
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co/Ni-polyoxotungstate photocatalysts as precursor materials for electrocatalytic water oxidation†
Robin Güttinger‡
 ,
Giann Wiprächtiger‡,
Olivier Blacque and
Greta R. Patzke*
,
Giann Wiprächtiger‡,
Olivier Blacque and
Greta R. Patzke*
Department of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: greta.patzke@chem.uzh.ch; Web: [http://www.patzke.ch]
First published on 18th March 2021
Abstract
An open-core cobalt polyoxometalate (POM) [(A-α-SiW9O34)Co4(OH)3(CH3COO)3]8− Co(1) and its isostructural Co/Ni-analogue [(A-α-SiW9O34)Co1.5Ni2.5(OH)3(CH3COO)3]8− CoNi(2) were synthesized and investigated for their photocatalytic and electrocatalytic performance. Co(1) shows high photocatalytic O2 yields, which are competitive with leading POM water oxidation catalysts (WOCs). Furthermore, Co(1) and CoNi(2) were employed as well-defined precursors for heterogeneous WOCs. Annealing at various temperatures afforded amorphous and crystalline CoWO4- and Co1.5Ni2.5WO4-related nanoparticles. CoWO4-related particles formed at 300 °C showed substantial electrocatalytic improvements and were superior to reference materials obtained from co-precipitation/annealing routes. Interestingly, no synergistic interactions between cobalt and nickel centers were observed for the mixed-metal POM precursor and the resulting tungstate catalysts. This stands in sharp contrast to a wide range of studies on various heterogeneous catalyst types which were notably improved through Co/Ni substitution. The results clearly demonstrate that readily accessible POMs are promising precursors for the convenient and low-temperature synthesis of amorphous heterogeneous water oxidation catalysts with enhanced performance compared to conventional approaches. This paves the way to tailoring polyoxometalates as molecular precursors with tuneable transition metal cores for high performance heterogeneous electrocatalysts. Our results furthermore illustrate the key influence of the synthetic history on the performance of oxide catalysts and highlight the dependence of synergistic metal interactions on the structural environment.
1. Introduction
Sunlight-driven splitting of water into hydrogen and oxygen, also known as artificial photosynthesis, is among the most direct and elegant one-step concepts for renewable energy sources.1 However, the development of efficient and noble-metal free water oxidation catalysts (WOCs) for this complex four electron transfer process is still a crucial bottleneck of artificial photosynthesis.2 A variety of approaches to WOC design have been reported, and in many of them the cuboidal {CaMn4O5} core of nature's photosystem II is a central motif.3 Recently, several transition metal WOCs with cubane-related cores have been reported.4–10Polyoxometalates (POMs) are promising WOC candidates, because they combine robustness and structural versatility with the capability of undergoing rapid, reversible, and stepwise multi-electron transfer reactions, and we also refer to key review articles here.11–16 Among the attractive and low cost 3d transition metal water splitting catalysts, cobalt-based homogeneous catalysts and POMs keep attracting intense attention.17–22 Specifically, Co-POMs have recently been applied as co-catalysts on photoanodes or for enhanced performance in composite systems.23–28.
The coordination of multiple metal centers between two or more lacunary POM units has proven a powerful and quite flexible catalytic motif, such as in the OEC-related [MnIII3MnIVO3(CH3COO)3(SiW9O34)]6− with a mixed-valent {Mn4} core.29 Although many POMs have wide operational stability windows, they can undergo leaching of heteroatoms/transition metals to form active nanoscale oxide catalysts, especially during electrochemical water oxidation.30,31 This gives rise to ongoing and challenging speciation studies.32
Recently, the performance of an amorphous sandwich-type Co-based POM WOC was first enhanced by annealing at 400 °C to form CoWO4 nanoparticles, which outperformed analogous electrocatalysts obtained via precipitation routes.33 Along these lines, mixed-metal manganese cubane34 and Ni–Zn cubane-like35 precursors were further applied to enhance the performance of oxide WOCs. These findings show that synthetic methods and underlying mechanisms36 are an important step in WOC optimization,37–39 and that a certain extent of pre-organization of the metal centers in the precursor is beneficial for higher catalytic activity.40 To this end, we systematically explored synthesis-activity relationships with a series of studies on spinel-type Co3O4 catalysts, starting with in situ PXRD monitoring of temperature-dependent hydrothermal Co3O4 formation mechanisms.41 Next, we revealed the preparation-dependent properties of Co3O4 WOCs in different test assays,42 and studies of their microwave-assisted synthesis further confirmed the crucial role of synthetic pathways for the catalytic performance.43
POM as oxide precursors have enabled the efficient synthesis of CoOx electrocatalysts44 or of ultrafine transition metal-clusters,45 as well as inspirational studies on transformation pathways of pre-organized metal centers into structural features of the resulting multinary heterogeneous catalysts.46,47 Recent trends further employed POMs as versatile metal sources for carbide-, phosphide- and sulphide-based water splitting electrocatalysts.48–50 All in all, the complexity of such precursor-properties relations remains to be fully explored and understood for efficient WOC design.
Another crucial principle in the optimization of transition metal catalysts are synergistic effects between mixed metal centers, such as the widely studied Ni/Fe interactions in WOCs.51–53 In comparison, Co/Ni-interactions in oxygen evolution and other catalysts are far more diverse and controversial. Mixed heterogeneous Co/Ni-oxide electrocatalysts have been studied for several decades54 and were frequently reported to be favourable over binary systems.55–57 However, later studies on mixed Co/Ni-hydroxides pointed to either productive58 or adverse59 effects, or to no significant interactions in the case of oxides at all.60 Although Co/Ni synergisms were recently observed for sulphide61 and phosphide62 water splitting electrocatalysts, their understanding is still empirical to a large extent and modelling studies are now being undertaken.62 Even less is known about the effect of Co/Ni-substitution on molecular WOCs and other catalysts. In our own work, for example, we have observed drastic contrasts between the notable improvement of solid CoNCN WOCs through Ni-doping63 vs. detrimental effects on molecular {Co(II)4O4} cubane WOCs.64 Similar adverse Co/Ni-interactions have been reported for other molecular systems.65 Generally, a wide range of further studies is now needed to explore the role of materials type, preparative method and test conditions (photo- vs. electrochemistry) in the performance of Co/Ni-based catalysts. These widely unresolved questions concerning the prediction and explanation of Co/Ni-interactions inspired us herein to first investigate molecular photocatalyst performance with respect to Ni introduction, followed by its effect on the solid electrocatalysts obtained from such molecular precursors.
To this end, we selected Co/Ni-POMs as attractive and comprehensive models to investigate (a) Co/Ni interactions in molecular WOCs vs. (b) those in oxide-based catalysts, while (c) exploring the benefits of POM precursors for oxide WOCs.
To this end, we targeted CoWO4 with favourable 500–650 nm light absorption properties that was also reported as an effective and noble metal-free WOC at low overpotential.66 Furthermore, CoWO4 performance was found to depend on crystallinity67 with amorphous CoWO4 being superior to its crystalline form, along the lines of self-repairing CoPi films.68–70 However, little is still known about the electrocatalytic performance and other applications of mixed Co/Ni-tungsten oxides.71,72 Binary CoWO4 and NiWO4, for example, display better electrochemical performance in water oxidation than NiCo2O4 spinels.72 While mixed (Co, Ni)WO4 materials are attractive for supercapacitor development,73,74 no synergistic benefits were reported for their use in photocatalytic methylene blue degradation.75
We therefore newly investigated the influence of Ni-doping on the performance of CoWO4-related water oxidation catalysts, which were obtained from crystallographically well-defined, bio-inspired M4-POMs with an exposed metal core architecture. While Ni-containing POMs were reported to be stable and active for water oxidation,76,77 their potential as mixed metal precursors for heterogeneous WOCs still needs to be explored. To this end, we synthesized and characterized [(A-α-SiW9O34)Co4(OH)3(CH3COO)3]8− Co(1) together with its isostructural analogue [(A-α-SiW9O34)Co1.5Ni2.5(OH)3(CH3COO)3]8− CoNi(2).78 First, both POMs were compared with respect to their respective photo- and electrocatalytic water oxidation activity. Moreover, they were used as annealing precursors to form CoWO4- and mixed (Co,Ni)WO4-related electrocatalysts, and Co(1) was found to be an efficient precursor at temperatures as low as 300 °C.
2. Experimental
2.1 Materials
All chemicals were used as purchased without purification. The lacunary precursor Na10[A-α-SiW9O34]·19H2O was synthesized as previously described.792.2 Physical methods
Attenuated total reflectance Fourier-transform (ATR-FT-IR) spectra were recorded on a Bruker Vertex 70 spectrometer equipped with a Platinum ATR accessory containing a diamond crystal. UV/Vis spectra were recorded on a Lambda 650 S PerkinElmer UV-visible spectrometer in the range of 200–800 nm using a Quartz SUPRASIL precision cell (10 mm). Raman spectroscopy was recorded with a Renishaw inVia Qontor confocal Raman microscope equipped with a diode laser (785 nm). Thermogravimetric analyses were performed on a Netzsch STA 449C between 24 and 600 °C with a heating rate of 10 K min−1 in N2 atmosphere and Al2O3 crucible. PXRD patterns were recorded on a STOE STADI P diffractometer (transmission mode, Ge monochromator) with Cu or Mo radiation. XPS analysis was performed using a PhI 5000 VersaProbe spectrometer (ULVAC-PHI, Inc.) equipped with a 180° spherical capacitor energy analyzer and a multi-channel detection system with 16 channels. Spectra were acquired at a base pressure of 5 × 10−8 Pa using a focused scanning monochromatic Al-Kα source (1486.6 eV) with a spot size of 200 μm and 50 W. The instrument was run in the FAT analyzer mode with electrons emitted at 45° to the surface normal. Pass energy used for survey scans was 187.85 eV and 46.95 eV for detail spectra. Charge neutralisation utilizing both a cool cathode electron flood source (1.2 eV) and very low energy Ar+ ions (10 eV) was applied throughout the analysis.Visible-light-driven water oxidation was first monitored in solution using an oxygen sensor (OX-N) Clark electrode from Unisense. Constant temperature was maintained with a mineral insulated thermosensor (2 mm tip diameter, TP2000, Unisense). Second, O2 evolution was measured in the headspace of the vial using an Agilent Technologies 7820A gas chromatograph with helium as the carrier gas and a 3 m × 2 mm packed molecular sieve 13 × 80–100 column to separate O2 and N2. The oven was operated isothermally at 100 °C. The analysis of the headspace was performed by taking 100 μL samples with a Hamilton (1825 RN) gas-tight microliter syringe. Gases were detected using a thermal conductivity detector (Varian) operated at 200 °C.
