
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of 12 insect growth regulator residues in foods of different matrixes by modified QuEChERS and UPLC-MS/MS†
Ke Wang*ab,
Lingzhi Zhaoa,
Can Zhangc,
Hong Zhangab and
Kaoqi Lian *c
*c
aShijiazhuang Center for Disease Control and Prevention, Shijiazhuang 050011, China. E-mail: wkecdc@163.com
bShijiazhuang Technology Innovation Center for Chemical Poison Detection and Risk Early Warning, Shijiazhuang 050011, China
cHebei Key Laboratory of Environment and Human Health, School of Public Health, Hebei Medical University, Shijiazhuang, 050017, China. E-mail: liankq@hebmu.edu.cn
First published on 25th March 2021
Abstract
An analytical method was developed and validated for the simultaneous determination of 12 insect growth regulators (IGRs) (buprofezin, cyantraniliprole, flubendiamide, flonicamid, tolfenpyrad, chlorantraniliprole, RH-5849, methoxyfenozide, chromafenozide, tebufenozide, pyriproxyfen and fenoxycarb) in foods collected from different matrixes by modified QuEChERS and ultraperformance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). The samples were ultrasonically extracted with acetonitrile containing 0.5% formic acid, and different QuEChERS purification conditions were optimized for different matrixes (vegetable oil, fruit and tea). 12 IGRs were separated on a Plus C18 column, and detected by MS/MS under multiple reaction monitoring (MRM) mode. The developed method was validated in terms of linearity, matrix effect, accuracy and precision. Acceptable recoveries of IGRs in three different substrates (vegetable oil, tea and fruit) at three spiked levels were in the range of 65.47–95.17%, 80.55–110.15%, and 62.02–96.50%, respectively, with RSDs less than 11.58%. The method showed a good linearity (R2 ≥ 0.9994) for all analytes in the range of 0.2–200 μg L−1. The LODs (S/N = 3) and LOQs (S/N = 10) of the method were 0.04–0.40 μg kg−1, and 0.13–1.24 μg kg−1, respectively. Owing to the advantages of simple operation, high accuracy and sensitivity, this method is suitable for the rapid and simultaneous detection of 12 IGRs in vegetable oil, tea and fruit.
1. Introduction
Insect growth regulators (IGRs) are chemicals that disrupt the life cycle of insects and lead to their death. According to the insecticidal mechanism, IGRs are divided in three major classes including juvenile hormone analogs, ecdysteroid agonists, and chitin synthase inhibitors.1,2 Because of their mode of action, IGRs can efficiently control many arthropod pests in many crops (fruits and vegetables, tea, cereals, etc.), and they are particularly effective against many lepidopteran pests while exhibiting good selectivity with low toxicity to nontarget arthropods. They act by hindering the biosynthesis and deposition of chitin, reducing the hardness of new epidermis, and hindering the growth and development of chitin.3,4 IGRs are advantageous over conventional insecticides as they are more potent, less toxic to mammals, and biodegradable in soil and water.5 Pyriproxyfen and fenoxycarb are two new insecticides of the class juvenile hormone analogs; their characteristics include high efficiency, low dosage, and long duration.2,6,7 RH-5849, tebufenozide, methoxyfenozide and chromafenozide are new generation of diacylhydrazide IGRs and ecdysone analogs. They mainly induce lepidoptera pests to molt and mature prematurely by reducing or inhibiting the feeding ability of larva and adult, resulting in incomplete development of insects and reduction in oviposition; thus, the pests die in a few days, thus achieving insecticidal effect by hindering insect reproduction.8,9 Buprofezin, chloroanilide, flubendiamide, flonicamid, tolfenpyrad, and cyananilide are all chitin synthesis inhibitors that activate ryanodine receptors, stimulating the uncontrolled release of calcium ions from the sarcoplasmic reticulum of muscle cells and resulting in feeding cessation, lethargy, muscle paralysis, and death.10,11 With the wide application of IGRs as insecticides, it has been found that RH-5849 is a moderately toxic insecticide, with a certain degree of damage to the genetic material of human peripheral blood lymphocytes;12 excessive use of fenoxycarb will produce a certain reproductive toxicity to animals and lead to anemia in mammals.13,14To ensure food safety and human health, many countries and regions have set the maximum residue limit (MRL) standards for IGRs in food. The United States, EU, and Japan have published thousands of maximum residue limits (MRLs) for pesticides.15 The European Union has set the MRLs for methoxyfenozide, chromafenozide and flonicamid in tea as 50, 50, and 100 μg kg−1, respectively.16 The Japanese positive list provides the MRLs for methoxyfenozide, chromafenozide, tebufenozide in vegetables as 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 5000, and 20000 μg kg−1, respectively.17 Chinese national food safety standard GB 2763-2019 stipulates the MRLs for buprofezin, tebufenozide, tolfenpyrad, cyantraniliprole, pyriproxyfen, chlorantraniliprole, and flonicamid in foods (fruits, vegetables, tea, cereals, etc.) as 10–10000, 10–20000, 500–50000, 30–20000, 10–3000, 10–40000 and 200–1000 μg kg−1, respectively.18 Therefore, it is very important to determine the level of IGR residues in agricultural products and monitor the safety of agricultural products.
000, 5000, and 20000 μg kg−1, respectively.17 Chinese national food safety standard GB 2763-2019 stipulates the MRLs for buprofezin, tebufenozide, tolfenpyrad, cyantraniliprole, pyriproxyfen, chlorantraniliprole, and flonicamid in foods (fruits, vegetables, tea, cereals, etc.) as 10–10000, 10–20000, 500–50000, 30–20000, 10–3000, 10–40000 and 200–1000 μg kg−1, respectively.18 Therefore, it is very important to determine the level of IGR residues in agricultural products and monitor the safety of agricultural products.
Several analytical methods have been developed to determine IGRs by in food and environmental samples by high-performance liquid chromatography (HPLC),19,20 HPLC coupled with tandem mass spectrometry (HPLC-MS/MS),4,21 gas chromatography (GC),10 gas chromatography coupled with tandem mass spectrometry (GC-MS/MS),22 and enzyme-linked immunosorbent assays (ELISAs).23 Among them, because of the advantages of HPLC-MS/MS and ultraperformance liquid chromatography with tandem mass spectrometry (UPLC-MS/MS) in multiple reaction monitoring (MRM) mode, such as high sensitivity, selectivity, and specificity, it has been widely applied to detect pesticide residues. For sample preparation, various pretreatment methods were used for pesticide analysis, such as liquid–liquid extraction (LLE),22,24 solid-phase extraction (SPE),25 magnetic solid-phase extraction (MSPE),26,27 dispersive liquid–liquid microextraction (DLLME)28 and solid-phase microextraction (SPME),29 and so on. Compared with other pretreatment technique, quick, easy, cheap, rugged, effective, and safe (QuEChERS)21,30–32 method has the advantages of simple and fast operation, requirement of less solvent in the extraction process, and less environmental pollution. This is followed by a salting-out step, causing the partitioning of two liquid phases. A clean-up step, usually dispersive solid-phase extraction, is then added to remove interferences.
Currently, studies on IGRs have focused on samples of fruits, vegetables, and grains, often testing for one or more pesticides, there are few reports of simultaneous detection of multiple IGRs in different matrices. For QuEChERS pretreatment, although the standard operating procedures were used in most cases, further study should be conducted on the pretreatment modification and quantitative evaluation, especially for complex sample matrix. The aim of this study was to develop simultaneous determination of 12 IGRs involving four ecdysteroid agonists (RH-5849, methoxyfenozide, chromafenozide, and tebufenozide), six chitin synthase inhibitors (buprofezin, cyantraniliprole, flubendiamide, flonicamid, tolfenpyrad, and chlorantraniliprole), and two juvenile hormone analogs (pyriproxyfen and fenoxycarb) in vegetable oil, tea, and fruit by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). To guarantee the accuracy and sensitivity of detection of different substrates, this study explored the improvement of QuEChERS method, optimization of detection conditions, and improvement of the pretreatment conditions for different matrices. A UPLC-MS/MS method was developed for the simultaneous determination of 12 IGRs in different substrates by using modified QuEChERS and UPLC-MS/MS techniques. This method might provide technical support for food safety and risk assessment.
2. Material and methods
2.1. Chemicals and reagents
Standards of flonicamid (99% purity; CAS Registry no. 158062-67-0), RH5849 ((1,2-dibenzoyl-1-tert-butylhydrazine), 99.0% purity; CAS Registry no. 112225-87-3), chlorantraniliprole (97.8% purity; CAS Registry no. 500008-45-7), methoxyfenozide (99.0% purity; CAS Registry no. 161050-58-4), chromafenozide (99.9% purity; CAS Registry no. 143807-66-3), fenoxycarb (99.5% purity; CAS Registry no. 72490-01-8), tebufenozide (99.6% purity; CAS Registry no. 112451-23-8), flubendiamide (99.2% purity; CAS Registry no. 272451-65-7), buprofezin (99.9% purity; CAS Registry no. 69327-76-0), tolfenpyrad (99.3% purity; CAS Registry no. 129558-76-5), pyriproxyfen (99.9% purity; CAS Registry no. 95737-68-1) were obtained from Dr Ehrenstorfer GmbH (Augsburg, Germany); cyantraniliprole was purchased from ANPEL Laboratory Technologies (Shanghai) Inc. HPLC-grade acetonitrile (ACN) was obtained from Merck (Darmstadt, Germany), methanol, acetone and ethyl acetate were purchased from Thermo Fisher Co., Ltd. (USA). Hexane, formic acid and ammonium acetate were purchased from Dikma (USA). Superior pure petroleum ether was purchased from Tianjin Kemiou Chemical Reagent Co., Ltd (China). Sodium chloride (NaCl) and magnesium sulfate anhydrous were purchased from Tianjin Yongda Chemical Reagent Co., Ltd (Tianjin, China) and were of analytical grade. Primary secondary amine (PSA) and graphitized carbon black (GCB) were supplied by Dikma (USA). C18 was purchased from Welch (40–50 μm, USA). Ultra-pure water was prepared using a Millipore Milli-Q Gradient Water Purification System (USA).2.2. Standard solution preparation
Individual standard solutions of 12 IGRs were prepared in ACN at a concentration of 100 μg mL−1. A working solution (1 μg mL−1) was prepared by mixing a certain amount of individual standard solution and methanol. The solvent calibration standard solutions were prepared by mixing the working standard solutions with additional methanol to reach the different concentration. Matrix-matched calibration standard solutions were prepared in blank sample containing different analytes contents. All standards solutions were stored at −20 °C in a bottle and freshly prepared solvent or matrix standards were used before each analysis.2.3. UPLC-MS/MS analysis conditions
A UPLC-MS/MS instrument (Exion-TRILPLE QUAD 5500, AB SCIEX, USA) was used to analyze the 12 target compounds. The LC unit was used to separate the target compounds using an Agilent ZORBAX Eclipse Plus-C18 column (2.1 mm × 100 mm, 1.8 μm) held at 40 °C. The injection volume and flow rate were 3 μL and 0.3 mL min−1, respectively. The mobile phase was composed of 0.1% formic acid solution (A) and methanol (B), and a linear-gradient elution program was set as follows: 45% B (0–0.1 min), 45–71% B (0.1–0.2 min), 71% B (0.2–2.8 min), 71–63% B (2.8–2.9 min), 63% B (2.9–3.8 min), 63–82% B (3.8–3.9 min), 82% (3.9–6.9 min), 82–45% (6.9–7.0 min), and 45% B (7.0–10.0 min). Ionization was carried out using an electrospray ionization (ESI) source in the positive mode, and mass spectrometric analysis was carried out in MRM mode. The parameters for MS/MS were set as follows: spray voltage, 5500 V; vaporizer temperature, 550 °C; nebulizing gas pressure, 55 psi; auxiliary gas pressure, 55 psi; and curtain gas pressure, 40 psi. Table 1 showed the optimized MRM data acquisitions.| Compound | Retention time (min) | Precursor ion (m/z) | Declustering potential (V) | Product ion (m/z) | Collision energy (eV) |
|---|---|---|---|---|---|
| a Quantifying ions. | |||||
| Flonicamid | 1.23 | 230.2 | 130.2 | 203.1 | 23.8 |
| 81.9 | 176.2 | 32.1 | |||
| Cyantraniliprole | 2.73 | 475.1 | 71.0 | 286.0a | 18.9 |
| 71.9 | 443.9 | 22.6 | |||
| RH-5849 | 2.87 | 297.2 | 73.3 | 105.1 | 24.8 |
| 62.1 | 241.2 | 11.0 | |||
| Chlorantraniliprole | 3.25 | 484.2 | 75.1 | 453.0a | 26.1 |
| 69.0 | 286.1 | 19.2 | |||
| Methoxyfenozide | 3.96 | 369.4 | 54.1 | 149.2a | 23.6 |
| 67.1 | 313.2 | 11.7 | |||
| Chromafenozide | 4.41 | 395.2 | 87.9 | 175.2a | 20.9 |
| 81.8 | 339.3 | 10.7 | |||
| Fenoxycarb | 5.57 | 302.1 | 80.7 | 116.0a | 15.3 |
| 95.1 | 256.1 | 17.7 | |||
| Tebufenozide | 5.79 | 353.3 | 66.0 | 133.2a | 23.0 |
| 63.6 | 297.4 | 11.7 | |||
| Flubendiamide | 6.07 | 700.2 | 63.0 | 408.0a | 18.9 |
| 65.9 | 273.7 | 42.1 | |||
| Buprofezin | 7.38 | 306.2 | 60.8 | 201.0a | 16.8 |
| 60.1 | 116.2 | 22.4 | |||
| Tolfenpyrad | 7.82 | 384.3 | 77.2 | 197.2a | 34.1 |
| 77.2 | 170.8 | 28.7 | |||
| Pyriproxyfen | 8.12 | 322.2 | 90.4 | 227.0a | 20.4 |
| 89.3 | 185.1 | 28.9 | |||
2.4. Sample preparation
Vegetable oil, fruit, and tea samples were purchased from local supermarkets of Shijiazhuang. For fruit, the edible part of sample was uniformly crushed using a proofer and placed in a closed container, then it was stored in a refrigerator at 4 °C away from light. The tea sample was ground in a mortar and placed in a clean self-sealing bag until use. Blank samples were used for validation studies and matrix-matched standard calibrations. Samples for recovery studies were spiked with a known amount of fortification standard and left for 10 min before the extraction.The general procedures for extraction and purification were presented separately as follows.
Extraction: 2.00 g samples were accurately weighed in a 50 mL centrifuge tube, 2 mL water was added, vortexed for 30 s. Then, 10 mL acetonitrile containing 0.5% formic acid was added. After vortex mixing and ultrasonication for 10 min, 800 mg NaCl was added and mixed by vortexing for 30 s. The mixture was centrifuged at 4000 rpm for 9 min and subjected to purification.
Purification: 8 mL of the supernatant was transferred to a 15 mL centrifuge tube containing different sorbents. Then, the mixture was vortexed for 30 s, left standing for 2 min, centrifuged for 5 min at 8000 rpm, and 5 mL of the supernatant was evaporated to dryness under nitrogen at 40 °C. The residue was dissolved in 1 mL methanol, vortexed for 30 s, and passed through a 0.20 μm nylon filter membrane before LC-MS/MS analysis.
3. Results and discussion
3.1. Optimization of MS/MS conditions
A tandem mass detector has high selectivity and sensitivity and provides effective detection. First, a needle pump was used for continuous sampling. A parent ion scan (Q1 MS) was performed to obtain the parent ion of target, and then the fragment ion was obtained through product ion scan. A detection ion pair was formed by the parent ion and two fragment ions with strong response signals. In addition to the [M + H]+ of flubendiamide with weak response signal, other 11 IGRs showed the [M + H]+ ion as the precursor ion. The intensities of addition peaks [M + Na]+ and [M + NH4]+ of flubendiamide were relatively good. However, because the stability of addition peaks of [M + Na]+ was significantly affected by the matrix, the addition ions [M + NH4]+ were selected as the parent ions. The parent ions, product ions, declustering potential, and collision energy of IGRs using MRM were shown in Table 1.3.2. Optimization of UPLC conditions
Despite the high specificity of MS/MS detection, chromatographic separation (resolution) is decisive for improving the sensitivity and peak shape of analytes.4 The chromatographic conditions of compounds were affected by the packing, particle size, and mobile phase. To obtain the best chromatographic conditions for the target compounds, the separation effects of chromatographic columns with three different specifications were compared. It was found that 12 types of target compounds could not be well separated using Zorbax Eclipse XDB-C18 (2.1 × 50 mm, 1.8 μm) and Zorbax Eclipse XDB-C18 (4.6 × 50 mm, 1.8 μm) column. The separation effect was better on Zorbax Eclipse Plus C18 (4.6 × 100 mm, 1.8 μm) column, and the response of each substance was higher.Regarding the optimization of chromatographic conditions, mobile phase was another important factor affecting chromatographic peak-to-peak type and their response. In the experiment, two mobile phases A and B were optimized. Methanol and acetonitrile were selected and compared as the organic phase B. Flonicamid showed poor peak shape and split peak in acetonitrile as the mobile phase. While methanol was used as the mobile phase, flonicamid exhibited good peak shape and 4 times higher response than that in acetonitrile; meanwhile, chlorantraniliprole showed about 2 times higher response. In addition, the other nine types of compounds showed better response in methanol except buprofezin. Therefore, methanol was selected as the organic phase of mobile phase B. Because the mass spectrum peak of flubendiamide showed an additional ammonia peak, two aqueous phases were compared: 2 mmol L−1 ammonium acetate, 0.1% formic acid aqueous solution containing 2 mmol L−1 ammonium acetate. The [M + NH4]+ signal intensity of flubendiamide did not increase significantly after adding ammonium acetate to aqueous phase, and the response signals of other some compounds were suppressed. When the aqueous phase A contained 0.1% formic acid, the separation effect of 12 IGRs was better, the response signal had a higher intensity, and the peak shape was sharp. Therefore, 0.1% formic acid aqueous solution and methanol was finally selected as the mobile phase, and separation was carried out by gradient elution procedure. Within 9 min, 12 compounds were well separated (Fig. 1).
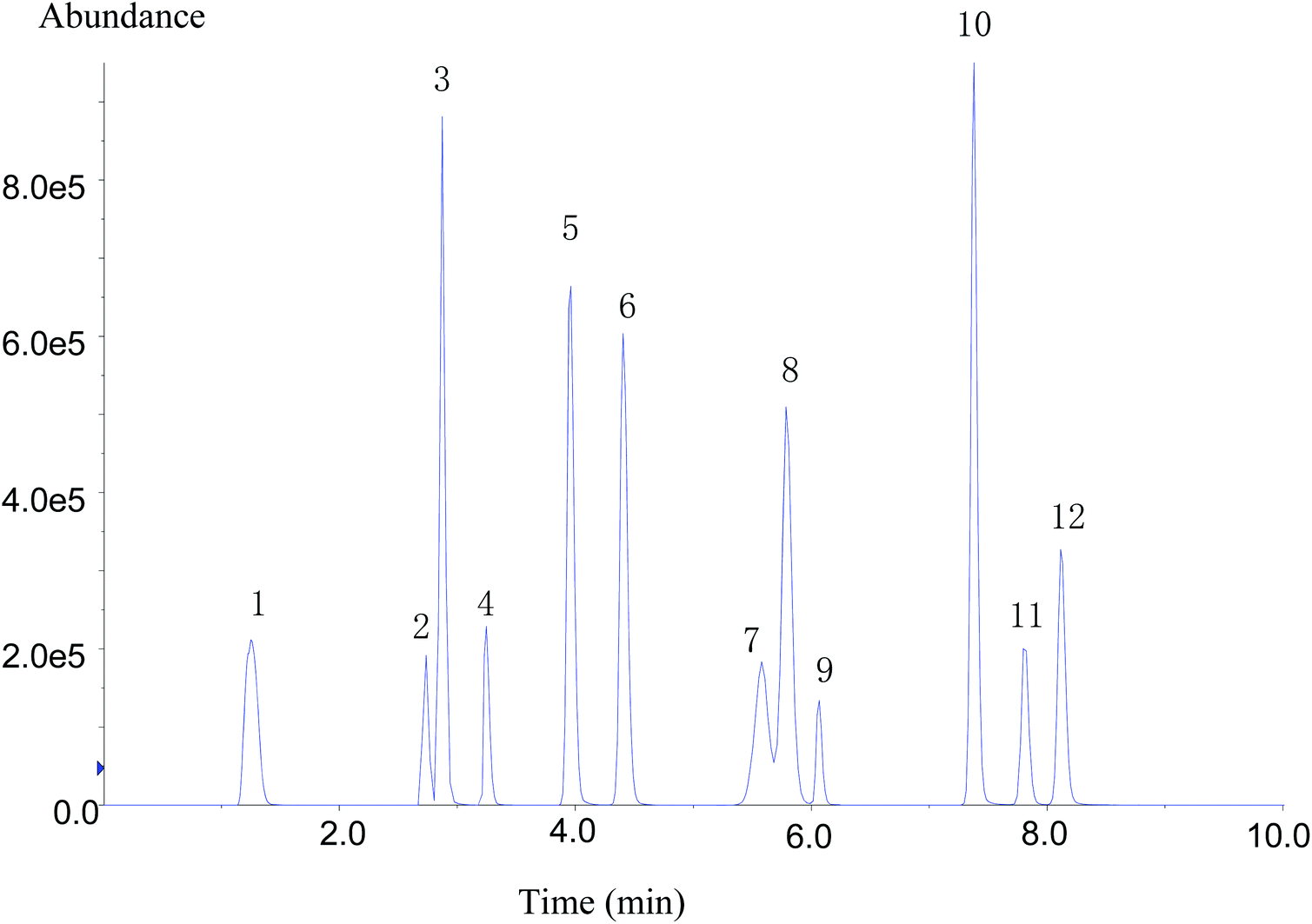 | ||
| Fig. 1 Chromatograms of 12 kinds of IGRs (50 μg L−1). 1. Flonicamid, 2. cyantraniliprole, 3. RH-5849, 4. chlorantraniliprole, 5. methoxyfenozide, 6. chromafenozide, 7. fenoxycarb, 8. tebufenozide, 9. flubendiamide, 10. buprofezin, 11. tolfenpyrad, 12. pyriproxyfen. | ||
3.3. Optimization of modified QuEChERS for different food matrix samples
In this experiment, soybean oil was used as a vegetable oil substrate. Various extraction conditions such as the extractant, extractant volume, ultrasonic extraction time were examined, 3 replicates were done for every test. Because of the complexity of sample matrix, the extractant should not only solubilize the target compound well, but also avoid the extraction of impurities. Currently, acetonitrile, acetone and ethyl acetate are the most used solvents for extraction in QuEChERS. Moreover, acidified MeCN showed good extraction efficiency for many analytes.33 Therefore, acetonitrile, acetone, acetonitrile with different concentrations of formic acid were investigated in the experiment.
Firstly, 2.00 g blank vegetable oil was spiked at the level of 10 μg kg−1, the extraction and purification procedure was done as shown in section 2.4 with different extractant, and the 0.1 g PSA + 0.3 g MgSO4 was used as purifying sorbent. When acetone was used as an extractant, more impurities were obtained after drying under nitrogen, and the recoveries of most of the substances were below 50% (Fig. 2). The recoveries of 12 IGRs were between 40.99% and 62.21% when pure acetonitrile was used as an extraction reagent. The recoveries of 12 IGRs increased with the amount of formic acid added, and the extraction effect was the best when the amount of formic acid added was 0.5%. The recoveries of 12 IGRs were 50.11–108.07%; most of them were over 80% (Fig. 2).
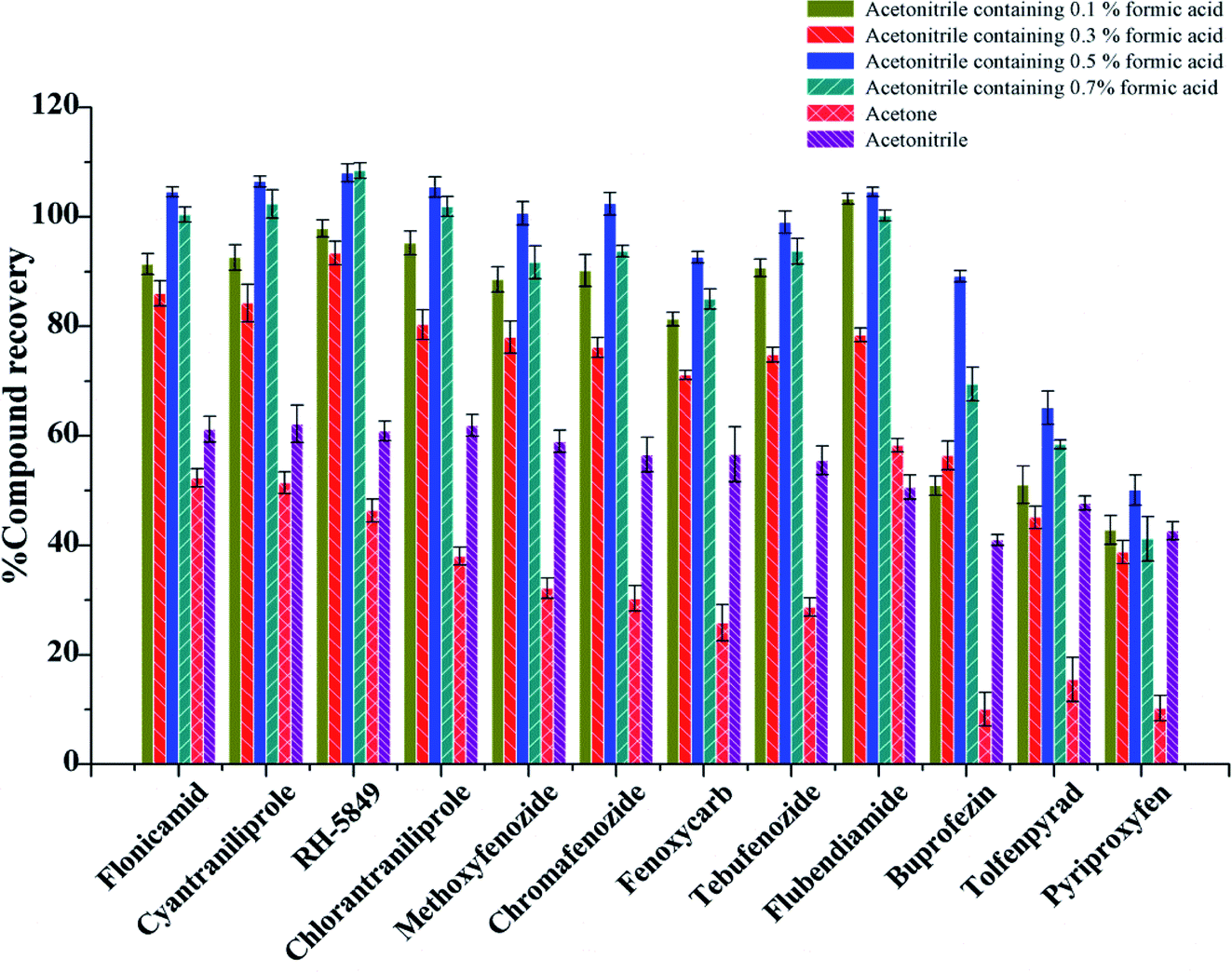 | ||
| Fig. 2 Recoveries of 12 kinds of IGRs with different extractant. | ||
Furthermore, different volume of acetonitrile containing 0.5% formic acid were examined. The recoveries of 12 IGRs were 55.83–105.98%, when the extractant volume was 10 mL, which was better than 6 mL, 8 mL, 12 mL and 14 mL (Fig. 3).
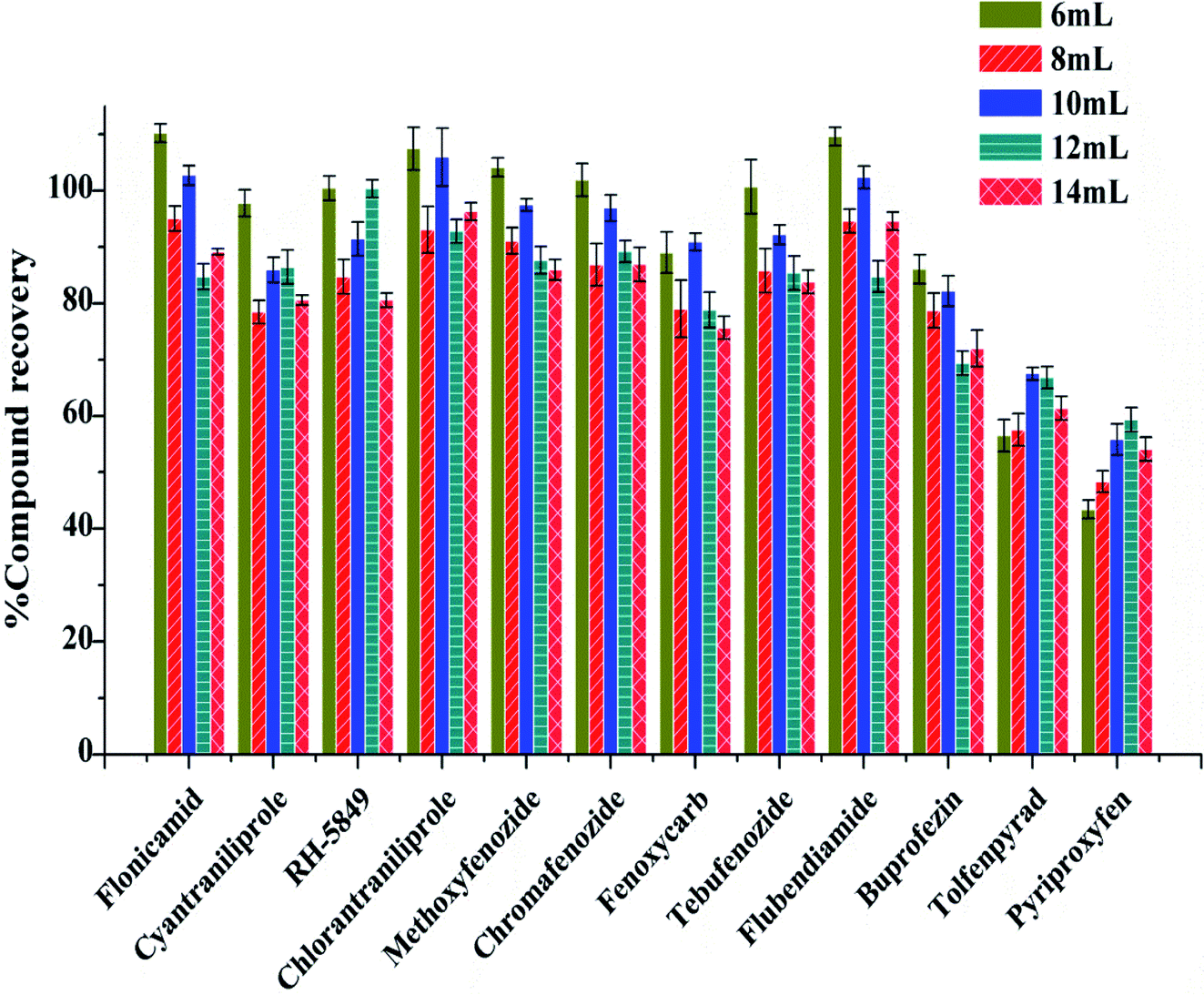 | ||
| Fig. 3 Recoveries of 12 kinds of IGRs with different extractant volume. | ||
The extraction was carried out with the help of ultrasonication in this experiment. The effect of ultrasonication time on the recovery was evaluated at 5 min, 10 min, 15 min, and 20 min. The results show that with the increase of ultrasonication time, the average recoveries increased and reached the best at 10 min with the recoveries of 61.10–93.71% (Fig. 4). Therefore, 10 min was the best ultrasonication time for extraction.
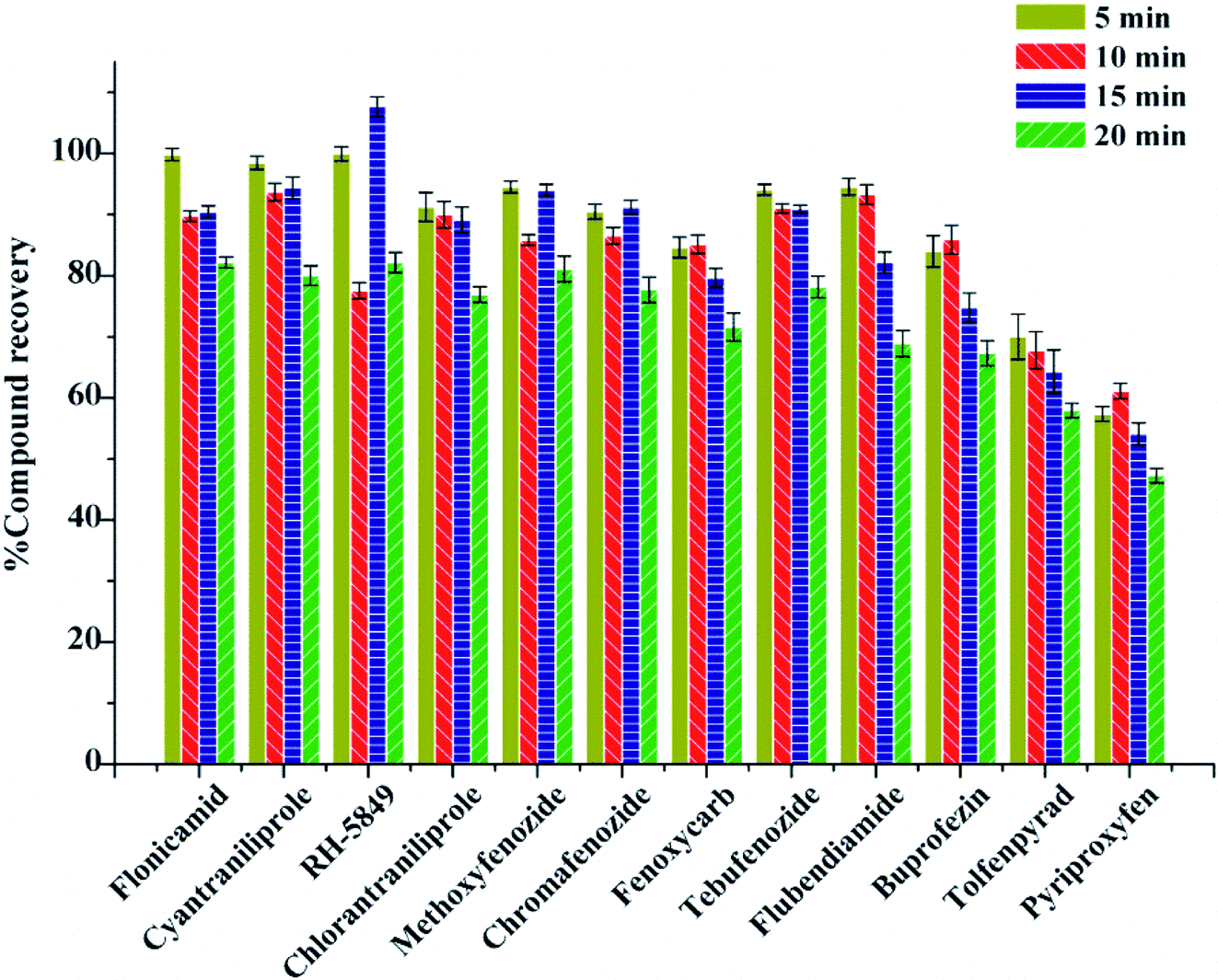 | ||
| Fig. 4 Recoveries of 12 kinds of IGRs under different ultrasonic extraction time. | ||
Moreover, for most QuEChERS method, the combination of NaCl and MgSO4 was used in extraction to make salting-out effect,33 causing the partitioning of water and acetonitrile. However, no substantial differences were observed for recoveries of 12 IGRs in vegetable oil, when only NaCl or the combination of NaCl and MgSO4 were used. As a result, only NaCl was used for partitioning of water and acetonitrile.
For vegetable oil, fruit, and tea samples, the sample characteristic showed obvious difference, the optimal extraction procedure for vegetable oil was also investigated and adjusted for fruit and tea. The experiment showed that addition of water in the extraction procedure made more impurities for fruit and tea, which reduced extraction efficiency. Meanwhile, when 10 mL extractant was examined, 2 g tea make it hard to remove 8 mL supernatant, then 1 g tea was chosen for the extraction procedure. For fruit and tea, no addition of water in the extraction procedure, then less amount of NaCl (500 mg) was used for fruit, and NaCl was not added for tea.
In the purification experiment, the sorbents and their amount were discussed with vegetable oil, fruit and tea, respectively. The blank sample was spiked at the level of 10 μg kg−1, after extraction with the optimal procedure, the purifying procedure was explored according to Section 2.4 with different sorbents, and the recoveries was calculated by matrix-matched standard curves, the average recoveries based on 5 replicates were used to evaluate the purifying efficiency.
For the fruit sample, the efficiencies of six sets of sorbents (0.1 g PSA + 0.3 g MgSO4; 0.1 g C18 + 0.3 g MgSO4; 0.1 g PSA + 0.1 g C18 + 0.3 g MgSO4; 0.1 g GCB + 0.1 g C18 + 0.1 g PSA + 0.3 g MgSO4; 0.1 g GCB + 0.1 g C18 + 0.3 g MgSO4; 0.1 g GCB + 0.1 g PSA + 0.3 g MgSO4) were compared by purifying the sample extracts spiked with 10 μg kg−1 of the target 12 IGRs. The results are shown in Table S1.† Graphitized carbon black (GCB) showed a strong adsorption capacity to cyanoformamide and chlorantraniliprole. The recoveries of cyanoformamide and chlorantraniliprole were 16.58–27.49% using the purified sorbents containing GCB; at the same time, GCB also adsorbed tolfenpyrad and pyriproxyfen, which can be attributed to the planar structures of analytes.34 The other four sorbents without GCB showed better purification effect on 12 IGRs, and the recovery of each substance was above 79.9%. According the results shown in Table S1,† the sorbent C18 made better recovery for 12 IGRs than PSA or PSA + C18. Then, a mixture of 0.1 g C18 and 0.3 g MgSO4 was selected as the sorbent, and the corresponding recoveries of 12 IGRs were 90.12–107.08%.
Based on the above results, the purification efficiency of PSA, C18, MgSO4, and their different combinations was separately evaluated for vegetable oil and tea. As shown in Table S2,† a mixture of 0.1 g PSA and 0.3 g MgSO4 was the most effective sorbent for the purification of vegetable oil with the recoveries of 60.34–105.85%. For tea, the mixture of 0.1 g PSA, 0.2 g C18 and 0.1 g MgSO4 was the optimal purification sorbent, which can obtain the best recoveries of 12 IGRs with 62.97–110.81% (Table S3†).
3.4. Method validation
| Compound | Spiked level (μg kg−1) | Vegetable oil (%) | Fruit (%) | Tea (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Flonicamid | 10 | 50 | 100 | 86.45 | 80.16 | 83.46 | 37.52 | 37.46 | 43.42 | 10.19 | 10.05 | 9.98 |
| Cyantraniliprole | 10 | 50 | 100 | 89.96 | 87.88 | 96.88 | 52.81 | 54.72 | 63.24 | 81.17 | 85.70 | 87.76 |
| RH-5849 | 10 | 50 | 100 | 90.34 | 86.74 | 89.20 | 80.58 | 88.93 | 90.47 | 80.89 | 85.76 | 87.88 |
| Chlorantraniliprole | 10 | 50 | 100 | 98.79 | 94.68 | 98.18 | 78.88 | 79.96 | 87.36 | 86.89 | 91.42 | 92.82 |
| Methoxyfenozide | 10 | 50 | 100 | 91.11 | 87.09 | 89.24 | 102.44 | 100.01 | 111.89 | 91.03 | 94.45 | 97.20 |
| Chromafenozide | 10 | 50 | 100 | 93.62 | 89.76 | 84.95 | 103.26 | 100.61 | 118.31 | 93.01 | 94.64 | 95.79 |
| Fenoxycarb | 10 | 50 | 100 | 93.12 | 91.81 | 94.57 | 87.36 | 90.40 | 103.32 | 90.62 | 93.09 | 94.32 |
| Tebufenozide | 10 | 50 | 100 | 91.67 | 90.79 | 89.86 | 105.17 | 101.24 | 112.00 | 90.95 | 96.80 | 96.50 |
| Flubendiamide | 10 | 50 | 100 | 112.97 | 119.19 | 115.32 | 121.01 | 113.26 | 122.91 | 87.69 | 102.93 | 94.16 |
| Buprofezin | 10 | 50 | 100 | 90.16 | 86.35 | 83.98 | 96.37 | 100.35 | 116.94 | 96.43 | 97.07 | 98.39 |
| Tolfenpyrad | 10 | 50 | 100 | 109.74 | 96.02 | 99.10 | 91.54 | 96.18 | 111.34 | 97.79 | 95.09 | 94.99 |
| Pyriproxyfen | 10 | 50 | 100 | 90.23 | 88.07 | 91.03 | 91.67 | 96.88 | 113.90 | 90.39 | 90.28 | 92.78 |
| Compound | Linear range (μg L−1) | Calibration curve | R2 | LOD (μg kg−1) | LOQ (μg kg−1) |
|---|---|---|---|---|---|
| Flonicamid | 0.2–200 | y = 157355.59x+ 826.70 |
0.9999 | 0.11 | 0.36 |
| Cyantraniliprole | 0.2–200 | y = 7307.53x + 112.64 | 0.9998 | 0.37 | 1.12 |
| RH-5849 | 0.2–200 | y = 61441.90x + 22201.13 |
0.9997 | 0.10 | 0.30 |
| Chlorantraniliprole | 0.2–200 | y = 3566.99x +774.37 | 0.9999 | 0.04 | 0.13 |
| Methoxyfenozide | 0.2–200 | y = 63663.93x + 6348.65 |
0.9999 | 0.05 | 0.17 |
| Chromafenozide | 0.2–200 | y = 78091.30x + 10905.83 |
0.9999 | 0.08 | 0.26 |
| Fenoxycarb | 0.2–200 | y = 51842.30x + 2040.27 |
0.9994 | 0.40 | 1.24 |
| Tebufenozide | 0.2–200 | y = 63738.42x +1748.25 |
0.9999 | 0.23 | 0.74 |
| Flubendiamide | 0.2–200 | y = 3869.97x + 108.77 | 0.9999 | 0.21 | 0.71 |
| Buprofezin | 0.2–200 | y = 140217.13x+ 6772.69 |
0.9999 | 0.04 | 0.15 |
| Tolfenpyrad | 0.2–200 | y = 30751.13x + 948.79 |
0.9999 | 0.28 | 0.94 |
| Pyriproxyfen | 0.2–200 | y = 26817.48x + 2904.63 |
0.9999 | 0.09 | 0.31 |
| Compound | Spiked level (μg kg−1) | Recovery (%) | RSD (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Flonicamid | 10 | 50 | 100 | 83.20 | 67.82 | 65.47 | 3.16 | 8.63 | 3.49 |
| Cyantraniliprole | 10 | 50 | 100 | 88.02 | 95.19 | 85.03 | 3.27 | 3.63 | 3.34 |
| RH-5849 | 10 | 50 | 100 | 78.45 | 77.33 | 80.27 | 11.09 | 8.10 | 6.95 |
| Chlorantraniliprole | 10 | 50 | 100 | 77.08 | 78.11 | 79.87 | 4.38 | 3.62 | 3.45 |
| Methoxyfenozide | 10 | 50 | 100 | 80.22 | 83.25 | 81.58 | 5.41 | 3.28 | 3.79 |
| Chromafenozide | 10 | 50 | 100 | 78.39 | 80.28 | 82.53 | 4.26 | 5.51 | 4.06 |
| Fenoxycarb | 10 | 50 | 100 | 76.56 | 78.27 | 80.51 | 3.54 | 5.69 | 1.93 |
| Tebufenozide | 10 | 50 | 100 | 81.26 | 80.92 | 83.28 | 6.24 | 4.63 | 4.01 |
| Flubendiamide | 10 | 50 | 100 | 78.47 | 78.01 | 76.03 | 11.06 | 9.40 | 4.35 |
| Buprofezin | 10 | 50 | 100 | 71.94 | 70.68 | 74.12 | 2.60 | 5.68 | 1.92 |
| Tolfenpyrad | 10 | 50 | 100 | 63.89 | 66.09 | 65.15 | 2.44 | 6.21 | 4.29 |
| Pyriproxyfen | 10 | 50 | 100 | 67.35 | 62.75 | 65.06 | 3.07 | 3.21 | 2.97 |
| Compound | Spiked level (μg kg−1) | Recovery (%) | RSD (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Flonicamid | 10 | 50 | 100 | 88.70 | 102.81 | 96.75 | 3.65 | 7.66 | 3.18 |
| Cyantraniliprole | 10 | 50 | 100 | 88.02 | 103.19 | 85.03 | 2.95 | 3.88 | 3.84 |
| RH-5849 | 10 | 50 | 100 | 85.82 | 102.36 | 88.12 | 9.68 | 7.31 | 8.57 |
| Chlorantraniliprole | 10 | 50 | 100 | 99.70 | 105.53 | 92.02 | 4.75 | 3.40 | 5.07 |
| Methoxyfenozide | 10 | 50 | 100 | 93.76 | 101.49 | 89.32 | 6.05 | 3.41 | 4.14 |
| Chromafenozide | 10 | 50 | 100 | 80.55 | 99.53 | 95.78 | 3.56 | 5.93 | 4.16 |
| Fenoxycarb | 10 | 50 | 100 | 86.03 | 94.03 | 84.45 | 3.76 | 5.53 | 2.79 |
| Tebufenozide | 10 | 50 | 100 | 90.88 | 100.74 | 102.78 | 6.54 | 4.47 | 5.40 |
| Flubendiamide | 10 | 50 | 100 | 89.85 | 110.15 | 90.95 | 11.07 | 8.91 | 4.37 |
| Buprofezin | 10 | 50 | 100 | 89.65 | 103.08 | 95.88 | 3.22 | 6.61 | 3.02 |
| Tolfenpyrad | 10 | 50 | 100 | 84.67 | 96.14 | 86.57 | 2.52 | 5.64 | 4.40 |
| Pyriproxyfen | 10 | 50 | 100 | 84.57 | 95.66 | 83.33 | 3.21 | 4.30 | 3.14 |
| Compound | Spiked level (μg kg−1) | Recovery (%) | RSD (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Flonicamid | 10 | 50 | 100 | 68.29 | 62.02 | 65.13 | 9.35 | 4.46 | 3.18 |
| Cyantraniliprole | 10 | 50 | 100 | 71.14 | 69.07 | 67.86 | 6.95 | 2.44 | 4.19 |
| RH-5849 | 10 | 50 | 100 | 71.52 | 65.49 | 68.45 | 6.12 | 3.38 | 4.31 |
| Chlorantraniliprole | 10 | 50 | 100 | 78.62 | 76.45 | 74.82 | 11.58 | 2.05 | 1.91 |
| Methoxyfenozide | 10 | 50 | 100 | 80.54 | 81.66 | 91.05 | 8.93 | 3.59 | 4.19 |
| Chromafenozide | 10 | 50 | 100 | 64.95 | 84.86 | 84.28 | 7.59 | 2.48 | 3.57 |
| Fenoxycarb | 10 | 50 | 100 | 95.94 | 81.24 | 77.98 | 10.21 | 1.30 | 2.49 |
| Tebufenozide | 10 | 50 | 100 | 92.60 | 85.47 | 96.50 | 9.09 | 2.47 | 3.23 |
| Flubendiamide | 10 | 50 | 100 | 68.69 | 83.36 | 91.16 | 3.49 | 3.31 | 6.03 |
| Buprofezin | 10 | 50 | 100 | 89.50 | 81.97 | 85.64 | 10.03 | 3.29 | 3.06 |
| Tolfenpyrad | 10 | 50 | 100 | 97.99 | 104.86 | 98.79 | 8.47 | 11.29 | 4.12 |
| Pyriproxyfen | 10 | 50 | 100 | 86.75 | 79.98 | 81.67 | 4.50 | 3.55 | 1.17 |
The average recoveries at three spiked levels in three different substrates were in the range of 62.75–95.19% for vegetable oil, 80.55–110.15% for fruit, and 62.02–104.86% for tea, respectively, with relative standard deviations (RSDs, n = 7) of 1.17–11.58%. Owing to the advantages of simple operation and high accuracy and sensitivity, this method was suitable for rapid and simultaneous detection of 12 IGRs in vegetable oil, fruit, and tea.
3.5. Application to actual sample analysis
The developed method in this study was applied to the identification and quantification of 12 IGRs in 17 vegetable oils, 16 fruits, and 24 tea samples separately. The samples were obtained from local markets and supermarkets in Shijiazhuang and analyzed following the preparation procedure. Flonicamid (2.34 μg kg−1) and RH-5849 (0.20 μg kg−1) were observed in one fruit sample, cyantraniliprole (3.28 μg kg−1) was observed in one fruit sample, chlorantraniliprole (0.14–0.19 μg kg−1) was observed in four fruit samples, buprofezin (0.32–1.26 μg kg−1) was observed in four fruit samples, tolfenpyrad (1.63 μg kg−1) was observed in one fruit sample (Table S6†). The contents of chlorantraniliprole, buprofezin, and tolfenpyrad, in 24 tea samples are 0.74–84.72 μg kg−1, 0.18–38.60 μg kg−1 and 0.34–1125.15 μg kg−1 (Table S7†). The contents of 12 IGRs in edible oil samples were all below the LODs. According to the Chinese GB2763-2019 “National food safety standard—maximum residue limits for pesticides in food”,18 the above IGRs residue contents in the tested samples did not exceed the standard.3.6. Method performance comparison
Analytical performances of the developed QuEChERS-UPLC-MS/MS method for the determining IGRs were mainly compared with the reported LC-MS/MS methods. As shown in Table 7, SPE and QuEChERS were the most used pretreatment methods for the detection of IGRs. However, compared with SPE, QuEChERS was more convenient and fast. In these reported methods, most of them were developed to test one or two IGRs,9,25,37–39 and/or the testing sample matrix was single. Only a method for 8 IGRs in honey was established,4 and another method was used to detect 6 IGRs in water, sediment and aquatic products.21 In comparison, the new QuEChERS-UPLC-MS/MS method was developed for the determination of 12 IGRs in three different sample matrixes with suitable recoveries and the low LOQs, which showed high throughout and high sensitivity.| Analytical method | Pretreatment method | Analytes | Sample | Recovery (%) | LOQ (μg kg−1) | Ref. |
|---|---|---|---|---|---|---|
| LC-MS/MS | DPX (disposable pipette extraction) | 8 IGRs | Honey | 75–109 | 1.0–10 | 4 |
| LC-MS/MS | SPE | Tebufenozide | Vegetable | 70–110 | 4.0 | 9 |
| LC-MS/MS | SPE | Pyriproxyfen, tolfenpyrad | Citrus | 80.6–113 | 5.0 | 25 |
| LC-MS/MS | LLE, QuEChERS | 6 IGRs | Water, sediment, aquatic products | 80–99.7 | 1.27–3.20 | 21 |
| LC-MS/MS | QuEChERS | Flonicamid | Paprika | 94–101.7 | 10 | 37 |
| LC-MS/MS | QuEChERS | Flonicamid | Vegetable | 86.8–94.8 | 4.0 | 38 |
| LC-MS/MS | QuEChERS | Chlorantraniliprole, buprofezin | Fruit, vegetable | 64–115 | 0.03–0.51 | 39 |
| LC-MS/MS | QuEChERS | 12 IGRs | Vegetable, fruit, tea | 62.02–110.15 | 0.11–3.91 | This work |
4. Conclusion
A modified QuEChERS method coupled with UPLC-MS/MS analysis was successfully developed to determine 12 IGRs in foods of different matrixes. The samples were extracted with acetonitrile containing 0.5% formic acid, purified with different sorbents, detected by UPLC-MS/MS with ESI in positive ion mode under MRM mode, and quantified using the external standard method. The extraction and purification methods were optimized for different food matrix samples, and then the method was validated regarding its analytical range, precision, recovery, and ME. Based on the experimental results, the method is simple, fast, sensitive, which can be applied in accurate and high throughout detection of IGRs, providing technical support for relevant food risk assessment. However, flonicamid and cyantraniliprole still show obvious matrix effect in fruit and tea, the further improvement for pretreatment method would be explored to eliminate the matrix effect.Author contributions
Ke Wang: data curation, formal analysis, methodology, writing – original draft, funding acquisition. Lingzhi Zhao: data curation. Can Zhang: formal analysis, methodology, writing – original draft. Hong Zhang: methodology, validation. Kaoqi Lian: conceptualization, funding acquisition, project administration, resources, writing – review & editing. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Hebei Medical Applicable Technology Tracking Project (GZ2020042), the Science Technology Research and Development Guidance Program of Shijiazhuang (191460733), and the Natural Science Foundation of Hebei Province (Nos H 2018206122).Notes and references
- E. A. Gamal, E. E. Anwar, M. E. Ferial, M. E. Hamdi and M. E. Hesham, Pest Manage. Sci., 2003, 69, 95–102 Search PubMed.
- G. C. Zhang, H. Zou, N. N. Geng, N. Ding, Y. J. Wang, J. Zhang and C. Zou, Pestic. Biochem. Physiol., 2020, 163, 64–75 CrossRef CAS PubMed.
- J. D. Fine, C. A. Mullin, M. T. Frazier and R. D. Reynolds, J. Econ. Entomol., 2017, 110, 1993–2001 CrossRef CAS PubMed.
- S. Song, C. Zhang, Z. Chen, F. He, J. Wei, H. Tan and X. Li, J. Chromatogr. A, 2018, 1557, 51–61 CrossRef CAS PubMed.
- M. R. Qian, L. Q. Wu, H. Zhang, M. F. Xu, R. Li, X. Y. Wang and C. X. Sun, Anal. Bioanal. Chem., 2012, 402, 2451–2462 CrossRef CAS PubMed.
- D. Šatínský, L. Naibrtová, C. Fernández-Ramos and P. Solich, Talanta, 2015, 142, 124–130 CrossRef PubMed.
- X. L. Hu, J. J. Niu, Q. Meng, Y. H. Chai, K. H. Chu and K. M. Chan, Environ. Pollut., 2019, 253, 89–99 CrossRef CAS PubMed.
- J. L. Jiang, Z. J. Shan, X. R. Wang, Y. X. Zhu and J. Y. Zhou, Environ. Sci. Pollut. Res., 2018, 25, 10730–12739 CrossRef CAS PubMed.
- L. Dai, J. Z. Xu, T. Ding, J. J. Zhu, B. Wu, C. Y. Shen and Y. Jiang, Chin. J. Anal. Chem., 2008, 36, 87–90 CAS.
- T. Liu, M. Dong, F. J. Zhou, D. Yang and X. M. Zhang, Food Sci. Hum. Well., 2019, 8, 362–367 CrossRef.
- A. Kadala, M. Charretona and C. Colleta, J. Insect Physiol., 2020, 125, 104086 CrossRef CAS PubMed.
- S. L. Feng, Z. M. Kong, W. X. Wang, X. M. Wang and P. A. Peng, Teratog., Carcinog., Mutagen., 2002, 14, 210–213 Search PubMed.
- M. Liu, H. Yang, H. Liu, P. Han, X. Wang, S. Zhang and Y. Wu, J. Sci. Food Agric., 2008, 88, 62–67 CrossRef CAS.
- S. Navis, A. Waterkeyn, L. De Meester and L. Brendonck, Ecotoxicology, 2018, 27, 627–634 CrossRef CAS PubMed.
- G.-F. Pang, Chapter 1: Introduction, Analytical Methods for Food Safety by Mass Spectrometry, Academic Press, 2018, pp. 1–9 Search PubMed.
- Regulation (EC) No. 396/2005 of the European Parliament and of the Council of 23 February 2005 on maximum residue levels of pesticides in or on food and feed of plant and animal origin and amending Council Directive 91/414/EEC (text with EEA relevance).
- Ministry of Health, Labour and Welfare, The Japanese positive list system for agricultural chemical residues in foods, Ministry of Health, Labour and Welfare, Tokyo, 2006 Search PubMed.
- National Food Safety Standard, Maximum Residue Limits for Pesticides in Food: GB 2763-2019, Standards Press of China, Beijing, 2019 Search PubMed.
- F. M. Malhat, Food Anal. Methods, 2012, 5, 1492–1496 CrossRef.
- S. Mohapatra, A. K. Ahuja, M. Deepa, G. K. Jagadish, N. Rashmi and D. Sharma, J. Environ. Sci. Health, Part B, 2011, 46, 264–271 CrossRef CAS PubMed.
- J. Gan, H. Liu, Y. Chen, J. Peng, T. Liu, J. Chen and L. He, Ecotoxicol. Environ. Saf., 2021, 210, 111853–111861 CrossRef CAS PubMed.
- M. Saitta, G. Di Bella, M. R. Fede, V. Lo Turco, A. G. Potorti, R. Rando, M. T. Russo and G. Dugo, Food Addit. Contam., Part A, 2017, 34, 800–808 CAS.
- Y. L. Cui, K. C. Liu, C. Xu, F. Liu, Q. X. Li, S. Z. Liu and B. M. Wang, Food Chem., 2014, 143, 293–299 CrossRef CAS PubMed.
- G. P. de Pinho, A. A. Neves, M. E. L. R. de Queiroz and F. O. Silvério, Food Control, 2010, 21, 1307–1311 CrossRef CAS.
- M. Dong, G. Wen, H. Tang, T. Wang, Z. Zhao, W. Song, W. Wang and L. Zhao, Food Chem., 2018, 269, 136–141 CrossRef CAS PubMed.
- J. Ma, Z. Yao, L. Hou, W. Lu, Q. Yang, J. Li and L. Chen, Talanta, 2016, 161, 686–692 CrossRef CAS PubMed.
- J. Ma, L. Hou, G. Wu, L. Wang, X. Wang and L. Chen, Materials, 2020, 13, 5729–5742 CrossRef CAS PubMed.
- M. A. Farajzadeh, M. R. A. Mogaddam and H. Ghorbanpour, J. Chromatogr. A, 2014, 1347, 8–16 CrossRef CAS PubMed.
- J. Beltran, F. J. Lopez and F. Hernandez, J. Chromatogr. A, 2000, 885, 389–404 CrossRef CAS.
- Y. Wen, Z. Niu, Y. Ma, J. Ma and L. Chen, J. Chromatogr. A, 2014, 1368, 18–25 CrossRef CAS PubMed.
- S. Kammoun, B. Mulhauser, A. Aebi, E. A. D. Mitchell and G. Glauser, Environ. Pollut., 2019, 247, 964–972 CrossRef CAS PubMed.
- T. Xu, X. Feng, L. Pan, J. Jing and H. Zhang, RSC Adv., 2018, 8, 35485–35495 RSC.
- R. Perestrelo, P. Silva, P. Porto-Figueira, J. A. M. Pereira, C. Silva, S. Medina and J. S. Camara, Anal. Chim. Acta, 2019, 1070, 1–28 CrossRef CAS PubMed.
- L. C. da Cabrera, S. S. Caldas, O. D. Prestes, E. G. Primel and R. Zanella, J. Sep. Sci., 2016, 39, 1945–1954 CrossRef PubMed.
- X. Li, J. Chen, X. He, Z. Wang, D. Wu, X. Zheng, L. Zheng and B. Wang, Chemosphere, 2019, 234, 224–231 CrossRef CAS PubMed.
- P. P. Vazquez, A. Lozano, S. Ucles, M. M. Ramos and A. R. Fernandez-Alba, J. Chromatogr. A, 2015, 1426, 161–173 CrossRef CAS PubMed.
- A. Y. Ko, A. M. Abd El-Aty, M. M. Rahman, J. Jang, S. W. Kim, J. H. Choi and J. H. Shim, Food Chem., 2014, 157, 413–420 CrossRef CAS PubMed.
- Y. Xu, L. F. Shou and Y. L. Wu, J. Chromatogr. A, 2011, 1218, 6663–6666 CrossRef CAS PubMed.
- H. Li, C. Wang, Q. Zhu, H. Du, S. Guan, F. Wang, W. Zhang, W. Fan, Z. Chen, G. Yang and H. Y. Aboul-Enein, Anal. Methods, 2016, 8, 5061–5069 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00046b |
| This journal is © The Royal Society of Chemistry 2021 |