
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanical behaviors and probabilistic multiphase network model of polyvinyl alcohol hydrogel after being immersed in sodium hydroxide solution
Zeyu Zuo†
a,
Yongrou Zhang†b,
Licheng Zhou*a,
Zejia Liua,
Zhenyu Jianga,
Yiping Liua and
Liqun Tang *ac
*ac
aSchool of Civil Engineering and Transportation, South China University of Technology, No. 381, Wushan Road, Guangzhou, Guangdong, China. E-mail: ctlczhou@scut.edu.cn; lqtang@scut.edu.cn
bInstitute of Intelligent Manufacturing, Guangdong Academy of Sciences, No. 100, Xianliezhong Road, Guangzhou, China
cState Key Laboratory of Subtropical Building Science, South China University of Technology, No. 381, Wushan Road, Guangzhou, Guangdong, China
First published on 19th March 2021
Abstract
Because of the advantages of a uniform distribution of reinforcing particles and in situ preparation, in situ precipitation has become an important way to prepare magnetic and other smart hydrogels. An important step in this process is to immerse hydrogels in alkaline solution to implant magnetic particles. Previous studies generally have ignored the effect of this process on the network structure and mechanical properties of hydrogels. In this study, we immersed polyvinyl alcohol (PVA) hydrogel samples in sodium hydroxide solutions of different concentrations to study changes in mechanical properties, such as stress–strain relationship, self-recovery, and fracture failure. The results showed that after the immersion process, the hydrogel's tensile and compressive properties changed significantly, and the failure behavior changed from brittle fracture to ductile fracture. Through a microscopic mechanism, the alkaline solution caused a high degree of phase separation and crystallization within the polymer network, thereby changing the PVA hydrogel network from a single phase to a multiphase. Hence, we used a continuous multiphase network model with a certain probability distribution to describe this tensile behavior. This model well described the stress–strain relationship of the hydrogel from stretching to fracture and revealed that the macroscopic failure corresponded to the peak of fracture distribution. Studies have shown that attention should be paid to the influence of the in situ precipitation on the mechanical properties, and the probabilistic multiphase network model can be used to predict the mechanical behavior of hydrogels with multiple phase separation.
Introduction
Hydrogels consist of a three-dimensional network of hydrophilic polymer chains. The water content and hardness of some hydrogels are similar to that of soft tissues in the human body;1 hence, hydrogels have wide application prospects in the biomechanical field. As a synthetic material with better mechanical properties,2 physical crosslinked hydrogels often have better biocompatibility than chemical crosslinked hydrogels,3 thus becoming the focus of biomechanical research.Physically crosslinked polyvinyl alcohol (PVA) hydrogels have attracted wide attention because of their good physical properties and biocompatibility.4,5 Peppas and Merrill first reported the physical crosslinked PVA hydrogel preparation in 1976.6 The PVA solution was frozen and thawed to prepare hydrogels with a stable structure, high water content, and good biocompatibility. Some researchers have reported that physical PVA hydrogel was used as a base material to obtain intelligent PVA hydrogels with both biocompatibility and environmental sensitivity. Among these hydrogels, magnetic PVA hydrogel has great values. It has the ability to respond to the external magnetic field remotely, like other magnetically controlled nanocarriers, and has been shown to be an effective drug delivery system. Use of the thermal and magnetic response of the hydrogel under alternating magnetic field to control drug release can greatly overcome the lack of spatial and temporal control existing in conventional methods.7,8 Researchers have demonstrated that magnetic PVA hydrogels have good responses to an external magnetic field and have investigated applying the material in biomechanics, including the use of magnetic hydrogels to move drugs to the target area or regulate the release rate of the drugs by adjusting the external magnetic field.8,9
The preparation of traditional magnetic PVA hydrogel is to mix PVA solution and magnetic particles directly (i.e., γ-Fe2O3/Fe3O4). This process is simple and effective, but it has the shortcoming of uneven particle distribution: because the polymer network in the early freezing stage was not completely formed, magnetic particles could not be fixed in the fiber network; thus, they would sink to the bottom under gravity. Therefore, its magnetism is obviously uneven along the vertical direction. In addition, the mechanical properties and stability of magnetic PVA hydrogel are restricted by the base material, which limits its applications in some aspects. For example, when magnetic PVA hydrogel was used in thermal anticancer therapy,10,11 the hydrogen bonds of physical crosslinked PVA hydrogels would break at a high temperature, which led to the collapse of the polymer network.12 As a result, the hydrogels not only could not work properly, but also may have caused problems such as magnetic particle exudation and biological toxicity.13 If a PVA hydrogel was used for bearing or driving, it would face problems of insufficient hardness (0.1–0.2 MPa) or toughness (102 to 103 J m−2).14 Researchers have proposed some methods, such as dry-anneal,15 cast-dry16 or cyclic pre-stretching.17 Although the mechanical properties of the material were improved, it took a longer preparation cycle and introduced more complex procedures. The preparation of magnetic hydrogels by an in situ precipitation method could address these problems. To obtain the magnetic PVA hydrogel through in situ precipitation, the PVA hydrogel was immersed in an Fe2+/Fe3+ solution and sodium hydroxide (NaOH) aqueous solution successively to produce nano Fe3O4 particles (Fig. 1(a) and (b)). This method has several advantages: first, relying on the intact three-dimensional network structure, the magnetic particles produced are evenly distributed. Second, the mechanical properties of PVA hydrogels were obviously improved. By comparing the compression curves of hydrogel immersed in NaOH aqueous solution, magnetic hydrogel, and pristine hydrogel, we found that its immersing process affected the properties of hydrogel (Fig. 1(c)). This effect was often neglected in the studies of magnetic hydrogel. We found that the improvement of mechanical properties was mainly due to the process of PVA hydrogel immersing in an alkaline solution, and the degree of enhancement was directly related to the concentration.
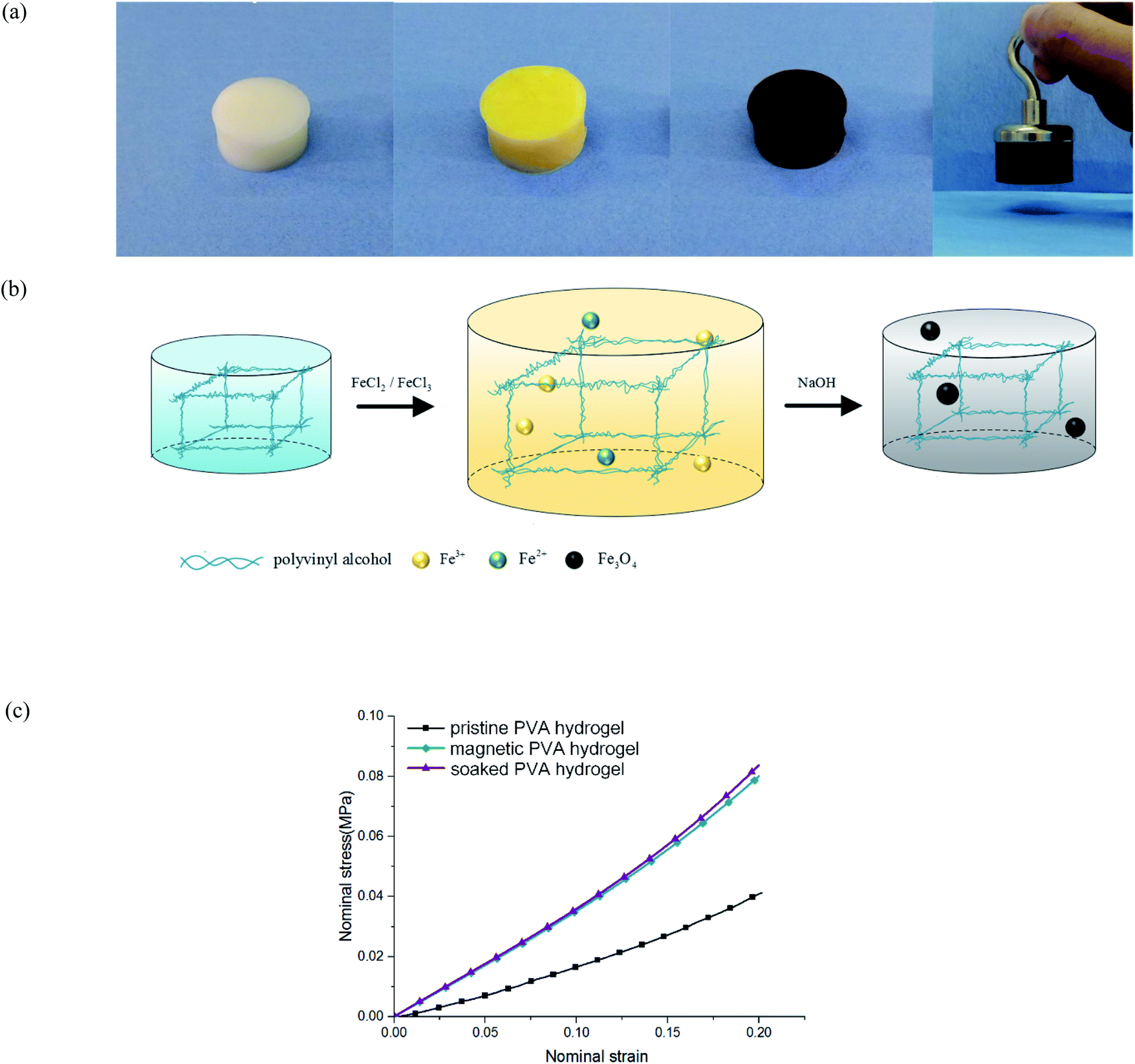 | ||
| Fig. 1 Magnetic PVA hydrogel: (a) magnetic PVA hydrogel prepared by in situ precipitation method, hydrogels exhibited good magnetic properties; (b) schematic of the in situ precipitation preparation of magnetic hydrogels; and (c) stress curves of different hydrogels under uniaxial compression. | ||
The modification of hydrogels after being immersed in solution has attracted much attention in the past few years. Studies have shown that hydrogels may produce salting-out under immersion and may affect the macroscopic properties of materials.18 When polyacrylamide hydrogel was immersed in a high concentration of dimethylamide solution, it lost water and shrank, the hardness was increased, and the transparency was reduced.19 Poly-dimethylpropionamide (PDMA) hydrogel had similar behavior in NaOH aqueous solution, which could be used to prepare a high-toughness magnetic PDMA hydrogel.20 Polyvinyl alcohol/sodium alginate (PVA/SA) composite hydrogel had better mechanical properties and higher conductivity after being immersed in saturated sodium chloride (NaCl) solution.21 Darabi et al. discussed the reaction between PVA and NaOH solution, and revealed that the acid–base reaction was the primary reason that led to the hydrogen bonding and crystallite formation inside the polymer. Therefore, the physical and chemical properties of the material would be changed obviously.22 Sato et al. thought that when the hydrogel was immersed in good solution, it would absorb water and swell, and that in poor solution, it would lose water. They found, however, that the mechanical properties were enhanced, and this enhancement was attributed to the phase separation of the polymer network.19 Few researchers, however, have established a mechanical model to analyze this mechanism.