Cyclic voltammetry (CV) measurements were performed on a Metrohm 797 VA Computrace instrument with a platinum electrode (Metrohm AG, 2 mm diameter) as a working electrode, Ag/AgCl reference electrode (sat. KCl, 0.197 V vs. NHE) and platinum plate (Metrohm AG) counter electrode. Prior to all measurements, solutions were deaerated with Ar for 15 min. The platinum working electrode was polished between runs with alumina slurry, thoroughly rinsed with water and dried under ambient conditions. The platinum plate was washed in a nitric acid/hydrogen peroxide (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution for 5 min and dried with N2. The working electrodes were produced by dispersing 5 mg of the sample in 100 μL of H2O, applying 40 μL of this dispersion on 1 cm2 fluorine doped tin oxide (FTO), and drying the electrodes at 80 °C for 30 min before covering with 10 μL Nafion 1% solution.
:1) solution for 5 min and dried with N2. The working electrodes were produced by dispersing 5 mg of the sample in 100 μL of H2O, applying 40 μL of this dispersion on 1 cm2 fluorine doped tin oxide (FTO), and drying the electrodes at 80 °C for 30 min before covering with 10 μL Nafion 1% solution.
2.3 K5Na3[(A-α-SiW9O34)Co4(OH)3(CH3COO)3]·15H2O (1)78
Co(CH3COO)2·4H2O (0.712 g, 2.86 mmol) was dissolved in an aqueous solution of potassium acetate (0.5 M, 16 mL), adjusted to pH 8 with HCl and stirred for 15 min. Na10[A-α-SiW9O34]·19H2O (1.977 g, 0.7 mmol) was added and stirred for 45 min at 40 °C. The mixture was then cooled to room temperature and placed in the fridge for 10 min. The purple suspension was centrifuged, filtered and left at room temperature for slow evaporation. After three weeks purple crystals were collected and analysed by FT-IR and Raman spectroscopy, powder X-ray diffraction, ICP-MS, EDX and ESI-MS. (yield 0.34 g, 15% based on tungsten). FT-IR:![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1598 (m), 1552 (m), 1415 (m), 1350 (w), 979 (w), 931 (m), 883 (s), 792 (s), 665 (s), 514 (s), 451 cm−1(m). ESI-MS: 879.3938 [M-(CH3COO) + 5H+]3−.
= 1598 (m), 1552 (m), 1415 (m), 1350 (w), 979 (w), 931 (m), 883 (s), 792 (s), 665 (s), 514 (s), 451 cm−1(m). ESI-MS: 879.3938 [M-(CH3COO) + 5H+]3−.
2.4 K5Na3[(A-α-SiW9O34)Co1.5Ni2.5(OH)3(CH3COO)3]·16H2O (2)
Co(CH3COO)2·4H2O (0.352 g, 1.41 mmol) and Ni(CH3COO)2·4H2O (0.366 g, 1.47 mmol) were dissolved in an aqueous solution of potassium acetate (1 M, 16 mL), adjusted to pH 8 with HCl and stirred for 15 min. Na10[A-α-SiW9O34]·19H2O (1.513 g, 0.53 mmol) was added and stirred for 45 min at 40 °C. The mixture was then cooled to room temperature and placed in the fridge for 10 min. The purple suspension was centrifuged, filtered and left at room temperature for slow evaporation. After two weeks light purple crystals were collected and analysed by FT-IR and Raman spectroscopy, powder X-ray diffraction, ICP-MS, EDX and ESI-MS (yield 0.23 g, 13% based on tungsten). FT-IR: = 1606 (m), 1556 (m), 1421 (m), 1350 (w), 979 (w), 941 (m), 883 (s), 802 (s), 665 (s), 514 (s), 451 cm−1 (m). ESI-MS: 876.7113 [M-(CH3COO) + 5H+]3−.
= 1560 (m), 1404 (m), 1338 (w), 1116 (w), 987 (w), 939 (m), 865 (s), 779 (s), 661 (s), 524 cm−1 (m). = 1564 (w), 1404 (w), 1340 (w), 1128 (w), 939 (m), 852 (s), 771 (s), 702 (s), 538 cm−1 (m). = 1128 (m), 929 (w), 823 (s), 790 (s), 599 (s), 518 (s), 460 cm−1 (m). = 1114 (m), 931 (w), 819 (s), 599 (s), 516 (s), 462 cm−1 (s). = 1562 (m), 1407 (m), 1344 (w), 1122 (w), 981 (w), 933 (m), 858 (s), 798 (s), 671 (s), 520 cm−1 (m). = 1569 (w), 1404 (w), 1128 (w), 933 (m), 838 (s), 788 (s), 711 (s), 540 cm−1 (m). = 1107 (w), 941 (m), 858 (s), 813 (s), 773 (s), 725 cm−1 (s). = 1116 (m), 933 (w), 821 (s), 605 (s), 509 (s), 466 cm−1 (m).3. Results and discussion
3.1 Synthesis and analytical characterization
[(A-α-SiW9O34)Co4(OH)3(CH3COO)3]8− Co(1) was synthesized by mixing stoichiometric amounts of the precursor80 Na10[A-α-SiW9O34] and cobalt acetate in potassium acetate (0.5 M, pH 8) solution with moderate heating. After cooling to room temperature, the mixture was filtered and any insoluble residue was removed, whereupon crystals were obtained after slow evaporation.FT-IR analysis shows characteristic bands for the bidentate bridging acetate ligands in the range of 1650 to 1400 cm−1. Additional bands related to the Keggin structure appear around 934 (νas(W–Od)), 883 (νas(W–Ob)) and 740 cm−1 (νas(W–Oc)) (Fig. S1†).81 Raman spectra show representative peaks at 959 cm−1 (νas(W–Od)), 939 cm−1 (νas(W–Ob–W)) and 891 cm−1 (νas(W–Ob–W)) of Keggin-type POMs (see below, Fig. 3).80,82 Further analysis with powder X-ray diffraction (PXRD) of Co(1) (Fig. S13†) showed crystalline purity when compared to the calculated pattern (CCDC-619251).
The all-cobalt POM Co(1) was further mixed with nickel acetate in a stoichiometric 1:1 ratio under slightly changed reaction conditions. The filtration process after the synthesis had to be extended, but phase pure crystals were obtained after slow evaporation and yielded [(A-α-SiW9O34)Co1.5Ni2.5(OH)3(CH3COO)3]8− CoNi(2). FT-IR analysis confirms the presence of the bridging acetate ligands as well as the characteristic bands of the Keggin-type POM at around 941 (νas(W–Od)), 883 (νas(W–Ob)) and 748 cm−1 (νas(W–Oc)) (Fig. S2†).81 The PXRD pattern confirmed phase purity of CoNi(2) and its isostructural relation to Co(1), and only small peak shifts of the peaks towards higher angles are visible which corresponds to a smaller unit cell, as expected (Fig. S16†). Raman spectra show the same representative peaks as observed for Co(1) (see below, Fig. 3).
UV-Vis monitoring of Co(1) in borate buffer (0.1 M, pH 8) during 24 h showed no significant changes in the spectra. This indicates that Co(1) is stable and does not leach any Co2+ ions into the solution under these operational conditions (Fig. S6†). According to previous studies,78 the tetracobalt core is stabilized by an all-inorganic tungstosilicate, as well as bridged by three μ2-acetate ligands (Fig. 1). All CoII centers of the {CoII4O3} core are in an octahedral environment and the whole unit displays CS symmetry, with a mirror plane through the Co3, Co2 and Si atoms. Three CoII centers are connected to the lacunary side of the [α-SiW9O34]10− POM. The Co–Co distances fall in the range of 2.978(1)–3.711(2) Å and the Co–O distances range from 2.043(0) to 2.117(7) Å, respectively.78 We confirmed the presence of these structural features in Co(1) with single crystal X-ray diffraction analyses giving rise to analogous values (data not shown). The tetracobalt core displays features related to the natural OEC with Mn–Mn distances in the range of 2.8–3.3 Å,3 and the acetate-bridged cobalt centers relate it to previously reported {CoII4O4} cubanes.6,83,84
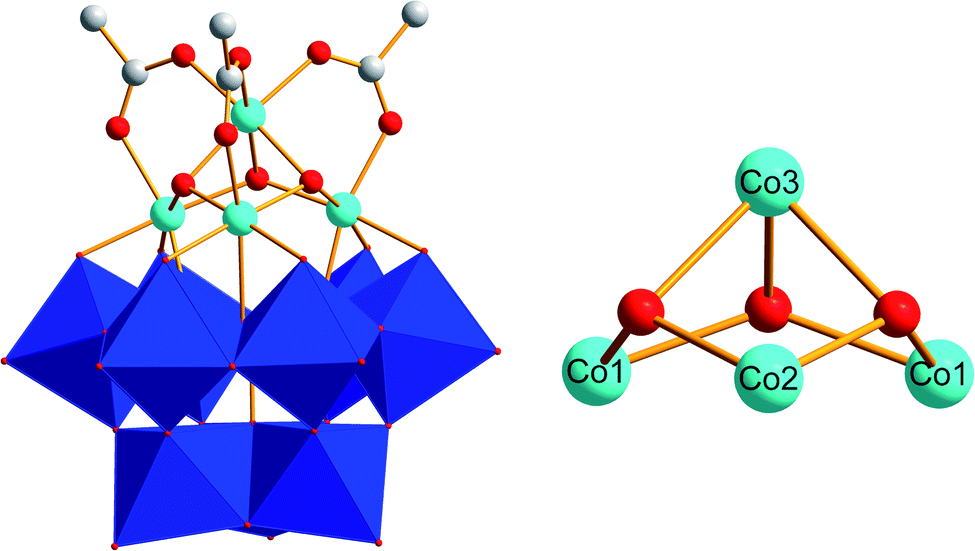 | ||
| Fig. 1 Polyhedral and ball-and-stick representation of the [(A-α-SiW9O34)Co4(OH)3(CH3COO)3]8− polyanion Co(1) (blue octahedra: {WO6}; light blue spheres: Co; white spheres: C; red spheres: O; image derived from CCDC-619251).71 | ||
In addition, mixed nickel/cobalt acetate precursors yielded the iso-structural CoNi(2) with a Co:Ni ratio around 1.5:2.5. This ratio was confirmed with EDX, ICP-MS as well as with XPS measurements (Fig. S20, Tables S3, S11 and S12†). The measured Co/Ni:W as well as Co/Ni:Si ratios correspond to the respective calculated ratios of 4:9 and 4:1 (Tables S11 and S12†). Further HR-ESI-MS analyses showed slightly different masses for the corresponding [M-(CH3COO) + 5H+]3− fragment which correspond to the isotopic distributions of Co and Ni (Fig. S29–S32†). Both POMs display good agreement between experimental PXRD patterns and the respective calculated data (Fig. S13, S15, Tables S1 and S2†). The PXRD pattern of CoNi(2) showed a slight shift of the peaks compared to the calculated reference pattern of Co(1) (CCDC-619251). Further comparison with the calculated PXRD pattern (Fig. S14†) of the lacunary Ni-analogue [(A-α-SiW9O34)Ni4(CH3COO)3]5– that crystallizes in a different space group85 (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 1c other than P21/m for Co(1)78) clearly showed that the phase pure CoNi(2) sample is isostructural with Co(1), which was also confirmed by Rietveld refinement results (Fig. S15†).
1c other than P21/m for Co(1)78) clearly showed that the phase pure CoNi(2) sample is isostructural with Co(1), which was also confirmed by Rietveld refinement results (Fig. S15†).
CoWO4 nanoparticles keep attracting intense interest as target for synthetic studies, e.g. via precipitation,86,87 hydrothermal88,89 or spray pyrolysis routes.90 Here, we newly used both Co(1) and CoNi(2) as precursors for annealing in air at temperatures ranging from 200 to 500 °C. With a ramping rate of 5 °C min−1 and an annealing time of 1 h, amorphous and crystalline nanoparticles were formed. PXRD patterns show the presence of an amorphous material up to 300 °C, while at temperatures of 400 °C and above a crystalline material emerges from both precursor types (Fig. 2). The majority of the peaks in patterns recorded with MoKα radiation can be assigned to monoclinic CoWO4 (PDF 01-072-0479) and its Ni-doped analogue, in line with previous studies.86,89,90 While a preceding study on the use of sandwich-type [Co4(H2O)2(PW9O34)2]10− POMs for cobalt tungstate catalysts reported on the formation of Na2W2O7 as a secondary phase at annealing temperatures of 500 °C,33 we did not find any indication for major W- or Co-based side products. Extensive database search provided SiO2 (PDF No. 12-0711) as the closest match to account for the minority phases observed here.
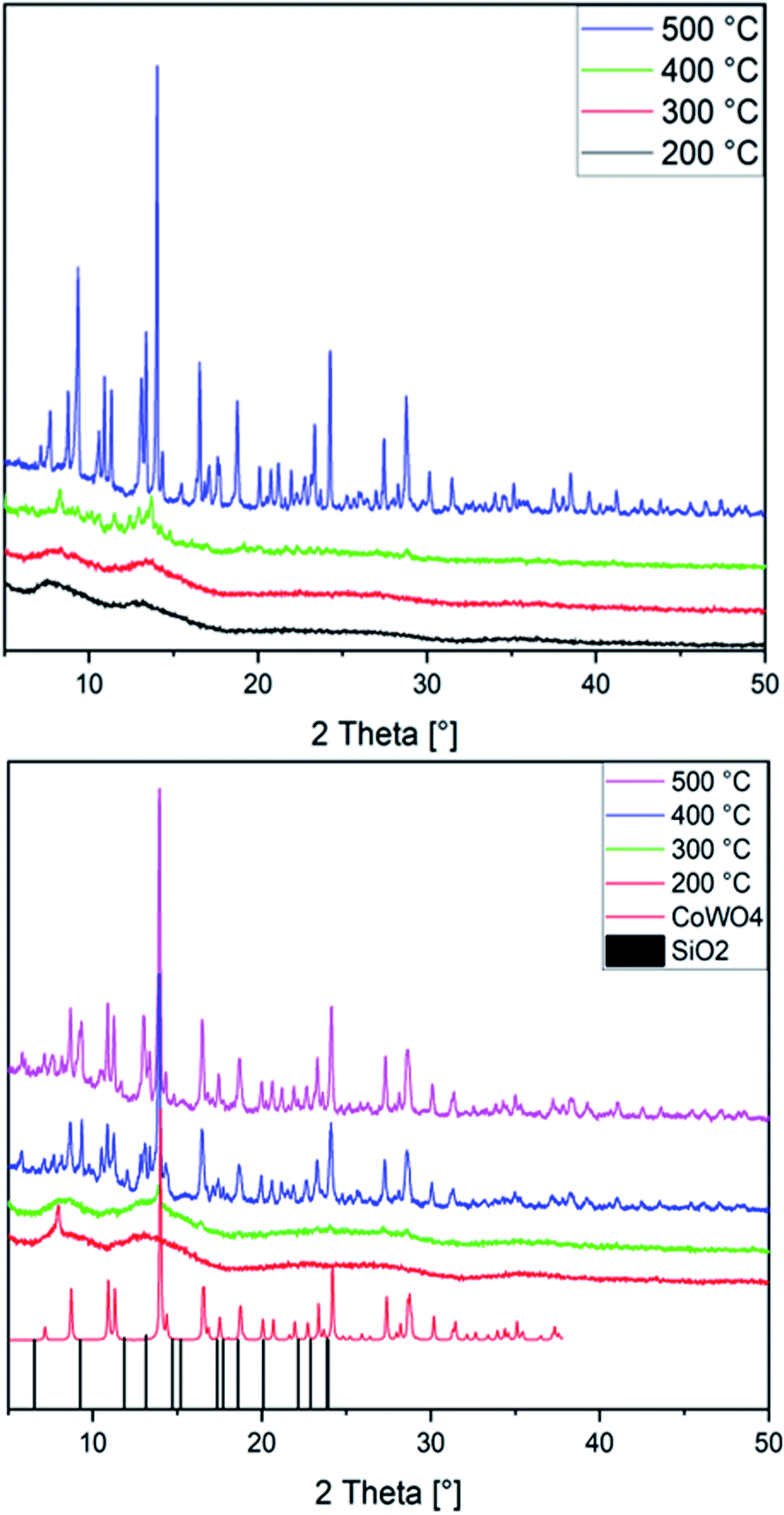 | ||
| Fig. 2 PXRD patterns of the CoNiX00 (top) and CoWX00 (bottom) series annealed at 200 °C (black) up to 500 °C (purple; CoWO4: CCDC-619251, SiO2: PDF No. 12-0711). | ||
Given that SiO2 is widely known as a catalyst support material rather than as active phase, we further considered cobalt tungstosilicates a more reasonable precursor choice than tungstophosphates. The latter may eventually give rise to highly catalytically active cobalt phosphate-related side products. Indeed, phosphorus peaks had been shown in the EDS spectra of the most active amorphous cobalt tungstate catalyst obtained from [Co4(H2O)2(PW9O34)2]10− at 400 °C in the above-mentioned study, but no further discussion of the influence of P heteroatoms on the structure or catalytic performance was provided.33
The Raman spectra of crystalline CoW300/400/500 are in good agreement with the reported pattern of CoWO4 (Fig. 3).91 The most intense band located at 885 cm−1 corresponds to the stretching W–O vibration and is shifted to higher frequencies upon mixing with Ni. The small band around 929 cm−1 can be attributed to the symmetric stretching mode of the terminal (W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bond.66,92 The amorphous CoW300 and CoNi300/400 samples show a small blue shift of the main W–O stretching vibration compared to the crystalline samples. This is a sign of compressive stress, indicating that the respective Co–Co and Co–Ni distances are smaller compared to the crystalline samples.92 The weak peak around 500 cm−1 can be assigned to the minority phase related to SiO2.93
O) bond.66,92 The amorphous CoW300 and CoNi300/400 samples show a small blue shift of the main W–O stretching vibration compared to the crystalline samples. This is a sign of compressive stress, indicating that the respective Co–Co and Co–Ni distances are smaller compared to the crystalline samples.92 The weak peak around 500 cm−1 can be assigned to the minority phase related to SiO2.93
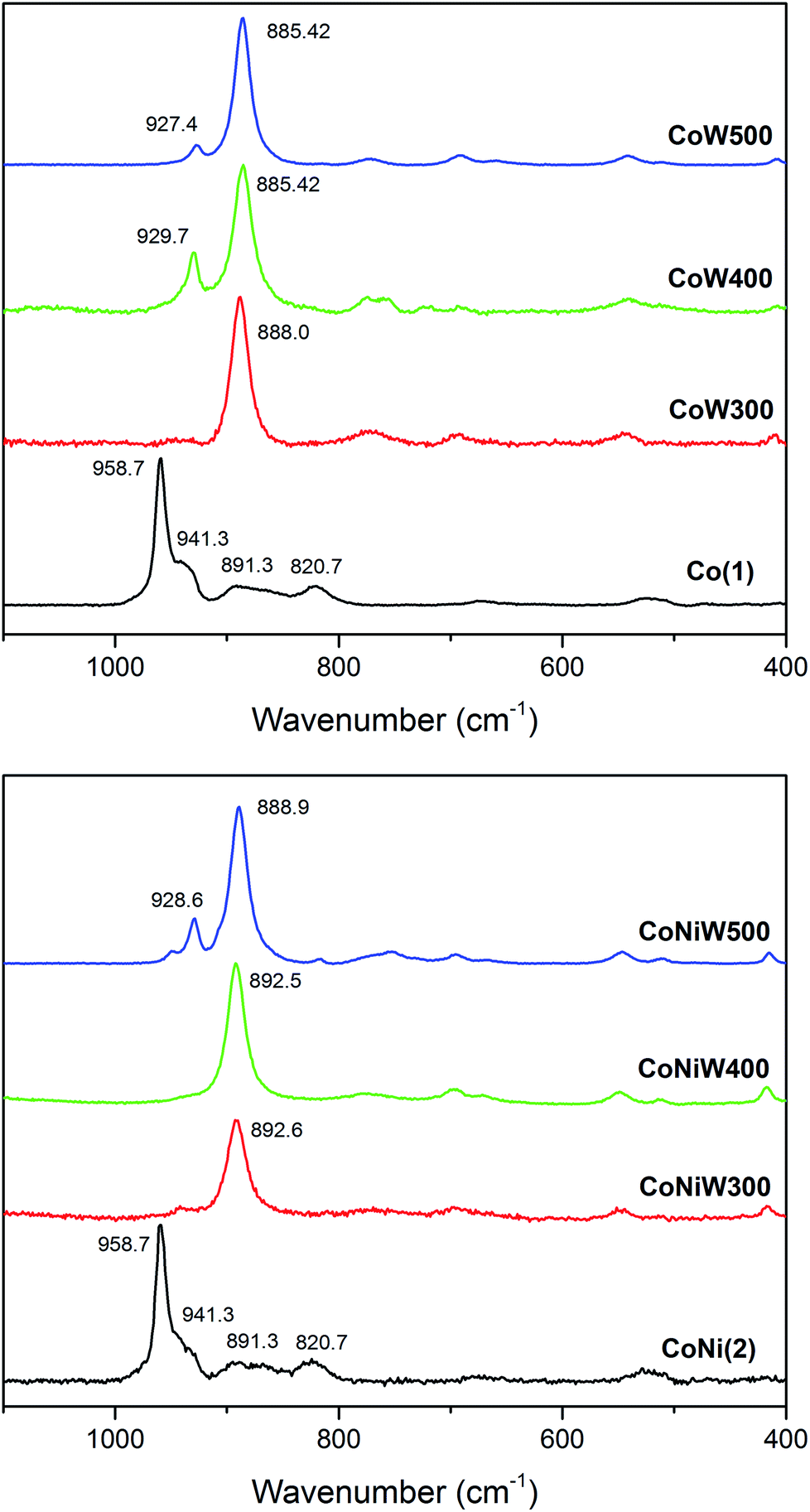 | ||
| Fig. 3 Raman spectra of Co(1) together with the CoW300/400/500 series (top; black/red/green/blue) and of compound CoNi(2) with the CoNiW300/400/500 series (bottom; black/red/green/blue). | ||
EDX mappings of the different CoWX00 and CoNiWX00 tungsten oxides show a homogenous distribution of Co/Ni, W and O in all samples (Fig. S23–S28 and Tables S5–S10†) and the elemental ratios of the CoWX00 series are in line with CoWO4.
3.2 Photo- and electrocatalytic water oxidation activity of Co(1) and CoNi(2)
The general mechanism of photocatalytic water oxidation using the [Ru(bpy)3]2+/S2O82− assay has been studied in numerous works, which are summarized in recent topical reviews,94 including POM water oxidation catalysts.15 In short, a wide range of studies confirmed that the photoexcited state [Ru(bpy)3]2+* is quenched by S2O82− to generate [Ru(bpy)3]3+ along with 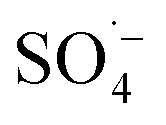 , which can bring forward another molecule of [Ru(bpy)3]3+. To finally generate O2, four holes are first transferred to the POM-WOC via four [Ru(bpy)3]3+ equivalents, and the so oxidized POM catalyst can then further oxidize two water molecules. The precise local mechanisms at the active transition metal centers of different POM-WOCs are subject to advanced theoretical studies and further investigations, and they may vary individually for each POM type.95
, which can bring forward another molecule of [Ru(bpy)3]3+. To finally generate O2, four holes are first transferred to the POM-WOC via four [Ru(bpy)3]3+ equivalents, and the so oxidized POM catalyst can then further oxidize two water molecules. The precise local mechanisms at the active transition metal centers of different POM-WOCs are subject to advanced theoretical studies and further investigations, and they may vary individually for each POM type.95
For photocatalytic performance evaluation, first Co(1) was tested in different buffer solutions and pH values to explore the optimal water oxidation conditions. Borate buffer (0.1 M, pH 8) led to the best performance ahead of borate buffer (0.1 M, pH 9) and phosphate buffer (0.1 M, pH 7). No activity was observed in acetate buffer (0.1 M, pH 4.75) (Fig. S33†).
Second, concentration screening of Co(1) was performed to further optimize the working conditions (Fig. 4, S34 and S35†). Although water oxidation is generally thermodynamically favorable at higher pH values, performances at pH 8 were found to be superior to pH 9,96 in line with other studies.6 The maximum O2 yield of 63% was achieved with 40 μM of Co(1). Compared to other reported Co- or Ni-based POMs, this O2 yield is competitive for the applied photocatalytic assay (Table 1). In comparison, the Mn-based analogue [MnIII3MnIVO3(CH3COO)3(A-α-SiW9O34)]6− exhibited a rather low photocatalytic performance with 3% oxygen yield.29
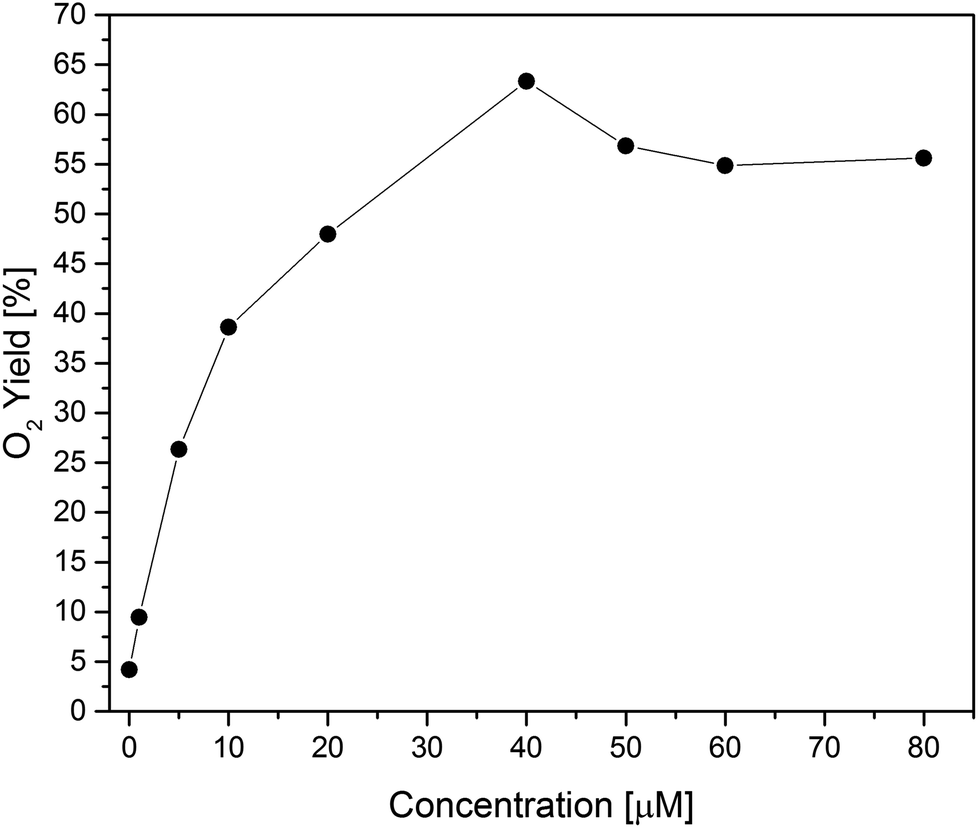 | ||
| Fig. 4 Photocatalytic oxygen yield vs. WOC concentration for Co(1). | ||
| Catalyst | TON | TOF | O2 yield/% | Ref. |
|---|---|---|---|---|
| a 2 μM cat., borate buffer (80 mM, pH 9).b 0.5 μM cat., borate buffer (80 mM, pH 9).c 25 μM cat., borate buffer (0.2 M, pH 8).d [Ru(bpy)3]3+ used as oxidant.e 1.27 μM cat., borate buffer (80 mM, pH 8).f 4 μM cat., borate buffer (80 mM, pH 8.5). | ||||
| Co(1) | 40 | 0.5 | 63 | This work |
| CoNi(2) | 16 | 0.2 | 26 | This work |
| a[(SiW9O34)2Co8(OH)6(H2O)2(CO3)3]16− | 545 | 3.1 | 44 | 4 |
| b[(SiW9O34)2Co8(OH)6(H2O)2(CO3)3]16− | 1436 | 10 | 29 | 4 |
| c[CoII5CoIII2(mdea)4(N3)2(CH3CN)6(OH)2(H2O)2]4− | 88 | 1.75 | 24 | 98 |
| d[Co4(H2O)2(PW9O34)2]10− | 75 | 5 | 64 | 99 |
| e[Co6(H2O)30{Co9Cl2(OH)3(H2O)9(SiW8O31)3}]5− | 100 | 0.042 | — | 100 |
| f[{Co2Sb2(H2O)10(B-β-(SbW9O33)}2]4− | 193 | 5.3 | 31 | 18 |
| [{β-SiNi2W10O36(OH)2(H2O)}4]24 | 335 | 1.7 | 27 | 77 |
In Table 1, TON, TOF and O2 yields are compared to several previously reported POM-WOCs. TONs were increasing with reduced catalyst concentrations, reaching a value of 235 at a catalyst concentration of 1 μM (Table S13†). In the absence of Co(1), a background O2 evolution of 0.83 μmol was detected, which corresponds to 4% O2 yield. Additionally, a reference WOC test with the same concentration of cobalt centers was performed for cobalt acetate (40 μM based on Co), and the obtained 54% O2 yield was lower compared to Co(1).
Recycling experiments showed further activity of the catalytic system after adding additional Na2S2O8 and adjusting the pH back to 8. The 2nd and 3rd cycle showed O2 yields of 25% and 11%, respectively (Table S14 and Fig. S36†). Throughout the recycling, a continuous color change was observed from bright orange to dark green, suggesting a slow decomposition of the photosensitizer [Ru(bpy)3]2+. This shows that low photosensitizer stability associated with the formation of sulfate radicals from Na2S2O8 are the main reasons for the decline of O2 evolution.
CoNi(2) showed reduced photocatalytic oxygen evolution performance compared to pure Co(1). O2 yields decreased from 63% to 26% at a catalyst concentration of 40 μM (Table 1). The calculated O2 yield per Co center (for 40 μM catalyst) is 16% for Co(1) and 15% for CoNi(2), respectively. This underscores further that the Ni centers are most likely inactive. Our previous work on the molecular cubane water oxidation catalyst [Co4(dpy-C{OH}O)4(OAc)2(H2O)2](ClO4)2 showed a comparable trend towards lower O2 yields upon introducing Ni into the cobalt sites. This may imply that an intramolecular O–O coupling pathway between two Co–OHn ligands prevails for O2 evolution from such oxocluster WOCs, as reported for Co3O4.97 CoNi(2) furthermore displays a larger band gap (2.66 eV) than Co(1) (2.41 eV, see Fig. S7 and S8†).
The expected formation of a solid POM–PS complex was observed after the catalytic O2 evolution tests.101 FT-IR analysis shows the presence of both photosensitizer and Co(1) in the precipitate (Fig. 5). For the POM–PS complex, characteristic bands are observed at 989, 939 (νasW–Od) and 874 cm−1 (νasW–Ob) compared to 980, 932 and 884 cm−1 for the pristine Co(1). Additional, three bands at 1463, 1444 and 1423 cm−1 can be attributed to [Ru(bpy)3]2+, in line with previous reports on POM–PS complex formation.102
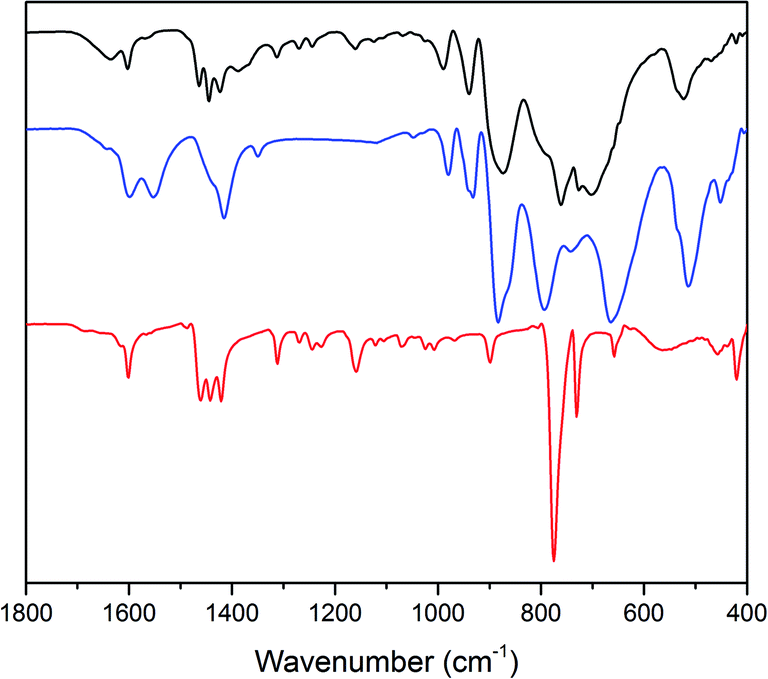 | ||
| Fig. 5 FT-IR spectra of the Co(1)–POM–PS complex (black), pristine Co(1) (blue) and [Ru(bpy)3]Cl2 (red). | ||
This precipitation is a general phenomenon of POM-WOCs in such photocatalytic assays due to the electrostatic interactions between negatively charged POMs and positively charged photosensitizer molecules. Only at low catalyst concentrations <2 μM no measureable precipitation was detected, although such amounts most likely fall below the detection limit of frequently used DLS devices. Note that those devices were designed to quantify size distribution of large amounts of nanoparticles rather than for evidencing their absence.103
Further EDX analyses of the POM–PS complex show a homogeneous distribution of Ru, Co and W and calculated ratios of 2.5:1 [Ru(bpy)3]2+:Co(1) (Table S4,† based on the N:W ratio, Fig. S22†). Notably, lyophilisation of the remaining solution showed no presence of cobalt, which further supports that Co(1) does not undergo leaching of Co2+ ions into the solution (Fig. S21†). A subsequent WOC test with the filtered solution and additional Na2S2O8 showed no further activity (Fig. S37†).
Photocatalytic tests with the recovered POM–PS complex, revealed its continuous activity with 34% O2 yield, which is superior to the direct recycling run of the pristine POM in solution. This shows that the POM–PS complex is still active as a catalyst and that the photocatalytic assay ([Ru(bpy)3]2+/Na2S2O8) causes the fast decline of O2 formation for pristine Co(1).
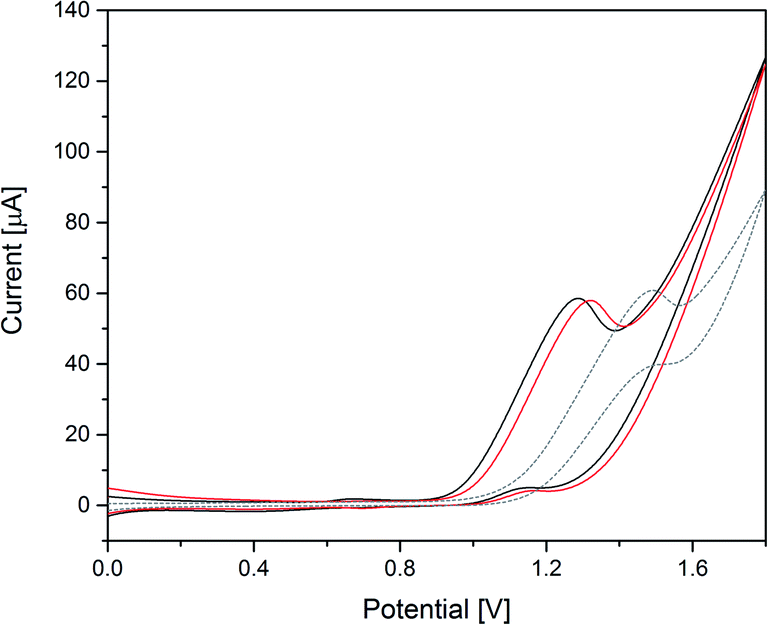 | ||
| Fig. 6 Cyclic voltammograms of 50 μM Co(1) (black) and 50 μM CoNi(2) (red) in 0.1 M borate buffer pH 8 and blank measurements (grey, dashed; V vs. Ag/AgCl, scan rate: 20 mV s−1, 3rd scan is shown). | ||
3.3. Co(1) and CoNi(2) as precursors for heterogeneous WOCs
Next, the heterogeneous tungstate WOCs emerging from Co(1) and CoNi(2) as precursors were investigated for their electrocatalytic performance.The significant influence of the annealing temperature is quite evident from the cyclic voltammetry results (Fig. 7). Amorphous CoW300 has an onset potential of 0.79 V vs. Ag/AgCl in borate buffer (0.1 M, pH 8) solution and maintains its catalytic performance over the measured eight cycles.
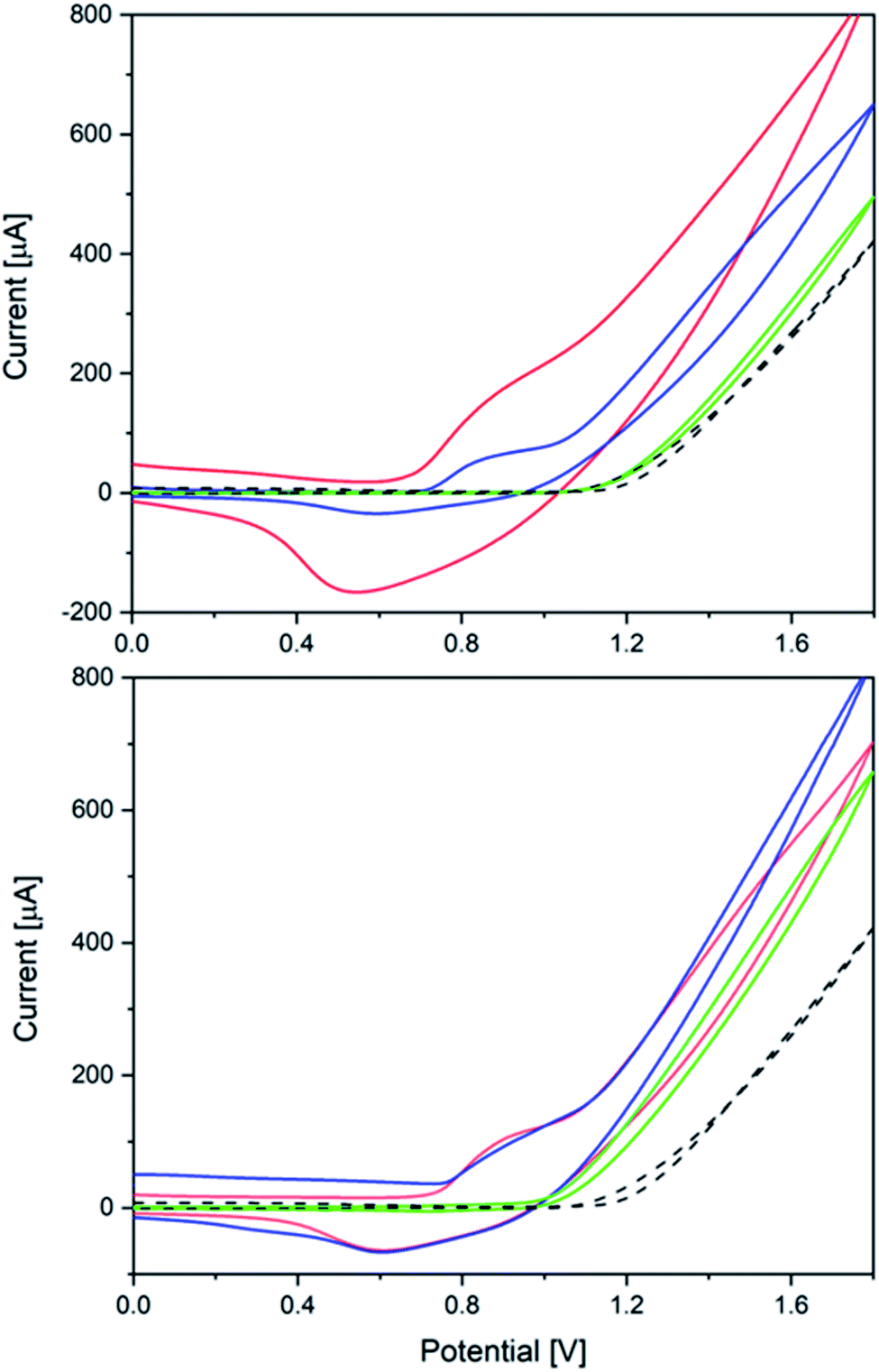 | ||
| Fig. 7 Top: Cyclic voltammograms of CoW300 (red), CoW400 (blue) and CoW500 (green); bottom: CVs of CoNi300 (red), CoNi400 (blue) and CoNi500 (green); all measurements on FTO in 0.1 M borate buffer pH 8 vs. blank measurements (black, dashed; V vs. Ag/AgCl, scan rate: 20 mV s−1, 3rd scan is shown). | ||
A broad cathodic peak at 0.55 V can be seen in the backward scan, which moves slowly to higher potentials in the subsequent scans (0.07 V during eight cycles, cf. Fig. S47†). The amorphous sample shows constant catalytic performance over several cycles with stable anodic and cathodic peaks. Previously reported amorphous CoWO4 showed a substantial change in the anodic and cathodic peak after the first cycle.66
The onset potential of the crystalline samples CoW400 and CoW500 gradually increases to 0.94 V and 1.18 V vs. Ag/AgCl, respectively (Fig. S45†). This is in line with previous observations for amorphous and crystalline CoWO4 and their onset potentials.68,69 Interestingly, the onset potential of CoW500 is almost the same as of the blank FTO electrode.
The onset potentials for the Ni-doped analogues are shifted to higher potentials (Fig. 7). Other than the pure Co samples, the onset potential values for CoNi300 and CoNi400 are closer, namely 0.91 V and 0.98 V, respectively (Fig. S46†). On the other hand, CoNi500 has a better onset potential (1.08 V) compared to its binary analogue CoW500 (1.18 V).
As mentioned above, the blue shift of the {WO4} Raman peak of CoW300 suggests a reduced distance between Co2+ ions, which can further influence the mechanism of the electrocatalytic water oxidation.66,92 This was previously reported and confirmed with EXAFS analyses for amorphous and crystalline CoWO4,33 where the outstanding performance of the amorphous CoWO4 was attributed to the shorter Co–Co distances.
Although the water oxidation reaction is a complex process, it was reported in previous studies that its mechanism strongly depends on the distance between the active sites of heterogeneous electrocatalysts.33 Along these lines, it is assumed that closer intermetallic distances in the range of the O–O bond distance of dioxygen are favoring the “dual-site” Langmuir–Hinshelwood (LH) mechanism. In this mechanism, oxygen species are first adsorbed on adjacent sites, followed by their formation of molecular oxygen. In the case of longer distances between the active metal sites, however, the “single-site” Eley–Rideal (ER) mechanism may take place instead.
The dual-site LH mechanism requires a lower overpotential than the single-site ER mechanism, because the latter includes the formation of a peroxo intermediate at the single active metal center. This is considered a thermodynamically less favorable step for the overall water oxidation.66,107 In the present case, the observed decrease of the Co–Co distances in CoW300 may therefore facilitate the bridging of two terminal oxo groups to generate dioxygen via the LH mechanism.66,108 This agrees with the observed lowest onset potential for CoW300 among the tungstate catalyst series.
CoW300 as best performing member of the tungstate series was further compared to a reference sample obtained from a conventional solution co-precipitation method (annealed at 300 °C) for CoWO4 and to RuO2 as a well-established benchmark WOC (Fig. 8).66 CoW300 showed a lower onset potential (0.79 V) than the as-synthesized reference sample (0.84 V).
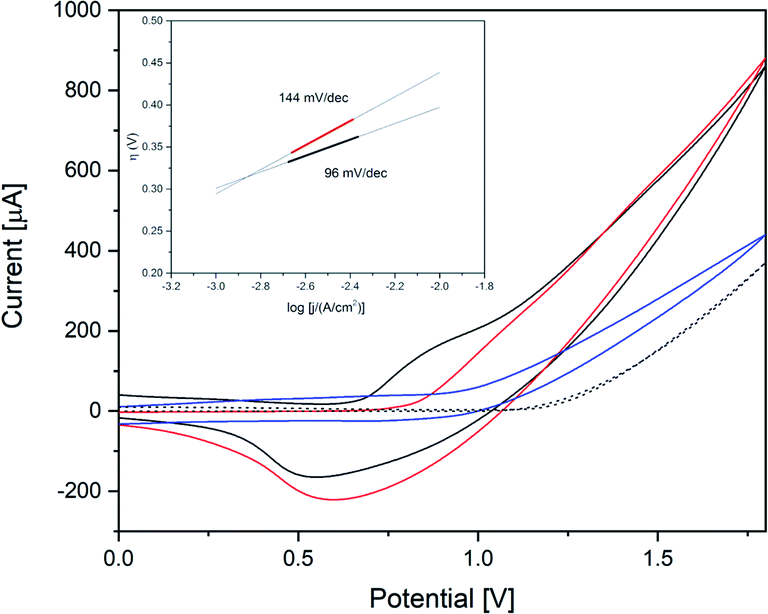 | ||
| Fig. 8 Cyclic voltammograms of CoW300 (black); CoWO4 (red); RuO2 (blue) and reference FTO (dotted) in 0.1 M borate buffer pH 8 (scan rate: 20 mV s−1); inset: Tafel plot of CoW300 (black) and CoWO4 (red). | ||
Furthermore, chronoamperometry measurements of CoW300 and the conventionally synthesized reference material were performed. The superior performance of CoW300 is clearly evident from the lower Tafel slope (inset Fig. 8) of 96 mV dec−1, compared to 144 mV dec−1 for the reference material obtained from Na2WO4 and Co(NO3)2.66
4. Conclusions
A bio-inspired polyoxometalate with an open Co4-core architecture, K5Na3[(A-α-SiW9O34)Co4(OH)3(CH3COO)3] Co(1), was synthesized as a model system to investigate crucial questions of water oxidation catalyst (WOC) design, namely (1) the controversially discussed effect of Co/Ni-synergisms in molecules vs. solids, (2) the influence of preparative history and precursor choice on WOC activity and (3) the role of amorphous features in solid WOC performance.First, the open Co4-POM Co(1) displayed competitive photocatalytic activity with an oxygen yield of 63% for the optimal catalyst concentration of 40 μM. Furthermore, its new mixed Co/Ni isostructural analogue CoNi(2) was synthesized, analyzed and tested for water oxidation activity. Co/Ni substitution did not exert a productive influence on the water oxidation activity of the mixed-metal POMs, in contrast to widely reported Co/Ni synergisms in solid WOCs.
Next, to investigate the effect of mixed metal molecular precursors on cobalt tungstate-related WOCs as attractive target materials, Co(1) and CoNi(2) were subjected to thermal treatment. Both compounds afforded CoWO4- as well as (Co, Ni)WO4-related phases with increasing degrees of crystallinity upon higher annealing temperatures.
Concerning the influence of crystallinity on the performance, cyclic voltammetry measurements clearly showed that amorphous CoW300 obtained from annealing Co(1) at 300 °C showed the lowest onset potential among both series of tungsten oxides obtained from POM precursors. Most importantly, CoW300 displayed a lower onset potential than a representative reference sample of CoWO4 that was synthesized via a conventional co-precipitation/annealing method.
In line with the catalytic trends for the Co(1) and CoNi(2) precursor POMs, introduction of nickel centers did not exert a productive effect on the heterogeneous tungstate catalysts either. This is in stark contrast to the growing number of literature reports on Co/Ni synergisms in a wide range of oxide and non-oxide heterogeneous electrocatalysts. Further systematic studies are now required to understand the dependence of such metal–metal interactions on the catalyst matrix and its preparative history, as well as on the applied performance test conditions.
Our results demonstrate that readily accessible POMs are promising precursors with pre-organized metal centers for the convenient synthesis of amorphous heterogeneous water oxidation catalysts, which outperform products of conventional high temperature approaches starting from simple binary educts. Interestingly, neither the molecular precursors nor their heterogeneous WOC products were responsive to widely employed synergistic Co/Ni doping strategies. This highlights the complexity and matrix dependence of such mixed metal optimization strategies, which are in the focus of forefront catalytic endeavours.
To fully transfer the tunable potential of polynuclear molecular precursors into high performance amorphous catalysts with optimal near-range order properties, in-depth monitoring and theoretical studies of mixed metal interactions in different settings are now required.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the University of Zurich Research Priority Program (URPP) for Solar Light to Chemical Energy Conversion (LightChEC) and by the Swiss National Science Foundation (Sinergia Grant No. CRSII2_160801/1). R. G. thanks the University of Zurich for support of his PhD thesis work with a Forschungskredit Candoc (FK-16-084) grant. The authors thank S. Esmael Balaghi (Department of Chemistry, UZH) for his support with powder diffraction methods and CV measurements.References
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS.
- F. Song, K. Al-Ameed, M. Schilling, T. Fox, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2019, 141, 8846–8857 CrossRef CAS PubMed.
- J. Wei, Y. Feng, P. Zhou, Y. Liu, J. Xu, R. Xiang, Y. Ding, C. Zhao, L. Fan and C. Hu, ChemSusChem, 2015, 8, 2630–2634 CrossRef CAS PubMed.
- F. Evangelisti, R. Güttinger, R. Moré, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2013, 135, 18734–18737 CrossRef CAS PubMed.
- G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935–1943 CrossRef CAS PubMed.
- B. Gao, T. Wang, Y. Li, X. Fan, H. Gong, C. Jiang, P. Li, X. Huang and J. He, Chem. Commun., 2020, 56, 4244–4247 RSC.
- S. Tandon, J. Soriano-López, A. C. Kathalikkattil, G. Jin, P. Wix, M. Venkatesan, R. Lundy, M. A. Morris, G. W. Watson and W. Schmitt, Sustainable Energy Fuels, 2020, 4, 4464–4468 RSC.
- A. Dey, V. Kumar, S. Pal, A. Guha, S. Bawari, T. N. Narayanan and V. Chandrasekhar, Dalton Trans., 2020, 49, 4878–4886 RSC.
- (a) Q. Yin and C. L. Hill, Nat. Chem., 2018, 10, 6–7 CrossRef CAS; (b) W. Liu, W. Mu, M. Liu, X. Zhang, H. Cai and Y. Deng, Nat. Commun., 2014, 5, 3208 CrossRef PubMed; (c) H. G. T. Ly, G. Absillis, R. Janssens, P. Proost and T. N. Parac-Vogt, Angew. Chem., Int. Ed. Engl., 2015, 54, 7391–7394 CrossRef CAS PubMed; (d) F. Lucarini, M. Pastore, S. Vasylevskyi, M. Varisco, E. Solari, A. Crochet, K. M. Fromm, F. Zobi and A. Ruggi, Chem.–Eur. J., 2017, 23, 6768–6771 CrossRef CAS PubMed.
- D. Gao, I. Trentin, L. Schwiedrzik, L. González and C. Streb, Molecules, 2019, 25, 157–177 CrossRef PubMed.
- (a) Y. V. Geletii, B. Botar, P. Kögerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., Int. Ed., 2008, 47, 3896–3899 CrossRef CAS; (b) A. Sartorel, M. Carraro, G. Scorrano, R. de Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5007 CrossRef CAS PubMed; (c) G. Zhu, Y. V. Geletii, P. Kögerler, H. Schilder, J. Song, S. Lense, C. Zhao, K. I. Hardcastle, D. G. Musaev and C. L. Hill, Dalton Trans., 2012, 41, 2084–2090 RSC.
- D. Nesterov and O. Nesterova, Catalysts, 2018, 8, 602 CrossRef.
- Q. Han and Y. Ding, Dalton Trans., 2018, 47, 8180–8188 RSC.
- X.-B. Han, C. Qin, X.-L. Wang, Y.-Z. Tan, X.-J. Zhao and E.-B. Wang, Appl. Catal., B, 2017, 211, 349–356 CrossRef CAS.
- X.-B. Han, D.-X. Wang, E. Gracia-Espino, Y.-H. Luo, Y.-Z. Tan, D.-F. Lu, Y.-G. Li, T. Wågberg, E.-B. Wang and L.-S. Zheng, Chin. J. Catal., 2020, 41, 853–857 CrossRef CAS.
- Q. Han, D. Sun, J. Zhao, X. Liang and Y. Ding, Chin. J. Catal., 2019, 40, 953–958 CrossRef CAS.
- J. Lin, X. Meng, M. Zheng, B. Ma and Y. Ding, Appl. Catal., B, 2019, 241, 351–358 CrossRef CAS.
- S. D. Adhikary, A. Tiwari, T. C. Nagaiah and D. Mandal, ACS Appl. Mater. Interfaces, 2018, 10, 38872–38879 CrossRef CAS PubMed.
- F. Lucarini, J. Fize, A. Morozan, M. Marazzi, M. Natali, M. Pastore, V. Artero and A. Ruggi, Sustainable Energy Fuels, 2020, 4, 589–599 RSC.
- F. Song, Y. Ding, B. Ma, C. Wang, Q. Wang, X. Du, S. Fu and J. Song, Energy Environ. Sci., 2013, 6, 1170–1184 RSC.
- K. Fan, H. Chen, B. He and J. Yu, Chem. Eng. J., 2020, 392, 123744 CrossRef CAS.
- S. Bae, H. Kim, D. Jeon and J. Ryu, ACS Appl. Mater. Interfaces, 2019, 11, 7990–7999 CrossRef CAS PubMed.
- W. Fang, R. Tao, Z. Jin, Z. Sun, F. Li and L. Xu, J. Alloys Compd., 2019, 797, 140–147 CrossRef CAS.
- Y. Choi, D. Jeon, Y. Choi, D. Kim, N. Kim, M. Gu, S. Bae, T. Lee, H.-W. Lee, B.-S. Kim and J. Ryu, ACS Nano, 2019, 13, 467–475 CrossRef CAS.
- L. Zhang, X. Ding, M. Cong, Y. Wang and X. Zhang, Int. J. Hydrogen Energy, 2019, 44, 9203–9209 CrossRef CAS.
- X. Cao, Y. Wang, J. Lin and Y. Ding, J. Mater. Chem. A, 2019, 7, 6294–6303 RSC.
- R. Al-Oweini, A. Sartorel, B. S. Bassil, M. Natali, S. Berardi, F. Scandola, U. Kortz and M. Bonchio, Angew. Chem., Int. Ed., 2014, 53, 11182–11185 CrossRef CAS PubMed.
- J. Stracke and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 14872–14875 CrossRef CAS PubMed.
- (a) J. J. Stracke and R. G. Finke, ACS Catal., 2013, 3, 1209–1219 CrossRef CAS; (b) S. Goberna-Ferrón, J. Soriano-López, J. R. Galán-Mascarós and M. Nyman, Eur. J. Inorg. Chem., 2015, 2015, 2833–2840 CrossRef.
- K. P. Sullivan, M. Wieliczko, M. Kim, Q. Yin, D. L. Collins-Wildman, A. K. Mehta, J. Bacsa, X. Lu, Y. V. Geletii and C. L. Hill, ACS Catal., 2018, 8, 11952–11959 CrossRef CAS.
- Y. Han, K. Choi, H. Oh, C. Kim, D. Jeon, C. Lee, J. H. Lee and J. Ryu, J. Catal., 2018, 367, 212–220 CrossRef CAS.
- S. Suseno, C. C. L. McCrory, R. Tran, S. Gul, J. Yano and T. Agapie, Chem.–Eur. J., 2015, 21, 13420–13430 CrossRef CAS PubMed.
- J. Pfrommer, A. Azarpira, A. Steigert, K. Olech, P. W. Menezes, R. F. Duarte, X. Liao, R. G. Wilks, M. Bär, T. Schedel-Niedrig and M. Driess, ChemCatChem, 2017, 9, 672–676 CrossRef.
- R. Deshmukh and M. Niederberger, Chem.–Eur. J., 2017, 23, 8542–8570 CrossRef CAS PubMed.
- L. Warmuth, C. Ritschel and C. Feldmann, RSC Adv., 2020, 10, 18377–18383 RSC.
- L. Warmuth and C. Feldmann, ACS Omega, 2019, 4, 13400–13407 CrossRef CAS PubMed.
- A. L. Luna, F. Matter, M. Schreck, J. Wohlwend, E. Tervoort, C. Colbeau-Justin and M. Niederberger, Appl. Catal., B, 2020, 267, 118660 CrossRef CAS.
- (a) X. Xing, Y. Gui, G. Zhang and C. Song, Electrochim. Acta, 2015, 157, 15–22 CrossRef CAS; (b) F. Ahmadi, M. Rahimi-Nasrabadi, A. Fosooni and M. Daneshmand, J. Mater. Sci.: Mater. Electron., 2016, 27, 9514–9519 CrossRef CAS; (c) K. Adib, M. Rahimi-Nasrabadi, Z. Rezvani, S. M. Pourmortazavi, F. Ahmadi, H. R. Naderi and M. R. Ganjali, J. Mater. Sci.: Mater. Electron., 2016, 27, 4541–4550 CrossRef CAS.
- L. Reith, K. Lienau, D. S. Cook, R. Moré, R. I. Walton and G. R. Patzke, Chem.–Eur. J., 2018, 24, 18424–18435 CrossRef CAS PubMed.
- L. Reith, K. Lienau, C. A. Triana, S. Siol and G. R. Patzke, ACS Omega, 2019, 4, 15444–15456 CrossRef CAS PubMed.
- K. Lienau, C. A. Triana, L. Reith, S. Siol and G. R. Patzke, Front. Chem., 2020, 8, 473 CrossRef CAS PubMed.
- Y.-H. Lai, C.-Y. Lin, Y. Lv, T. C. King, A. Steiner, N. M. Muresan, L. Gan, D. S. Wright and E. Reisner, Chem. Commun., 2013, 49, 4331–4333 RSC.
- X.-B. Han, X.-Y. Tang, Y. Lin, E. Gracia-Espino, S.-G. Liu, H.-W. Liang, G.-Z. Hu, X.-J. Zhao, H.-G. Liao, Y.-Z. Tan, T. Wagberg, S.-Y. Xie and L.-S. Zheng, J. Am. Chem. Soc., 2019, 141, 232–239 CrossRef CAS PubMed.
- P. Beato, A. Blume, F. Girgsdies, R. E. Jentoft, R. Schlögl, O. Timpe, A. Trunschke, G. Weinberg, Q. Basher, F. A. Hamid, S. B. A. Hamid, E. Omar and L. Mohd Salim, Appl. Catal., A, 2006, 307, 137–147 CrossRef CAS.
- S. Bommineni, M. D. Skoglund, A. R. Morris, E. J. Doskocil and J. H. Holles, Appl. Catal., A, 2013, 467, 202–210 CrossRef CAS.
- Y. Huang, Y. Sun, X. Zheng, T. Aoki, B. Pattengale, J. Huang, X. He, W. Bian, S. Younan, N. Williams, J. Hu, J. Ge, N. Pu, X. Yan, X. Pan, L. Zhang, Y. Wei and J. Gu, Nat. Commun., 2019, 10, 982 CrossRef CAS PubMed.
- Y. Huang, J. Ge, J. Hu, J. Zhang, J. Hao and Y. Wei, Adv. Energy Mater., 2018, 8, 1701601 CrossRef.
- J.-S. Li, Y. Wang, C.-H. Liu, S.-L. Li, Y.-G. Wang, L.-Z. Dong, Z.-H. Dai, Y.-F. Li and Y.-Q. Lan, Nat. Commun., 2016, 7, 11204 CrossRef CAS PubMed.
- Y. Zhao, W. Wan, Y. Chen, R. Erni, C. A. Triana, J. Li, C. K. Mavrokefalos, Y. Zhou and G. R. Patzke, Adv. Energy Mater., 2020, 2002228 CrossRef CAS.
- L. Zhang, Q. Fan, K. Li, S. Zhang and X. Ma, Sustainable Energy Fuels, 2020, 4, 5417–5432 RSC.
- L. Lei, D. Huang, M. Cheng, R. Deng, S. Chen, Y. Chen and W. Wang, Coord. Chem. Rev., 2020, 418, 213372 CrossRef CAS.
- S. M. Jasem and A. C. C. Tseung, J. Electrochem. Soc., 1979, 126, 1353–1360 CrossRef CAS.
- (a) R. P. Antony, A. K. Satpati, K. Bhattacharyya and B. N. Jagatap, Adv. Mater. Interfaces, 2016, 3, 1600632 CrossRef; (b) I. Abidat, C. Morais, C. Comminges, C. Canaff, J. Rousseau, N. Guignard, T. W. Napporn, A. Habrioux and K. B. Kokoh, J. Mater. Chem. A, 2017, 5, 7173–7183 RSC; (c) H. Shi and G. Zhao, J. Phys. Chem. C, 2014, 118, 25939–25946 CrossRef CAS; (d) R. Chen, H.-Y. Wang, J. Miao, H. Yang and B. Liu, Nano Energy, 2015, 11, 333–340 CrossRef CAS.
- Y. Yang, H. L. Fei, G. D. Ruan, C. S. Xiang and J. M. Tour, ACS Nano, 2014, 8, 9518–9523 CrossRef CAS PubMed.
- A. QayoomMugheri, A. Tahira, U. Aftab, M. IshaqAbro, S. R. Chaudhry, L. Amaral and Z. H. Ibupoto, Electrochim. Acta, 2019, 306, 9–17 CrossRef CAS.
- J. W. Nai, H. J. Yin, T. T. You, L. R. Zheng, J. Zhang, P. X. Wang, Z. Jin, Y. Tian and J. Z. Liu, Adv. Energy Mater., 2015, 5, 1401880 CrossRef.
- L. Wang, C. Lin, D. K. Huang, F. X. Zhang, M. K. Wang and J. Jin, ACS Appl. Mater. Interfaces, 2014, 6, 10172–10180 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed.
- Y. Tang, H. Yang, J. Sun, M. Xia, W. Guo, L. Yu, J. Yan, J. Zheng, L. Chang and F. Gao, Nanoscale, 2018, 10, 10459–10466 RSC.
- R. Wu, B. Xiao, Q. Gao, Y.-R. Zheng, X.-S. Zheng, J.-F. Zhu, M.-R. Gao and S.-H. Yu, Angew. Chem., Int. Ed., 2018, 57, 15445–15449 CrossRef CAS PubMed.
- R. J. Müller, J. Lan, K. Lienau, R. Moré, C. A. Triana, M. Iannuzzi and G. R. Patzke, Dalton Trans., 2018, 47, 10759–10766 RSC.
- F. Song, R. Moré, M. Schilling, G. Smolentsev, N. Azzaroli, T. Fox, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2017, 139, 14198–14208 CrossRef CAS PubMed.
- S. Zhang, Q. Xing and W.-H. Sun, RSC Adv., 2016, 6, 72170–72176 RSC.
- H. Jia, J. Stark, L. Q. Zhou, C. Ling, T. Sekito and Z. Markin, RSC Adv., 2012, 2, 10874–10881 RSC.
- C. Ling, L. Q. Zhou and H. Jia, RSC Adv., 2014, 4, 24692–24697 RSC.
- A. Indra, P. W. Menezes, N. R. Sahraie, A. Bergmann, C. Das, M. Tallarida, D. Schmeißer, P. Strasser and M. Driess, J. Am. Chem. Soc., 2014, 136, 17530–17536 CrossRef CAS PubMed.
- D. Chen, C.-L. Dong, Y. Zou, D. Su, Y.-C. Huang, L. Tao, S. Dou, S. Shen and S. Wang, Nanoscale, 2017, 9, 11969–11975 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- S. L. González-Cortés, T.-C. Xiao, P. M. Costa, S. M. Rodulfo-Baechler and M. L. Green, J. Mol. Catal. A: Chem., 2005, 238, 127–134 CrossRef.
- V. Srirapu, A. Kumar, P. Srivastava, R. N. Singh and A. Sinha, Electrochim. Acta, 2016, 209, 75–84 CrossRef CAS.
- B. Huang, H. Wang, S. Liang, H. Qin, Y. Li, Z. Luo, C. Zhao, L. Xie and L. Chen, Energy Storage Mater., 2020, 32, 105–114 CrossRef.
- A. S. Rajpurohit, N. S. Punde, C. R. Rawool and A. K. Srivastava, Chem. Eng. J., 2019, 371, 679–692 CrossRef CAS.
- S. M. El-Sheikh and M. M. Rashad, J. Cluster Sci., 2015, 26, 743–757 CrossRef CAS.
- X.-B. Han, Y.-G. Li, Z.-M. Zhang, H.-Q. Tan, Y. Lu and E.-B. Wang, J. Am. Chem. Soc., 2015, 137, 5486–5493 CrossRef CAS PubMed.
- L. Yu, Y. Ding, M. Zheng, H. Chen and J. Zhao, Chem. Commun., 2016, 52, 14494–14497 RSC.
- L. Lisnard, P. Mialane, A. Dolbecq, J. Marrot, J. M. Clemente-Juan, E. Coronado, B. Keita, P. de Oliveira, L. Nadjo and F. Sécheresse, Chem.–Eur. J., 2007, 13, 3525–3536 CrossRef CAS PubMed.
- A. P. Ginsberg, Inorganic Syntheses, John Wiley & Sons, Inc, Hoboken, NJ, USA, 1990 Search PubMed.
- C. Rocchiccioli-Deltcheff, M. Fournier, R. Franck and R. Thouvenot, Inorg. Chem., 1983, 22, 207–216 CrossRef CAS.
- J. Niu, Z. Wang and J. Wang, J. Coord. Chem., 2004, 57, 1271–1277 CrossRef CAS.
- R. Thouvenot, M. Fournier, R. Franck and C. Rocchiccioli-Deltcheff, Inorg. Chem., 1984, 23, 598–605 CrossRef CAS.
- F. Song, K. Al-Ameed, M. Schilling, T. Fox, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2019, 141, 8846–8857 CrossRef CAS PubMed.
- F. Song, R. Moré, M. Schilling, G. Smolentsev, N. Azzaroli, T. Fox, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2017, 139, 14198–14208 CrossRef CAS PubMed.
- C. Pichon, P. Mialane, A. Dolbecq, J. Marrot, E. Rivière, B. S. Bassil, U. Kortz, B. Keita, L. Nadjo and F. Sécheresse, Inorg. Chem., 2008, 47, 11120–11128 CrossRef CAS PubMed.
- T. Montini, V. Gombac, A. Hameed, L. Felisari, G. Adami and P. Fornasiero, Chem. Phys. Lett., 2010, 498, 113–119 CrossRef CAS.
- S. J. Naik and A. V. Salker, Solid State Sci., 2010, 12, 2065–2072 CrossRef CAS.
- T.-D. Nguyen, C.-T. Dinh and T.-O. Do, Nanoscale, 2011, 3, 1861–1873 RSC.
- T. You, G. Cao, X. Song, C. Fan, W. Zhao, Z. Yin and S. Sun, Mater. Lett., 2008, 62, 1169–1172 CrossRef CAS.
- P. K. Pandey, N. S. Bhave and R. B. Kharat, J. Mater. Sci., 2007, 42, 7927–7933 CrossRef CAS.
- R. L. Moreira, R. M. Almeida, K. P. F. Siqueira, C. G. Abreu and A. Dias, J. Phys. D: Appl. Phys., 2015, 49, 45305 CrossRef.
- Q. Chen, T.-W. Huang, M. Baldini, A. Hushur, V. Pomjakushin, S. Clark, W. L. Mao, M. H. Manghnani, A. Braun and T. Graule, J. Phys. Chem. C, 2011, 115, 24021–24027 CrossRef CAS.
- V. K. Malinovsky, V. N. Novikov, N. V. Surovtsev and A. P. Shebanin, Phys. Solid State, 2000, 42, 65–71 CrossRef CAS.
- S. Fukuzumi, J. Jung, Y. Yamada, T. Kojima and W. Nam, Chem.–Asian J., 2016, 11, 1138–1150 CrossRef CAS PubMed.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC.
- X.-B. Han, Z.-M. Zhang, T. Zhang, Y.-G. Li, W. Lin, W. You, Z.-M. Su and E.-B. Wang, J. Am. Chem. Soc., 2014, 136, 5359–5366 CrossRef CAS.
- S. Koroidov, M. F. Anderlund, S. Styring, A. Thapper and J. Messinger, Energy Environ. Sci., 2015, 8, 2492–2503 RSC.
- J.-H. Xu, L.-Y. Guo, H.-F. Su, X. Gao, X.-F. Wu, W.-G. Wang, C.-H. Tung and D. Sun, Inorg. Chem., 2017, 56, 1591–1598 CrossRef CAS PubMed.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- M. Natali, I. Bazzan, S. Goberna-Ferrón, R. Al-Oweini, M. Ibrahim, B. S. Bassil, H. Dau, F. Scandola, J. R. Galán-Mascarós, U. Kortz, A. Sartorel, I. Zaharieva and M. Bonchio, Green Chem., 2017, 19, 2416–2426 RSC.
- P.-E. Car, M. Guttentag, K. K. Baldridge, R. Alberto and G. R. Patzke, Green Chem., 2012, 14, 1680–1688 RSC.
- F. Evangelisti, P.-E. Car, O. Blacque and G. R. Patzke, Catal. Sci. Technol., 2013, 3, 3117–3129 RSC.
- J. Li, R. Güttinger, R. Moré, F. Song, W. Wan and G. R. Patzke, Chem. Soc. Rev., 2017, 46, 6124–6147 RSC.
- S. J. Folkman, J. Soriano-Lopez, J. R. Galán-Mascarós and R. G. Finke, J. Am. Chem. Soc., 2018, 140, 12040–12055 CrossRef CAS PubMed.
- (a) R. Chen, C. Yang, W. Cai, H.-Y. Wang, J. Miao, L. Zhang, S. Chen and B. Liu, ACS Energy Lett., 2017, 2, 1070–1075 CrossRef CAS; (b) P. P. Lopes, D. Strmcnik, D. Tripkovic, J. G. Connell, V. Stamenkovic and N. M. Markovic, ACS Catal., 2016, 6, 2536–2544 CrossRef CAS.
- S. Cherevko, A. R. Zeradjanin, G. P. Keeley and K. J. J. Mayrhofer, J. Electrochem. Soc., 2014, 161, H822–H830 CrossRef.
- Y.-H. Fang and Z.-P. Liu, J. Am. Chem. Soc., 2010, 132, 18214–18222 CrossRef CAS.
- L.-P. Wang and T. van Voorhis, J. Phys. Chem. Lett., 2011, 2, 2200–2204 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10792a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |