for building target THPs (see a summary in Table 2). We hope that this bibliographic update presented herein may become a useful tool for the synthetic community to continue being inspired by the fascinating world of marine macrolides.
Table 2 Summary of the strategies employed to the building of the 4-O-2,3,4,5-tetrasubstituted THPs
| NP |
Group |
Strategy |
| 1 |
Masamune's (1997)17 |
Intramolecular nucleophilic addition |
| 1 |
Evans' (1999)15 |
Intramolecular heteroconjugate addition |
| 1 |
Trost's (2008)29 |
(1) Ru-catalyzed alkene–alkyne coupling, (2) Pd-catalyzed allylic alkylation |
| 2a |
Markó's (2009)42 |
— |
| 2a |
Lee's (2010)44 |
Intramolecular Prins cyclisation |
| 2a |
Sasaki's (2012)55 |
Catalytic asymmetric hetero Diels–Alder reaction |
| 2a |
Fürstner's (2013)66 |
Intramolecular oxy-Michael reaction |
| 2a/2b |
Sasaki's (2017)73 |
Catalytic asymmetric hetero Diels–Alder reaction |
| 2a |
Kadari & Yerrabelly's (2018)77 |
— |
| 3a |
Jennings' (2006)88 |
(1) Molander–Reformatsky intramolecular aldol addition, (2) alkylation, (3) reduction |
| 3a |
Jennings' (2007)95 |
— |
| 3a |
Ghosh's (2007)98 |
Jacobsen catalytic asymmetric hetero-Diels–Alder |
| 3a |
Ghosh's (2008)98a |
— |
| 3a |
Ghosh's (2012)107 |
Jacobsen catalytic asymmetric hetero-Diels–Alder |
| 3a |
Trost's (2014)113 |
Simultaneous Hoveyda–Grubbs 2nd generation catalyzed cross metathesis/intramolecular oxa-Michael addition |
| 3a |
Trost's (2018)123 |
Intramolecular oxa-Michael addition |
| 3a |
Hong's (2018)124 |
Intramolecular oxa-Michael addition |
| 4a |
Raghavan's (2017)134 |
— |
| 4a |
Krische's (2019)143 |
Intramolecular alkoxypalladation/carbonylation of alkenes |
| 5a |
Paterson's (2013)150 |
Intramolecular Michael cyclisation |
| 5a |
Carter's (2016)158 |
Silver catalysed cyclisation of propargyl benzoate |
| 5a |
Lee's (2016)162 |
Ir-catalysed visible light induced radical cyclisation |
Abbreviations
| 2,2-DMP | 2,2-Dimethoxypropane |
| 2,6-lut | 2,6-Lutidine |
| 9-BBN | 9-Borabicyclo[3.3.1]nonane |
| Ac | Acetate |
| BAIB | Bis(acetoxy)iodobenzene |
| BINAP | 2,2′-Bis(diphenylphosphino)-1,1′-binaphthyl |
| Bn | Benzyl |
| Boc | tert-Butoxycarbonyl |
| brsm | Based on recovered starting material |
| Bz | Benzoyl |
| c-hex | Cyclohexyl |
| Cp | Pentamethylcyclopentadienyl |
| CSA | Camphorsulfonic acid |
| dba | Dibenzylideneacetone |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCC | N,N′-Dicyclohexylcarbodiimide |
| DCM | Dichloromethane |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| DET | Diethyl tartrate |
| (DHQ)2PYR | Hydroquinine 2,5-diphenyl-4,6-pyrimidinediyl diether |
| DIAD | Diisopropyl azodicarboxylate |
| DIBAL-H | Diisobutylaluminum hydride |
| DIPEA | N,N-Diisopropylethylamine |
| DIPT | N,N-Diisopropyltryptamine |
| DMAc | Dimethylacetamide |
| DMAP | 4-Dimethylaminopyridine |
| DMD | Dimethyldioxirane |
| DME | Dimethoxyethane |
| DMF | Dimethylformamide |
| DMP | Dess–Martin periodinane |
| DMSO | Dimethyl sulphoxide |
| dppf | 1,1′-Bis(diphenylphosphino)ferrocene |
| dr | Diastereomeric ratio |
| EDAC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| ee | Enantiomeric excess |
| Et | Ethyl |
| HMDS | Hexamethyldisilazane |
| HMPA | Hexamethylphosphoramide |
| HWE | Horner–Wadsworth–Emmons |
| IC50Half maximal inhibitory concentrationImid | Half maximal inhibitory concentrationImidImidazole |
| Ipc | Isopinocampheyl |
| ir | Isomeric ratio |
| LAH | Lithium aluminium hydride |
| LCLo | Lethal concentration low |
| LDA | Lithium diisopropylamide |
| m-CPBA | meta-Chloroperoxybenzoic acid |
| Me | Methyl |
| Mes | Mesityl |
| Ms | Mesyl |
| MS | Molecular sieves |
| MTM | Methylthiomethyl |
| NADP | Nicotinamide adenine dinucleotide phosphate |
| naph | Naphthalene |
| NBS | N-Bromosuccinimide |
| NCS | N-Chlorosuccinimide |
| NMO | N-Methylmorpholine N-oxide |
| NP | Natural product |
| PCC | Pyridinium chlorochromate |
| PDC | Pyridinium dichromate |
| Piv | Pivaloyl |
| PMB | p-Methoxybenzyl |
| PNP | p-Nitrophenol |
| PPTS | Pyridinium p-toluenesulfonate |
| PTSA | p-Toluenesulfonic acid |
| py | Pyridine |
| RCAM | Ring closing alkyne metathesis |
| RCM | Ring-closing metathesis |
| sia | sec-Isoamyl |
| TBA | Tetra-n-butylammonium |
| TBDPS | tert-Butyldiphenylsilyl |
| TBHP | tert-Butyl hydroperoxide |
| TBS | tert-Butyldimethylsilyl |
| TDMPP | Tris(2,6-dimethoxyphenyl)phosphine |
| TEA | Triethyl amine |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl |
| TES | Triethyl silane |
| Tf | Triflate |
| TFA | Trifluoroacetic acid |
| THP | Tetrahydropyran |
| THT | Tetrahydrothiophene |
| TIPS | Triisopropylsilyl |
| TMG | 1,1,3,3-Tetramethylguanidine |
| TMS | Trimethylsilyl |
| TPAP | Tetrapropylammonium perruthenate |
| Ts | Tosyl |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Ministerio de Ciencia, Innovación y Universidades (MCIU) of Spain-European Regional Development Fund (ERDF) (PGC2018-094503-B-C22) and ACIISI (Canary Islands Government, ProID2017010118).
Notes and references
-
(a) G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, 2013, 1830, 3670–3695 CrossRef CAS;
(b) A. L. Harvey, Drug Discovery Today, 2008, 13, 894–901 CrossRef CAS;
(c) A. Ganesan, Curr. Opin. Chem. Biol., 2008, 12, 306–317 CrossRef CAS.
- R. Montaser and H. Luesch, Future Med. Chem., 2011, 3, 1475–1489 CrossRef CAS.
- D.-X. Kong, Y.-Y. Jiang and H.-Y. Zhang, Drug Discovery Today, 2010, 15, 884–886 CrossRef.
- A. L. Lane and B. S. Moore, Nat. Prod. Rep., 2011, 28, 411–428 RSC.
- S. R. Park, A. R. Han, Y.-H. Ban, Y. J. Yoo, E. J. Kim and Y. J. Yoon, Appl. Microbiol. Biotechnol., 2010, 85, 1227–1239 CrossRef CAS.
- K.-S. Yeung and I. Paterson, Chem. Rev., 2005, 105, 4237–4313 CrossRef CAS.
-
(a) X. Song, G. Yuan, P. Li and S. Cao, Molecules, 2019, 24, 3913 CrossRef CAS;
(b) M. Wang, J. Zhang, S. He and X. Yan, Mar. Drugs, 2017, 15, 126 CrossRef;
(c) D. J. Newman and G. M. Cragg, in Macrocycles in Drug Discovery, ed. J. Levin, The Royal Society of Chemistry, Cambridge, 2014, ch. 1, pp. 1–36 Search PubMed.
-
(a) H. Fuwa, Mar. Drugs, 2016, 14, 65 CrossRef;
(b) N. M. Nasir, K. Ermanis and P. A. Clarke, Org. Biomol. Chem., 2014, 12, 3323–3335 RSC;
(c) P. A. Clarke and S. Santos, Eur. J. Org. Chem., 2006, 2045–2053 CrossRef CAS.
- Q. Lu, D. S. Harmalkar, Y. Choi and K. Lee, Molecules, 2019, 24, 3778 CrossRef.
-
(a) S. J. Álvarez-Méndez, M. Fariña-Ramos, M. L. Villalba, M. D. Perretti, C. García, L. M. Moujir, M. A. Ramírez and V. S. Martín, J. Org. Chem., 2018, 83, 9039–9066 CrossRef;
(b) N. M. Raju, J. M. Babu and B. V. Rao, Asian J. Chem., 2017, 29, 2771–2775 CrossRef CAS;
(c) A. Millán, J. R. Smith, J. L.-Y. Chen and V. K. Aggarwal, Angew. Chem., Int. Ed., 2016, 55, 2498–2502 CrossRef;
(d) A. Mahmood, J. R. Suárez, S. P. Thomas and V. K. Aggarwal, Tetrahedron Lett., 2013, 54, 49–51 CrossRef CAS;
(e) C. E. Bennett, R. Figueroa, D. J. Hart and D. Yang, Heterocycles, 2006, 70, 119–128 CrossRef CAS;
(f) C. St, J. Barry, S. R. Crosby, J. R. Harding, R. A. Hughes, C. D. King, G. D. Parker and C. L. Willis, Org. Lett., 2003, 5, 2429–2432 CrossRef.
- T. Higa, J. Tanaka, M. Komesu, D. G. Gravalos, J. L. Fernandez Puentes, G. Bernardinelli and C. W. Jefford, J. Am. Chem. Soc., 1992, 114, 7587–7588 CrossRef CAS.
- L. Barriault, S. L. Boulet, K. Fujiwara, A. Murai, L. A. Paquette and M. Yotsu-Yamashita, Bioorg. Med. Chem. Lett., 1999, 9, 2069–2072 CrossRef CAS.
- P. A. Horton, F. E. Koehn, R. E. Longley and O. J. McConnell, J. Am. Chem. Soc., 1994, 116, 6015–6016 CrossRef CAS.
- P. L. Winder, PhD thesis, Florida Atlantic University, 2009.
- D. A. Evans, D. H. B. Ripin, D. P. Halstead and K. R. Campos, J. Am. Chem. Soc., 1999, 121, 6816–6826 CrossRef CAS.
- P. Kollár, J. Rajchard, Z. Balounová and J. Pazourek, Pharm. Biol., 2014, 52, 237–242 CrossRef.
- T. Yoshimitsu, J. J. Song, G.-Q. Wang and S. Masamune, J. Org. Chem., 1997, 62, 8978–8979 CrossRef CAS.
- A. Abiko, J.-F. Liu and S. Masamune, J. Am. Chem. Soc., 1997, 119, 2586–2587 CrossRef CAS.
-
(a) S. Saito, T. Ishikawa, A. Kuroda, K. Koga and T. Moriwake, Tetrahedron, 1992, 48, 4067–4086 CrossRef CAS;
(b) A. Meyers and J. P. Lawson, Tetrahedron Lett., 1982, 23, 4883–4886 CrossRef CAS.
- A. J. Mancuso and D. Swern, Synthesis, 1981, 165–185 CrossRef CAS.
- R. Appel, Angew. Chem., Int. Ed., 1975, 14, 801–811 CrossRef.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- P. Brownbridge, T. H. Chan, M. A. Brook and G. J. Kang, Can. J. Chem., 1983, 61, 688–693 CrossRef CAS.
- D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127–2129 CrossRef CAS.
- K. Shimoji, H. Taguchi, K. Oshima, H. Yamamoto and H. Nozaki, J. Am. Chem. Soc., 1974, 96, 1620–1621 CrossRef CAS.
-
(a) W. S. Wadsworth and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733–1738 CrossRef CAS;
(b) L. Horner, H. M. R. Hoffmann, H. G. Wippel and G. Klahre, Chem. Ber., 1959, 92, 2499–2505 CrossRef CAS.
- N. Kornblum, B. Taub and H. E. Ungnade, J. Am. Chem. Soc., 1954, 76, 3209–3211 CrossRef CAS.
- M. Nitta and T. Kobayashi, J. Chem. Soc., Chem. Commun., 1982, 877–878 RSC.
- B. M. Trost and B. L. Ashfeld, Org. Lett., 2008, 10, 1893–1896 CrossRef CAS.
- B. M. Trost and A. J. Frontier, J. Am. Chem. Soc., 2000, 122, 11727–11728 CrossRef CAS.
- B. M. Trost and M. Machacek, Angew. Chem., Int. Ed. Engl., 2002, 41, 4693–4697 CrossRef CAS.
- B. M. Trost, H. Yang and G. Wuitschik, Org. Lett., 2005, 7, 4761–4764 CrossRef CAS.
-
(a) A. K. Chatterjee, T. L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS;
(b) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS.
- M. Yotsu-Yamashita, R. L. Haddock and T. Yasumoto, J. Am. Chem. Soc., 1993, 115, 1147–1148 CrossRef CAS.
- M. Yotsu-Yamashita, T. Seki, V. J. Paul, H. Naoki and T. Yasumoto, Tetrahedron Lett., 1995, 36, 5563–5566 CrossRef CAS.
- M. Yotsu-Yamashita, K. Abe, T. Seki, K. Fujiwara and T. Yasumoto, Tetrahedron Lett., 2007, 48, 2255–2259 CrossRef CAS.
- M. Yotsu-Yamashita, T. Yasumoto, S. Yamada, F. F. A. Bajarias, M. A. Formeloza, M. L. Romero and Y. Fukuyo, Chem. Res. Toxicol., 2004, 17, 1265–1271 Search PubMed.
- G. Navarro, S. Cummings, J. Lee, N. Moss, E. Glukhov, F. A. Valeriote, L. Gerwick and W. H. Gerwick, Environ. Sci. Technol. Lett., 2015, 2, 166–170 Search PubMed.
- E. Cagide, M. C. Louzao, I. R. Ares, M. R. Vieytes, M. Yotsu-Yamashita, L. A. Paquette and T. Yasumoto, Cell. Physiol. Biochem., 2007, 19, 185–194 CrossRef CAS.
- K. Fujiwara, A. Murai, M. Yotsu-Yamashita and T. Yasumoto, J. Am. Chem. Soc., 1998, 120, 10770–10771 CrossRef CAS.
- L. A. Paquette and M. Yotsu-Yamashita, in Phycotoxins: Chemistry and Biochemistry, ed. L. M. Botana, Blackwell Publishing, Iowa, 2007, ch. 15, pp. 275–296 Search PubMed.
- P. Jourdain, F. Philippart, R. Dumeunier and I. E. Markó, Tetrahedron Lett., 2009, 50, 3366–3370 CrossRef CAS.
- C. Pérez-Balado and I. E. Markó, Tetrahedron, 2006, 62, 2331–2349 CrossRef.
- S. K. Woo and E. Lee, J. Am. Chem. Soc., 2010, 132, 4564–4565 CrossRef CAS.
- J. Nokami, M. Ohga, H. Nakamoto, T. Matsubara, I. Hussain and K. Kataoka, J. Am. Chem. Soc., 2001, 123, 9168–9169 CrossRef CAS.
- C. H. Kim, H. J. An, W. K. Shin, W. Yu, S. K. Woo, S. K. Jung and E. Lee, Angew. Chem., Int. Ed., 2006, 45, 8019–8021 CrossRef CAS.
- P. R. Blakemore, C. C. Browder, J. Hong, C. M. Lincoln, P. A. Nagornyy, L. A. Robarge, D. J. Wardrop and J. D. White, J. Org. Chem., 2005, 70, 5449–5460 CrossRef CAS.
-
(a) D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277–7287 CrossRef CAS;
(b) D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- S. Bhunia, K.-C. Wang and R.-S. Liu, Angew. Chem., Int. Ed., 2008, 47, 5063–5066 CrossRef CAS.
- W. P. Griffith, S. V. Ley, G. P. Whitcombe and A. D. White, J. Chem. Soc., Chem. Commun., 1987, 1625–1627 RSC.
- K. Takai, K. Nitta and K. Utimoto, J. Am. Chem. Soc., 1986, 108, 7408–7410 CrossRef CAS.
- K. C. Nicolaou, S. P. Seitz and D. P. Papahatjis, J. Am. Chem. Soc., 1983, 105, 2430–2434 CrossRef CAS.
- K. Fujiwara, S. Amano and A. Murai, Chem. Lett., 1995, 24, 855–856 CrossRef.
- L. A. Paquette, L. Barriault, D. Pissarnitski and J. N. Johnston, J. Am. Chem. Soc., 2000, 122, 619–631 CrossRef CAS.
- Y. Kasai, T. Ito and M. Sasaki, Org. Lett., 2012, 14, 3186–3189 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- E. P. Boden and G. E. Keck, J. Org. Chem., 1985, 50, 2394–2395 CrossRef CAS.
- J. Robertson, J. W. P. Dallimore and P. Meo, Org. Lett., 2004, 6, 3857–3859 CrossRef CAS.
- A. G. Dossetter, T. F. Jamison and E. N. Jacobsen, Angew. Chem., Int. Ed., 1999, 38, 2398–2400 CrossRef CAS.
-
(a) A. L. Gemal and J. L. Luche, J. Am. Chem. Soc., 1981, 103, 5454–5459 CrossRef CAS;
(b) J. L. Luche, J. Am. Chem. Soc., 1978, 100, 2226–2227 CrossRef CAS.
- E. Lee, Y. R. Lee, B. Moon, O. Kwon, M. S. Shim and J. K. J. Yun, J. Org. Chem., 1994, 59, 1444–1456 CrossRef CAS.
- M. Kurosu, M.-H. Lin and Y. Kishi, J. Am. Chem. Soc., 2004, 126, 12248–12249 CrossRef CAS.
- T. M. Hansen, G. J. Florence, P. Lugo-Mas, J. Chen, J. N. Abrams and C. J. Forsyth, Tetrahedron Lett., 2003, 44, 57–59 CrossRef CAS.
- M. Sasaki and H. Fuwa, Nat. Prod. Rep., 2008, 25, 401–426 RSC.
-
(a) L. J. van den Bos, J. D. C. Codée, J. C. van der Toorn, T. Boltje, J. H. van Boom, H. Overkleeft and G. A. van der Marel, Org. Lett., 2004, 6, 2165–2168 CrossRef CAS;
(b) J. B. Epp and T. S. Widlanski, Org. Chem., 1999, 64, 293–295 CrossRef CAS.
- L. Brewitz, J. Llaveria, A. Yada and A. Fürstner, Chem.–Eur. J., 2013, 19, 4532–4537 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS.
- M. A. McGowan, C. P. Stevenson, M. A. Schiffler and E. N. Jacobsen, Angew. Chem., Int. Ed., 2010, 49, 6147–6150 CrossRef CAS.
- M. Yamaguchi and I. Hirao, Tetrahedron Lett., 1983, 24, 391–394 CrossRef CAS.
- P. Knochel, M. C. P. Yeh, S. C. Berk and J. Talbert, J. Org. Chem., 1988, 53, 2390–2392 CrossRef CAS.
-
(a) K. J. Ralston and A. N. Hulme, Synthesis, 2012, 44, 2310–2324 CrossRef CAS;
(b) D. A. Evans and A. H. Hoveyda, J. Am. Chem. Soc., 1990, 112, 6447–6449 CrossRef CAS.
-
(a) J. Heppekausen, R. Stade, A. Kondoh, G. Seidel, R. Goddard and A. Fürstner, Chem.–Eur. J., 2012, 18, 10281–10299 CrossRef CAS;
(b) J. Heppekausen, R. Stade, R. Goddard and A. Fürstner, J. Am. Chem. Soc., 2010, 132, 11045–11057 CrossRef CAS.
- K. Iwasaki, S. Sasaki, Y. Kasai, Y. Kawashima, S. Sasaki, T. Ito, M. Yotsu-Yamashita and M. Sasaki, J. Org. Chem., 2017, 82, 13204–13219 CrossRef CAS.
-
(a) S.-I. Kiyooka and M. A. Hena, J. Org. Chem., 1999, 64, 5511–5523 CrossRef CAS;
(b) S.-I. Kiyooka, Y. Kaneko, M. Komura, H. Matsuo and M. Nakano, J. Org. Chem., 1991, 56, 2276–2278 CrossRef.
- K. C. Nicolaou, J.-J. Liu, Z. Yang, H. Ueno, E. J. Sorensen, C. F. Claiborne, R. K. Guy, C.-K. Hwang, M. Nakada and P. G. Nantermet, J. Am. Chem. Soc., 1995, 117, 634–644 CrossRef CAS.
-
(a) A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243–251 CrossRef CAS;
(b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS.
- S. Kadari, H. Yerrabelly, T. Gogula, Y. G. Erukala, J. R. Yerrabelly and P. K. Begari, J. Heterocycl. Chem., 2018, 55, 1986–1990 CrossRef CAS.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS.
- P. A. Horton, F. E. Koehn, R. E. Longley, O. J. McConnell and S. A. Pomponi, US Pat., 5684036, 1997 Search PubMed.
-
(a) H. Y. Song, J. M. Joo, J. W. Kang, D.-S. Kim, C.-K. Jung, H. S. Kwak, J. H. Park, E. Lee, C. Y. Hong, S. Jeong, K. Jeon and J. H. Park, J. Org. Chem., 2003, 68, 8080–8087 CrossRef;
(b) E. Lee, H. Y. Song, J. M. Joo, J. W. Kang, D. Kim, C. K. Jung, C. Y. Hong, S. Jeong and K. Jeon, Bioorg. Med. Chem. Lett., 2002, 12, 3519–3520 CrossRef CAS;
(c) E. Lee, H. Y. Song, J. W. Kang, D. Kim, C. Jung and J. M. Joo, J. Am. Chem. Soc., 2002, 124, 384–385 CrossRef CAS.
- A. E. Wright, Y. Chen, P. L. Winder, T. P. Pitts, S. A. Pomponi and R. E. Longley, J. Nat. Prod., 2004, 67, 1351–1355 CrossRef CAS.
- J. M. Joo, H. S. Kwak, J. H. Park, H. Y. Song and E. Lee, Bioorg. Med. Chem. Lett., 2004, 14, 1905–1908 CrossRef CAS.
- R. A. Isbrucker, E. A. Guzmán, T. P. Pitts and A. E. Wright, J. Pharmacol. Exp. Ther., 2009, 331, 733–739 CrossRef CAS.
- Y.-W. Zhang, A. K. Ghosh and Y. Pommier, Cell Cycle, 2012, 11, 4424–4435 CrossRef CAS.
- R. Jossé, Y.-W. Zhang, V. Giroux, A. K. Ghosh, J. Luo and Y. Pommier, Mar. Drugs, 2015, 13, 3625–3639 CrossRef.
-
(a) R. Dubey, C. E. Stivala, H. Q. Nguyen, Y.-H. Goo, A. Paul, J. E. Carette, B. M. Trost and R. Rohatgi, Nat. Chem. Biol., 2020, 16, 206–213 CrossRef CAS;
(b) Z. Li, S. N. S. Morris and J. A. Olzmann, Nat. Chem. Biol., 2020, 16, 111–112 CrossRef CAS.
- M. Shindo, in Marine Natural Products. Topics in Heterocyclic Chemistry, ed. H. Kiyota, Springer, Berlin, 2006, vol. 5, pp. 179–254 Search PubMed.
- K. B. Sawant, F. Ding and M. P. Jennings, Tetrahedron Lett., 2006, 47, 939–942 CrossRef CAS.
