
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and self-assembly of a penta[60]fullerene bearing benzo[ghi]perylenetriimide units†
Clément Droua,
Théo Merlandb,
Antoine Busseaua,
Sylvie Dabos-Seignon a,
Antoine Goujona,
Piétrick Hudhommea,
Lazhar Benyahiab,
Christophe Chassenieux*b and
Stéphanie Legoupy*a
a,
Antoine Goujona,
Piétrick Hudhommea,
Lazhar Benyahiab,
Christophe Chassenieux*b and
Stéphanie Legoupy*a
aLaboratoire MOLTECH-Anjou, UMR CNRS 6200, Univ. Angers, SFR Matrix, 2 Bd Lavoisier, 49045 Angers Cedex, France. E-mail: Stephanie.Legoupy@univ-angers.fr
bInstitut des Molécules et Matériaux du Mans, UMR CNRS 6283, Le Mans Univ., 1, Av Olivier Messiaen, 72085 Le Mans Cedex 9, France. E-mail: christophe.chassenieux@univ-lemans.fr
First published on 3rd February 2021
Abstract
A benzo[ghi]perylenetriimide (BPTI) derivative bearing a terminal azido group on the expanded π-conjugated backbone has been synthesized and characterized. This promising photo- and electroactive BPTI motif has been used to obtain an original penta(organo)fullerene as a promising multi-electron acceptor system. Our studies show its self-assembly resulting from aggregation via π–π stacking interaction in solution and in the solid state.
Introduction
Perylenediimide (PDI) derivatives are well-known organic dyes with outstanding thermal, chemical and photostability properties.1 Thanks to their strong electron-accepting ability, ease of functionalization,2,3 and high electron mobility,4,5 PDIs are promising n-type organic semiconductors which have been widely used in organic electronic and optoelectronic devices.6–9 They have also gained interest as building blocks for supramolecular polymers.10A new class of acceptor molecules has recently emerged with benzo[ghi]perylenetriimide (BPTI) derivatives. In comparison with the structure of PDI, their π-conjugated system is laterally expanded by a five-membered imide ring. BPTI chromophore shows a blue-shifted absorption and a stronger solid state fluorescence11 compared to that of PDI. This π-extended BPTI backbone has very recently been studied in perovskite solar cells as electron transporting layer,12 organic–lithium redox-flow batteries,13 photocatalytic degradation of pollutants,14 single component organic solar cells (SCOSC)15 and devices for artificial photosynthesis in water splitting.16 Moreover a BPTI derivative has been used in supramolecular chemistry affording a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 supramolecular complex with zinc porphyrin through hydrogen bonds and an ultrafast photo-induced electron transfer occurred between the two columns of donors and BPTI acceptors.17
:1 supramolecular complex with zinc porphyrin through hydrogen bonds and an ultrafast photo-induced electron transfer occurred between the two columns of donors and BPTI acceptors.17
Fullerene C60 thanks to its tridimensional structure and excellent electron accepting properties18 has been widely used in the formation of supramolecular polymeric nano-network. In particular, the pentasubstituted fullerenes developed by Nakamura and co-workers are well suited. The conical structure obtained by regioselective pentafunctionalization of C60 offers remarkable self-assembly capabilities.19–24 Recently, we reported the syntheses of penta(organo)[60]fullerenes using five tetrathiafulvalene25 (TTF) or (Zn)porphyrin26 fragments as electroactive recognition units which led, thanks to π–π and electronic interactions, to supramolecular arrangements similar to shuttlecocks nested into each other. With the aim of developing new supramolecular assembly based on penta(organo)[60]fullerenes, we were interested in the use of the self-aggregative BPTI motifs to assemble fullerene derivatives.
Results and discussion
We first synthesized a BPTI derivative bearing an azido terminal group to be clicked to a penta(organo)[60]fullerene by using the highly efficient so-called CuAAC (copper-catalysed azide–alkyne cycloaddition) reaction.27BPTI azide 3 was synthesized in two steps starting from benzo[ghi]perylenediimide (BPDI) 1 prepared according to reported procedure.28 The imidization reaction was carried out using 1-aminododecanol on the anhydride function of BPDI 1 affording BPTI 2. Then, a nucleophilic substitution on the hydroxyl group using sodium azide after activation with diphenylphosphoryl azide29 led to BPTI azide 3 (Scheme 1).
 | ||
| Scheme 1 Synthesis of BPTI-azide 3. | ||
BPTI 3 was then clicked to penta(TMS)[60]fullerene304 under CuAAC conditions. Compound 4 was first desilylated in situ with tetrabutylammonium fluoride (TBAF)31 to provide the corresponding terminal alkyne and reaction with excess of azide 3 afforded desired penta(BPTI)[60]fullerene 5 in a remarkable 84% yield, resulting from five one-pot cycloadditions. The latter can be assimilated to a central fullerene surrounded by five BPTI “arms” (shuttlecock type) (Scheme 2).
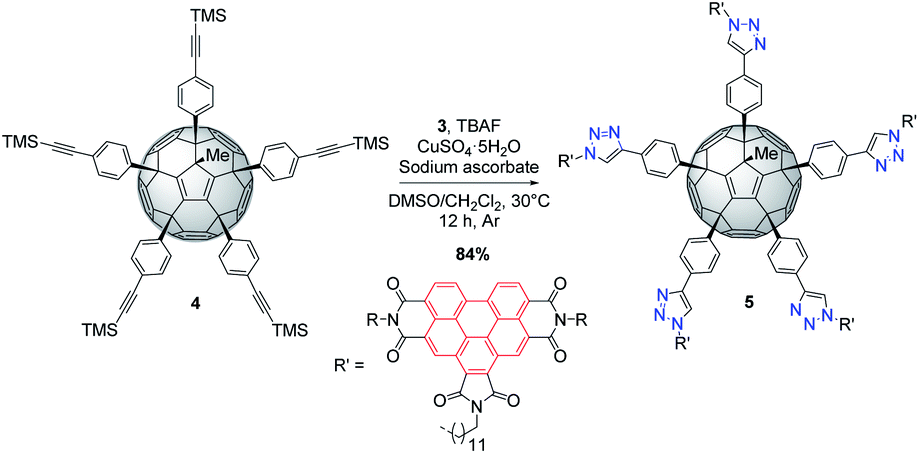 | ||
| Scheme 2 Synthesis of penta(BPTI)[60]fullerene 5. | ||
Reference molecule 6 was also synthesized using CuAAC click reaction between BPTI azide 3 and phenylacetylene (Scheme 3).
 | ||
| Scheme 3 Synthesis of the reference molecule 6. | ||
The structure of compounds 5 and 6 were confirmed by their 1H, 1H–1H COSY and 13C NMR spectra (Fig. S1–S10†) as well as by HR MALDI-TOF mass spectrometry (Fig. S16–S19†). The purity of penta(BPTI)[60]fullerene could be estimated to be higher than 99.5% thanks to HPLC analysis (Fig. S20†).
The self-assembly of these derivatives has been investigated, both in solution and solid state, by NMR spectroscopy, electrochemical analyses, mass spectrometry, atomic force microscopy (AFM) and combined dynamic/static light scattering (DLS/SLS).
1H NMR spectra of compound 5 was recorded at different concentrations between 0.16 and 10 mM (CDCl3, 25 °C; Fig. 1 and S11†). A downfield shift of the aromatic protons is observed when the concentration increases (−0.1 ppm), suggesting the aggregation of BPTI units. On the contrary, no change could be observed in the chemical shift of the triazole protons. As expected by the introduction of the dodecyl spacer length, the triazole ring seems not to be involved in such an aggregation phenomenon (Fig. 1).
 | ||
| Fig. 1 1H NMR spectra of compound 5 at different concentrations (0.16–10 mM) between 7.1 and 10.1 ppm (phenyl aromatic region) in CDCl3 at 25 °C. | ||
Complementary 1H DOSY NMR experiments on compound 5 were performed between 0.31 and 5 mM in CDCl3 (Fig. 2). A decrease of the self-diffusion coefficient Ds was observed when the concentration increases which is in agreement with the formation of aggregates.
 | ||
| Fig. 2 1H DOSY NMR spectrum (300 MHz, CDCl3, 298 K) of penta(BPTI)[60]fullerene 5 at different concentrations. | ||
Using the Stokes–Einstein equation (eqn (S8)†), the hydrodynamic radius of the objects was estimated to be between 2 and 3 nm depending on the concentration (Table 1).
| [C] (mM) | D s (m2 s−1) | r (Å) |
|---|---|---|
| 0.31 | 2.01 × 10−10 | 20 |
| 0.62 | 1.85 × 10−10 | 22 |
| 1.25 | 1.72 × 10−10 | 23 |
| 2.5 | 1.53 × 10−10 | 26 |
| 5 | 1.34 × 10−10 | 30 |
Similar analysis was carried out with reference molecule 6 (Fig. 3 and S13†). 1H NMR spectra at different concentrations (between 0.33 mM and 42 mM) showed a downfield shift of the aromatic protons five times higher than the one obtained for pentaadduct 5. This suggests that π–π stacking governs the weak interaction involved in the self-assembly of reference 6. A downfield shift of the third imide proton (−0.02 ppm) was also noticed while an upfield shift of the protons of the symmetrical diimide (+0.04 ppm) was observed (Fig. S12†).
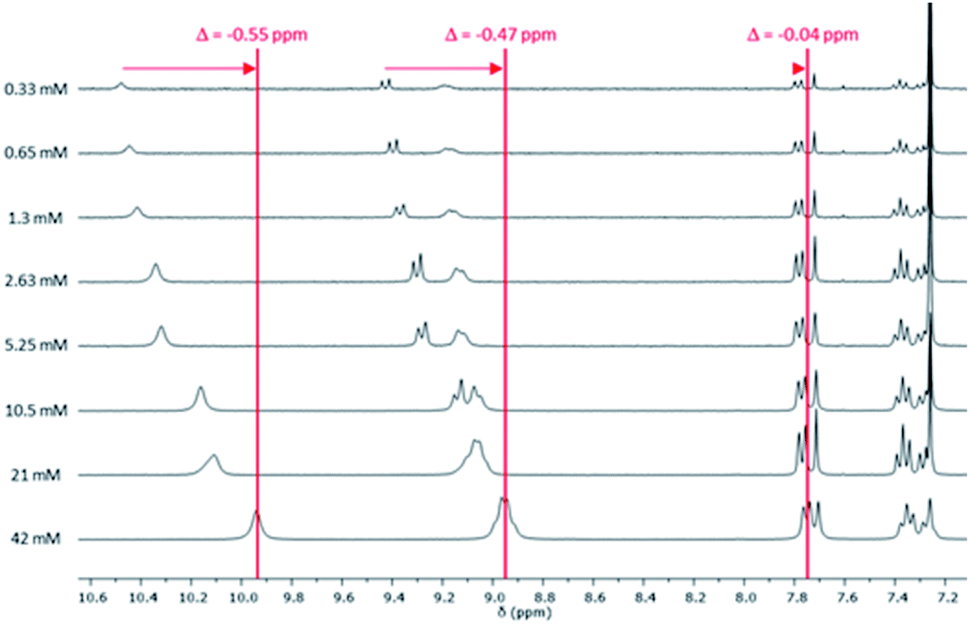 | ||
| Fig. 3 1H NMR spectra of reference 6 at different concentrations (0.33–42 mM) between 7.1 and 10.5 ppm (phenyl aromatic region) in CDCl3 at 25 °C. | ||
These results allowed us to propose a hypothesis concerning the interactions arising in the assembly of pentaadduct 5. Indeed, we can assume that the BPTI cores are aggregating via π–π stacking and that the fullerene unit is not involved in such assembly. Additional NMR experiments were performed to confirm this assumption. Indeed, the mixture of compound 6 with fullerene C60 or with compound 4 did not display a significant shift of the aromatic protons even at increased concentrations (Fig. S14 and S15†).
Aggregation of penta(BPTI)[60]fullerene 5 in solution was confirmed by both static and dynamic light scattering measurements. Fig. 4a displays the concentration dependence of the cooperative diffusion coefficient (Dc) for compound 5 measured by DLS which probed a single diffusive mode of relaxation (Fig. S25†). The Dc value is in close agreement with the one of Ds obtained from DOSY NMR measurements from both the point of view of its magnitude and concentration dependence. Upon increasing the concentration of penta(BPTI)[60]fullerene 5, there is a clear shift of the distribution of sizes derived from DLS (Fig. 4b and S24†) towards higher values (from 2 up to 4 nm) and also a broadening of these distributions, both facts accounting for aggregation of compound 5 in solution. However we noticed that this association led to aggregates of small size over the whole concentration range investigated.
 | ||
Fig. 4 (a) Diffusion coefficients (filled symbols) and hydrodynamic radii (empty symbols) of compound 5 calculated from DLS ( ) and DOSY NMR ( ) and DOSY NMR ( ) measurements. (b) Distribution of relaxation times measured at 90° for various concentrations of compound 5 in chloroform as labelled on the figure. ) measurements. (b) Distribution of relaxation times measured at 90° for various concentrations of compound 5 in chloroform as labelled on the figure. | ||
An estimation of the aggregation number of compound 5 thanks to SLS measurements may be attempted. First, it should be mentioned that solutions of C60 and compound 4 did not scatter light in chloroform which meant that their contrast is very small and that only BPTI units contribute to the scattered intensity for solutions of compounds 5 and 6 in the same solvent. Solutions of penta(BPTI)[60]fullerene 5 scatter much more than solutions of reference 6 at the same concentration (Fig. 5).
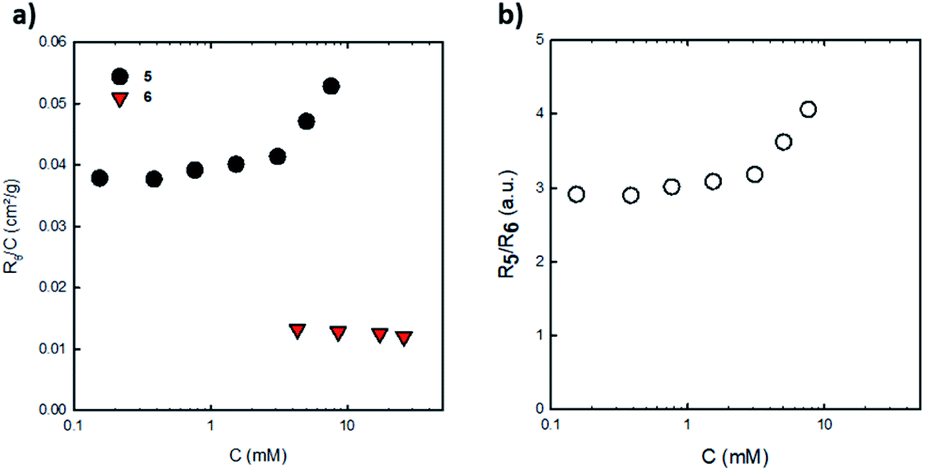 | ||
| Fig. 5 (a) Rayleigh ratios normalized by concentration for solutions of penta(BPTI)[60]fullerene 5 and reference 6 in CHCl3 at 20 °C. (b) Ratio of Rayleigh ratios of 5 and 6 against 5 concentration. | ||
The ratio in their scattering intensities is then related to the number of BPTI units involved which should be five if both compounds 5 and 6 were not aggregated in chloroform. The ratio of scattered intensity between compounds 5 and 6 ranges from three up to four meaning that 5 in chloroform is at least three times more aggregated than reference 6. However, the aggregation number of 6 in chloroform is unknown but as evidenced from NMR, 6 is far from being at the unimer state in the concentration range investigated. It can then be concluded that the penta(BPTI)[60]fullerene 5 is aggregated in chloroform and that its aggregation number is at least three times higher than the reference BPTI 6 at the same concentration.
AFM studies were carried out to investigate the self-assembly of penta(BPTI)[60]fullerene 5 in solid state. Solutions with concentration ranging from 0.2 to 0.6 mM were spin coated onto glass substrate. The AFM images showed aggregates which height is mainly 3–4 nm (Fig. 6, S28 and S29†), with some up to 9 nm, in line with the self-assembly of several molecules as evidenced from other techniques.
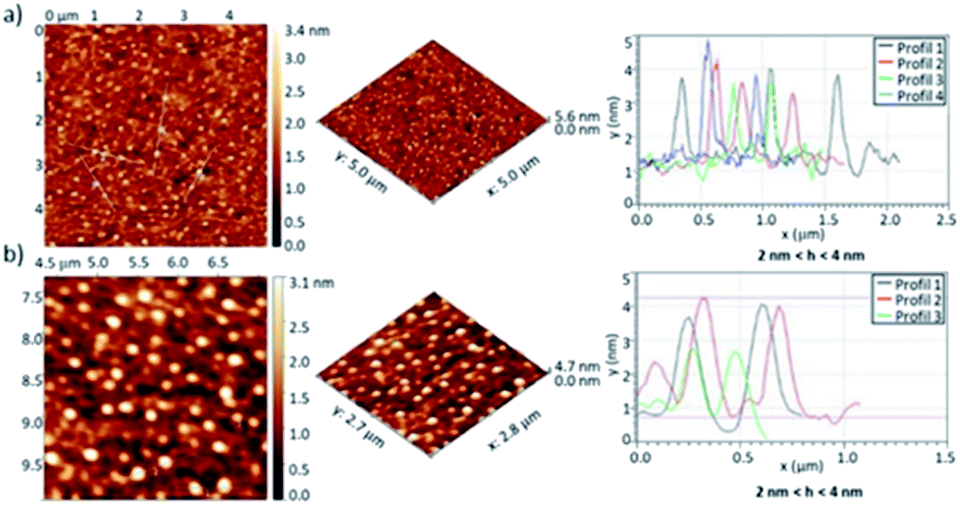 | ||
| Fig. 6 AFM images of penta(BPTI)[60]fullerene 5 in 2D and 3D at 0.4 mM, spin coated onto glass substrate and associated profiles. (a) 5 μm × 5 μm scan (b) magnification of (a). | ||
In the case of reference molecule 6 no obvious assembly was observed, as shown in Fig. S30.† AFM succeeded in proving the self-assembly of penta(BPTI)[60]fullerene 5 in solid state but results for reference 6 were less conclusive.
The emission spectra of 5 in chloroform were measured at different concentrations (Fig. 7a). From 1 μM to 11 μM the emission intensity increased with the concentration. Interestingly, an extinction of the luminescence of penta(BPTI)[60]fullerene 5 was then observed until 96 μM. Since the fullerene is not involved in the assembly, this quenching can be related to the aggregation of the molecule via BPTI–BPTI interaction.
 | ||
| Fig. 7 Concentration-dependent fluorescence spectra of (a) penta(BPTI)[60]fullerene 5 (addition of 5 μM per spectrum), region 450–750 nm and (b) reference 6 (addition of 10 μM per spectrum until 201 μM and then 20 μM per spectrum), region 450–650 nm, CHCl3, λexc = 467 nm, 298 K, 1 cm cuvette. Pink curves: concentration corresponding to the maximum of emission intensity. | ||
Identical measurements were performed on reference molecule 6 (Fig. 7b). The same behaviour was observed except that the concentration at which the extinction appears was much higher. Indeed, if we compare in terms of BPTI units (i.e. BPTI concentration in solution), the extinction concentration in penta(BPTI)[60]fullerene 5 was 2.2 μM against 21 μM for reference 6. Thus, the luminescence quenching is facilitated in compound 5.
In order to confirm the difference between compounds 5 and 6, additional analyses were performed by mixing reference 6 with compound 4 according to a ratio 5/1 which simulated 5 (Fig. 8). The resulting spectrum was comparable to this from reference 6 alone, suggesting that fullerene does not govern the formation of the assembly. On the contrary, the conformation of the BPTI arms in 5 favours the aggregation. Indeed, the pentafullerene scaffold, with the five arms sufficiently close to each other, seems to enhance the BPTI–BPTI interactions.
 | ||
| Fig. 8 Concentration-dependent fluorescence spectra of compounds 4 + 6 (in a molar ratio 1:5 of 4 and 6, respectively), region 400–800 nm, CHCl3, λexc = 467 nm, 298 K, 1 cm cuvette, addition of 10 μM of compound 4 and 50 μM of reference 6 per spectrum. Pink curve: concentration corresponding to the maximum of emission intensity. The legend displays concentration of 4. | ||
The absorption properties of both compounds 5 and 6 were also studied (Fig. S21–S23†). Concerning reference 6, the band in the 400–500 nm region is typically assigned to the S0 → S1 optical transition of BPTI and the band in the 350–400 nm region is attributed to the S0 → S2.11,15 The absorption spectrum of penta(BPTI)[60]fullerene 5 showed absorption bands corresponding to the addition of both fullerene and BPTI moieties, but the self-assembly phenomenon could not be observed by UV-Vis spectroscopy.
The electronic properties were investigated using cyclic voltammetry technique. Cyclic voltammograms of compounds 5 and 6 were recorded at room temperature in o-dichlorobenzene (o-DCB)/MeCN containing 0.1 M nBu4NPF6. The voltammogram of penta(BPTI)[60]fullerene 5 consists in an average between the electronic contribution of fullerene C60 and reference 6 (Fig. S26 and Table S1†). Successive redox couples, corresponding to the multiple reduction process of the respective BPTI unit and C60 fragment, were observed. As shown in Fig. S27 and Table S2,† an increase of the concentration of 5 produced no shift in reduction waves. Then no electronic communication between C60 and BPTI could be observed. Thus, C60 can be self-assembled while preserving its electronic properties.
Conclusions
We have described in this work the synthesis of a benzo[ghi]perylenetriimide (BPTI) derivative bearing an azido group which was clicked using the CuAAC reaction to a penta(organo)fullerene. Using different techniques, we have shown the existence of π–π stacking interactions between the BPTI arms leading to aggregation of the pentaadduct. This aggregation is concentration dependent as evidenced from LS, AFM, fluorescence and NMR measurements. Moreover, this original penta(BPTI)[60]fullerene presents multi-redox accepting properties opening the way to innovative applications in materials science.Experimental section
General procedure for the synthesis of compound 2
A solution of BPDI 1 (800 mg, 0.94 mmol) and 1-aminododecanol (380 mg, 1.88 mmol) in DMF (20 mL) was refluxed for 5 hours. The solvent was evaporated under vacuum and the crude was purified by chromatography (silica gel, CHCl3 followed by 2% v/v MeOH in CHCl3). A second chromatography column (neutral aluminium oxide, CHCl3 followed by 2% v/v MeOH in CHCl3) was necessary for completing purification and gives the desired product as an orange solid (700 mg, 72% yield). 1H NMR (300 MHz, CDCl3) δ ppm: 10.30 (s, 2H), 9.26 (d, J = 8.3 Hz, 2H), 9.11 (d, J = 8.0 Hz, 2H), 5.31 (s, 2H), 3.98 (t, J = 5.9 Hz, 2H), 3.63 (t, J = 6.60 Hz, 2H), 2.50–2.26 (m, 4H), 2.06–1.82 (m, 6H), 1.71–1.18 (m, 51H), 0.92–0.77 (m, 12H). 13C NMR (125 MHz, CDCl3) δ ppm: 168.1, 132.8, 129.9, 127.4, 127.3, 127.1, 125.4, 124.4, 123.7, 122.9, 120.2, 63.2, 55.5, 38.8, 33.0, 32.6, 32.0, 29.5, 28.8, 17.3, 27.1, 25.9, 22.8, 14.2. HRMS (MALDI-TOF, negative mode): calcd for C66H85N3O7 1031.6388 [M]−; found 1031.6385.General procedure for the synthesis of compound 3
A solution of compound 2 (300 mg, 0.29 mmol) in dry DMF (30 mL) was stirred under argon bubbling for 5 minutes. DPPA (0.62 mL, 1.45 mmol) and DBU (45 μL, 0.29 mmol) were added and the solution was stirred at room temperature for 5 hours under argon atmosphere. NaN3 (96 mg, 1.45 mmol) was added and the mixture was stirred at 70 °C for 24 hours. The product was extracted with CHCl3 (200 mL), then the organic layer was washed with brine (3 × 200 mL), dried over MgSO4 and concentrated under reduced pressure. The crude mixture was purified by two successive chromatography columns using respectively silica gel (CHCl3) and neutral aluminium oxide (CHCl3 followed by 2% v/v MeOH in CHCl3) affording the desired product as an orange solid (204 mg, 66% yield). 1H NMR (300 MHz, CDCl3) δ ppm: 10.36 (s, 2H), 9.31 (d, J = 8.58 Hz, 2H), 9.14 (d, J = 7.70 Hz, 2H), 5.32 (s, 2H), 3.98 (t, J = 7.25 Hz, 2H), 3.24 (t, J = 6.95 Hz, 2H), 2.48–2.26 (m, 4H), 2.12–1.8 (m, 6H), 1.67–1.14 (m, 50H), 0.94–0.75 (m, 12H). 13C NMR (125 MHz, CDCl3) δ ppm: 168.4, 133.3, 130.8, 130.4, 130.1, 129.6, 127.7, 124.9, 124.0, 123.4, 55.4, 51.6, 38.8, 32.6, 32.0, 31.6, 30.4, 29.9, 29.8, 29.7, 29.6, 29.4, 29.3, 29.0, 28.9, 27.2, 27.1, 26.9, 22.8, 14.2. HRMS (MALDI-TOF, negative mode): calcd for C66H84N6O6 1056.6452 [M]−; found 1056.6467.General procedure for the synthesis of compound 5
Tetrabutylammonium fluoride (TBAF; 1 M in THF, 0.12 mL, 0.14 mmol) was added to a solution of penta(TMS)-2-methyl[60]fullerene 4 (ref. 32) (8 mg, 5 μmol), freshly prepared azido compound 3 (42 mg, 40 μmol), CuSO4·5H2O (0.25 mg, 0.9 μmol), and sodium ascorbate (3.6 mg, 18 μmol) in 3 mL of DMSO/CH2Cl2 (1:2, v/v). The mixture was stirred at room temperature for 24 hours. Then the solution was diluted with CHCl3 (100 mL), washed with a saturated NH4Cl aqueous solution (1 × 100 mL) and water (2 × 100 mL), dried with MgSO4, filtered, and concentrated under vacuum. The crude product was purified by chromatography (silica gel, CHCl3 followed by 2% v/v MeOH in CHCl3) and centrifuged in CHCl3/MeCN to afford penta(BPTI)[60]fullerene 5 as a bright orange solid (27 mg, 84% yield). 1H NMR (300 MHz, CDCl3) δ ppm: 9.71 (s, 10H), 8.89 (m, 10H), 8.71 (m, 10H), 7.86–7.79 (m, 20H), 7.69–7.46 (m, 5H), 5.31 (s, 10H), 4.54–4.25 (m, 10H), 3.99 (m, 10H), 3.64 (s, 1H), 2.50–2.32 (m, 20H), 2.15–1.82 (m, 40H), 1.62–1.20 (m, 242H), 0.93–0.80 (m, 60H). 13C NMR (125 MHz, CDCl3) δ ppm: 167.6, 164.3, 163.6, 163.3, 162.4, 161.8, 160.5, 159.7, 157.0, 152.9, 151.8, 148.8, 148.7, 148.6, 148.5, 148.4, 148.3, 147.9, 147.8, 147.4, 147.2, 146.9, 145.7, 145.5, 144.9, 144.5, 144.4, 144.2, 144.0, 143.8, 142.9, 142.7, 139.4, 139.0, 137.8, 132.0, 130.7, 130.6, 130.5, 129.3, 128.8, 127.0, 126.7, 126.4, 126.2, 126.1, 126.0, 125.6, 125.3, 124.8, 123.6, 123.5, 123.1, 122.8, 122.1, 120.2, 120.1, 119.7, 62.6, 62.5, 61.0, 58.2, 55.5, 50.7, 50.6, 38.9, 33.6, 32.6, 32.0, 30.6, 29.8, 29.7, 29.5, 29.4, 29.2, 28.8, 27.3, 26.8, 26.7, 24.9, 22.8, 14.3. HRMS (MALDI-TOF, negative mode): calcd for C431H448N30O30 6523.4453 [M]−; found 6523.4926. HPLC analysis: retention time = 3.96 min (eluent: toluene, flow rate: 1 mL min−1, wavelength: 320 nm).
General procedure for the synthesis of compound 6
CuSO4·5H2O (1 mg, 4 μmol) and sodium ascorbate (10 mg, 50 μmol) were added to a solution of (phenylethynyl)trimethylsilane (24 μL, 0.22 mmol) and freshly prepared compound 3 (117 mg, 0.11 mmol) in a mixture of DMSO (2 mL) and CH2Cl2 (4 mL). The solution was stirred at room temperature overnight. The mixture was subsequently diluted with CH2Cl2 and washed with a saturated NH4Cl aqueous solution (1 × 100 mL), brine (1 × 100 mL) and water (1 × 100 mL). The organic phase was dried (MgSO4), filtered and concentrated under vacuum. The crude was purified by gel chromatography (silica gel, 2% v/v MeOH in CHCl3) to obtain the desired compound as an orange solid (108 mg, 84% yield). 1H NMR (300 MHz, CDCl3) δ ppm: 9.95 (s, 2H), 8.97 (s, 4H), 7.76 (d, J = 7.35 Hz, 2H), 7.71 (s, 1H), 7.35 (t, J = 7.20 Hz, 2H), 7.3–7.26 (m, 1H), 5.32 (s, 2H), 4.38 (t, J = 7.28 Hz, 2H), 4.00 (t, J = 7.37, Hz, 2H), 2.48–2.30 (m, 4H), 2.11–1.99 (m, 4H), 1.98–1.83 (m, 4H), 1.60–1.20 (m, 48H), 0.88–0.82 (m, 12H). 13C NMR (125 MHz, CDCl3) δ ppm: 167.9, 147.8, 132.5, 130.8, 128.9, 128.1, 127.2, 127.1, 126.8, 125.7, 124.1, 132.6, 122.7, 119.4. HRMS (MALDI-TOF, negative mode): calcd for C74H90N6O6 1158.6922 [M]−; found 1158.6923.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge CNRS, Université d'Angers and Lumomat (DecaSupra project) for financial support. They also thanks the SFR Matrix (Univ. Angers) for its assistance in spectroscopic analyses and AFM studies. Stéphanie Legoupy acknowledges to Prof. Eiichi Nakamura and coworkers for fruitful discussions, as well as the Japan Society for Promotion of Science for an invitation fellowship.Notes and references
- H. Langhals, Heterocycles, 1995, 40, 477–500 CrossRef CAS.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS.
- A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC.
- Y. Liu, Y. Wang, L. Ai, Z. Liu, X. Ouyang and Z. Ge, Dyes Pigm., 2015, 121, 363–371 CrossRef CAS.
- S. Naqvi, M. Kumar and R. Kumar, ACS Omega, 2019, 4, 19735–19745 CrossRef CAS PubMed.
- Z. Guo, X. Zhang, Y. Wang and Z. Li, Langmuir, 2019, 35, 342–358 CrossRef CAS.
- S. Chen, P. Slattum, C. Wang and L. Zang, Chem. Rev., 2015, 115, 11967–11998 CrossRef CAS.
- A. L. Briseno, S. C. B. Mannsfeld, C. Reese, J. M. Hancock, Y. Xiong, S. A. Jenekhe, Z. Bao and Y. Xia, Nano Lett., 2007, 7, 2847–2853 CrossRef CAS PubMed.
- W. Huang, J. C. Markwart, A. L. Briseno and R. C. Hayward, ACS Nano, 2016, 10, 8610–8619 CrossRef CAS.
- C. Jarrett-Wilkins, X. He, H. E. Symons, R. L. Harniman, C. F. J. Faul and I. Manners, Chem.–Eur. J., 2018, 24, 15556–15565 CrossRef CAS PubMed.
- H. Langhals and S. Kirner, Eur. J. Org. Chem., 2000, 365–380 CrossRef CAS.
- P. Karuppuswamy, H.-C. Chen, P.-C. Wang, C.-P. Hsu, K.-T. Wong and C.-W. Chu, ChemSusChem, 2018, 11, 415–423 CrossRef CAS.
- L. Li, H.-X. Gong, D.-Y. Chen and M.-J. Lin, Chem.–Eur. J., 2018, 24, 13188–13196 CrossRef CAS PubMed.
- S. Zhang, H. Guo, W. Hou, X. Ji and H. Zhang, Mater. Lett., 2018, 221, 38–41 CrossRef CAS.
- F. Yang, J. Li, C. Li and W. Li, Macromolecules, 2019, 52, 3689–3696 CrossRef CAS.
- H.-C. Chen, C.-P. Hsu, J. N. H. Reek, R. M. Williams and A. M. Brouwer, ChemSusChem, 2015, 8, 3639–3650 CrossRef CAS.
- H. Sakai, K. Ohkubo, S. Fukuzumi and T. Hasobe, Chem.–Asian J., 2016, 11, 613–624 CrossRef CAS PubMed.
- Q. Xie, E. Perez-Cordero and L. Echegoyen, J. Am. Chem. Soc., 1992, 114, 3978–3980 CrossRef CAS.
- Y. Matsuo, T. Ichiki and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 9932–9937 CrossRef CAS PubMed.
- Y. Matsuo, K. Morita and E. Nakamura, Chem.–Asian J., 2008, 3, 1350–1357 CrossRef CAS.
- Y.-W. Zhong, Y. Matsuo and E. Nakamura, J. Am. Chem. Soc., 2007, 129, 3052–3053 CrossRef CAS.
- C. J. Tassone, A. L. Ayzner, R. D. Kennedy, M. Halim, M. So, Y. Rubin, S. H. Tolbert and B. J. Schwartz, J. Phys. Chem. C, 2011, 115, 22563–22571 CrossRef CAS.
- T. Niinomi, Y. Matsuo, M. Hashiguchi, Y. Sato and E. Nakamura, J. Mater. Chem., 2009, 19, 5804–5811 RSC.
- Y. Matsuo, A. Muramatsu, Y. Kamikawa, T. Kato and E. Nakamura, J. Am. Chem. Soc., 2006, 128, 9586–9587 CrossRef CAS.
- A. Busseau, C. Villegas, S. Dabos-Seignon, C. Cabanetos, P. Hudhomme and S. Legoupy, Chem.–Eur. J., 2016, 22, 8452–8456 CrossRef CAS.
- A. Busseau, C. Villegas, S. Dabos-Seignon, P. Hudhomme and S. Legoupy, Eur. J. Org. Chem., 2018, 4860–4866 CrossRef CAS.
- E. Haldón, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC.
- L. C. K. Viswanath, J. Bernhardt, K. Kumar Gnanasekaran, C. Want, J. Frank, J. Faulkner and S. Krishnan, Dyes Pigm., 2016, 134, 453–458 CrossRef CAS.
- A. S. Thompson, G. R. Humphrey, A. M. DeMarco, D. J. Mathre and E. J. J. Grabowski, J. Org. Chem., 1993, 58, 5886–5888 CrossRef CAS.
- Y.-W. Zhong, Y. Matsuo and E. Nakamura, Org. Lett., 2006, 8, 1463–1466 CrossRef CAS.
- J. Iehl and J.-F. Nierengarten, Chem.–Eur. J., 2009, 15, 7306–7309 CrossRef CAS.
- Y. W. Zhong, Y. Matsuo and E. Nakamura, Org. Lett., 2006, 8, 1463–1466 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00287b |
| This journal is © The Royal Society of Chemistry 2021 |