DOI:
10.1039/D1RA00244A
(Paper)
RSC Adv., 2021,
11, 11771-11778
Grignard coupling-based synthesis of vinyl-substituted hydridopolycarbosilane: effect of starting material and polymerization behavior
Received
12th January 2021
, Accepted 16th March 2021
First published on 23rd March 2021
Abstract
Despite the multitude of available alternatives, the Grignard coupling-based synthesis of polycarbosilanes remains attractive, offering the benefit of easy structural design. Moreover, this method allows one to obtain a polymer precursor with the stoichiometric Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C ratio required for SiC ceramic production and is also suited for the synthesis of polymers containing curable functional groups (e.g., allyl and vinyl). Herein, vinyl-substituted hydridopolycarbosilane was synthesized from three different starting materials (Cl3SiCH2Cl, (MeO)3SiCH2Cl, and (EtO)3SiCH2Cl) using Grignard coupling, and the effects of starting material on polymer growth behavior were investigated. The alkoxysilane starting materials had the advantage of being safe to handle, but had a limitation in polymer growth. The largest molecular weight was observed for Cl3SiCH2Cl, although side reactions occurred. This behavior was ascribed to the retention of unreacted –SiCH2Cl groups contributing to polymer growth via coupling with neighboring polymer chains.
:C ratio required for SiC ceramic production and is also suited for the synthesis of polymers containing curable functional groups (e.g., allyl and vinyl). Herein, vinyl-substituted hydridopolycarbosilane was synthesized from three different starting materials (Cl3SiCH2Cl, (MeO)3SiCH2Cl, and (EtO)3SiCH2Cl) using Grignard coupling, and the effects of starting material on polymer growth behavior were investigated. The alkoxysilane starting materials had the advantage of being safe to handle, but had a limitation in polymer growth. The largest molecular weight was observed for Cl3SiCH2Cl, although side reactions occurred. This behavior was ascribed to the retention of unreacted –SiCH2Cl groups contributing to polymer growth via coupling with neighboring polymer chains.
Introduction
Since their development 50 years ago, preceramic polymers have been studied and modified to fabricate ceramics with variable morphology (e.g., fibers, composites, and thin films), as exemplified by the widespread use of polycarbosilanes as SiC (especially SiC fiber) precursors. Although SiC fiber and related materials are most commonly prepared using the one-pot Yajima method, polycarbosilanes can be synthesized through various routes, with each method giving unique characteristics to the resulting polymer. In the Yajima method, polydimethylsilane is placed in a sealed container and heated at 350–470 °C to afford a polymer with a carbosilane structure.1,2 Although this method is advantageous for mass production in batch-type reactors, it is severely limited in that only methyl and phenyl groups are tolerated due to the high reaction temperature.
Another notable polymerization method relies on Grignard coupling, generally affording polymers in a liquid phase and allowing the synthesis of polymers featuring a 1:1 Si:C ratio (hydridopolycarbosilane) or containing various functional groups (e.g., allyl and vinyl).3,4 While polycarbosilane is originally known to be a precursor to SiC fibers, these modified polymers are mainly applied to additive manufacturing or PIP (polymer infiltration and pyrolysis) process.5–7 As the industrial field to which the polymer derived ceramics (PDC) technology applied is rapidly advanced, interest in preceramic polymer is growing again. In order to develop application-specific processes, polycarbosilane with proper properties is needed, but there is a limit to the use of commercial products. In PDC technology, the main properties of carbosilane-based polymers are melting point, viscosity, and thermal decomposition yield. As these depend on the polymer size, the molecular weight of the polymer must be adequately controlled.
The Grignard coupling-based synthesis of polycarbosilanes has already been investigated in terms of mechanism, structural characteristics, kinetics, and side reactions;8–11 however, out current understanding of polycarbosilane growth via this method is very limited.12,13 For example, Guodong and Jang analyzed and discussed the change in the properties of polycarbosilane with the growth behavior of the polymer, but it is limited to the Yajima method.14,15 In other cases, molecular weight was analyzed only to evaluate individual synthesis conditions, and the studies related to polymer growth were very insignificant.16–18,27 Therefore, in order to prepare the suitable precursor for the required properties of each different field, it is necessary to understand growth behavior of the polymer and to control the reaction condition accordingly.
Chlorosilanes such as Cl3SiCH2Cl is the most commoly used starting materials for polycarbosilane synthesis. Although chlorosilanes are toxic and may cause side reactions, they remain the most commonly used reagents because of their high reactivity. Considering that these silanes are converted to alkoxysilane intermediates in some reactions, increasing attention has been drawn to using relatively safe alkoxysilanes as alternative starting materials.19–21 Despite the fact that reaction conditions such as starting material type can affect polymer formation and growth, many studies have mainly focused on the reaction properties and structural characteristics of individual synthetic conditions to form polymers, thus leaving polycarbosilane growth behavior largely unexplored. Herein, vinyl-substituted hydridopolycarbosilane (VHPCS) is synthesized using a Grignard reaction, and the effects of reaction conditions and starting material (i.e., Cl3SiCH2Cl, (MeO)3SiCH2Cl, and (EtO)3SiCH2Cl) on molecular weight are analyzed.
Experimental
Materials
Cl3SiCH2Cl (98%), (MeO)3SiCH2Cl (98%), and (EtO)3SiCH2Cl (98%) were purchased from JSI Silicone and used as received. Vinyl magnesium bromide (ViMgBr, 1.6 M in tetrahydrofuran (THF), Sigma-Aldrich), magnesium turnings (Mg, 99%, Alfa-Aesar), LiAlH4 (95%, Alfa-Aesar), and BrCH2CH2Br (98%, Alfa-Aesar) were used without purification. THF was distilled over sodium benzophenone ketyl. Moisture- or air-sensitive compounds were handled under an Ar atmosphere using standard Schlenk techniques.
VHPCS synthesis
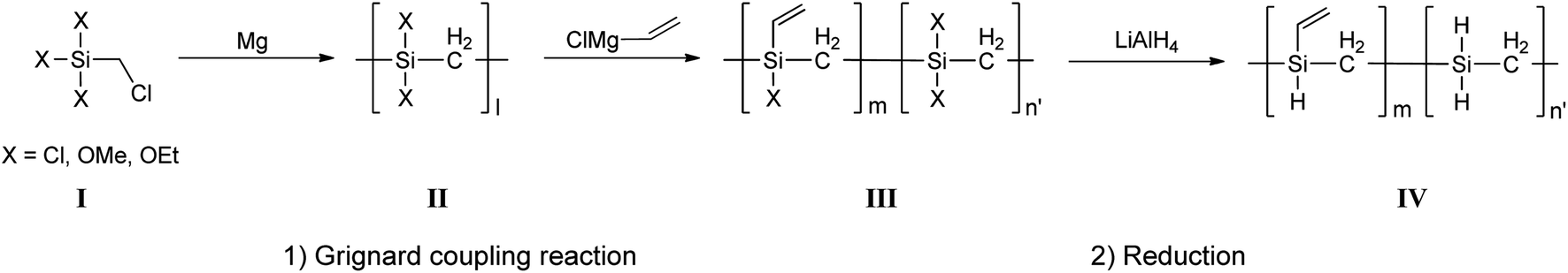
All VHPCS samples were synthesized using the same method, with a representative example for Cl3SiCH2Cl described in detail below (Fig. 1). A three-neck flask equipped with a dropping funnel, magnetic stirrer, and a reflux condenser was charged with dry THF (30 mL) and magnesium turnings (2.2 g, 90 mmol). Several drops of BrCH2CH2Br were added for magnesium activation and Cl3SiCH2Cl (14.7 g, 80 mmol) diluted with a small amount of THF was then added dropwise using a dropping funnel. The reaction mixture containing the MgCl2 precipitate was stirred at 60 °C for 1–3 days, and then warmed to room temperature (∼20 °C). Thereafter, ViMgCl (1.6 M in THF, 10 mL, 16 mmol) was added into filtered solution, and the mixture was stirred for 12 h. The suspension was then cooled to room temperature as about 20 °C, and LiAlH4 (1.5 g, 40 mmol) was added. The resulting slurry was then slowly warmed to 60 °C and stirred overnight at this temperature. The slurry was then cooled in an ice bath and hydrolyzed with 4 M HCl and hexane. The organic THF/hexane phase was separated from the water phase using separation funnel. Than, the solution was dried over anhydrous MgSO4 (≥99.5%, Sigma-Aldrich), and concentrated under vacuum to afford a viscous yellow oil.
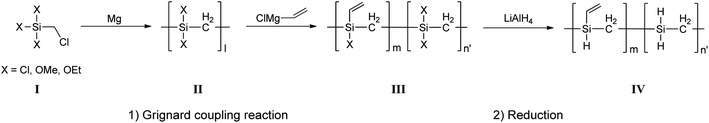 |
| | Fig. 1 Mechanism of Grignard reaction-induced VHPCS chain growth. | |
Instrumentation
1H NMR and 13C NMR spectra were recorded on Avance II DRX 400 MHz (Bruker Corp.) and Avance III HD 300 MHz spectrometers (Bruker Corp.), respectively. Chemical shifts were reported relative to the residual peak of CDCl3 (7.26 ppm) and expressed in δ (ppm). 29Si NMR spectra were acquired on a Bruker ADVANCE III 400 MHz solid-state NMR spectrometer at the Korea Basic Science Institute, Western Seoul Center. Fourier-transform infrared (FT-IR) spectra were recorded on a VERTEX 70 FT-IR spectrometer (Bruker Corp.) in attenuated total reflection mode. Molecular weight distribution patterns were obtained by gel permeation chromatography (GPC; Agilent 1200 series, Agilent Technologies, Inc.) using a toluene mobile phase (1.0 mL min−1, HPLC-grade toluene, J. T. Baker™), and the average molecular weight was determined using a polystyrene calibration kit (Agilent PS-H EasiVial standards kit, Agilent Technologies, Inc.).
Results and discussion
Spectroscopic analysis of VHPCS
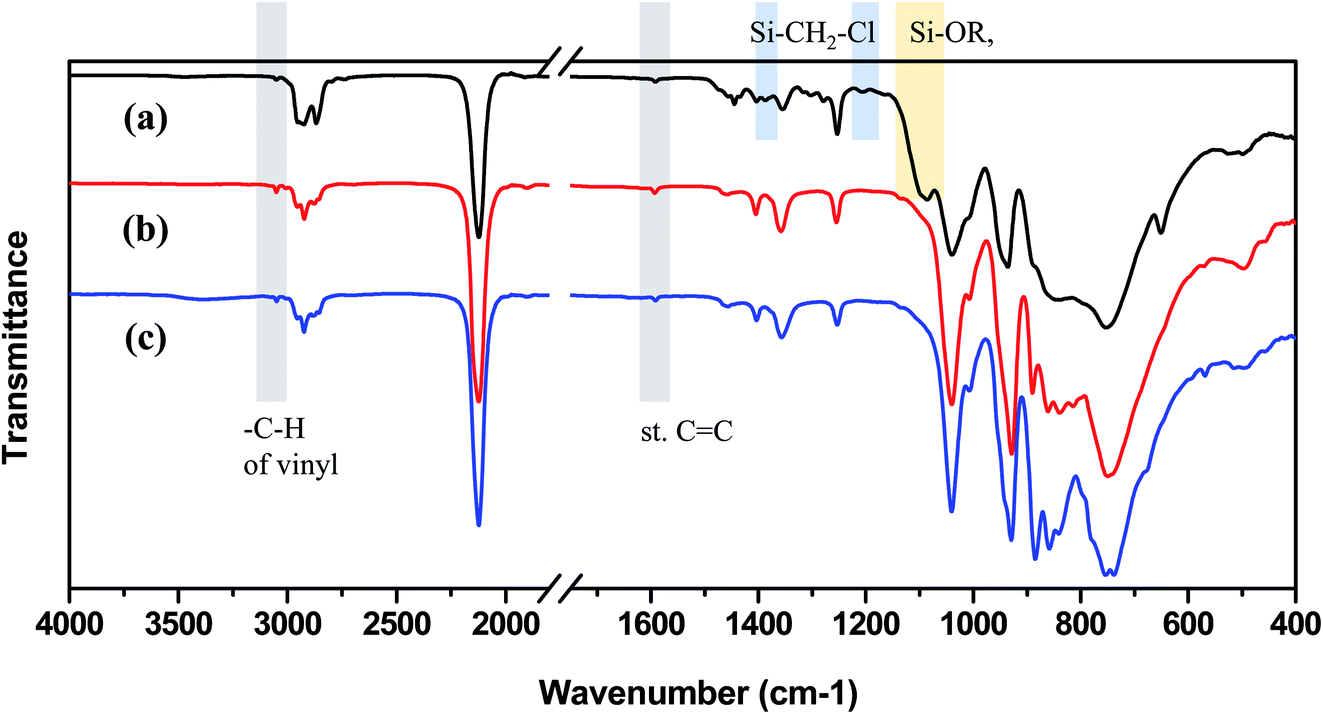
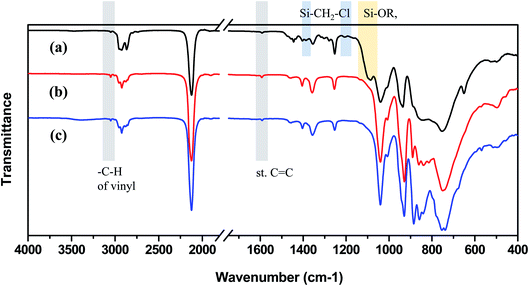
Samples synthesized from all three starting materials were obtained as viscous yellow liquids. The skeleton of VHPCS comprises Si–C bonds, and the polymer network can be probed by spectroscopic (e.g., FT-IR and NMR) analyses. As shown in Fig. 2, the FT-IR spectra of all samples featured strong peaks at 1040, 855, and 750 cm−1, which were ascribed to the bending and stretching vibrations of Si–CH2–Si.22,23 The other strong peaks at 2150 and 950 cm−1 were attributed to the stretching and bending vibrations of Si–H bonds, respectively. The two peaks at >3000 and 1600 cm−1 were attributed to Calkene–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond vibrations, respectively, indicating the presence of vinyl groups.24 Finally, two very weak peaks at 3050 and 1600 cm−1 were observed for all three samples. Thus, a carbosilane chain functionalized with vinyl groups was formed in all cases.
C bond vibrations, respectively, indicating the presence of vinyl groups.24 Finally, two very weak peaks at 3050 and 1600 cm−1 were observed for all three samples. Thus, a carbosilane chain functionalized with vinyl groups was formed in all cases.
 |
| | Fig. 2 FT-IR spectra of VHPCS samples obtained from (a) Cl3SiCH2Cl (P1-1), (b) (MeO)3SiCH2Cl (P2), and (c) (EtO)3SiCH2Cl (P3). | |
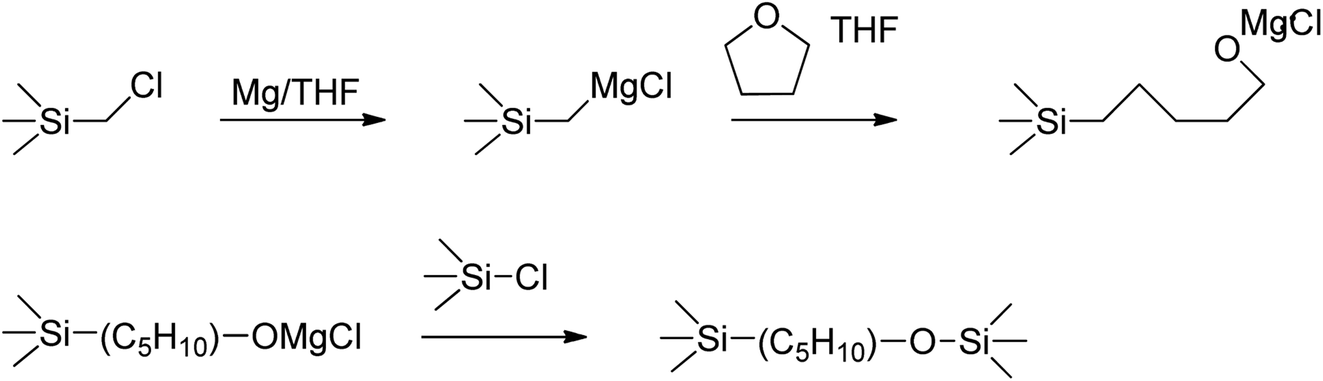
However, the spectrum of the polymer prepared from Cl3SiCH2Cl (P1-1) differed in some regions from those of the polymers prepared from alkoxysilanes, especially at 1500–1200 cm−1 and the emergence of a shoulder at 1100 cm−1. The shoulder at 1100 cm−1 was ascribed to the Si–O bond vibration, in line with the fact that silanes easily undergo oxidation to form Si–O–Si networks or Si–OR units. The rate of silane Grignard coupling is affected by solvent type, with the most common solvents being THF and diethyl ether.25 However, when Cl3SiCH2Cl is used, these solvents may be involved in side reactions to form Si–OR units at Si–Cl sites.26 In this study, THF was used as the solvent, so the side reaction was estimated as shown in Fig. 3. In the case of alkoxysilanes, the shoulder at 1100 cm−1 was not observed (Fig. 2(b) and (c)), which indicates that these species were more suitable starting materials when performing the Grignard coupling in THF. The peaks at 1500–1200 cm−1 generally correspond to the vibrations of Calkyl single bonds, such as C–O and C–H, but are not easy to index. Therefore, NMR spectroscopy was used to gain further insights.
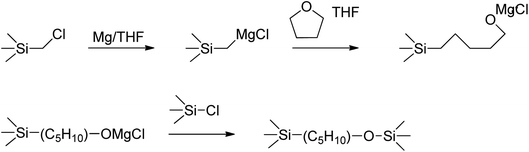 |
| | Fig. 3 Side reaction induced by chlorosilane with THF in Grignard coupling reaction. | |
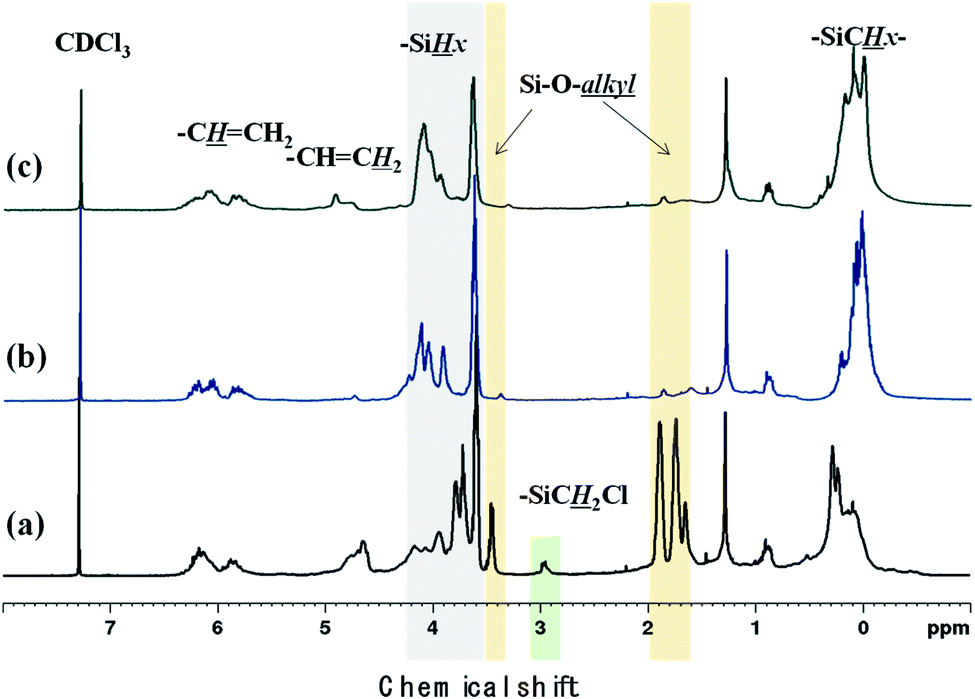
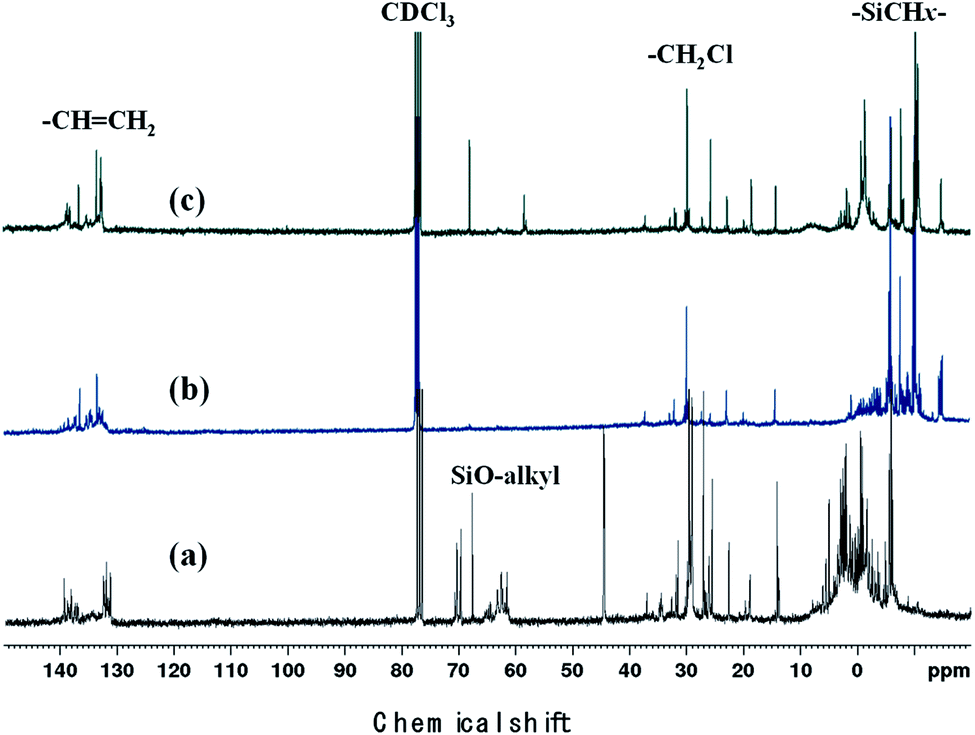
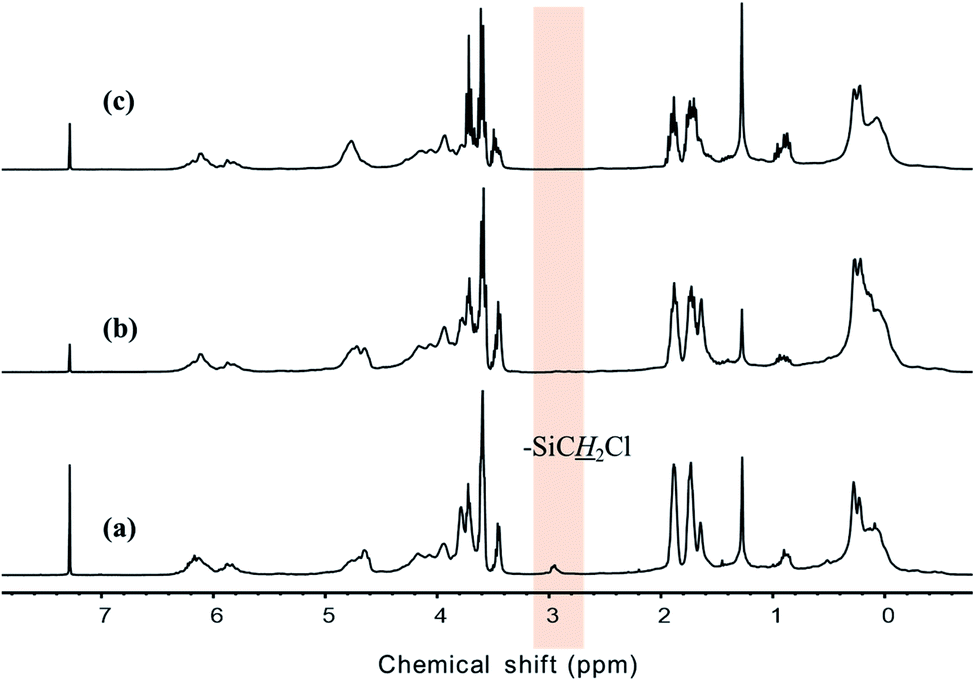
Polycarbosilane hydrogen exists in the forms of Si–C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) x and Si–x, featuring 1H NMR chemical shifts of 0 and 4 ppm, respectively. The vinyl group can be identified based on the appearance of –CHC2 and –CCH2 signals at 4.5–5 and ∼6 ppm, respectively. All of these defining peaks were observed for all three samples (Fig. 4), with the vinyl group peaks being more pronounced than those of the corresponding FT-IR spectra. However, when Cl3SiCH2Cl was used, the 1H NMR spectrum contained additional strong peaks at 1.5–4 ppm (Fig. 3(a)) that were not observed (or were very weak) when the other starting materials were used. Although the NMR patterns in Fig. 4(a) and (b) were close to those expected for the theoretical structure of VHPCS,19,27,28 the pattern shown in Fig. 4(a) had also been reported as VHPCS elsewhere.7,29–32 However, there was no agreement regarding the origin of the peaks at 1.5–4 ppm; Zhong et al. indexed the peak at 3.4 ppm to –C2Cl.33 Sawai et al. and Whitmarsh et al. assigned the peak at ∼2 ppm to –C2– units in the scaffold and SiC2Cl units, respectively.34,35 Herein, based on the 1H NMR spectra of (CH3)3SiCH2Cl and (CH3)SiOCH2CH3,36,37 we confirmed that the peaks observed at 1.5–4 ppm originate from Si–O–alkyl and –SiC2Cl units (Fig. 4).
x and Si–x, featuring 1H NMR chemical shifts of 0 and 4 ppm, respectively. The vinyl group can be identified based on the appearance of –CHC2 and –CCH2 signals at 4.5–5 and ∼6 ppm, respectively. All of these defining peaks were observed for all three samples (Fig. 4), with the vinyl group peaks being more pronounced than those of the corresponding FT-IR spectra. However, when Cl3SiCH2Cl was used, the 1H NMR spectrum contained additional strong peaks at 1.5–4 ppm (Fig. 3(a)) that were not observed (or were very weak) when the other starting materials were used. Although the NMR patterns in Fig. 4(a) and (b) were close to those expected for the theoretical structure of VHPCS,19,27,28 the pattern shown in Fig. 4(a) had also been reported as VHPCS elsewhere.7,29–32 However, there was no agreement regarding the origin of the peaks at 1.5–4 ppm; Zhong et al. indexed the peak at 3.4 ppm to –C2Cl.33 Sawai et al. and Whitmarsh et al. assigned the peak at ∼2 ppm to –C2– units in the scaffold and SiC2Cl units, respectively.34,35 Herein, based on the 1H NMR spectra of (CH3)3SiCH2Cl and (CH3)SiOCH2CH3,36,37 we confirmed that the peaks observed at 1.5–4 ppm originate from Si–O–alkyl and –SiC2Cl units (Fig. 4).
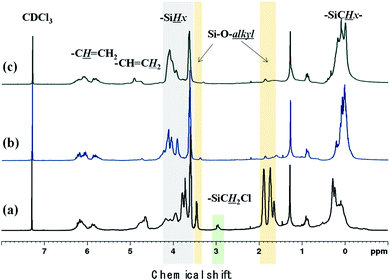 |
| | Fig. 4 1H NMR spectra of (a) P1-1, (b) P2, and (c) P3. | |
The –SiC2Cl unit, which is responsible for the formation of the polycarbosilane basic unit or polymer chain, was present in all starting materials. However, detection of this unit in the polymer suggested that the coupling reaction was not complete. As shown in the 13C NMR spectra (Fig. 5), the –SiC2Cl signal was observed at 30 ppm for all three polymers, with the strongest signal observed for the polymer produced from Cl3SiCH2Cl (Fig. 5(a)). Thus, even under the same reaction conditions, the reaction rate and polymer structure depended on the starting material.
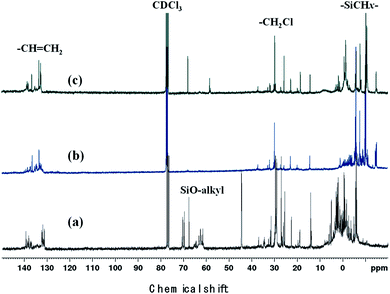 |
| | Fig. 5 13C NMR spectra of (a) P1-1, (b) P2, and (c) P3. | |
VHPCS polymerization behavior
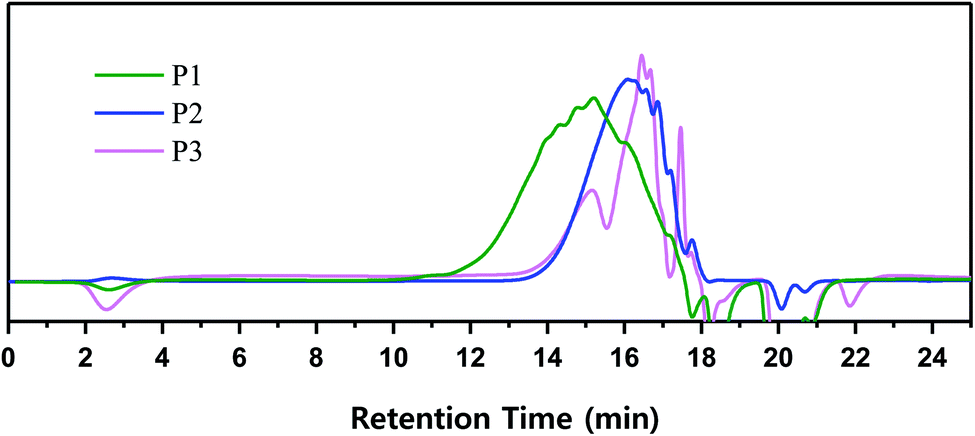
All three types of polymers exhibited the VHPCS structure and showed no specific structural differences except for the occurrence of a side reaction. Polymerization behavior was characterized in terms of GPC-determined molecular weight distribution. GPC separates polymers according to their size, with molecular weight being inversely correlated to retention time. The signal of P1-1 appeared at 11 min, whereas the signals of P2 and P3 appeared only after 13 min (Fig. 6). When the molecular weight distribution was converted using the polystyrene standard, the weight-average molecular weight (Mw) of P1-1 was estimated as 2500 g mol−1, while that of P2 (∼900 g mol−1) was only one-third of this value (Table 1). Thus, even though these polymers were obtained under the same reaction conditions, their molecular weights were different.
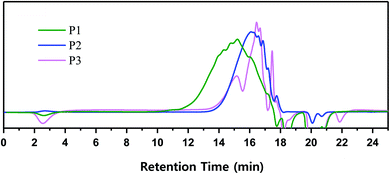 |
| | Fig. 6 Molecular weight distribution of VHPCS obtained from different starting materials. | |
Table 1 Parameters of VHPCS formed under different conditions
| Sample name |
Starting material |
Reaction time (day) |
Production yield (%) |
GPC analysis results |
| RTa (min) |
Mn (g mol−1) |
Mw (g mol−1) |
PDI |
| In this table the RT (retention time) means the initial time of signal detected. |
| P1-1 |
Cl3SiCH2Cl |
1 |
85 |
11 |
850 |
2500 |
3.0 |
| P1-2 |
2 |
11 |
1000 |
2600 |
2.5 |
| P1-3 |
3 |
11 |
1260 |
3500 |
2.8 |
| P2 |
MeO3SiCH2Cl |
1 |
62 |
13 |
400 |
900 |
1.3 |
| P3 |
EtO3SiCH2Cl |
3 |
65 |
13 |
500 |
600 |
1.2 |
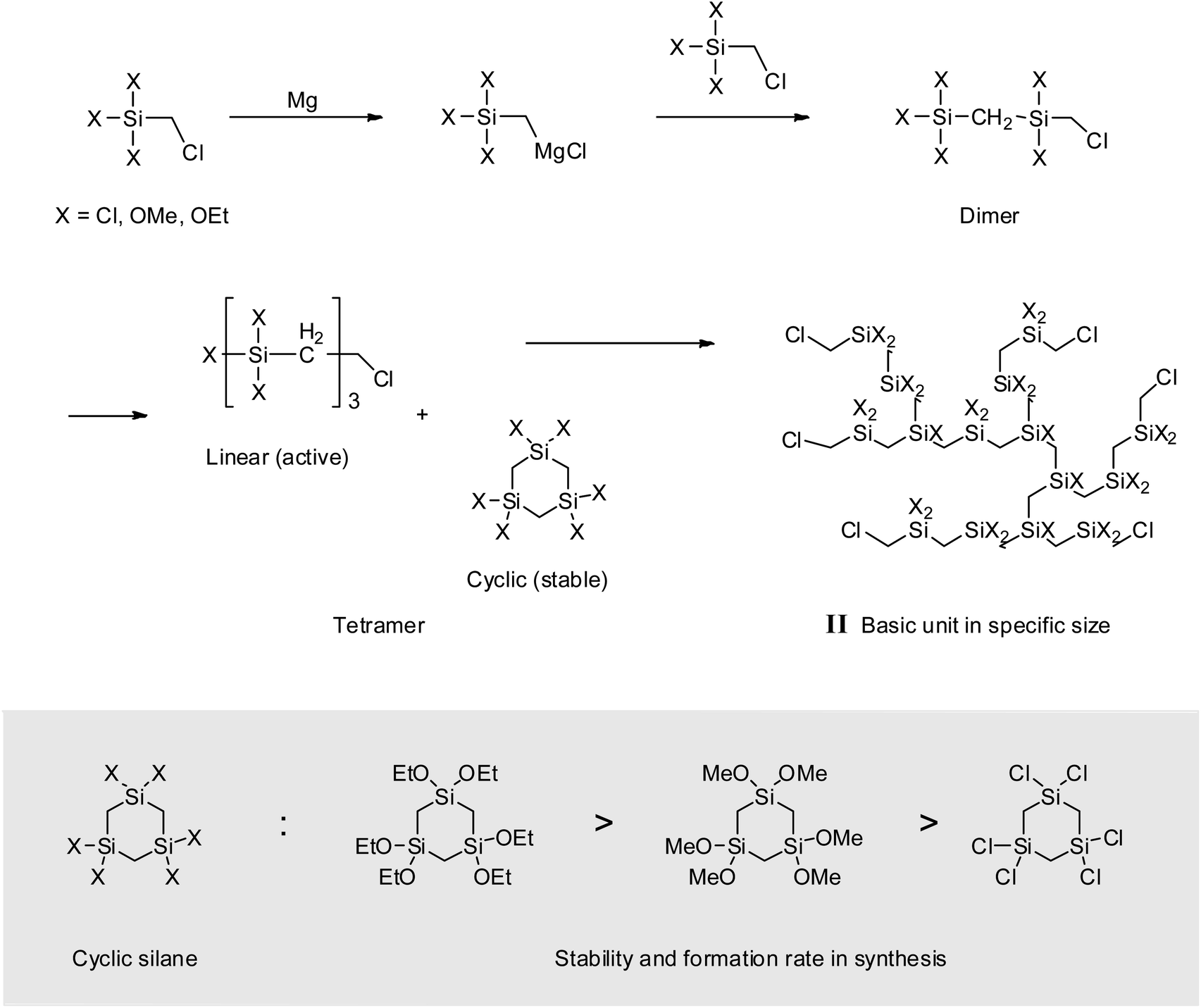
When alkoxysilanes were used as starting materials, polymerization behavior was affected by the nature of the alkoxide (–OR) substituent. For example, P2 and P3 featured Mw values of 900 and 600 g mol−1, respectively (Table 1 and Fig. 6). In the case of P3, although the reaction time was three times longer than for P2, the molecular weight did not increase considerably. Grignard coupling induces polymerization by chain growth, in which the polymer is formed by the linking of monomers to afford a linear structure, although cyclic or branched structures may also be produced as shown in Fig. 7;8,38 the formation of relatively stable cyclic silanes hinders growth of the carbosilane polymer. Brondani et al. reported that alkoxysilanes preferentially formed six-membered ring structures relative to linear polymers, with the cyclic silane formation rate observed for ethoxysilane exceeding that of methoxysilane.39 Herein, VHPCS derived from alkoxysilanes had a low molecular weight and was obtained in ∼60% yield, thus indicating that the growth of molecular weight is related to the reactivity of the starting material. The polymerization and the formation of the cyclic silane according to the type of starting material are summarized in Fig. 7.
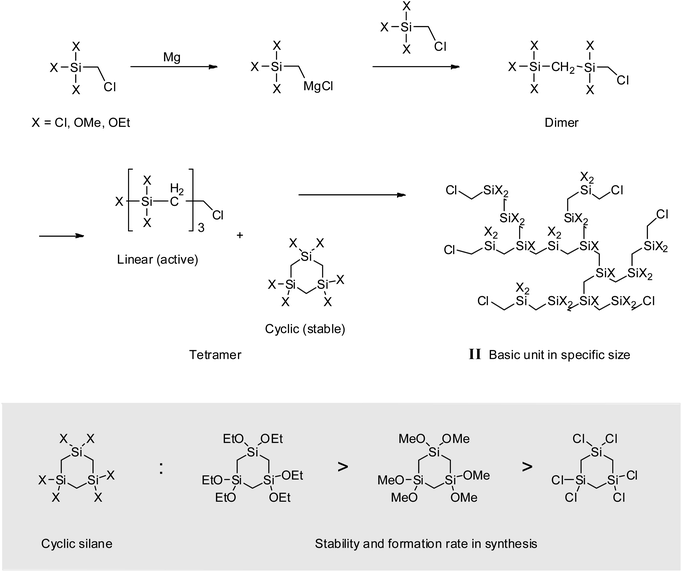 |
| | Fig. 7 Schematic of polymerization in a basic unit. | |
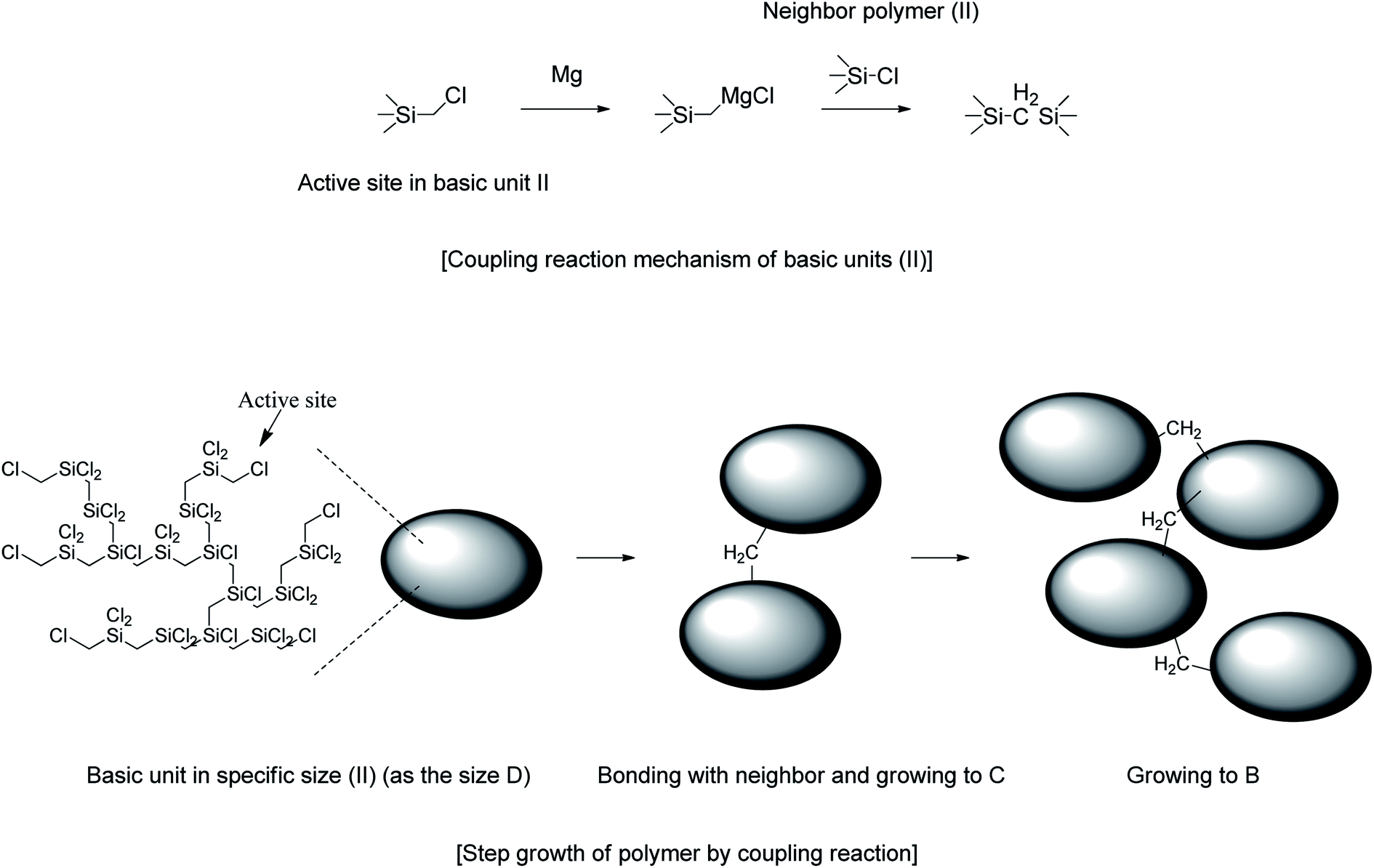
Growth via unit-size polymer coupling
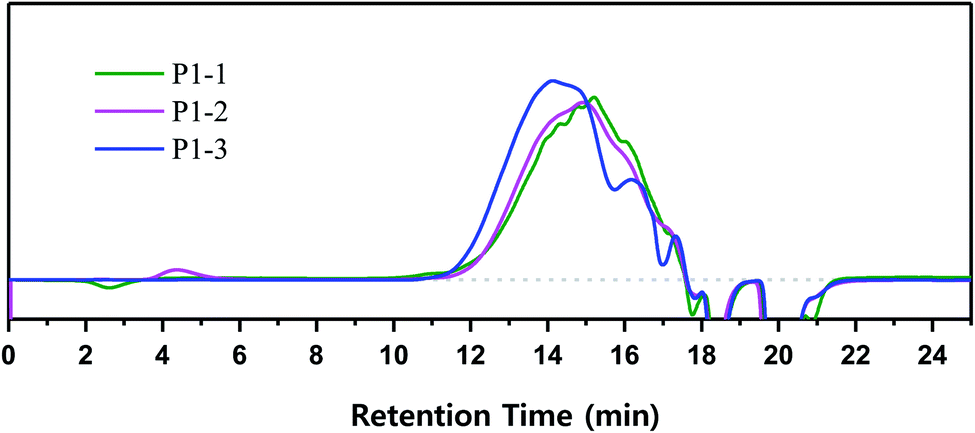
Cl3SiCH2Cl afforded VHPCS with the highest molecular weight but promoted side reactions, and the polymer weight increased with increasing reaction time. According to GPC results, upon going from a reaction time of 1 day to 2 days, the retention time decreased slightly, and Mw increased from 2500 g mol−1 (P1-1) to 2600 g mol−1 (P1-2), but no significant differences were observed (Fig. 8 and Table 1). However, when the reaction time was extended to 3 days, the GPC pattern remarkably shifted to higher molecular weights, and Mw increased to 3500 g mol−1. Herein, the reaction time was the time during which the reactive state (i.e., the stirring state) was maintained after the starting material had been added. Therefore, even without further addition of starting material, the reaction continued for 3 days to increase the molecular weight.
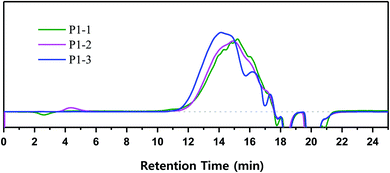 |
| | Fig. 8 Time-dependent polymerization behavior of VHPCS followed for 3 days. | |
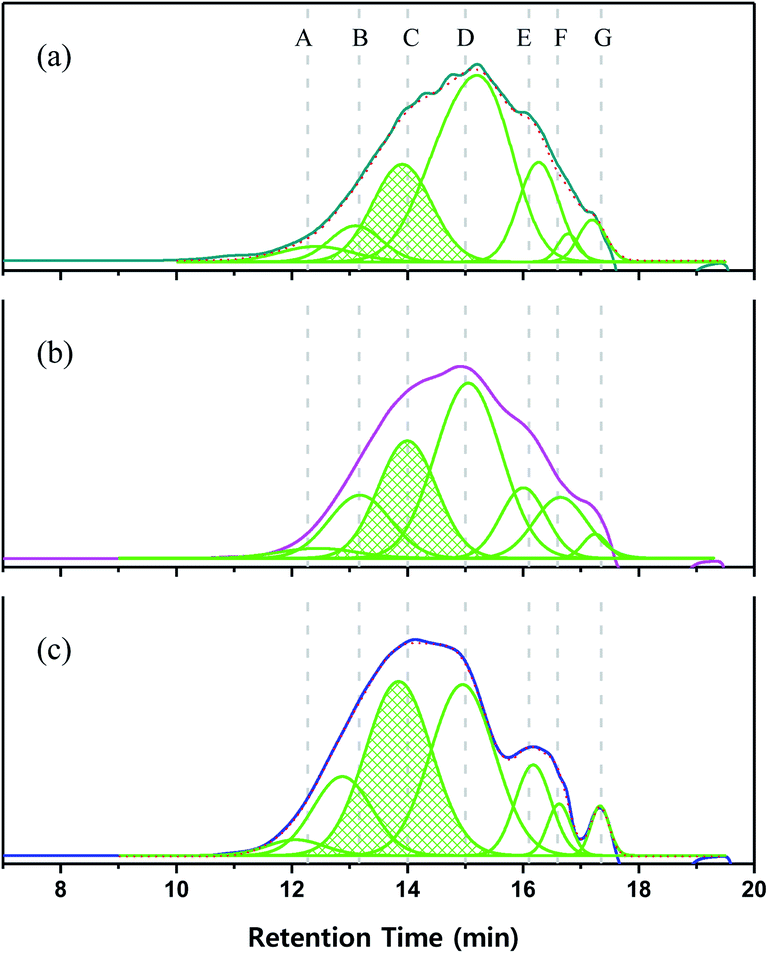
To shed further light on polymer growth behavior, the GPC curves were divided into seven regions according to detection time, which depends on polymer size or length (Fig. 9). The area ratio of each molecular weight region changed with increasing reaction time, especially in zones A–D. In the case of P1-1, the areas of high-molecular-weight (A–C) and low-molecular-weight (E–G) regions were symmetrically distributed around D (retention time = 15 min). With increasing reaction time, the area of D decreased, and those of B and C increased. After 3 days, the area of D was markedly reduced, which resulted in rearrangement of the overall molecular weight distribution. These phenomena suggested that the polymer in region D was transformed to C, and then from C to B as the reaction time increased.
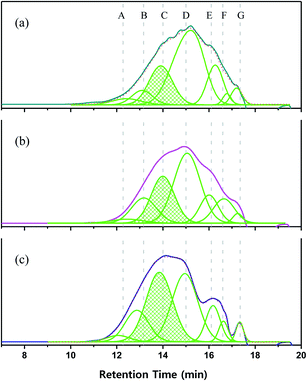 |
| | Fig. 9 GPC curves and the related fits of (a) P1-1, (b) P1-2, and (c) P1-3. | |
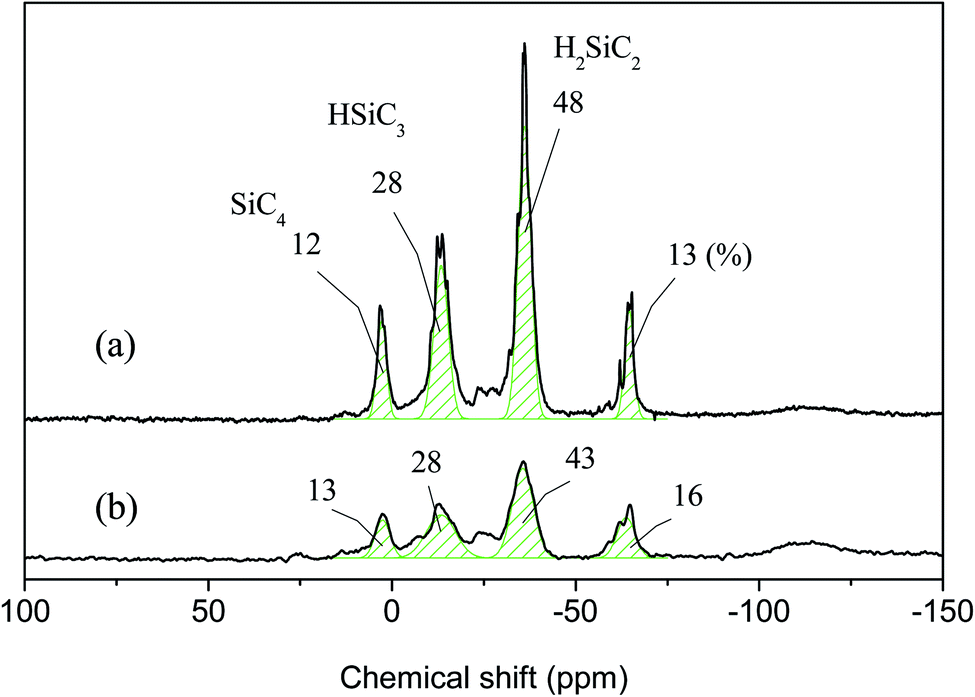
The results of 1H NMR analysis revealed that P1-1 contained residual –SiCH2Cl units (Fig. 4(a)), the proportion of which decreased with increasing reaction time and approached zero after 3 days (Fig. 10). In many cases, polymers are grown in a stepwise manner. In the first step, polymers with a specific size (e.g., corresponding to D) were formed by chain growth (Fig. 7), and subsequent rapid growth, then occurred via the coupling of –SiCH2Cl and –Si–Cl units in neighboring chains (Fig. 11). The average molecular weights corresponding to regions B, C, and D were estimated as 3850, 1820, and 830 g mol−1, respectively (Table 2). For these sizes, assuming that the vinyl group substitution ratio (m:n′ in structure IV) was 1:1 or 1:2, the number of unit chains was estimated as 114–158, 68–73, and 32–35, respectively, i.e., an approximately two-fold decrease upon going from B to C and from C to D. Fig. 12 compares the results of 29Si NMR analysis of the initial reaction mixture (1 day) and VHPCS obtained after 3 days, revealing that with increasing polymer size, the peak broadened and lost intensity. Although the area of the peak at −65 ppm (ascribed to –SiH3 (ref. 27)) increased, no significant differences were observed between the two spectra. As the polymerization reaction at only one site causes the polymer to double in size, no considerable changes to the 29Si NMR spectra were expected.
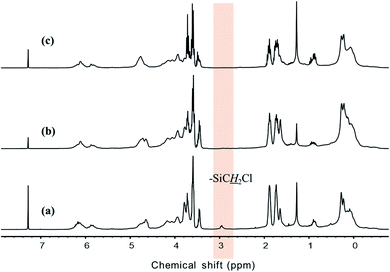 |
| | Fig. 10 1H-NMR spectra of VHPCS obtained at reaction times of (a) 1 day (P1-1), (b) 2 days (P1-2) and (c) 3 days (P1-3). | |
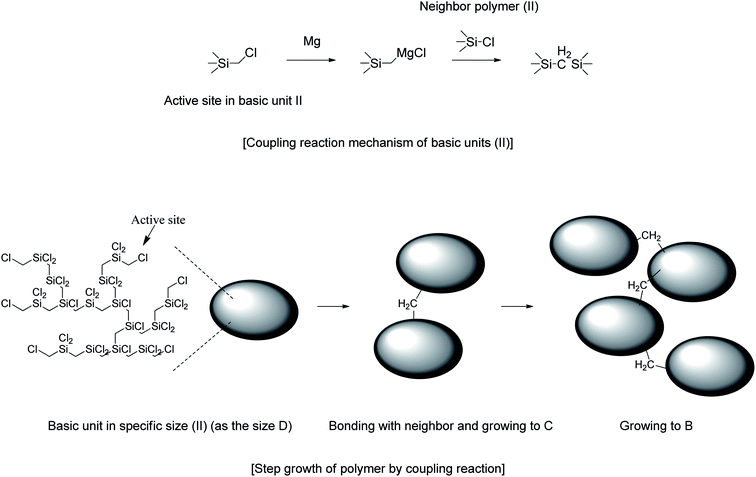 |
| | Fig. 11 Growth of VHPCS via the coupling of basic units. | |
Table 2 Molecular weights obtained from GPC retention times using the polystyrene standard and by matching the number of monomer units
| Position |
Retention time (min) |
Estimated MW (g mol−1) |
Estimated number of mer (n + m)a |
| It is calculated based on the ratio of n′/m of the structure IV as 1 or 2. |
| A |
12.3 |
10200 |
114–158 |
| B |
13.2 |
3850 |
68–73 |
| C |
14.0 |
1820 |
32–35 |
| D |
15.0 |
830 |
15–16 |
| E |
16.1 |
430 |
7.5–8 |
| F |
16.6 |
340 |
6–6.5 |
| G |
17.4 |
<150 |
2.5–3 |
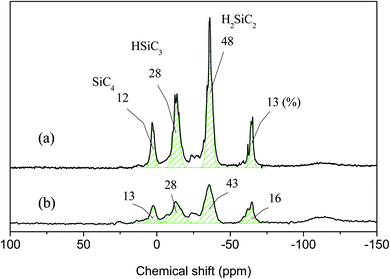 |
| | Fig. 12 29Si NMR spectra of (a) P1-1 and (b) P1-3. | |
Conclusions
Three silanes were used as starting materials for carbosilane polymer synthesis via Grignard coupling, and the effects of starting material on polymerization behavior were probed. According to spectroscopic data, the VHPCS structure was formed in all cases, but a distinct difference was observed between the chlorosilane and alkoxysilanes. In the former case, the presence of –SiCl units resulted in side reactions with THF to form –Si–OR moieties, and –SiCH2Cl units were partially retained. In contrast, no such side reactions were observed for the alkoxysilanes. However, using alkoxysilane, the growth in molecular weight was limited because the cyclic silane was the predominant reaction product. The residual –SiCH2Cl groups in chlorosilane-derived VHPCS acted as active sites to significantly increase the polymer molecular weight at prolonged reaction times. Polymer growth was characterized by the change in molecular weight distribution obtained by GPC analysis. As the molecular weight of each region (A–D) detected in 12–15 min of retention time was nearly twice as large as the smaller polymer, this indicates that the VHPCS grew by step growth polymerization, thus growth was determined by reaction time.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Korea Institute of Ceramic Engineering & Technology (KPP18002) and the Basic Science Research Program through the National Research Foundation of Korea, which was funded by the Ministry of Education (NRF-2019R1I1A2A01062609).
Notes and references
- S. Yajima, K. Okamura, J. Hayashi and M. Omori, Synthesis of continuous SiC fibers with high tensile strength, J. Am. Ceram. Soc., 1976, 59, 324 CrossRef CAS.
- Y. Hasegawa and K. Okamura, Synthesis of continuous silicon carbide fiber, J. Mater. Sci. Technol., 1983, 18, 3633 CAS.
- L. V. Interrante, C. W. Whimarsh, W. Sherwood, H.-J. Wu, R. Lewis, and G. Maciel, Hydridopolycarbosilane Precursors to Silicon Carbide, in Applications of Organometallic Chemistry in the Preparation and Processing of Advanced Materials, Springer, Dordrecht, 1995, pp. 173–183 Search PubMed.
- Advanced SiC/SiC ceramic composites: developments and applications in energy systems, ed. Akira Kohyama, Wiley-American Ceramic Society, 2002, vol. 144 Search PubMed.
- Z. C. Eckel, C. Zhou, J. H. Martin, A. J. Jacobsen, W. B. Carter and T. A. Schaedler, Additive manufacturing of polymer-derived ceramics, Science, 2016, 351, 58 CrossRef CAS PubMed.
- R. Sujith, S. Jothi, A. Zimmermann, F. Aldinger and R. Kumar, Mechanical behavior of polymer derived ceramics – a review, Int. Mater. Rev., 2020, 1, 1–24 Search PubMed.
- S. Park, D.-H. Lee, H.-I. Ryoo, T.-W. Lim, D.-Y. Yang and D.-P. Kim, Fabrication of three-dimensional SiC ceramic microstructures with near-zero shrinkage via dual crosslinking induced stereolithography, Chem. Commun., 2009, 4880 RSC.
- L. Interrante and Q. Shen, Hyperbranched polycarbosilanes via nucleophilic substitution reactions, in Silicon-containing dendritic polymers, Springer, Dordrecht, 2009, pp. 315–343 Search PubMed.
- A. Tulmets, B. T. Nguyen, D. Panov, M. Sassian and J. Järv, Kinetics of the Grignard Reaction with Silanes in Diethyl Ether and Ether-Toluene Mixtures, J. Org. Chem., 2003, 68, 9933 CrossRef PubMed.
- K. J. Wynne and R. W. Rice, Ceramics via polymer pyrolysis, Annu. Rev. Mater. Sci., 1984, 14, 297 CrossRef CAS.
- C. K. Whitmarsh and L. V. Interrante, Synthesis and structure of a highly branched polycarbosilane derived from (chloromethyl)trichlorosilane, Organometallics, 1991, 10, 1336 CrossRef CAS.
- X. Liu, J. S. Rathore, G. Dubois and L. V. Interrante, Grignard condensation routes to 1,3-disilacyclobutane-containing cyclolinear polycarbosilane, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1547 CrossRef CAS.
- T. S. Key, G. B. Wilks, T. A. Parthasarathy, D. S. King, Z. D. Apostolov and M. K. Cinibulk, Process modeling of the low-temperature evolution and yield of polycarbosilanes for ceramic matrix composites, J. Am. Ceram. Soc., 2018, 101, 2809 CrossRef CAS.
- W. Guodong, S. Yongcai and L. Yongqiang, Evolution of molecular composition of polycarbosilane and its effect on spinnability, RSC Adv., 2018, 8, 21863 RSC.
- S. Jang, S. G. Bae, D.-G. Shin, K. Cho, Y. Lee and Y. Lee, Characteristics of polycarbosilanes produced under different synthetic conditions and their influence on SiC fibers: part I, Ceram. Int., 2020, 46, 5602 CrossRef CAS.
- J. Yang, X. Cheng, Y. Yu and Y. Zhang, Quantitative determinations in the molecular structures of polyaluminocarbosilane, Polymer, 2011, 52, 3811 CrossRef CAS.
- X. Liu, J. S. Rathore, G. Dubois and L. V. Interrante, Grignard condensation routes to 1,3-disilacyclobutane-containing cyclolinear polycarbosilanes, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1547 CrossRef CAS.
- X. Zhong, X. Pei, Y. Miao, L. He and Q. Huang, Accelerating the crosslinking process of hyperbranched polycarbosilane by UV irradiation, J. Eur. Ceram. Soc., 2017, 37, 3263 CrossRef CAS.
- Q. H. Shen and L. V. Interrante, Structural Characterization of Poly(silylenemethylene), Macromolecules, 1996, 29, 5788 CrossRef CAS.
- L. V. Interrante, I. Rushkin and Q. Shen, Linear and Hyperbranched Polycarbosilanes with Si-CH2-Si Bridging Groups: A Synthetic Platform for the Construction of Novel Functional Polymeric Materials, Appl. Organomet. Chem., 1998, 12, 695 CrossRef CAS.
- L. He, Z. Zhang, X. Yang, L. Jiao, Y. Li and C. Xu, Liquid polycarbosilanes: synthesis and evaluation as precursors for SiC ceramic, Polym. Int., 2015, 64, 979 CrossRef CAS.
- E. Bacque, J. P. Pillot, M. Birot and J. Dunogues, New polycarbosilane models. 1. Poly[(methylchlorosilylene)methylene], a novel, functional polycarbosilane, Macromolecules, 1988, 21, 30 CrossRef CAS.
- H. P. Martin, E. Muller, R. Richter, G. Roewer and E. Brendler, Conversion process of chlorine containing polysilanes into silicon carbide: part I synthesis and crosslinking of poly(chloromethyl)silanes–carbosilanes and their transformation into inorganic amorphous silicon carbide, J. Mater. Sci., 1997, 32, 1381 CrossRef CAS.
- D. Su, Y.-L. Li, H.-J. An, X. Liu, F. Hou, J. Y. Li and X. Fu, Pyrolytic transformation of liquid precursors to shaped bulk ceramics, J. Eur. Ceram. Soc., 2010, 30, 1503 CrossRef CAS.
- A. Tuulmets, B. T. Nguyen, D. Panov, M. Sassian and J. Järv, Kinetics of the Grignard Reaction with Silanes in Diethyl Ether and Ether - Toluene Mixtures, J. Org. Chem., 2003, 68, 9933 CrossRef CAS PubMed.
- C. K. Whitmarsh and V. I. Leonard, Synthesis and structure of a highly branched polycarbosilane derived from (chloromethyl)trichlorosilane, Organometallics, 1991, 10, 1336 CrossRef CAS.
- I. L. Rushkin, Q. Shen, S. E. Lehman and L. V. Interrante, Modification of a Hyperbranched Hydridopolycarbosilane as a Route to New Polycarbosilanes, Macromolecules, 1997, 30, 3141 CrossRef CAS.
- X. Zhong, X. Pei, Y. Miao, L. He and Q. Huang, Accelerating the crosslinking process of hyperbranched polycarbosilane by UV irradiation, J. Eur. Ceram. Soc., 2017, 37, 3263 CrossRef CAS.
- O. Funayama, T. Aoki, T. Kato, M. Okoda and T. Isoda, Synthesis of thermosetting copolymer of polycarbosilane and perhydropolysilazane, J. Mater. Sci., 1996, 31, 6369 CrossRef CAS.
- R. Sreeja, B. Swaminathan, A. Painuly, T. V. Sebastian and S. Packirisamy, Allylhydridopolycarbosilane (AHPCS) as matrix resin for C/SiC ceramic matrix composites, Mater. Sci. Eng., B, 2010, 168, 204 CrossRef CAS.
- L. He, Z. Zhang, X. Yang, L. Jiao, Y. Lia and C. Xua, Liquid polycarbosilanes: synthesis and evaluation as precursors for SiC ceramic, Polym. Int., 2015, 64, 979 CrossRef CAS.
- H. Li, L. Zhang, L. Cheng, Y. Wang, Z. Yu, M. Huang, H. Tu and H. Xia, Polymer–ceramic conversion of a highly branched liquid polycarbosilane for SiC-based ceramics, J. Mater. Sci., 2008, 43, 2806 CrossRef CAS.
- X. Zhong, X. Pei, Y. Miao, L. He and Q. Huang, Accelerating the crosslinking process of hyperbranched polycarbosilane by UV irradiation, J. Eur. Ceram. Soc., 2017, 37, 3263 CrossRef CAS.
- Y. Sawai, Y. Iwamoto, S. Okuzaki, Y. Yasutomi, K. Kikuta and S. Hirano, Synthesis of silicon carbide ceramics using chemically modified polycarbosilanes as a compaction binder, J. Am. Ceram. Soc., 1999, 82, 2121 CrossRef CAS.
- C. K. Whitmarsh and L. V. Interrante, Synthesis and Structure of a Highly Branched Polycarbosilane Derived from (Chloromethyl)trichlorosilane, Organometallics, 1991, 10, 1336 CrossRef CAS.
- X. Wang, Y. Chen, Y. Li and C. Xu, A new SC precursor with high ceramic yield: synthesis and characterization of CHxMeSiH2-containing poly(ethylsilane-carbosilane), J. Appl. Polym. Sci., 2019, 136, 47618 CrossRef.
- Sigma-Aldrich data base: Ethoxytrimethylsilane, https://www.sigmaaldrich.com/spectra/fnmr/FNMR004476.PDF.
- Q. H. Shen and L. V. Interrante, Structural Characterization of Poly(silylenemethylene), Macromolecules, 1996, 29, 5788 CrossRef CAS.
- D. J. Brondani, R. J. P. Corriu, S. El Ayoubi, J. J. E. Momau and M. W. C. Man, Polyfunctional Carbosilanes and Organosilicon Compounds Synthesis via Grignard Reactions, Tetrahedron Lett., 1993, 34, 2111 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b