
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free synthesis of 1,4-benzodiazepines and quinazolinones from hexafluoroisopropyl 2-aminobenzoates at room temperature†
Jiewen Chen‡
,
En Liang‡,
Jie Shi*,
Yinrong Wu,
Kangmei Wen,
Xingang Yao and
Xiaodong Tang *
*
Guangdong Provincial Key Laboratory of New Drug Screening, School of Pharmaceutical Sciences, Southern Medical University, 1023 South Shatai Road, Baiyun District, Guangzhou 510515, P. R. China. E-mail: shijie7542@163.com; tangxdong@smu.edu.cn
First published on 28th January 2021
Abstract
Herein, we describe the novel reactivity of hexafluoroisopropyl 2-aminobenzoates. The metal-free synthesis of 1,4-benzodiazepines and quinazolinones from hexafluoroisopropyl 2-aminobenzoates has been developed at room temperature. These procedures feature good functional group tolerance, mild reaction conditions, and excellent yields. The newly formed products can readily be converted to other useful N-heterocycles. Moreover, the products and their derivatives showed potent anticancer activities in vitro by MTT assay.
Benzodiazepines (BDZs), especially 1,4-benzodiazepines, are privileged motifs in pharmaceuticals.1 For examples, oxazepam is used to treat anxiety disorders or alcohol withdrawal symptoms; triazolam is used to treat insomnia (Scheme 1). Until now, some synthetic approaches to 1,4-benzodiazepine skeletons have been developed include isocyanide-based multicomponent reactions,2 cycloadditions,3 metal-catalyzed tandem reactions,4 and redox-neutral [5+2] annulation with o-aminobenzaldehydes.5 However, these procedures have some limitations involving unavailable materials, several steps, harsh reaction conditions and low yields. α-Haloamides are widely used to synthesize N-heterocycles.6 Recently, some groups reported the synthesis of 1,4-benzodiazepines with α-haloamides.7 Kim and coworkers developed a [4+3]-annulation reaction between α-haloamides and isatoic anhydrides for 1,4-benzodiazepines, but the reaction required 80 °C reaction temperature and provided an unsatisfactory yield.7a Singh's group reported a two-step method to construct 1,4-benzodiazepines from α-haloamides and anthranils, but the anthranils are not readily available substrates and the second step also required 80 °C reaction temperature.7b Quinazolinones are a significant class of heterocycles that widely occur in natural products and pharmaceuticals (Scheme 1).8 These compounds exhibit a range of biological activities including anticancer, antibacterial, antiinflammatory, antifungal, etc. Due to their significant value, the synthesis of quinazolinones has attracted considerable attention. The reported synthetic methods can be summarized as: (i) condensation of 2-aminobenzamides with carbonyl compounds;9 (ii) oxidative cyclization of primary alcohols with 2-aminobenzamides or 2-aminobenzonitriles;10 (iii) metal-catalyzed coupling/cyclization reactions;11 and (iv) palladium-catalyzed carbonylation reactions.12 But these synthetic methods also had some disadvantages. Thus, it is highly desirable to develop new available reagents for synthesizing the useful N-heterocycles such as benzodiazepines and quinazolinones with good yields under mild reaction conditions.
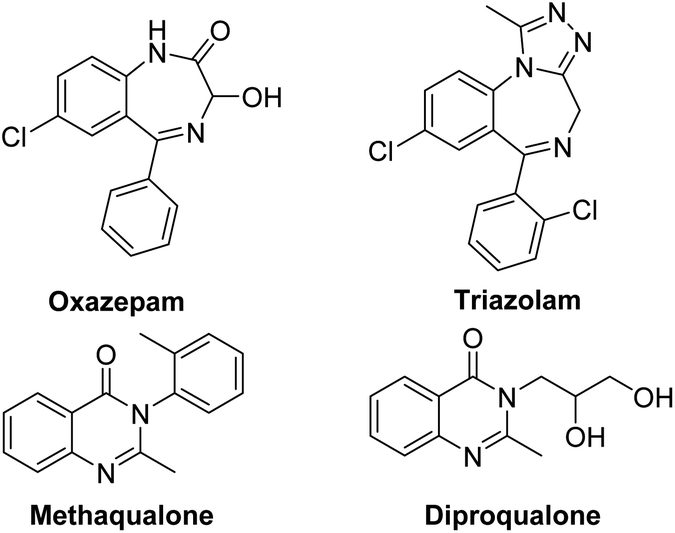 | ||
| Scheme 1 Structures of representative 1,4-benzodiazepine and quinazolinone drugs. | ||
In the past few years, 2-aminobenzoates have been used for the synthesis of N-heterocycles via [4+n] cyclization (Scheme 2a).13 However, harsh reaction conditions such as high reaction temperatures and strong bases or acids were required to effect alkoxy leaving. When we tried to synthesize benzodiazepines or quinazolinones with methyl or tert-butyl 2-aminobenzoates, we failed. Recently, hexafluoroisopropanol (HFIP) has attracted a lot of attention when used as solvent or substrate, due to its special properties.14 When we used isatoic anhydrides as substrates and NEt3 as base in HFIP at room temperature, we unexpectedly discovered that hexafluoroisopropyl 2-aminobenzoates were completely formed. We supposed hexafluoroisopropyl 2-aminobenzoates were good synthons for the synthesis of N-heterocycles. Herein, we report metal-free procedures for the synthesis of 1,4-benzodiazepines and quinazolinones from hexafluoroisopropyl 2-aminobenzoates at room temperature with excellent yields (Scheme 2b).
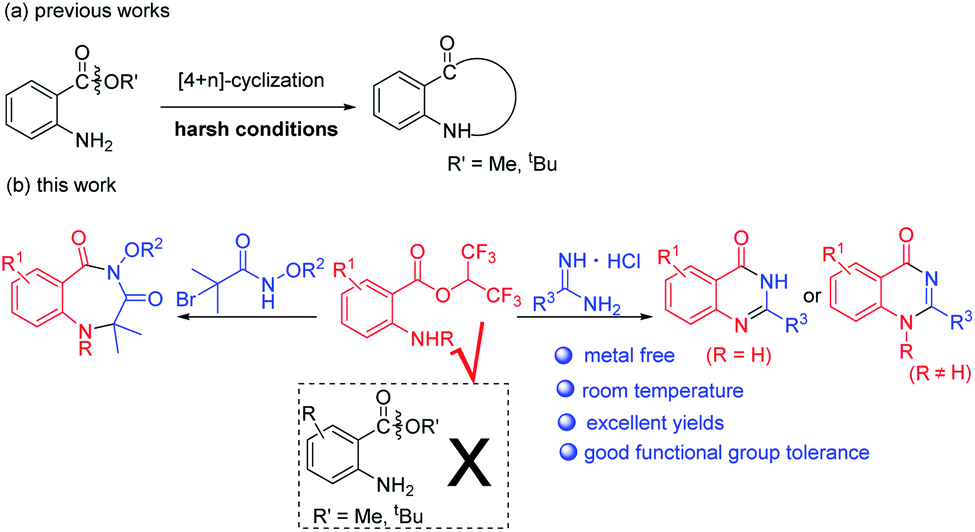 | ||
| Scheme 2 Synthesis of N-heterocycles from 2-aminobenzoates. | ||
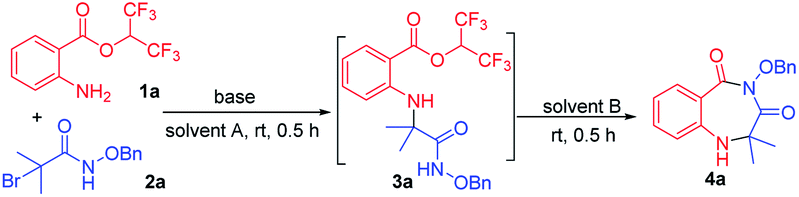
We examined the annulation reaction with hexafluoroisopropyl 2-aminobenzoate (1a) and α-bromoamide (2a) as the model substrates. Initially, when the reaction was performed with 1 equiv. of Et3N in HFIP at room temperature for 0.5 h, 3a was formed, but cyclization product 4a was not obtained. We thought the transformation from 3a to the product 4a needing to release one molecule of HFIP, and the transformation maybe be inhibited when HFIP was used as solvent. So we removed the solvent HFIP under vacuum and added 2.0 mL DMF to react for 0.5 h. Pleasingly, the desired product 4a was obtained in 68% yield (Table 1, entry 1). Then, a series of bases were checked, and Cs2CO3 seemed to be the best choice (Table 1, entries 2–7). When using NaHCO3 and K2CO3 as bases, 3a was obtained, but it cannot be converted to the product 4a. The reaction cannot take place without base (Table 1, entry 8). When we replaced the HFIP with another solvent (DMSO, DMA, MeCN, toluene), the reaction cannot occur (Table 1, entries 9–12). The transformation from 3a to product 4a with different solvents was also investigated; the results showed that other solvents, such as DMA, DMSO, MeCN and dioxane, NMP, toluene, were less effective (Table 1, entries 13–16) or ineffective (Table 1, entries 17 and 18). A gram-scale reaction was performed to give product 4a in 92% yield (Table 1, entry 19). We also tried to use a mixture of HFIP and other solvent in order to directly form the desired cyclic compound in a one-pot manner, but the yields was low.
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent A | Solvent B | Yield (%) |
| a Reaction conditions: unless otherwise noted, all reactions were performed with 1a (0.3 mmol), 2a (0.3 mmol), and base (0.3 mmol) in solvent A (3.0 mL) at room temperature for 0.5 h, then the solvent A was removed under vacuum and solvent B (2.0 mL) added to react for 0.5 h. Isolated yield.b Yield on a 3.0 mmol scale. | ||||
| 1 | Et3N | HFIP | DMF | 68 |
| 2 | Cs2CO3 | HFIP | DMF | 97 |
| 3 | NaHCO3 | HFIP | DMF | n.d. |
| 4 | K2CO3 | HFIP | DMF | n.d. |
| 5 | DBU | HFIP | DMF | 61 |
| 6 | NaOH | HFIP | DMF | 93 |
| 7 | DIPEA | HFIP | DMF | 63 |
| 8 | — | HFIP | DMF | 0 |
| 9 | Cs2CO3 | DMSO | — | 0 |
| 10 | Cs2CO3 | DMA | — | 0 |
| 11 | Cs2CO3 | MeCN | — | 0 |
| 12 | Cs2CO3 | Toluene | — | 0 |
| 13 | Cs2CO3 | HFIP | DMA | 71 |
| 14 | Cs2CO3 | HFIP | DMSO | 41 |
| 15 | Cs2CO3 | HFIP | MeCN | 40 |
| 16 | Cs2CO3 | HFIP | Dioxane | 38 |
| 17 | Cs2CO3 | HFIP | NMP | n.d. |
| 18 | Cs2CO3 | HFIP | Toluene | n.d. |
| 19b | Cs2CO3 | HFIP | DMF | 92 |
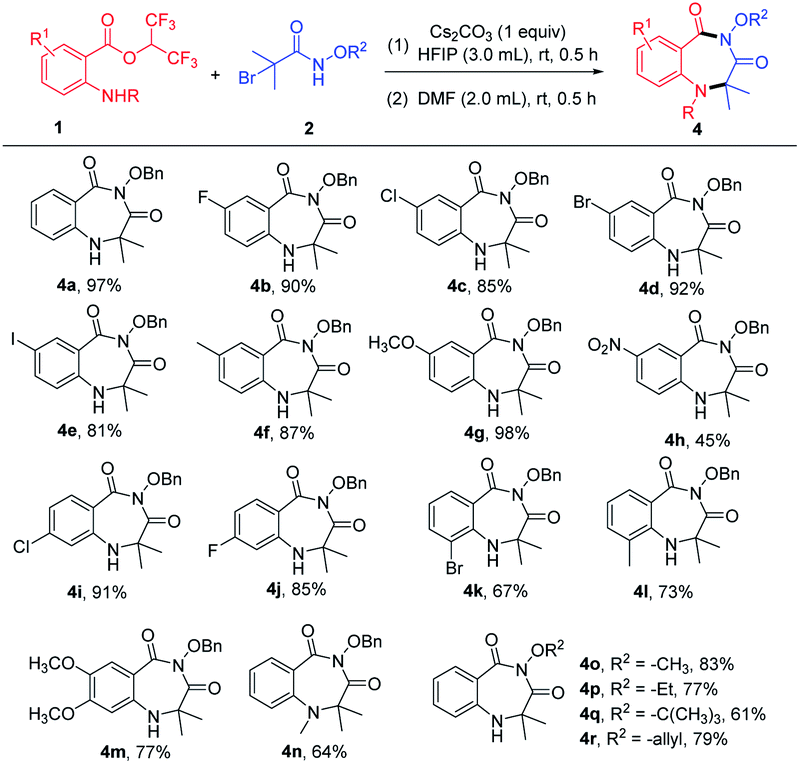
After determining the optimized reaction conditions, the scope of the cyclization reaction for 1,4-benzodiazepines was examined (Table 2). Various 5-substituted 2-aminobenzoates bearing halo groups (F, Cl, Br, I) and electron-donating groups (CH3, OCH3) smoothly underwent cyclization reaction to furnish desired products in good to excellent yields (Table 2, 4b–4g). The nitro group was tolerated in the transformation, but the yield was low (Table 2 and 4h). The reactions also proceeded in the case of 4-substituted 2-aminobenzoates with high yields (Table 2, 4i–4j). When 3-substituted 2-aminobenzoates were employed as substrates, the yields were relatively low because the steric hindrance was unfavorable in intramolecular nucleophilic attack (Table 2, 4k–4l). Furthermore, 4,5-dimethoxy 2-aminobenzoate and 2-(methylamino)benzoate afforded the expected products 4m and 4n in 77% and 64% yields, respectively. In addition, α-bromoamides with diverse N-protecting groups (–OCH3, –OEt, –OtBu, –allyloxy) showed good compatibility, delivering the corresponding products in 61–83% yields (Table 2, 4o–4r). Unfortunately, mono-substituted α-bromohydroxamates, unsubstituted α-bromohydroxamates, and N-alkylated bromoacetamides did not react under the current reaction conditions.
| a Reaction conditions: 1 (0.3 mmol), 2 (0.3 mmol), and Cs2CO3 (0.3 mmol) in solvent HFIP (3.0 mL) at room temperature for 0.5 h, then the HFIP was removed under vacuum and added DMF (2.0 mL) to continue to react for 0.5 h. Isolated yield. |
|---|
 |
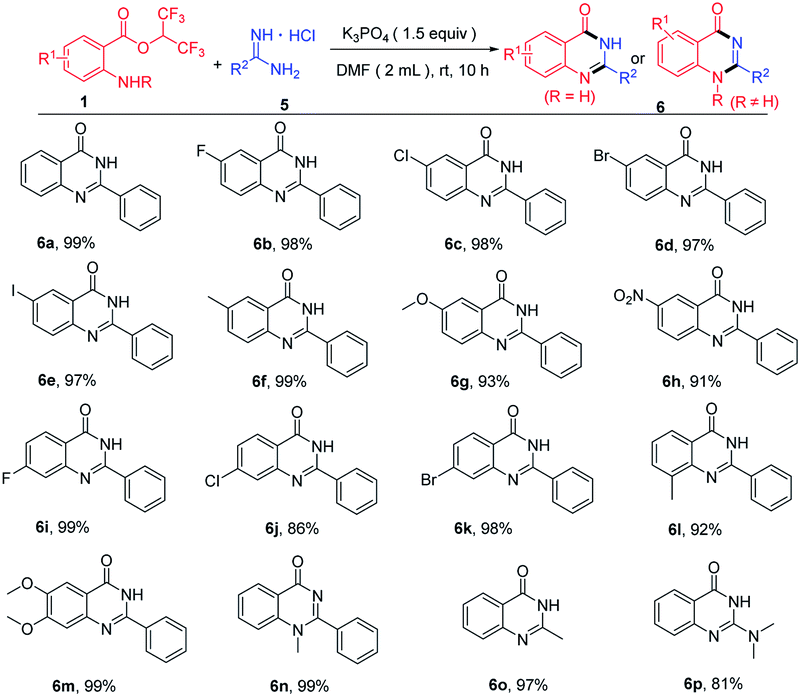
When hexafluoroisopropyl 2-aminobenzoates reacted with amidines hydrochloride in the presence of base, quinazolinones were produced. Then, we optimized the reaction conditions to enhance the yields of the quinazolinones (see the ESI† for more details). With optimum conditions in hand, substrate scope for the synthesis of quinazolinones was next investigated (Table 3). Hexafluoroisopropyl 2-aminobenzoates bearing diverse groups at the amino para-position, including F, Cl, Br, I, CH3, OCH3 and NO2 were all compatible with this procedure to afford the cyclization products in excellent yields (Table 3, 6b–6h). In addition, various 4-substituted, 3-substituted and 4,5-disubstituted 2-aminobenzoates reacted well with benzamidine hydrochloride, and the corresponding product yields ranged 86% to 99% (Table 3, 6i–6m). Notably, methyl protected 2-aminobenzoates were also transformed to the product 6n in 99% yield. To our delight, this protocol was also applied to acetamidine hydrochloride and 1,1-dimethylguanidine hydrochloride affording the target products in 97% and 81%, respectively (Table 3, 6o–6p).
| a Reaction conditions: 1 (0.3 mmol), 5 (0.36 mmol), K3PO4 (0.45 mmol), DMF (2.0 mL) at room temperature for 10 h. Isolated yields. |
|---|
 |
To probe the reaction mechanism, several preliminary experiments were conducted (Scheme 3). Under standard reaction conditions, methyl 2-aminobenzoates 7 reacted with 2a to provide compound 8 in 92% yield, and 4a was not detected. This control experiment indicated the importance of hexafluoroisopropyl (Scheme 3, eqn (1)). Treatment of methyl 2-aminobenzoates 7 with 5a in the presence of K3PO4 did not furnish any product 6a, and 3a did not convert at all (Scheme 3, eqn (2)). On the basis of the control experiments and previous reports, we propose a possible mechanism. First, aza-oxyallyl cation A is formed from α-bromoamide with Cs2CO3.15 Whereafter, aza-oxyallyl cation A combines with 1a to produce compound 3a.15b The product 4a is obtained via intramolecular nucleophilic substitution, releasing a molecule of hexafluoroisopropanol. The nucleophilic attack of 5a onto 1a provides the intermediate B. Subsequently, product 6a is formed by intramolecular nucleophilic addition/deamination cyclization.
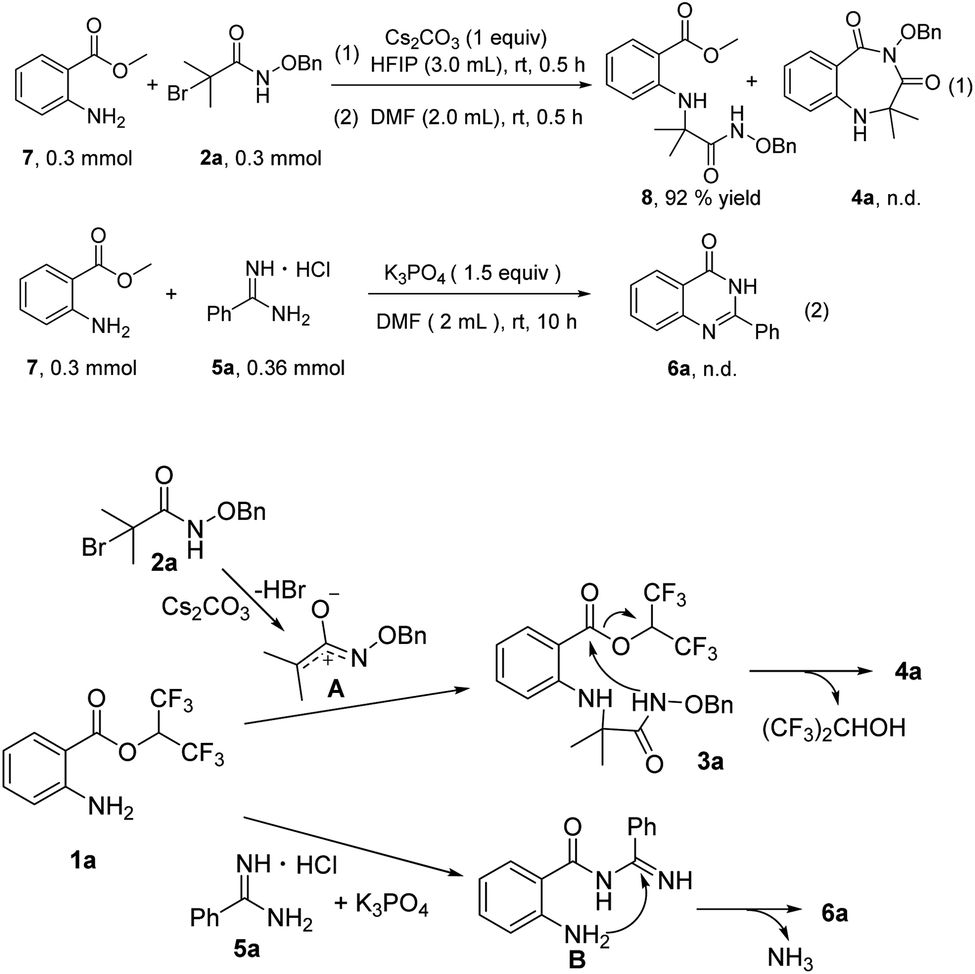 | ||
| Scheme 3 Control experiments and possible reaction mechanism. | ||
In order to address the potential synthetic application of our methods, the transformations of the obtained 1,4-benzodiazepines and quinazolinones were performed (Scheme 4). Compound 9 was formed from 4a through cleavage of the N–O bond with Mo(CO)6 (Scheme 4, eqn (1)). The quinazolinones can be transformed into substituted quinazolines with anilines or phenols as nucleophilic reagents in the presence of BOP and DBU (Scheme 4, eqn (2) and (3)).
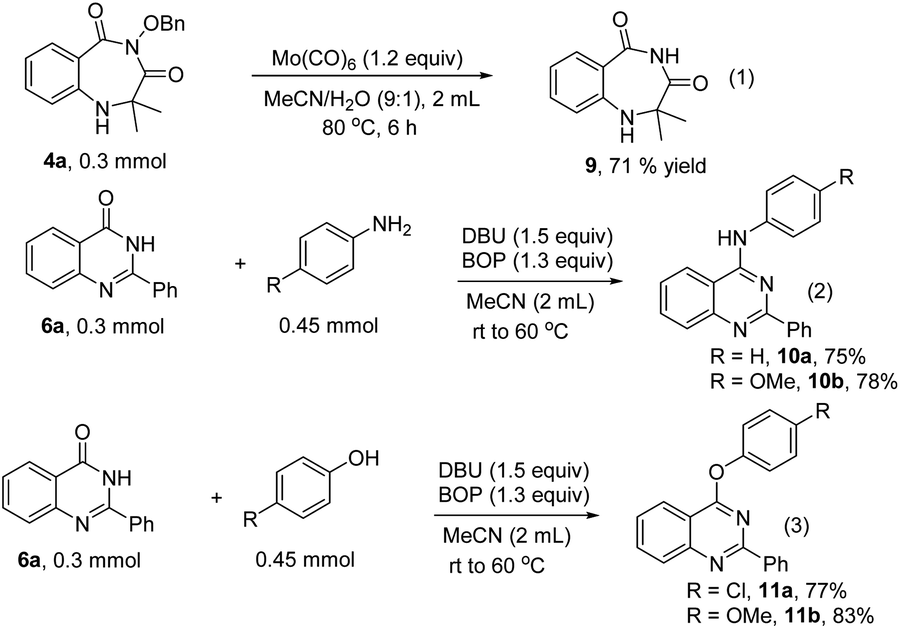 | ||
| Scheme 4 Derivatization of products. | ||
We next investigated the cytotoxicity of the products and their derivatives against cancer cell lines (A549, HCT116 and MCF7) by MTT assay, with 5-fluorouracil (5-FU) as the positive control. To our delight, some products and their derivatives exhibited potent inhibitory activities, and some of them showed better inhibitory activities than 5-Fu (Table 4). These results revealed that our methods had potential applications in discovering new lead compounds with anti-tumor activities.
| Compounds | IC50 (μM) | ||
|---|---|---|---|
| A549 | HCT116 | MCF7 | |
| 4e | 64.69 ± 7.35 | 33.27 ± 5.84 | 40.32 ± 0.49 |
| 6c | 35.11 ± 3.40 | 26.61 ± 1.26 | 58.12 ± 3.45 |
| 6d | 52.32 ± 2.85 | 23.58 ± 1.50 | 81.32 ± 2.80 |
| 6f | 82.89 ± 10.34 | 59.59 ± 1.60 | 38.52 ± 1.83 |
| 6g | 67.00 ± 8.24 | 32.90 ± 0.60 | 42.54 ± 3.79 |
| 6l | 19.56 ± 1.16 | 17.73 ± 2.32 | 25.00 ± 5.30 |
| 10a | 14.79 ± 1.15 | 26.31 ± 3.95 | 29.70 ± 0.09 |
| 10b | 5.98 ± 0.42 | 15.41 ± 4.41 | 21.12 ± 1.06 |
| 11a | 68.54 ± 3.70 | 17.84 ± 3.13 | 75.84 ± 2.50 |
| 11b | 94.76 ± 1.14 | 25.14 ± 5.31 | 67.13 ± 3.65 |
| 5-Fu | >100 | 13.03 ± 2.80 | 29.58 ± 12.86 |
In summary, we have developed novel and simple approaches for the synthesis of 1,4-benzodiazepines and quinazolinones from hexafluoroisopropyl 2-aminobenzoates with α-bromoamides or amidines hydrochloride. These protocols feature readily available starting materials, mild reaction conditions, good functional group tolerance, and excellent yields. In addition, the newly obtained products and their derivatives showed potent anticancer activities in vitro by MTT assay. Further studies on the synthesis of other N-heterocycles from hexafluoroisopropyl 2-aminobenzoates are in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21702096), and the High-level Talent Introduction Foundation of Southern Medical University (C1033520) for financial support.Notes and references
- (a) A. C. Araújo, A. P. Rauter, F. Nicotra, C. Airoldi, Ba. Costa and L. Cipolla, J. Med. Chem., 2011, 54, 1266 CrossRef PubMed; (b) A. K. Shah, J. Bariwal, S. Bansal, J. Chugh and J. B. Bariwal, Chem. Biol. Interface, 2012, 2, 12 CAS; (c) R. Siegrist, D. Pozzi, G. Jacob, C. Torrisi, K. Colas, B. Braibant, J. Mawet, T. Pfeifer, R. Kanter, C. Roch, M. Kessler, O. Corminboeuf and O. Bezencon, J. Med. Chem., 2016, 59, 10661 CrossRef CAS PubMed; (d) M. M. Poe, K. R. Methuku, G. Li, A. R. Verma, K. A. Teske, D. C. Stafford, L. A. Arnold, J. W. Cramer, T. M. Jones, R. Cerne, M. J. Krambis, J. M. Witkin, E. Jambrina, S. Rehman, M. Ernst, J. M. Cook and J. M. Schkeryantz, J. Med. Chem., 2016, 59, 10800 CrossRef CAS PubMed; (e) R. Ettari, S. Previti, S. Cosconati, S. Maiorana, T. Schirmeister, S. Grasso and M. Zappalà, Bioorg. Med. Chem. Lett., 2016, 26, 3453 CrossRef CAS PubMed.
- (a) R. S. Borisov, A. I. Polyakov, L. A. Medvedeva, V. N. Khrustalev, N. I. Guranova and L. G. Voskressensky, Org. Lett., 2010, 12, 3894 CrossRef CAS PubMed; (b) Y. Wang, M. Chen and M.-W. Ding, Tetrahedron, 2013, 69, 9056 CrossRef CAS; (c) H. Xie, J.-C. Liu and M.-W. Ding, Synthesis, 2016, 48, 4541 CrossRef CAS; (d) Y. Huang, K. Khoury, T. Chanas and A. Dömling, Org. Lett., 2012, 14, 5916 CrossRef CAS PubMed.
- (a) K. G. Guggenheim, H. Toru and M. J. Kurth, Org. Lett., 2012, 14, 3732 CrossRef CAS PubMed; (b) C. S. Chambers, N. Patel and K. Hemming, Tetrahedron Lett., 2010, 51, 4859 CrossRef CAS; (c) K. C. Majumdar and S. Ganai, Tetrahedron Lett., 2013, 54, 6192 CrossRef CAS; (d) J. Shin, J. Lee, D. Ko, N. De and E. J. Yoo, Org. Lett., 2017, 19, 2901 CrossRef CAS; (e) J. Feng, M. Zhou, X. Lin, A. Lu, X. Zhang and M. Zhao, Org. Lett., 2019, 21, 6245 CrossRef CAS.
- (a) P. Kundu, A. Mondal, B. Das and C. Chowdhury, Adv. Synth. Catal., 2015, 357, 3737 CrossRef CAS; (b) V. Murugesh, B. Harish, M. Adiseshu, J. B. Nanubolu and S. Suresh, Adv. Synth. Catal., 2016, 358, 1309 CrossRef CAS; (c) J. D. Neukom, A. S. Aquino and J. P. Wolfe, Org. Lett., 2011, 13, 2196 CrossRef CAS PubMed; (d) L. P. Tardibono and J. M. J. Miller, Org. Lett., 2009, 11, 1575 CrossRef CAS PubMed.
- (a) S. Wang, Y.-B. Shen, L.-F. Li, B. Qiu, L. Yu, Q. Liu and J. Xiao, Org. Lett., 2019, 21, 8904 CrossRef CAS PubMed; (b) S. Liu, T. Zhao, J. Qu and B. Wang, Adv. Synth. Catal., 2018, 360, 4094 CrossRef CAS.
- (a) A. Fantinati, V. Zanirato, P. Marchetti and C. Trapella, ChemistryOpen, 2020, 9, 100 CrossRef CAS PubMed; (b) A. E. Bouakher, A. Martel and S. Comesse, Org. Biomol. Chem., 2019, 17, 8467 RSC; (c) J. Xuan, X. Cao and X. Cheng, Chem. Commun., 2018, 54, 5154 RSC.
- (a) E. Kim, C. Y. Lee and S.-G. Kim, Adv. Synth. Catal., 2020, 362, 3594 CrossRef CAS; (b) A. J. Ansari, A. Yadav, A. Mukherjee, E. Sathish, K. Nagesh and R. Singh, Chem. Commun., 2020, 56, 4804 RSC.
- (a) H.-P. Buchstaller, U. Anlauf, D. Dorsch, D. Kuhn, M. Lehmann, B. Leuthner, D. Musil, D. Radtki, C. Ritzert, F. Rohdich, R. Schneider and C. Esdar, J. Med. Chem., 2019, 62, 7897 CrossRef CAS PubMed; (b) S. Gatadi, T. V. Lakshmi and S. Nanduri, Eur. J. Med. Chem., 2019, 170, 157 CrossRef CAS PubMed; (c) E. Pitta, O. Balabon, M. K. Rogacki, J. Gómez, F. Cunningham, J. Joosens, K. Augustyns, P. Veken and R. Bates, Eur. J. Med. Chem., 2017, 125, 890 CrossRef CAS PubMed; (d) C.-W. Yu, P.-T. Chang, L.-W. Hsin and J.-W. Chern, J. Med. Chem., 2013, 56, 6775 CrossRef CAS PubMed; (e) F. Rörsch, E. Buscató, K. Deckmann, G. Schneider, M. Schubert-Zsilavecz, G. Geisslinger, E. Proschak and S. Grösch, J. Med. Chem., 2012, 55, 3792 CrossRef PubMed.
- (a) F. Li, L. Lua and J. Ma, Org. Chem. Front., 2015, 2, 1589 RSC; (b) L. Cao, H. Huo, H. Zeng, Y. Yu, D. Lu and Y. Gong, Adv. Synth. Catal., 2018, 360, 4764 CrossRef CAS; (c) S. Mohammed, R. A. Vishwakarma and S. B. Bharate, J. Org. Chem., 2015, 80, 6915 CrossRef CAS PubMed.
- (a) J. Zhou and J. Fang, J. Org. Chem., 2011, 76, 7730 CrossRef CAS PubMed; (b) Z. Zhang, M. Wang, C. Zhang, Z. Zhang, J. Lu and F. Wang, Chem. Commun., 2015, 51, 9205 RSC; (c) A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, Org. Biomol. Chem., 2012, 10, 240 RSC; (d) K. Upadhyaya, R. K. Thakur, S. K. Shukla and R. P. Tripathi, J. Org. Chem., 2016, 81, 5046 CrossRef CAS PubMed; (e) S. Parua, S. Das, R. Sikari, S. Sinha and N. D. Paul, J. Org. Chem., 2017, 82, 7165 CrossRef CAS PubMed; (f) S. Das, S. Sinha, D. Samanta, R. Mondal, G. Chakraborty, P. Brandaõ and N. D. Paul, J. Org. Chem., 2019, 84, 10160 CrossRef CAS PubMed; (g) Y. Hu, S. Li, H. Li, Y. Li, J. Li, C. Duanmu and B. Li, Org. Chem. Front., 2019, 6, 2744 RSC; (h) H. Hikawa, Y. Ino, H. Suzuki and Y. Yokoyama, J. Org. Chem., 2012, 77, 7046 CrossRef CAS PubMed.
- (a) D. Yang, H. Fu, L. Hu, Y. Jiang and Y. Zhao, J. Comb. Chem., 2009, 11, 653 CrossRef CAS PubMed; (b) W. Xu and H. Fu, J. Org. Chem., 2011, 76, 3846 CrossRef CAS PubMed; (c) X. Zhang, D. Ye, H. Sun, D. Guo, J. Wang, H. Huang, X. Zhang, H. Jianga and H. Liu, Green Chem., 2009, 11, 1881 RSC.
- (a) B. Ma, Y. Wang, J. Peng and Q. Zhu, J. Org. Chem., 2011, 76, 6362 CrossRef CAS PubMed; (b) X.-F. Wu, L. He, H. Neumann and M. Beller, Chem.–Eur. J., 2013, 19, 12635 CrossRef CAS PubMed; (c) H. Li, L. He, H. Neumann, M. Beller and X.-F. Wu, Green Chem., 2014, 16, 1336 RSC; (d) J. Chen, K. Natte, H. Neumann and X.-F. Wu, Chem.–Eur. J., 2014, 20, 16107 CrossRef CAS PubMed.
- (a) J. Lei, G.-T. Song, Y.-F. Luo, D.-Y. Tang, W. Yan, H.-Y. Li, Z.-Z. Chen and Z.-G. Xu, Org. Chem. Front., 2020, 7, 737 RSC; (b) R. V. Gadhave and B. S. Kuchekar, Asian J. Chem., 2020, 32, 580 CAS; (c) A. V. Bogolubsky, S. V. Ryabukhin, A. S. Plaskon, S. V. Stetsenko, D. M. Volochnyuk and A. A. Tolmachev, J. Comb. Chem., 2008, 10, 858 CrossRef CAS PubMed; (d) Y. Chi, L. Xu, S. Du, H. Yan, W.-X. Zhang and Z. Xi, Chem.–Eur. J., 2015, 21, 10369 CrossRef CAS PubMed; (e) S. Kang, S. Park, K.-S. Kim, C. Song and Y. Lee, J. Org. Chem., 2018, 83, 2694 CrossRef CAS PubMed; (f) C. Lu, C. Gong, B. Zhao, L. Hu and Y. Yao, J. Org. Chem., 2018, 83, 1154 CrossRef CAS PubMed; (g) S. R. Narra, S. Avula, R. R. Kuchukulla, J. B. Nanubolu, N. Banda and R. Yadla, Tetrahedron, 2017, 73, 4730 CrossRef CAS.
- (a) W. Wang, X. Cao, W. Xiao, X. Shi, X. Zuo, L. Liu, W. Chang and J. Li, J. Org. Chem., 2020, 85, 7045 CrossRef CAS PubMed; (b) J. Chen, M. Li, J. Zhang, W. Sun and Y. Jiang, Org. Lett., 2020, 22, 3033 CrossRef CAS PubMed; (c) M. A. Boichenko, I. A. Andreev, A. O. Chagarovskiy, I. I. Levina, S. S. Zhokhov, I. V. Trushkov and O. A. Ivanova, J. Org. Chem., 2020, 85, 1146 CrossRef CAS PubMed; (d) A. Roth and S. E. Denmark, J. Am. Chem. Soc., 2019, 141, 13767 CrossRef CAS PubMed; (e) Z.-K. Wen, X.-M. Ge, Z.-K. Zhao and J.-B. Chao, Adv. Synth. Catal., 2019, 361, 983 CAS.
- (a) A. Acharya, K. Montes and C. S. Jeffrey, Org. Lett., 2016, 18, 6082 CrossRef CAS PubMed; (b) T. Bera, B. Singh, T. A. Hamlin, S. C. Sahoo and J. Saha, J. Org. Chem., 2019, 84, 15255 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, characterization of all compounds, copies of 1H and 13C NMR spectra for all target compounds. See DOI: 10.1039/d1ra00324k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |