DOI:
10.1039/D1RA00049G
(Paper)
RSC Adv., 2021,
11, 5325-5334
Polysorbate-80 surface modified nano-stearylamine BQCA conjugate for the management of Alzheimer's disease†
Received
4th January 2021
, Accepted 13th January 2021
First published on 28th January 2021
Abstract
Acetylcholinesterase (AChE) inhibitors such as donepezil, galantamine and rivastigmine are used for the management of dementia in Alzheimer's Disease (AD). These drugs elevate endogenous acetylcholine (ACh) levels at the M1 muscarinic receptor in the brain to achieve therapeutic benefits. However, their side effects, such as nausea, vomiting, dizziness, insomnia, loss of appetite, altered heart rate, etc., are related to non-specific peripheral activation of M2–M5 muscarinic subtypes. It is logical, therefore, to develop drugs that selectively activate brain M1 receptors. Unfortunately, the orthosteric site homology among the receptor subtypes does not permit this approach. An alternative approach is to use positive allosteric modulator (PAM) of M1 receptors like benzyl quinolone carboxylic acid (BQCA). PAMs although devoid of M1 agonist activity, however, when bound, enhance the binding affinity of orthosteric ligand, Ach. The current challenge with PAMS is their low brain half-life, permeability, and higher elimination rates. This study reports active targeting of brain M1 receptors using surface modified nano lipid–drug conjugates (LDC) of M1 PAM, BQCA, to treat AD. Polysorbate-80 (P-80) surface modified stearylamine (SA)-BQCA conjugated nanoparticles (BQCA-SA-P80-NPs) were prepared by conjugating BQCA to SA, followed by the formation of nanoparticles (NPs) using P-80 by solvent injection method. The BQCA-SA-P80-NPs are near-spherical with a particle size (PS) of 166.62 ± 1.24 nm and zeta potential (ZP) of 23.59 ± 0.37 mV. In the in vitro cytotoxicity (SH-SY5Y cells) and hemolysis assays, BQCA-SA-P80-NPs, show acceptable safety and compatibility. In mice, Alzheimer's model, BQCA-SA-P80-NPs significantly prevent STZ induced changes in memory, neuronal Aβ1–42, p-Tau, APP, NF-κB, and BACE levels and neuronal cell death, when compared to untreated disease control and naïve BQCA treated group. Further, BQCA-SA-P80-NPs significantly improve the therapeutic efficacy of AChE inhibitor, donepezil (DPZ), indicating its potentiating effects. In vivo biodistribution studies in mice show selective accumulation of BQCA-SA-P80-NPs in the brain, suggesting an improved brain bioavailability and reduced peripheral side effects of BQCA. The study results demonstrate that BQCA-SA-P80-NPs can improve brain bioavailability and therapeutic efficacy of BQCA in AD.
1. Background
Alzheimer's Disease (AD) is a neurodegenerative disorder which drastically affects memory, social and behavioural skills.1 It is the 6th leading cause of death in the United States and is the 6th foremost cause of mortality in older people aged above 65 years in the world. As per the reports of the Alzheimer's Association 2015, around 5.7 million people will be living with Alzheimer's related dementia in the year 2018, and this number will increase to 7.1 and 13.8 million by the year 2025 and 2030, respectively.2 In AD, β amyloid proteins and tau proteins accumulate, ventricles enlarge, and progressive degeneration of neurons of the basal forebrain cholinergic system and cortex is observed.3 The brain expression, cellular localization, and knockout mice model studies suggest that of all the muscarinic receptor subtypes M1 receptor is widely expressed in brain regions involved in memory and cognition, and these areas are significantly affected in AD.4–9 To date, the most efficient strategy to manage AD is through enhancing endogenous ACh levels. FDA approved acetylcholine esterase inhibitors (AChEIs) such as donepezil, rivastigmine, and galantamine10–12 serve this purpose. The inhibition of AChE by these agents results in a significant increase of ACh levels at both central and peripheral cholinergic receptor sites. This leads to non-selective activation of muscarinic ACh receptors (mAChRs).13,14 This non-selective activation is associated with several unacceptable peripheral side effects such as nausea, vomiting, dizziness, insomnia, loss of appetite, altered heart rate, etc.11,15,16 Specific brain targeting of these AChEIs will not help due to the presence of other mAChRs throughout the brain.17 Successful management of AD, therefore, requires agents that can specifically activate M1 receptors in the brain. Unfortunately, due to the high degree of receptor homology among mAChRs, it is challenging to develop M1 specific compounds.18,19 Alternately, positive allosteric modulators (PAMs) of M1 AChRs such as benzyl quinolone carboxylic acid (BQCA) can help in specific activation of M1 AChRs.20–22 PAMs through their selective binding at allosteric site of M1 mAChR lead to conformational changes in M1 mAChR. This facilitates enhanced binding of the orthosteric ligand, ACh, to the M1 mAChR, thus, selectively increasing the actions of endogenous ACh on M1 receptor over M2–M5 receptor subtypes. PAMs, however, suffer from low brain half-life and faster elimination rates.22–24 The brain bioavailability of PAMs can be significantly improved by formulating them as lipid drug conjugates (LDCs).25 LDCs improve the lipophilic characteristics of a drug and have high drug loading capacity.26,27 They offer ideal particle size (100–200 nm), permit surface modifications, improve stability of the drug, and sustain drug release.28,29 In this study, we report active targeting of brain M1 receptors using polysorbate 80 (P-80) surface modified nano-stearylamine-BQCA (BQCA-SA-P80-NPs) and also demonstrate that BQCA-SA-P80-NPs improve brain bioavailability and therapeutic efficacy of BQCA in mice model of AD.
2. Methods
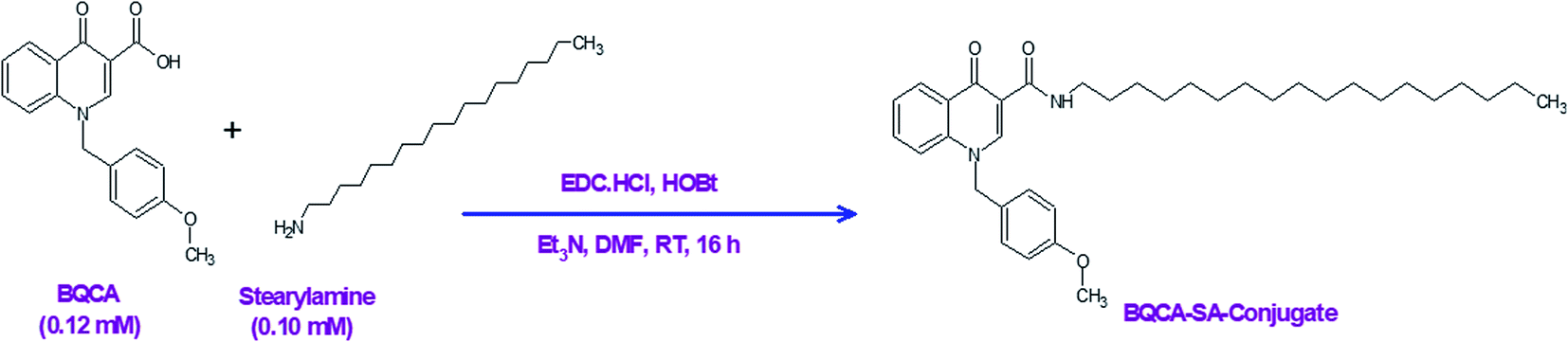
2.1. Synthesis of stearyl amine-BQCA conjugate (BQCA-SA-LDC)
In a three-necked round bottom flask, BQCA (0.37 g, 0.12 mmol) was mixed with dry DMF (3 ml) for 10 minutes on a magnetic stirrer. To this, EDC (0.03 g, 0.15 mmol) was added and stirred for 30 min at room temperature. Following this, HOBt (0.02 g, 0.15 mmol) was added and stirring continued for 1 h. After that, stearyl amine (0.27 g, 0.10 mmol) and triethylamine (0.36 mmol) were added, and the reaction mixture was stirred continuously for 16 h. The reaction was monitored by thin-layer chromatography (TLC). After completion of the reaction, water (10–20 ml) was added to precipitate the product. The precipitated product was filtered and washed 5-times with water (10 ml). The obtained solid was purified on a silica gel column (60–120 mesh silica gel), using chloroform and methanol (93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) as a mobile phase (Fig. 1).
:7) as a mobile phase (Fig. 1).
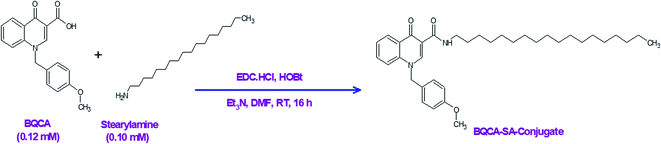 |
| | Fig. 1 Synthesis of the BQCA-SA-conjugate. | |
2.2. Preparation of BQCA-SA-P80 NPs
The solvent injection method was employed to prepared BQCA-SA-P80 NPs from BQCA-SA LDC.30 BQCA-SA LDC (50 mg) lipid bulk, surfactant, and a surface modifying agent P80, and isopropyl alcohol (IPA).30,31 Briefly, BQCA-SA LDC (50 mg) was dissolved in IPA (3 ml), and heated to 70 °C for 15 min on a water bath. This mixture was then rapidly injected (using a syringe) to an aqueous solution of P80 under stirring. The obtained mixture was homogenized (Ika-Ultra Turrax®, T25) at 1000 rpm for 10 min, to get a coarse emulsion. The coarse emulsion was probe sonicated (LABSONIC®M, Sartorius Ltd, Mumbai, India) to form BQCA-SA-P80 NPs. The resulting nanoemulsion was then stirred for 5 min and purged with inert nitrogen to evaporate the solvent. For imaging studies, rhodamine 6G (Rh) loaded stearyl amine-BQCA conjugate nanoparticles (Rh-BQCA-SA-P80-NPs) were prepared as above by adding Rh (0.4 mg) to lipid phase.32
2.3. Optimization of BQCA-SA-P80 NPs using box Behnken design
The BQCA-SA-P80 NPs formulation was optimized by employing a 3 factor and 3 level Box-Behnken Design (BBD), with 3 central points and 15 experimental runs using Minitab®18.1 statistical software (Minitab Ltd. Coventry, United Kingdom). The concentration of the surfactant P80 (% w/v) (A), homogenization amplitude (%) (B) and the homogenization time (min) (C) are independent variables at low (−1), medium (0) & high (+1) levels. Particle size (PS), and zeta potential (ZP) are dependent variables or responses (Table 1). By using the quadratic mathematical equation, the model was validated.33,34
Table 1 Independent variables and responses used for BBD
| Factors |
Levels |
| Independent variables |
Low level (−1) |
Medium level (0) |
High level (+1) |
| A: Surfactant concentration (% w/v) |
0.5 |
1.5 |
2.5 |
| B: Sonication amplitude (%) |
60 |
80 |
100 |
| C: Sonication time (min) |
3 |
6 |
9 |
| Dependent variables/responses |
Constraints |
| Particle size |
Minimize |
| Zeta potential |
Maximize |
2.4. Characterization of BQCA-SA-P80-NPs
2.4.1. Particle size (PS), zeta potential (ZP) and polydispersity index (PDI). PS, ZP, and PDI of BQCA-SA-P80-NPs were measured using Zetasizer (Anton Parr Litesizer™500, Kalliope software). The PS and PDI measured by analyzing the fluctuations in DLS by photon correlation spectroscopy technique, electrophoretic light scattering technique was used to determine ZP or surface charge of nanoparticles. All the measurements were performed three times, and the obtained results were expressed as the mean size of particle ± SD.35
2.4.2. Surface morphology. The surface morphology of the BQCA-SA-P80-NPs was characterized by scanning electron microscopy (SEM) (JSM-5510, JEOL Ltd, Tokyo, Japan) and transmission electron microscopy (TEM) (TEM, JEOL JEM-1200EX, Tokyo, Japan).
2.4.3. Determination of drug content. BQCA-SA-P80-NPs (10 mg) was mixed with amidase solution (10 μM, 10 ml), and incubated for 24 h. To the above mixture, 1 ml of acetone:methanol (1:1) solution was added to precipitate proteins. The mixture was then passed through membrane filter (0.25 μm), and suitably diluted and was analyzed using the developed LS-MS method.
2.5. In vitro drug release studies for an optimized batch of BQCA-SA-P80-NPs
The study was performed on the suspension of nanoparticles within 24 h of preparation using a dialysis membrane (molecular weight cut off 12000–14000 Daltons) (ESI†).34
2.6. Hemolysis assay
The hemocompatibility of BQCA-SA-P80-NPs was studied by hemolysis assay (ESI†).36
2.7. Cytotoxicity assay
Cytotoxicity of BQCA-SA-P80-NPs to SH-SY5Y neuronal cells was studied by SRB assay (ESI†).33
2.8. In vivo efficacy studies
2.8.1. Animals. Adult male Swiss albino mice (25–30 g) were maintained in the central animal house of JSS College of Pharmacy, Ooty. Animals were housed in standard laboratory conditions (22 ± 2 °C, light: dark cycle of 12:12 h) with normal pellet diet and water ad libitum. (IAEC Approval No. JSSCP/IAEC/OT/PhD/Ph.Cology/02/2017–18).
2.8.2. Induction of AD. Streptozotocin (STZ) was freshly prepared by dissolving in aCSF (30 mg ml−1). Mice were anesthetized with ketamine, xylazine cocktail [2 ml ketamine (50 mg ml−1) and 0.8 ml xylazine (20 mg ml−1), sterile water upto 10 ml; inject 0.1 ml per 10 g mice, i.p.]. Mice were then carefully placed in the stereotaxic frames, and the head was locked to prevent movement during the experiment. STZ (3 mg kg−1) was injected with Hamilton syringe at a flow rate of 0.5 ml min−1 using a stereotaxic apparatus. aCSF, instead of STZ was injected for the sham group using the same procedure.37,38
2.8.3. Treatment schedule. 14 days post-surgery; the animals were divided into 6 groups of 6 each. Groups 1 and 2 received treatment with the vehicle (saline, 1.0 ml kg−1 i.p.), group 3 received naïve Donepezil (DPZ) solution (3 mg kg−1 i.p.), group 4 received naïve BQCA (10 mg kg−1 i.p.), group 5 and 6 received BQCA-SA-P80-NPs 5, and 10 mg kg−1 i.p., respectively, group 7 received BQCA-SA-P80-NPs (5 mg kg−1, i.p.) and Donepezil solution (1 mg kg−1, i.p.). The treatment was given daily for 21 days. All the animals were subjected to the behavioural studies during the study period, and biochemical and histological studies were performed at the end of the study.
2.8.4. Morris water maze test (MWM) effect on acquisition and reversal learning. The apparatus consists of a large circular tank of (140 cm) diameter, painted black with non-toxic paint. The tank was filled with opaque water containing milk (10 ml in 100 ml) and was maintained at a temperature of 26 ± 1 °C.39 A platform of (14 cm diameter, 26.7 cm tall) was placed in the tank, such that it is 14 mm below the surface of the water after that, at the start of each trail of the acquisition phase, mice were placed in the tank at any one of the four entry points (East, West, North, and South) facing the wall of the tank, the trails were semi-randomized such that no trail had the same entry point as the earlier trail. However, the order was maintained constant across. In each trial, mice were given a time of 90 s (cut-off) to locate the platform. During which, if the mice fail to locate the platform, it was manually guided to the hidden platform, after which the mice were allowed to stay on the hidden platform for 20 s. Trails for the whole group were completed before the next trial began. Acquisition trails were carried out for 12 days. In addition to acquisition training, animals were tested for spatial memory using probe trails on day 6 and day 12, where the platform was removed, and the animals were allowed to explore the tank for 60 seconds to locate the platform. During the probe trial, the number of times the animal has crossed the hidden platform was noted. After 60 s time period, animals were returned to cages.40,41 After 3 days rest period after the acquisition phase, the reversal-learning task was performed for 5 days, where the platform location was changed to the opposite side of the acquisition phase, and thereby accessing the cognitive flexibility. A 3 day rest period was allowed between the acquisition phase and reversal. At the end of the reversal study, another probe trial was performed. During the entire period of study, the positions of cues remained unchanged, the location of the platform remained in the southwest quadrant throughout the acquisition phase. At the end of each day of training, the mice remained in warming cages for 10 min, allowing them to dry completely and then were returned to either their home cage.40–42
2.8.5. Biochemical and histopathological analysis. At the end of the study, animals were culled by isoflurane anaesthesia, and their brains were harvested.
2.8.5.1. Preparation of homogenate. The brain tissue (0.5 g) homogenized in ice-cold PBS (5.0 ml, 0.1 M, pH 7.4) to obtain 10% w/v homogenate. The homogenate was then centrifuged at 10000 rpm for 20 min at 4 °C. The supernatant was used for the biochemical estimations.
2.8.5.2. Estimation of Aβ42 levels. The levels of Aβ42 present in the mice brain were estimated using Aβ-42 ELISA kit (Thermofisher Scientific, Chennai, India)
2.8.5.3. Estimation by western blotting. The levels of APP, tau, NF-κB, BACE after treatment with BQCA-SA-P80-NPs was analyzed by western blotting.43
2.8.6. Histopathology studies. The brain tissue was fixed in a mixture of formaldehyde (40%), glacial acetic acid, and methanol (1:1:8). The tissues were cut into 3 mm thickness, and blocks were embedded in paraffin. Sections 4 and 5 μm thick were cut in the coronal plane and stained with hematoxylin and eosin (HE) and Cresyl Violet (CV) staining. The photomicrographs of the slides were recorded with a Motic Digital microscope assisted with Motic Images 2000 ver 1.3 software.
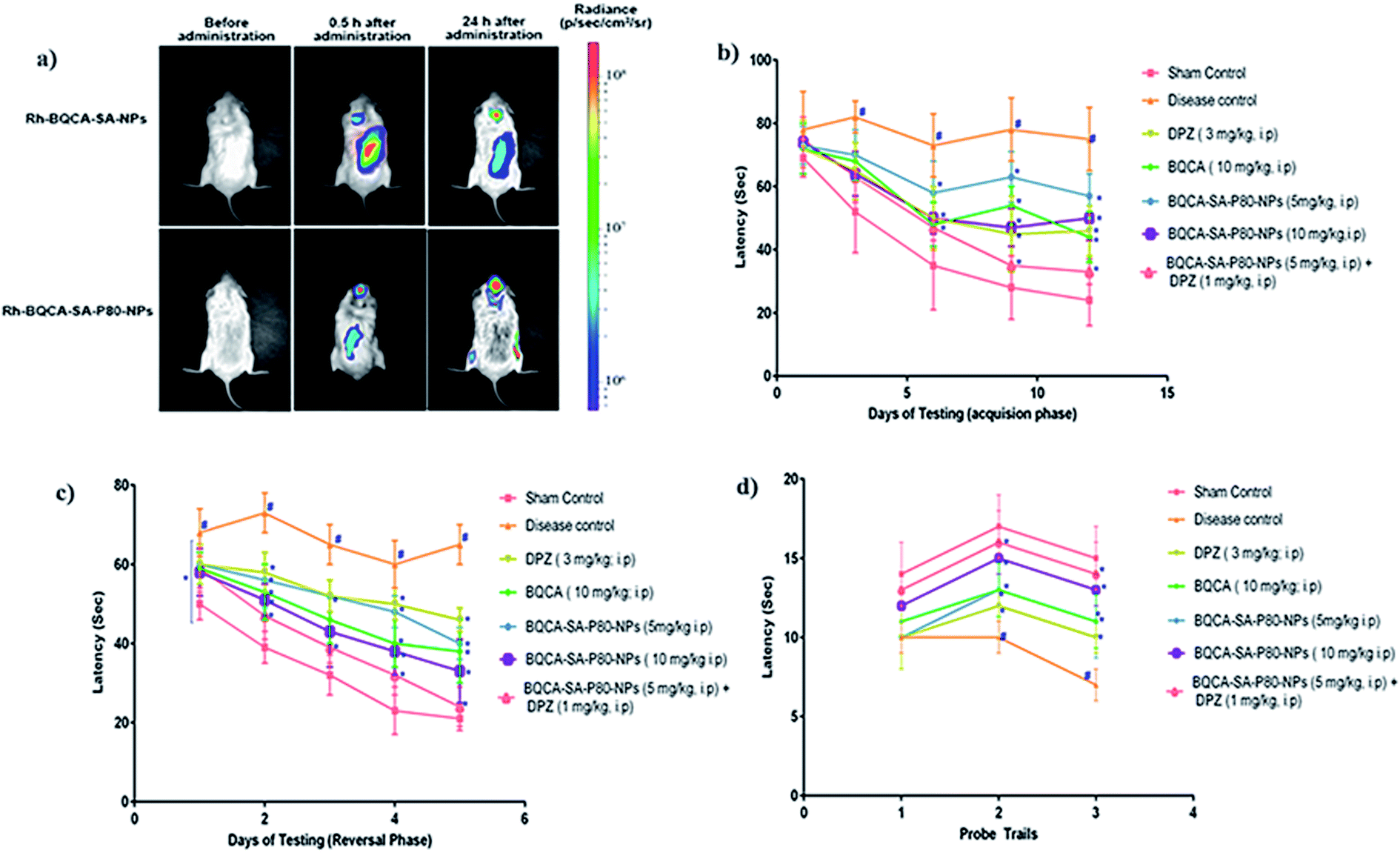
2.8.7. Bio-distribution study non-invasive imaging. Mice were injected with Rh-BQCA-SA-P80 (10 mg kg−1, i.p.) and Rh-BQCA-SA-NPs (10 mg kg−1, i.p.), at predetermined intervals of 0, 0.5 and 24 h fluorescent intensity was measured using IVIS, Massachusetts, USA. The images were analyzed using live imaging software (Xenogen, Alameda-ver 4.3.1, USA).
2.9. Statistical analysis
All the results were expressed as mean ± SEM. Statistical analysis was carried out using one way ANOVA followed by Dunnett's multiple comparison test by using graph pad prism. p < 0.05 was considered significant. (Graph Pad Prism, Version 6, Graph Pad Software Inc., La Jolla, USA).
3. Results
3.1. Structural characterization of BQCA-SA LDC
The structure of the synthesized BQCA-SA LDC (Fig. S2†) was confirmed by 1HNMR, 13CNMR, FTIR and LCMS (Fig. S4–S7†)
3.1.1. Appearance. White solid mp. 198–204 °C;1NMR (400 MHz, CDCl3): δ 10.04 (bs, 1H), 8.94 (s, 1H), 8.53 (d, J = 7.6 Hz, 1H), 7.61 (t, J = 7.5 Hz, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 7.9 Hz, 2H), 6.86 (d, J = 8.1 Hz, 2H), 5.41 (s, 2H), 8.77 (s, 3H), 3.47 (q, J = 6.4 Hz, 2H), 1.38–1.17 (m, 32H), 0.93–0.82 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ 176.8, 194.9, 159.7, 148.3, 139.3, 132.7, 128.1, 127.7, 127.3, 126.1, 125.0, 116.7, 114.7, 112.2, 57.2, 55.3, 39.4, 31.9, 29.7, 29.6, 29.4, 29.3, 27.2, 22.7, 14.1 ppm. FT-IR (KBr) 3048, 2955, 2922, 2852, 1741, 1658, 1609, 1522, 1495, 1468, 1242, 1177, 1045, 755, 720. HRMS: m/z calculated 561.4056 (M + 1), found 561.4048 (M + 1).
3.2. Design and optimization of BQCA-SA-P80-NPs
Experimental batches of BQCA-SA-P80-NPs for different combinations of surfactant concentration, sonication amplitude, and sonication time (independent variables) were prepared based on the Box-Behnken experimental design predicted by Minitab®18.1 statistical programs (Minitab Ltd. Coventry, United Kingdom). The results obtained were fed into the software and were analyzed. The effects of independent variables on PS (nm) and ZP (mV) were observed by response analysis of variance (ANOVA) table (Tables S2–S4†), Pareto charts, main effects plots and 3D surface plots (Fig. S8–S13†).
The selection of optimized formulation was achieved based on desirability criteria (near to 1), where the software predicted that surfactant concentration of 2.24% w/v, sonication amplitude of 75% and sonication time fo 9 min is required to achieve BQCA_SA-P80-NPs having PS of 163.85 nm (desirability value-1.000) and ZP of 22.90 mV (desirability value-0.8916). The composite desirability value was found to be 0.946 (Fig. S14†). Based on the predictions a new batch of BQCA-SA-P80-NPs formulation was prepared, where the experimental results gave BQCA-SA-P80-NPs with PS of 166.62 ± 1.24 nm and ZP of 23.59 ± 0.37 mV with a prediction error of 1.69 ± 0.75, and 3.03 ± 1.63 respectively (Table S5†).
3.3. Characterization of BQCA-SA-P80-NPs
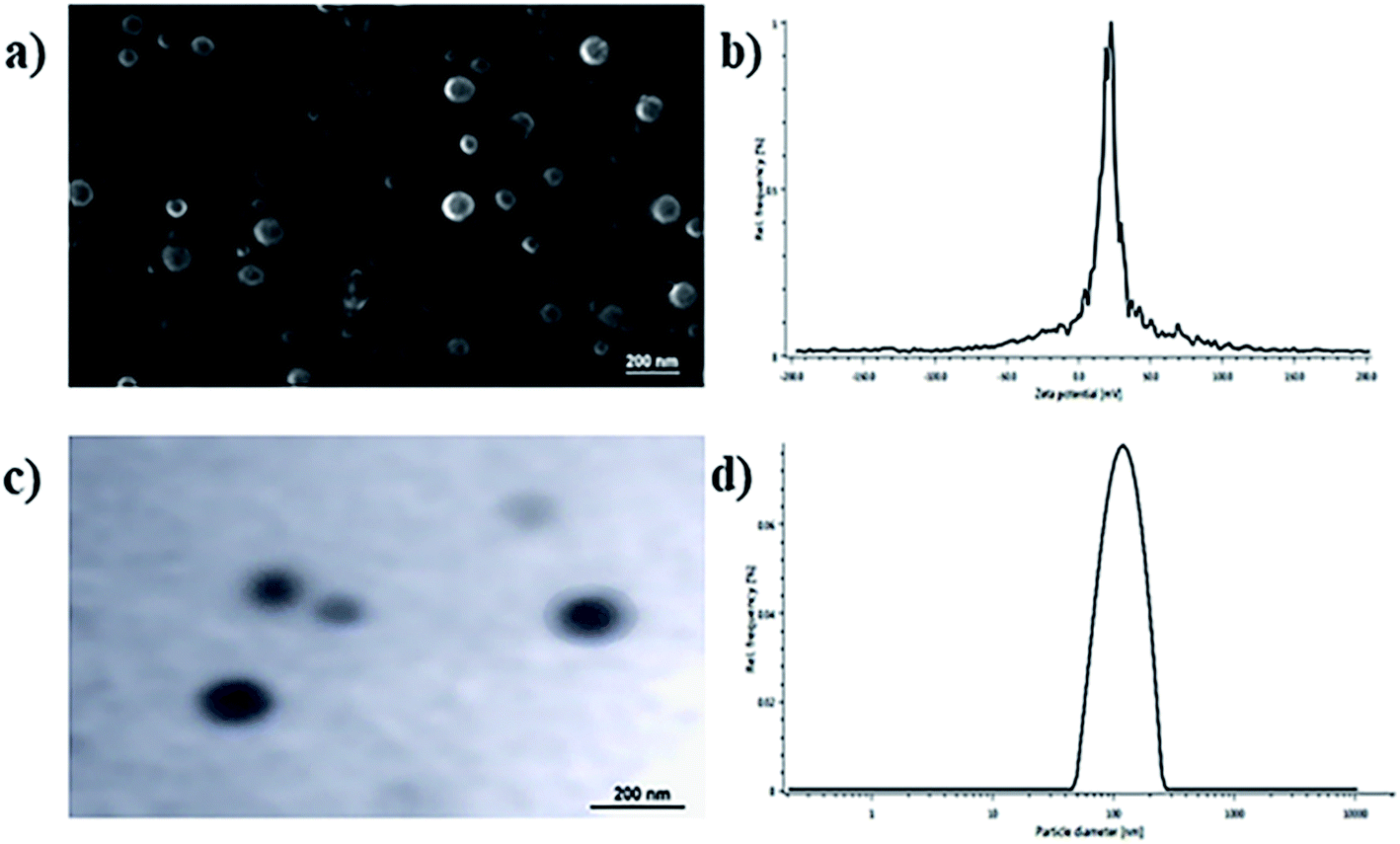
The physicochemical parameters of the optimized batch of BQCA-SA-P80-NPs was given Table 2. The particle size analysis, SEM and TEM studies revealed that BQCA-SA-P80-NPs are near monodisperse and have a spherical shape (Fig. 2).
Table 2 Physicochemical parameters of the optimized batch of BQCA-SA-P80-NPsa
| Parameter |
Value |
| The values are mean ± S.D., (n = 3). |
| PS (nm) |
166.62 ± 1.24 |
| ZP (mV) |
23.59 ± 0.37 |
| PDI |
0.397 ± 0.21 |
| Drug content (%) |
97.24 ± 2.14 |
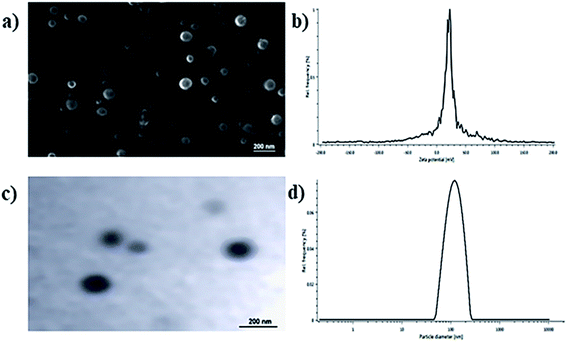 |
| | Fig. 2 (a) SEM image of optimized BQCA-SA-P80-NPs; (b) TEM image of optimized BQCA-SA-P80-NPs; (c) particle size distribution of optimized BQCA-SA-P80-NPs; (d) zeta potential curves for optimized BQCA-SA-P80-NPs. | |
3.4. In vitro drug release studies for an optimized batch of BQCA-SA-P80-NPs
In vitro drug release studies for the naïve BQCA and BQCA-SA-P80-NPs performed using aCSF as media, in the presence of amidase, in a dialysis bag. It was observed that the release of naïve BQCA from the solution was much faster than from BQCA-SA-P80-NPs. Naïve BQCA released almost completely (98.79 ± 1.55%) released from the solution by the end of 8th hour. However, the BQCA-SA-P80-NPs showed a biphasic release pattern, i.e., an initial burst release till the 4th h followed by sustained release up to 48 h (86.31 ± 2.27%) (Fig. S15†).The data obtained from dissolution studies were fitted into various release kinetic models (Fig. S16–S19†). Based on the R2 and ‘n’ values, it was found that the release of BQCA from BQCA-SA-P80-NPs follows first-order release kinetics and the non-Fickian mode of drug release (Table S8†).
3.5. Hemolysis assay
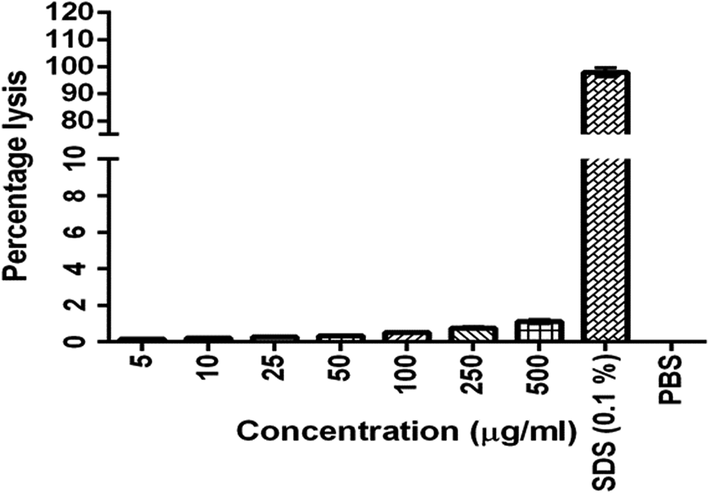
The results were given in Fig. 3. The results show that BQCA-SA-P80-NPs when incubated at concentrations of 5–500 μg ml−1, show minimal hemolysis. The highest incubated concentration of 500 μg ml−1, showed only a minimum of 1.12 ± 0.11% (mean ± SD) hemolysis. This is within the permitted limits of 5%.
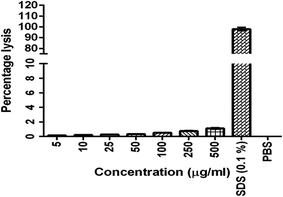 |
| | Fig. 3 In vitro hemolysis assay for optimized BQCA-SA-P80-NPs. | |
3.6. Cytotoxicity assay
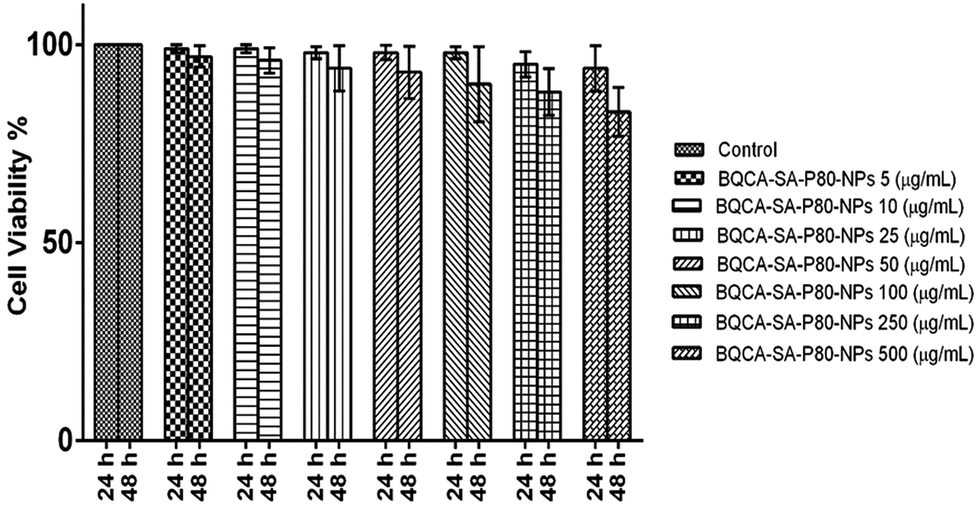
The results were given in Fig. 4. The results show that BQCA-SA-P80-NPs when incubated at concentrations of 5–500 μg ml−1 show minimal cytotoxicity to SH-SY5Y. BQCA-SA-P80-NPs show 85.0 ± 6.6% (mean ± SD), cell viability even at 500 μg ml−1 (48 h of incubation).
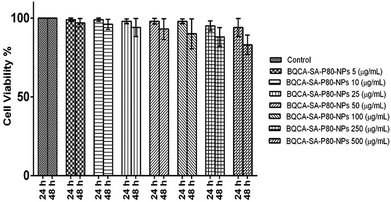 |
| | Fig. 4 In vitro cytotoxicity studies for optimized BQCA-SA-P80-NPs. | |
3.7. Bio-distribution studies using non-invasive imaging
The results were given in Fig. 5(a). From the bioluminescence studies, it was evident that Rh-BQCA-SA-P80-NPs accumulated explicitly in the brain compared to Rh-BQCA-SA-NPs formulation at 24 h.
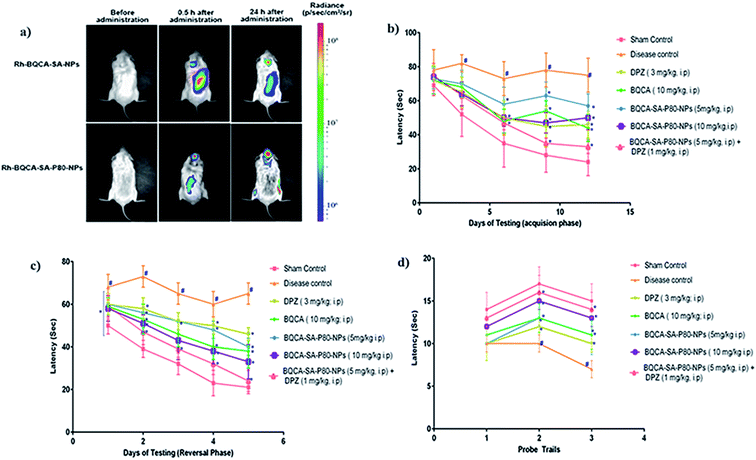 |
| | Fig. 5 (a) Biodistribution of Rh-BQCA-SA-P80-NPs before administration and 0.5 h, 24 h of in vivo administration; (b) latency to locate platform during acquisition phase performed for 12 days, #p < 0.05 vs. sham control; *p, 0.05 vs. disease control; (c) latency to locate platform during reversal phase performed for 5 days #p < 0.05 vs. sham control; *p, 0.05 vs. disease control; (d) passing times through the location of platform during probe trails; #p < 0.05 vs. sham control; *p, 0.05 vs. disease control. | |
3.8. In vivo efficacy studies
3.8.1. Morris water maze test-effect on acquisition and reversal learning latency. The results were given in Fig. 5(b–d). Administration of STZ to disease control animals has significantly increased the acquisition and reversal latency when compared to sham control animals (p < 0.05). All the treatment groups significantly reduced the STZ induced increase in the acquisition and reversal latency time, when compared to disease control animals (p < 0.05). Among the treatment groups, group 7 treated with a combination of BQCA-SA-P80-NPs (5 mg kg−1, i.p.) and DPZ (1 mg kg−1, i.p.) shows highest activity followed by group-6 and group-5 treated with BQCA-SA-LDC-NPs, high (10 mg kg−1, i.p.) and low (5 mg kg−1, i.p.) respectively. Group 3 and 4 treated with naïve DPZ (3 mg kg−1, i.p.) and naïve BQCA (10 mg kg−1, i.p.) show only a moderate effect.
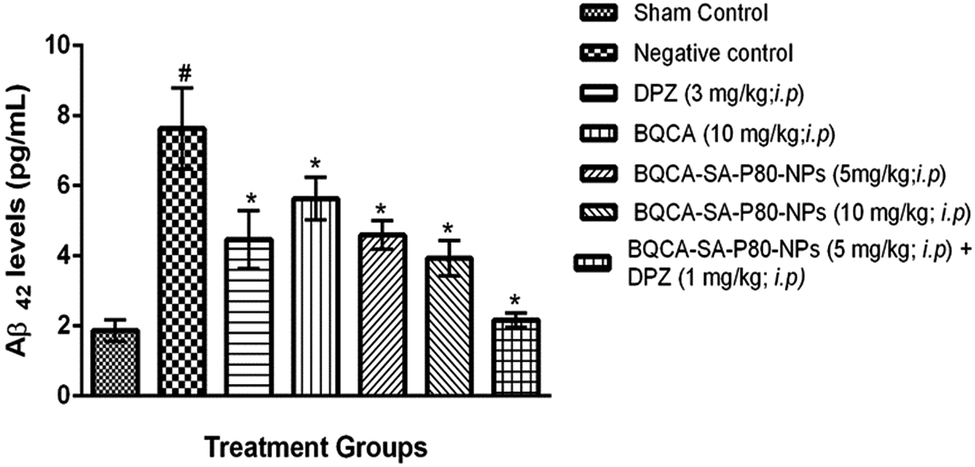
3.8.2. Estimation of levels of Aβ-42. The results were given in Fig. 6. STZ administration to disease control group has significantly increased the levels of Aβ-42 levels when compared to the sham control group (p < 0.05). All the treatment groups have shown a significant reduction in Aβ-42 levels in mice brains when compared to disease control (p < 0.05). Among the treatment groups, group 7 treated with combination of BQCA-SA-P80-NP s (5 mg kg−1, i.p.) and DPZ (1 mg kg−1, i.p.) shows highest activity followed by group-6 (BQCA-SA-LDC-NPs, 10 mg kg−1, i.p.), group-5 (BQCA-SA-LDC-NPs, 5 mg kg−1, i.p.), group 3 (DPZ 3 mg kg−1, i.p.) and group 4 (naïve BQCA 10 mg kg−1, i.p.).
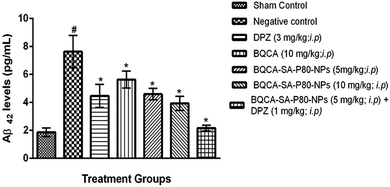 |
| | Fig. 6 Mean mouse Aβ42 levels in different treatment groups, #p < 0.05 vs. sham control, *p < 0.05 vs. disease control; the values are mean ± S.D. (n = 3). | |
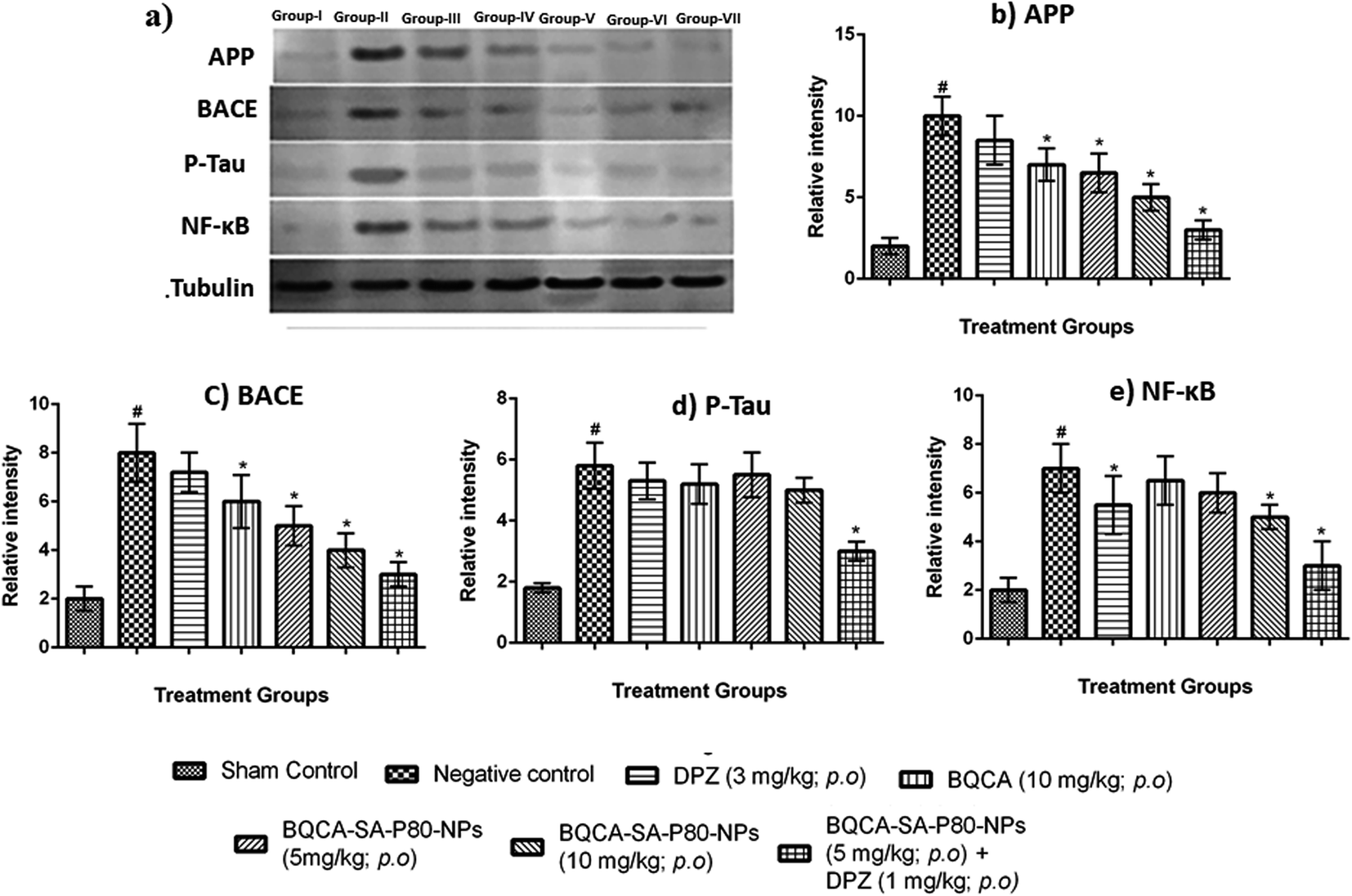
3.8.3. Estimation of APP, tau, NF-κB, BACE by western blotting. The results of western blot analysis were given in Fig. 7, and the results show that the treatment groups showed significant inhibition of the expression of APP, p-tau, NF-κB, and BACE, when compared to the disease control group (p < 0.05).
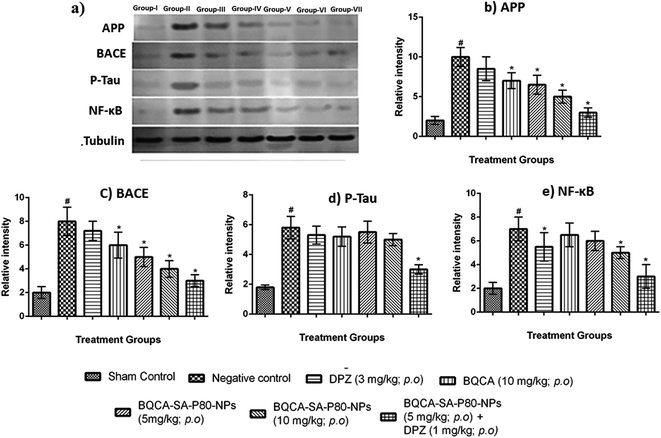 |
| | Fig. 7 (a) Western blot analysis for (a) qualitative and (b–e) quantitative measurement of APP, BACE, P-tau and NF-κB in different treatment groups respectively, #p < 0.05 vs. sham control, *p < 0.05 vs. disease control. The values are means ± S.D., n = 3. | |
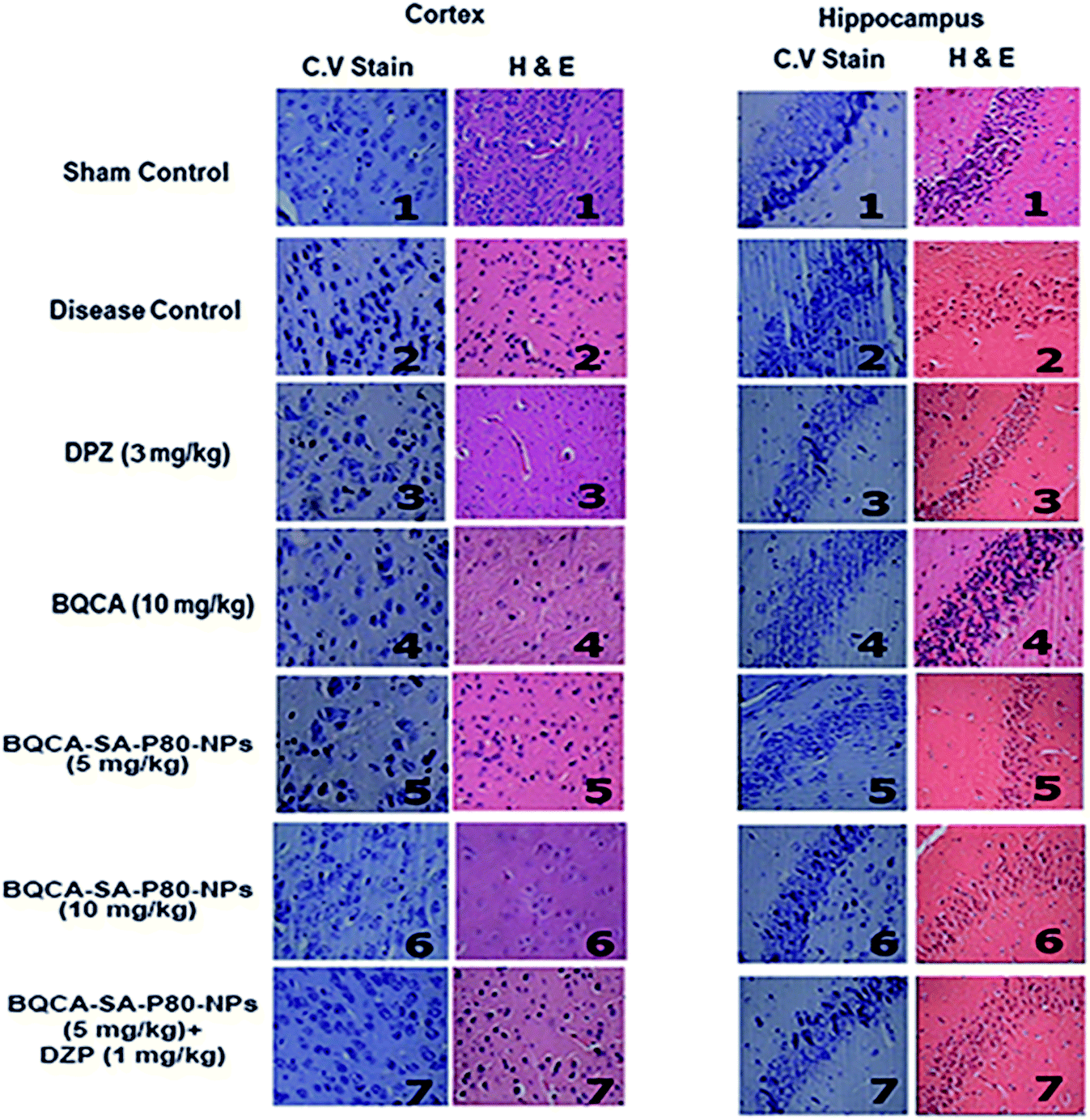
3.8.4. Histology. The results were given in Fig. 8. In the sham control group, the regular architecture of brain tissue was observed with well-defined pyramidal cells in the hippocampal regions. In the disease control group, brain sessions show significant neurodegeneration as indicated by reduced CV positive neurons in both cortex and hippocampus. All the treatment groups have shown significant protection against STZ induced neuronal changes.
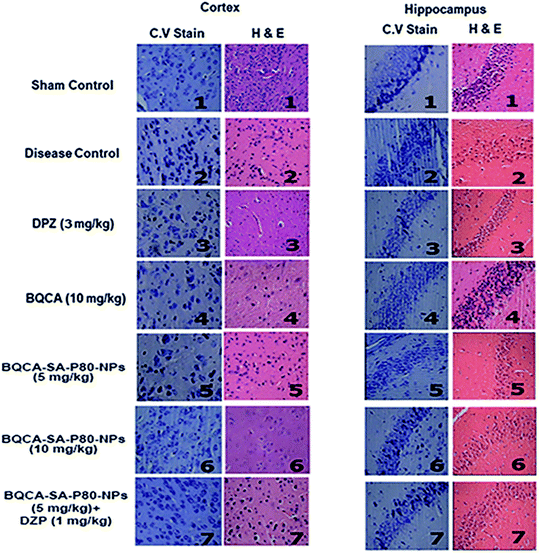 |
| | Fig. 8 CV stain and H & E staining of cortex and hippocampus of different treatment groups. (1) sham control showing the normal architecture of the brain; (2) disease control showing significant neurodegeneration; (3) DPZ (3 mg kg−1, i.p.) showing moderated neurodegeneration; (4, 5, 6, 7) BQCA (10 mg kg−1, i.p.), BQCA-SA-P80-NP (5 mg kg−1, i.p.), BQCA-SA-P80-NP (10 mg kg−1, i.p.), combination (BQCA-SA-P80-NP (5 mg kg−1, i.p. and DPZ (a mg kg−1, i.p.)), showing minimal neurodegeneration. | |
4. Discussion
In the present study, an attempt has been made to develop BQCA-SA-P80-NPs to improve the brain bioavailability and residence time of BQCA and to enhance the efficacy of endogenous ACh on M1 receptor in the brain for the better management of AD. BQCA-SA LDC lipid bulk was synthesized using BQCA and SA by an amide coupling reaction. The obtained BQCA-SA LDC was formulated into BQCA-SA-P80-NPs by the solvent injection method, using polysorbate 80 as a surfactant. PS plays a vital role in deciding the in vivo fate of nanoparticles. Particles of size 10–1000 nm were found to have accessibility in the body and are freely transported throughout the body by the circulatory system.44 ZP serves as the measure for the surface charge of the BQCA-SA-P80-NPs and plays an important role in the determination of stability of nanoparticulate systems.45 It is reported that particles with charge, +30 mV to −30 mV, tend to have excellent stability by repelling each other electrostatically and thereby prevent the formation of particle aggregates.46,47 In the present study, BQCA-SA-P80-NPs have PS range of 164.20 to 209.40 nm and ZP ranging from 25.99 to 28.13 mV. This indicates the excellent stability of prepared nanoparticles.
PDI serves as the measure of monodispersity of the formulation; it has a value ranging from 0.0–1.0. In the present study, the PDI of the optimized batch of BQCA-SA-P80-NPs was found to be 0.397 ± 0.21, indicating the near monodispersity of the formulation. Assessment of surface morphology of the BQCA-SA-P80-NPs was done by SEM and TEM analysis, and it revealed the near-spherical nature of the particles.
In vitro drug release studies of naïve BQCA and optimized BQCA-SA-P80-NPs was performed in aCSF using the dialysis bag method. Amidase was added into the release medium as it can mimic the lysosomal conditions and can aid in breaking the amide bond between BQCA and SA, thereby, releasing free BQCA.48,49 The release of naïve BQCA was found to be very fast from the dialysis bag (97.34% in 8 h); this may be attributed to the absence of nanoparticulate system or the barrier to regulate the drug release. However, BQCA-SA-P80-NPs were shown to release BQCA over a time of 48 h. BQCA-SA-P80-NPs were shown to produce biphasic release with an initial burst release up to 4 h, followed by a controlled release up to 48 h (86.31%). This can be attributed to the easy access of the dissolution fluid to the drug available on the surface of BQCA-SA-P80-NPs. However, as time progresses, the release of drug is hindered due to reduced access of the dissolution fluid to the matrix of BQCA-SA-P80-NPs due to their lipophilic nature.30 Drug release kinetics plots shown that the BQCA-SA-P80-NPs follow first-order release kinetics, and non-fickian mode of drug release.30,50
BQCA-SA-P80-NPs targeted to the brain should be biocompatible. The biocompatibility of BQCA-SA-P80-NPs was assessed by hemolytic and cytotoxicity assay. Available literature suggests that the hemolytic percentage should not exceed 5% for a formulation to be considered safe.51,52 In the present study, after 1 h of incubation, BQCA-SA-P80-NPs, even at a concentration of 500 μg ml−1 produced only 1.12 ± 1.2% hemolysis suggesting that the formulation as hemocompatible. However, we can expect a still lesser hemolytic percentage in vivo due to the dynamic movement of RBC in blood circulation. BQCA-SA-P80-NPs at concentrations of 5–500 μg ml−1, were exposed to SH-SY5Y, human neuroblastoma cell lines for 48 h. The observed CTC50 was found to be >500 μg; therefore, establishing the safety of the formulation.53,54 Biofluorescent studies were performed to accesses the brain localization of BQCA-SA-P80-NPs, using dye loaded Rh-BQCA-SA-P80-NPs. The results show that Rh-BQCA-SA-P80-NPs were significantly loaded in the brain by the end of 24 h. It is a well-established fact that uptake of P80 surface-modified nanoparticles to the brain was mediated through LDL receptor. Surface modification with P80 leads to adsorption of ApoE on the nanoparticles, which then facilitates BBB permeability via LDL receptor-mediated endocytosis55,56 and thereby, leads to increased brain uptake.57,58 BQCA-SA-P80-NPs were formulated using polysorbate 80 as a surfactant, where the polysorbate 80 forms a coating around the nanoparticles and, therefore, was predicted to increased brain uptake by the above mechanism. Reports suggest that BQCA has a lower brain half-life (2.04 h) than plasma half-life (3.3 h).59 In vivo fluorescence studies show that BQCA-SA-P80-NPs will be localized in the brain for a period of over 24 h and form the in vitro drug release studies BQCA show a controlled release over a period of 48 h. Therefore, the formulation of BQCA-SA-P80-NPs will significantly improve brain retention time and reduce elimination rates of BQCA.
M1 activation has been reported to enhance sAPPα generation and reduce Aβ production by promoting the activity of α-secretase and lowering the action of beta secretase (BACE).60,61 Some other studies show that M1 activation promotes the processing of amyloid precursor protein (APP) through the amyloidogenic pathway.60 Recent studies showed that M1 mAChR directly interacts with BACE and mediates its proteasomal degradation.62 In addition to inhibiting Aβ generation, activation of M1 mAChR counteracts Aβ-induced neurotoxicity through the Wnt signaling pathway, as Aβ impairs the Wnt pathway and M1 mAChR stimulation inactivates GSK-3β via PKC activation, stabilizes β-catenin, and induces the expression of Wnt-targeting genes engrailed and cyclin-D1 for neuron survival.63 As GSK-3β is a major kinase for tau phosphorylation, stimulation of M1 inhibits tau phosphorylation as well.62,64 The M1 mAChR cascade may also be involved in counteracting decreased cerebral blood flow, which is one of the most consistent characteristics in pathological conditions such as AD, ischemic brain injury, intracerebral hemorrhage, and cognitive dysfunction.65,66 In fact, Aβ has been shown to induce the uncoupling of M1 mAChR from G-protein, antagonizing the function of M1 mAChR under the pathological conditions of AD. In the present study, when compared to disease control, BQCA-SA-P80-NPs treatment show as significant dose-dependent reduction/prevention of STZ induced changes in memory levels of Aβ1–42, P-tau, APP, NF-κB and BACE levels) and also on neurodegeneration in cortex and hippocampus of the brain (p < 0.05). When compared with naïve BQCA treatment (group 4), the BQCA-SA-P80-NPs treated groups (group 5 and 6) show better activity in reversing STZ induced changes. This could be due to improved retention time of BQCA-SA-P80-NPs compared to naïve BQCA resulting in a prompt and efficient activation of M1 receptors. When combined with AChE inhibitor, DPZ (even at sub therapeutic dose of 1 mg kg−1, i.p.) (group 7), the activity significantly improves than the individual treatments (group 4, 5 & 6), therefore, indicating a possible synergistic or additive effect. The observed additive/synergism could be due to increased ACh levels (due to DPZ) and potentiation of ACh action on M1 receptors (due to BQCA), overall resulting in a prompt activation of M1 receptors.
5. Conclusion
In conclusion, the BQCA-SA-P80-NPs formulation improves the brain bioavailability and therapeutic efficacy of BQCA in the management of AD. Besides, the formulation also helps in reducing the therapeutic dose of AChE inhibitors such as DPZ and hence can reduce their associated peripheral side effects and overall help in improving patient compliance. Further development of this formulation could benefit millions of AD patients.
6. Ethical statement & approval
All animal procedures involved in the study were performed in accordance with the CPCSEA guidelines, Government of India and guidelines for care and use of laboratory animals, JSS College of Pharmacy, Ooty. A prior approval of the study plan has been obtained from the Institutional Animal Ethics Committee of JSS College of Pharmacy, Ooty (IAEC Approval No. JSSCP/IAEC/OT/PhD/Ph.Cology/02/2017–18).
Conflicts of interest
The authors declare that there is no conflict of interest.
Acknowledgements
The authors are thankful to the Department of Biotechnology (DBT), Government of India, New Delhi for financial support (DBT Sanction Oder No: BT/PR9979/NNT/28/705/2013).
References
- E. B. Larson, W. A. Kukull and R. L. Katzman, Annu. Rev. Public Health, 1992, 13, 431–449 CrossRef CAS PubMed.
- A. S. Association, Alzheimer's & Dementia, 2018, 14, 367–429 Search PubMed.
- M. Citron, Nat. Rev. Drug Discovery, 2010, 9, 387–398 CrossRef CAS PubMed.
- C. Geula, Neurology, 1998, 51, S18–S29 CrossRef CAS PubMed.
- A. Maeda, T. Kubo, M. Mishina and S. Numa, FEBS Lett., 1988, 239, 339–342 CrossRef CAS PubMed.
- R. Rodriguez-Puertas, J. Pascual, T. Vilaro and A. Pazos, Synapse, 1997, 26, 341–350 CrossRef CAS PubMed.
- R. Medeiros, M. Kitazawa, A. Caccamo, D. Baglietto-Vargas, T. Estrada-Hernandez, D. H. Cribbs, A. Fisher and F. M. LaFerla, Am. J. Pathol., 2011, 179, 980–991 CrossRef CAS PubMed.
- S. Jiang, Y. Li, C. Zhang, Y. Zhao, G. Bu, H. Xu and Y. W. Zhang, Neurosci. Bull., 2014, 30, 295–307 CrossRef CAS PubMed.
- H. C. Liu, C. J. Hong, T. Y. Liu, C. W. Chi and S. J. Tsai, Dementia Geriatr. Cognit. Disord., 2005, 19, 42–45 CrossRef CAS PubMed.
- M. Farlow, Int. Psychogeriatr., 2002, 14, 93–126 CrossRef PubMed.
- H. Imai, T. Hirai, R. Kumazawa, S. Nakagawa, A. Yonezawa, K. Matsubara and H. Nakao, PLoS One, 2020, 15, e0231226 CrossRef CAS PubMed.
- H. Akincioglu and I. Gulcin, Mini-Rev. Med. Chem., 2020, 20(8), 703–715 CrossRef CAS PubMed.
- S. Moriguchi, W. Marszalec, X. Zhao, J. Z. Yeh and T. Narahashi, J. Pharmacol. Exp. Ther., 2004, 310, 933–942 CrossRef CAS PubMed.
- D. Arsland, K. A. Engedal, H. A. Nygaard, J. Louhija, I. Ulstein and M. Holm, Tidsskr. Nor. Laegeforen., 2003, 123, 1500–1503 Search PubMed.
- B. Beermann, Acta Neurol. Scand., Suppl., 1993, 149, 53–54 CAS.
- L. E. Collins, N. E. Paul, S. F. Abbas, C. E. Leser, S. J. Podurgiel, D. J. Galtieri, J. J. Chrobak, Y. Baqi, C. E. Muller and J. D. Salamone, Pharmacol., Biochem. Behav., 2011, 99, 414–422 CrossRef CAS PubMed.
- J. Wei, E. A. Walton, A. Milici and J. J. Buccafusco, J. Neurochem., 1994, 63, 815–821 CrossRef CAS PubMed.
- A. C. Kruse, J. Hu, A. C. Pan, D. H. Arlow, D. M. Rosenbaum, E. Rosemond, H. F. Green, T. Liu, P. S. Chae, R. O. Dror, D. E. Shaw, W. I. Weis, J. Wess and B. K. Kobilka, Nature, 2012, 482, 552–556 CrossRef CAS PubMed.
- E. C. Hulme, E. Kurtenbach and C. A. Curtis, Biochem. Soc. Trans., 1991, 19, 133–138 CrossRef CAS PubMed.
- B. J. Melancon, J. C. Tarr, J. D. Panarese, M. R. Wood and C. W. Lindsley, Drug Discovery Today, 2013, 18, 1185–1199 CrossRef CAS PubMed.
- H. H. Nickols and P. J. Conn, Neurobiol. Dis., 2014, 61, 55–71 CrossRef PubMed.
- P. K. Chintamaneni, P. T. Krishnamurthy, P. V. Rao and S. S. Pindiprolu, Med. Hypotheses, 2017, 101, 17–22 CrossRef CAS PubMed.
- S. Schramm, L. Agnetta, M. Bermudez, H. Gerwe, M. Irmen, J. Holze, T. Littmann, G. Wolber, C. Trankle and M. Decker, ChemMedChem, 2019, 14, 1349–1358 CrossRef CAS PubMed.
- S. N. Mistry, C. Valant, P. M. Sexton, B. Capuano, A. Christopoulos and P. J. Scammells, J. Med. Chem., 2013, 56, 5151–5172 CrossRef CAS PubMed.
- S. Wohlfart, S. Gelperina and J. Kreuter, J. Controlled Release, 2012, 161, 264–273 CrossRef CAS PubMed.
- S. C. Yang, L. F. Lu, Y. Cai, J. B. Zhu, B. W. Liang and C. Z. Yang, J. Controlled Release, 1999, 59, 299–307 CrossRef CAS PubMed.
- C. Olbrich, A. Gessner, W. Schröder, O. Kayser and R. H. Müller, J. Controlled Release, 2004, 96, 425–435 CrossRef CAS PubMed.
- Q. R. Smith, The Blood-Brain Barrier: Biology and Research Protocols, 2003, pp. 193–208 Search PubMed.
- J. Kreuter, Adv. Drug Delivery Rev., 2014, 71, 2–14 CrossRef CAS PubMed.
- P. Sharma, B. Dube and K. Sawant, J. Biomed. Nanotechnol., 2012, 8, 928–937 CrossRef CAS PubMed.
- H. Vaghasiya, A. Kumar and K. Sawant, Eur. J. Pharm. Sci., 2013, 49, 311–322 CrossRef CAS PubMed.
- J. Emami, H. Mohiti, H. Hamishehkar and J. Varshosaz, Res. Pharm. Sci., 2015, 10, 17 CAS.
- V. T. Siddhartha, S. K. S. S. Pindiprolu, P. K. Chintamaneni, S. Tummala and S. Nandha Kumar, Artif. Cells, Nanomed., Biotechnol., 2017, 1–11, DOI:10.1080/21691401.2017.1313267.
- S. V. Talluri, G. Kuppusamy, V. V. S. R. Karri, K. Yamjala, A. Wadhwani, S. V. Madhunapantula and S. S. Pindiprolu, Artif. Cells, Nanomed., Biotechnol., 2016, 1–15 Search PubMed.
- K. Jain, S. Sood and K. Gowthamarajan, Drug Delivery, 2015, 22, 940–954 CrossRef CAS PubMed.
- R. J. Mudakavi, S. Vanamali, D. Chakravortty and A. M. Raichur, RSC Adv., 2017, 7, 7022–7032 RSC.
- H. Y. Kim, D. K. Lee, B.-R. Chung, H. V. Kim and Y. Kim, J. Visualized Exp., 2016, 109, 53308 Search PubMed.
- M. Nobakht, S. M. Hoseini, P. Mortazavi, I. Sohrabi, B. Esmailzade, N. R. Roosh and S. Omidzahir, Iran. Biomed. J., 2011, 15, 51 Search PubMed.
- W. M. Snow, P. S. Pahlavan, J. Djordjevic, D. McAllister, E. E. Platt, S. Alashmali, M. J. Bernstein, M. Suh and B. C. Albensi, Front. Mol. Neurosci., 2015, 8, 70 CrossRef PubMed.
- C. M. Walsh, V. Booth and G. R. Poe, Learn. Mem., 2011, 18, 422–434 CrossRef PubMed.
- D. Shah, M. Verhoye, A. Van der Linden and R. D’hooge, Cereb. Cortex, 2018, 8, 6264 Search PubMed.
- S. O. Nolan and J. N. Lugo, F1000Research, 2018, 7, 711 Search PubMed.
- S. K. S. Pindiprolu, P. K. Chintamaneni, P. T. Krishnamurthy and K. Ratna Sree Ganapathineedi, Drug Dev. Ind. Pharm., 2018, 1–25 Search PubMed.
- S. R. Mudshinge, A. B. Deore, S. Patil and C. M. Bhalgat, Saudi Pharm. J., 2011, 19, 129–141 CrossRef CAS PubMed.
- V. Mishra, K. Bansal, A. Verma, N. Yadav, S. Thakur, K. Sudhakar and J. Rosenholm, Pharmaceutics, 2018, 10, 191 CrossRef CAS PubMed.
- S. Honary and F. Zahir, Trop. J. Pharm. Res., 2013, 12, 255–264 Search PubMed.
- S. Honary and F. Zahir, Trop. J. Pharm. Res., 2013, 12, 265–273 Search PubMed.
- E. Romeo, S. Ponzano, A. Armirotti, M. Summa, F. Bertozzi, G. Garau, T. Bandiera and D. Piomelli, ACS Chem. Biol., 2015, 10, 2057–2064 CrossRef CAS PubMed.
- Q. Song, X. Wang, Y. Wang, Y. Liang, Y. Zhou, X. Song, B. He, H. Zhang, W. Dai and X. Wang, Mol. Pharmaceutics, 2016, 13, 190–201 CrossRef CAS PubMed.
- K. Soni, A. Mujtaba and K. Kohli, Int. J. Biol. Macromol., 2017, 103, 139–151 CrossRef CAS PubMed.
- L. Q. Chen, L. Fang, J. Ling, C. Z. Ding, B. Kang and C. Z. Huang, Chem. Res. Toxicol., 2015, 28, 501–509 Search PubMed.
- Z. r. Huang, S. c. Hua, Y. l. Yang and J. y. Fang, Acta Pharmacol. Sin., 2008, 29, 1094–1102 Search PubMed.
- J. Y. Choi, S. H. Lee, H. B. Na, K. An, T. Hyeon and T. S. Seo, Bioprocess Biosyst. Eng., 2010, 33, 21 Search PubMed.
- K. S. Kang, H. U. Lee, M. I. Kim, S. Y. Park, S.-J. Chang, J.-H. Park, Y. S. Huh, J. Lee, M. Yang and Y.-C. Lee, J. Nanobiotechnol., 2015, 13, 1–14 CrossRef PubMed.
- K. Prabhakar, S. M. Afzal, G. Surender and V. Kishan, Acta Pharm. Sin. B, 2013, 3, 345–353 CrossRef.
- S. Wagner, A. Zensi, S. L. Wien, S. E. Tschickardt, W. Maier, T. Vogel, F. Worek, C. U. Pietrzik, J. Kreuter and H. Von Briesen, PLoS One, 2012, 7, e32568 CrossRef CAS PubMed.
- B. Wilson, M. K. Samanta, K. Santhi, K. P. S. Kumar, N. Paramakrishnan and B. Suresh, Brain Res., 2008, 1200, 159–168 CrossRef CAS PubMed.
- X.-h. Tian, X.-N. Lin, F. Wei, W. Feng, Z.-C. Huang, P. Wang, L. Ren and Y. Diao, Int. J. Nanomed., 2011, 6, 445–452 CAS.
- J. K. Shirey, A. E. Brady, P. J. Jones, A. A. Davis, T. M. Bridges, J. P. Kennedy, S. B. Jadhav, U. N. Menon, Z. Xiang and M. L. Watson, J. Neurosci., 2009, 29, 14271–14286 CrossRef CAS PubMed.
- D. M. Müller, K. Mendla, S. A. Farber and R. M. Nitsch, J. Life Sci., 1997, 60, 985–991 CrossRef.
- S. Jiang, Y. Li, C. Zhang, Y. Zhao, G. Bu, H. Xu and Y.-W. Zhang, Neurosci. Bull., 2014, 30, 295–307 CrossRef CAS PubMed.
- A. Caccamo, S. Oddo, L. M. Billings, K. N. Green, H. Martinez-Coria, A. Fisher and F. M. LaFerla, Neuron, 2006, 49, 671–682 CrossRef CAS PubMed.
- G. G. Farías, J. A. Godoy, F. Hernández, J. Avila, A. Fisher and N. C. Inestrosa, Neurobiol. Dis., 2004, 17, 337–348 CrossRef PubMed.
- O. Forlenza, J. Spink, R. Dayanandan, B. Anderton, O. Olesen and S. Lovestone, J. Neural Transm., 2000, 107, 1201–1212 CrossRef CAS PubMed.
- H. Hanyu, T. Shimizu, Y. Tanaka, M. Takasaki, K. Koizumi and K. Abe, Dementia Geriatr. Cognit. Disord., 2003, 15, 177–182 CrossRef CAS PubMed.
- G. A. Bateman, C. R. Levi, P. Schofield, Y. Wang and E. C. Lovett, J. Clin. Neurosci., 2006, 13, 563–568 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00049g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Praveen Thaggikuppe Krishnamurthy
ab,
Praveen Thaggikuppe Krishnamurthy