- G. A. Molander, J. B. Etter, L. S. Harring and P. J. Thorel, J. Am. Chem. Soc., 1991, 113, 8036–8045 CrossRef CAS.
- E. J. Corey and J. A. Katzenellenbogen, J. Am. Chem. Soc., 1969, 91, 1851–1852 CrossRef CAS.
- M. Nakada, E. Kojima and H. Ichinose, Synth. Commun., 2000, 30, 863–868 CrossRef CAS.
- D. A. Evans, J. M. Takacs, L. R. McGee, M. D. Ennis, D. J. Mathre and J. Bartroli, Pure Appl. Chem., 1981, 53, 1109–1127 CAS.
- M. Prashad, D. Har, H.-Y. Kim and O. Repic, Tetrahedron Lett., 1998, 39, 7067–7070 CrossRef CAS.
- F. Ding and M. P. Jennings, Org. Lett., 2005, 7, 2321–2324 CrossRef CAS.
- K. B. Sawant, F. Ding and M. P. Jennings, Tetrahedron Lett., 2007, 48, 5177–5180 CrossRef CAS.
- K. Maruoka, M. Hasegawa, H. Yamamoto, K. Suzuki, M. Shimazaki and G. Tsuchihashi, J. Am. Chem. Soc., 1986, 108, 3827–3829 CrossRef CAS.
- L. Coppi, A. Ricci and M. Taddei, J. Org. Chem., 1988, 53, 911–913 CrossRef CAS.
-
(a) A. K. Ghosh and G. Gong, Chem.–Asian J., 2008, 3, 1811–1823 CrossRef CAS;
(b) A. K. Ghosh and G. Gong, Org. Lett., 2007, 9, 1437–1440 CrossRef CAS.
- K. Ando, J. Org. Chem., 1998, 63, 8411–8416 CrossRef CAS.
- Y. Gao, J. M. Klunder, R. M. Hanson, H. Masamune, S. Y. Ko and K. B. Sharpless, J. Am. Chem. Soc., 1987, 109, 5765–5780 CrossRef CAS.
- J. C. Medina, M. Salomon and K. S. Kyler, Tetrahedron Lett., 1988, 29, 3773–3776 CrossRef CAS.
- J. Schreiber, H. Maag, N. Hashimoto and A. Eschenmoser, Angew. Chem., Int. Ed., 1971, 10, 330–331 CrossRef CAS.
- X. Ma, R. Saksena, A. Chernyak and P. Kovac, Org. Biomol. Chem., 2003, 1, 775–784 RSC.
-
(a) P. R. Blakemore, W. J. Cole, P. J. Kocienski and A. Morley, Synlett, 1998, 26–28 CrossRef CAS;
(b) J. B. Baudin, G. Hareau, S. A. Julia and O. Ruel, Tetrahedron Lett., 1991, 32, 1175–1178 CrossRef CAS.
- J. Chen, T. Wang and K. Zhao, Tetrahedron Lett., 1994, 35, 2827–2828 CrossRef CAS.
- D. A. Evans, M. D. Ennis and D. J. Mathre, J. Am. Chem. Soc., 1982, 104, 1737–1739 CrossRef CAS.
- A. K. Ghosh and G.-B. Ren, J. Org. Chem., 2012, 77, 2559–2565 CrossRef CAS.
- P.-Y. Caron and P. Deslongchamps, Org. Lett., 2010, 12, 508–511 CrossRef CAS.
- J. R. Parikh and W. v. E. Doering, J. Am. Chem. Soc., 1967, 89, 5505–5507 CrossRef CAS.
- S. E. Denmark and B. J. Herbert, J. Org. Chem., 2000, 65, 2887–2896 CrossRef CAS.
- S. H. Kang, S. Y. Kang, H. Choi, C. M. Kim, H. Jun and J. Youn, Synthesis, 2004, 1102–1114 CrossRef CAS.
- D. R. Williams, D. A. Brooks and M. A. Berliner, J. Am. Chem. Soc., 1999, 121, 4924–4925 CrossRef CAS.
- B. M. Trost, C. E. Stivala, K. L. Hull, A. Huang and D. R. Fandrick, J. Am. Chem. Soc., 2014, 136, 88–91 CrossRef CAS.
- B. Trost, A. Fettes and B. T. Shireman, J. Am. Chem. Soc., 2004, 126, 2660–2661 CrossRef CAS.
- H. S. P. Rao, S. Rafi and K. Padmavathy, Tetrahedron, 2008, 64, 8037–8043 CrossRef.
- Y. Kawai, K. Takanobe and A. Ohno, Bull. Chem. Soc. Jpn., 1995, 68, 285–288 CrossRef CAS.
- J. M. Williams, R. B. Jobson, N. Yasuda, G. Marchesini, U.-H. Dolling and E. J. J. Grabowski, Tetrahedron Lett., 1995, 36, 5461–5464 CAS.
- C.-M. Yu, C. Kim and J. H. Kweon, Chem. Commun., 2004, 2494–2495 RSC.
- B. M. Trost, Z. T. Ball and T. Joge, Angew. Chem., Int. Ed., 2003, 42, 3415–3418 CrossRef CAS.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS.
- H. Fuwa, K. Noto and M. Sasaki, Org. Lett., 2010, 12, 1636–1639 CrossRef CAS.
- T. Hiyama and Y. Hatanaka, Pure Appl. Chem., 1994, 66, 1471–1478 CAS.
- B. M. Trost, C. E. Stivala, D. R. Fandrick, K. L. Hull, A. Huang, C. Pook and R. Kalkofen, J. Am. Chem. Soc., 2016, 138, 11690–11701 CrossRef CAS.
-
(a) L. Yang, Z. Lin, K. Zheng, L. Kong and R. Hong, Chin. J. Chem., 2020, 38, 725–736 CrossRef CAS;
(b) L. Yang and R. Hong, in Strategies and Tactics in Organic Synthesis, ed. M. Harmata, Academic Press, London, 2019, ch. 4, pp. 107–138 Search PubMed;
(c) L. Yang, Z. Lin, S. Shao, Q. Zhao and R. Hong, Angew. Chem., Int. Ed., 2018, 57, 16200–16204 CrossRef CAS , Angew. Chem., Int. Ed., 2019, 58, 4431.
- W. Li, L. Chen, Z. Lin, X. Tian, Y. Wang, S.-H. Huang and R. Hong, Tetrahedron Lett., 2016, 57, 603–606 CrossRef CAS.
- L. Yang, G. He, R. Yin, L. Zhu, X. Wang and R. Hong, Angew. Chem., Int. Ed., 2014, 53, 11600–11604 CrossRef CAS.
- S. H. Kang, S. Y. Kang, C. M. Kim, H.-W. Choi, H. S. Jun, B. M. Lee, C. M. Park and J. W. Jeong, Angew. Chem., Int. Ed., 2003, 42, 4779–4782 CrossRef CAS.
-
(a) J. Hu, M. Bian and H. Ding, Tetrahedron Lett., 2016, 57, 5519–5539 CrossRef CAS;
(b) H. Fuwa, Heterocycles, 2012, 85, 1255–1298 CrossRef CAS.
- M. Rafiee, K. C. Miles and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 14751–14757 CrossRef CAS.
- W. C. Still and C. Gennari, Tetrahedron Lett., 1983, 24, 4405–4408 CrossRef CAS.
- M. R. Rao and D. J. Faulkner, J. Nat. Prod., 2002, 65, 386–388 CrossRef CAS.
- K. L. Erickson, K. R. Gustafson, L. K. Pannell, J. A. Beutlerand and M. R. Boyd, J. Nat. Prod., 2002, 65, 1303–1306 CrossRef CAS.
- K. Lee, M. L. Lanier, J.-H. Kwak, H. Kim and J. Hong, Nat. Prod. Rep., 2016, 33, 1393–1424 RSC.
- S. Raghavan and R. K. Chiluveru, Tetrahedron Lett., 2017, 58, 2465–2467 CrossRef CAS.
- B. Reddy and H. M. Meshram, Tetrahedron Lett., 2010, 51, 4020–4022 CrossRef CAS.
- G. Solladie, G. Demailly and C. Greck, Tetrahedron Lett., 1985, 26, 435–438 CrossRef CAS.
- S. Raghavan, V. S. Babu and B. Sridhar, J. Org. Chem., 2011, 76, 557–565 CrossRef CAS.
- D. A. Evans, S. L. Bender and J. Morris, J. Am. Chem. Soc., 1988, 110, 2506–2526 CrossRef CAS.
- J. Drabowicz and S. Oae, Synthesis, 1977, 404–405 CrossRef CAS.
- K. Matsumara, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef.
- B. M. Trost, Z. T. Ball and K. M. Laemmerhold, J. Am. Chem. Soc., 2005, 127, 10028–10038 CrossRef CAS.
-
(a) K. Tamao and K. Maeda, Tetrahedron Lett., 1986, 27, 65–68 CrossRef CAS;
(b) K. Tamao, K. Maeda, T. Tanaka and Y. Ito, Tetrahedron Lett., 1988, 29, 6955–6956 CrossRef CAS.
- J. M. Cabrera and M. J. Krische, Angew. Chem., Int. Ed., 2019, 58, 10718–10722 CrossRef CAS.
- Y. Lu, I. S. Kim, A. Hassan, D. J. Del Valle and M. J. Krische, Angew. Chem., Int. Ed., 2009, 48, 5018–5021 CrossRef CAS.
-
(a) M. F. Semmelhack and C. Bodurow, J. Am. Chem. Soc., 1984, 106, 1496–1498 CrossRef CAS;
(b) D. M. Fenton and P. J. Steinwand, J. Org. Chem., 1972, 37, 2034–2035 CrossRef CAS.
-
(a) T. Kimura, M. Sekine, D. Takahashi and K. Toshima, Angew. Chem., Int. Ed., 2013, 52, 12131–12134 CrossRef CAS;
(b) D. J. Cox, M. D. Smith and A. J. Fairbanks, Org. Lett., 2010, 12, 1452–1455 CrossRef CAS.
- J. B. Son, S. N. Kim, N. Y. Kim and D. H. Lee, Org. Lett., 2006, 8, 661–664 CrossRef CAS.
- D. A. Evans, B. D. Allison, M. G. Yang and C. E. Masse, J. Am. Chem. Soc., 2001, 123, 10840–10852 CrossRef CAS.
- H. Nozaki, T. Aratani and T. Toraya, Tetrahedron Lett., 1968, 38, 4097–4098 CrossRef.
- I. Paterson and G. W. Haslett, Org. Lett., 2013, 15, 1338–1341 CrossRef CAS.
- I. Paterson, G. J. Florence, K. Gerlach, J. P. Scott and N. Sereinig, J. Am. Chem. Soc., 2001, 123, 9535–9544 CrossRef CAS.
-
(a) J. S. Dickschat, S. Wickel, C. J. Bolten, T. Nawrath, S. Schulz and C. Wittmann, Eur. J. Org. Chem., 2010, 2687–2695 CrossRef CAS;
(b) A. B. Smith III, C. M. Adams, S. A. Lodise Barbosa and A. P. Degnan, J. Am. Chem. Soc., 2003, 125, 350–351 CrossRef.
- H. Kim and J. Hong, Org. Lett., 2010, 12, 2880–2883 CrossRef CAS.
- M. C. Pirrung and N. J. G. Webste, J. Org. Chem., 1987, 52, 3603–3613 CrossRef CAS.
-
(a) M. W. Rathke and M. Nowak, J. Org. Chem., 1985, 50, 2624–2626 CrossRef CAS;
(b) M. A. Blanchette, W. Choy, J. T. Davis, A. P. Essenfeld, S. Masamune, W. R. Roush and T. Sakai, Tetrahedron Lett., 1984, 25, 2183–2186 CrossRef CAS.
- H. Fuwa, N. Ichinokawa, K. Noto and M. Sasaki, J. Org. Chem., 2012, 77, 2588–2607 CrossRef CAS.
-
(a) J. P. M. Nunes, L. Veiros, P. D. Vaz, C. A. M. Afonso and S. Caddick, Tetrahedron, 2011, 67, 2779–2787 CrossRef CAS;
(b) O. Achmatowicz and R. Bielski, Carbohydr. Res., 1977, 55, 165–176 CrossRef CAS.
- K. Watanabe, J. Li, N. Veerasamy, A. Ghosh and R. G. Carter, Org. Lett., 2016, 18, 1744–1747 CrossRef CAS.
-
(a) Y. Ni, R. Kassab, M. Chevliakov and J. Montgomery, J. Am. Chem. Soc., 2009, 131, 17714–17718 CrossRef CAS;
(b) Z. Huang and E. Negishi, Org. Lett., 2006, 8, 3675–3678 CrossRef CAS.
- S. Mahapatra and R. G. Carter, J. Am. Chem. Soc., 2013, 135, 10792–10803 CrossRef CAS.
- Y. Shi, J. Am. Chem. Soc., 1996, 118, 9806–9807 CrossRef.
- S. Hwang, I. Baek and C. Lee, Org. Lett., 2016, 18, 2154–2157 CrossRef CAS.
-
(a) T. Inoue, J. F. Liu, D. C. Buske and A. J. Abiko, J. Org. Chem., 2002, 67, 5250–5256 CrossRef CAS;
(b) A. Abiko, J. F. Liu and S. Masamune, J. Am. Chem. Soc., 1997, 119, 2586–2587 CrossRef CAS.
- M. Caballero, M. Garcia-Valverde, R. Pedrosa and M. Vicente, Tetrahedron: Asymmetry, 1996, 7, 219–226 CrossRef CAS.
- E. Lee, J. S. Tae, C. Lee and C. M. Park, Tetrahedron Lett., 1993, 34, 4831–4834 CrossRef CAS.
- B. Wu, M. Li and G. A. O'Doherty, Org. Lett., 2010, 12, 5466–5469 CrossRef CAS.
-
(a) R. S. Babu, M. Zhou and G. A. O'Doherty, J. Am. Chem. Soc., 2004, 126, 3428–3429 CrossRef CAS;
(b) A. C. Comely, E. Eelkema, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2003, 125, 8714–8715 CrossRef CAS;
(c) R. S. Babu and G. A. O'Doherty, J. Am. Chem. Soc., 2003, 125, 12406–12407 CrossRef CAS.
Footnote |
| † Present address: Sección de Ingeniería Agraria, Escuela Politécnica Superior de Ingeniería, Universidad de La Laguna, Carretera general de Geneto 2, 38200 La Laguna, Tenerife, Spain. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Celina García
ab,
Celina García