In this study, we first demonstrated the existence of an interaction between physical PVA hydrogel and NaOH solution. We then proved that NaOH solution caused this phase separation, which increased the microcrystalline in the polymer network. As a result, the hardness, toughness, and self-recovery of the hydrogel were improved. The relationship between mechanical properties and solution concentration was studied quantitatively. We also demonstrated that the essence of the effect of solution immersion on the mechanical properties was the energy dissipation mechanism. The phase separation significantly affected the mechanical behavior of the material during tensile failure, which resulted in the transformation from fast fracture to ductile fracture. Finally, we proposed a multiphase network model with continuous distribution to describe the tension behavior.
Experimental methods
Hydrogel synthesis
We prepared the PVA hydrogel according to the freeze–thaw method: PVA dry powder and deionized water were mixed with a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, and then placed in a high-temperature environment above 120 °C to obtain a transparent PVA aqueous solution. Then the solution was placed into the vacuum environment. After the air bubbles were removed, the solution was poured into molds. We performed five cycles of freezing and thawing for 12 h per stage (freezing at −20 °C and thawing at 25 °C). Finally, we obtained PVA hydrogels with 20% fiber content.
:4, and then placed in a high-temperature environment above 120 °C to obtain a transparent PVA aqueous solution. Then the solution was placed into the vacuum environment. After the air bubbles were removed, the solution was poured into molds. We performed five cycles of freezing and thawing for 12 h per stage (freezing at −20 °C and thawing at 25 °C). Finally, we obtained PVA hydrogels with 20% fiber content.
To obtain the magnetic PVA hydrogel, the hydrogel was immersed in the mixture solution of FeCl2 and FeCl3 at a concentration ratio of 1:2. After swelling to equilibrium, the opaque hydrogel turned orange. Finally, the Fe2+/Fe3+ loaded hydrogel was transferred to the NaOH aqueous solution to generate magnetic particles into the polymer network. Then, the following reaction occurred: 2Fe3+ + Fe2+ + 8OH− → Fe3O4(↓) + 4H2O.
To obtain the phase-separated PVA hydrogel with high mechanical property, we immersed the pristine PVA hydrogels directly in the NaOH aqueous solution until it reached the equilibrium. Afterward, we immersed the sample in deionized water to remove the excess ions. The volume and transparency of the hydrogel were tuned by changing the concentration of the NaOH solution.
Wide-angle X-ray diffraction measurement
We collected wide-angle X-ray powder diffraction (XRD) profiles at room temperature 25 °C in the degree 2θ range of 10°–50°.Tension test
The intact and pre-notched hydrogel samples of rectangular cross sections were fixed at both ends. We conducted quasi-static tensile tests using an INSTRON5567 testing machine. The strain rate was fixed as 0.01 s−1. For the uniaxial tensile test, we carried out the tensile loading until the material tore. For the cyclic loading experiment, when the tension reached the predetermined length, the machine would unload. The second loading would start when stress was reduced to 0, and the unloading rate was controlled to be the same.Compressive tests
We subjected cylindrical hydrogels to uniaxial compression. The strain rate was controlled at a constant 0.005 s−1 and the maximum compressive strain was 0.25. We recorded the experimental data by the testing machine.Confocal imaging of PVA hydrogels
To visualize the microstructures of the PVA hydrogels, we used a fluorescent dye (i.e., 5-[(4,6-dichlorotriazin-2yl)amino]) fluorescein hydrochloride [5-DTAF]) to label the PVA sides groups. PVA hydrogels were first immersed in a large volume of weak alkaline solution (pH = 9.0) for 12 h to equilibrate the pH within the samples. The 5 mg of 5-DTAF dissolved in 1.0 mL of anhydrous dimethyl sulfoxide was immersed in 100 mL of alkaline solution (pH = 9.0) to form a reactive dye solution. The previous PVA samples were immersed in the dye solution for 12 h at 5 °C in a dark environment to form conjugated fluorochromes. Then the PVA samples were rinsed several times with deionized water to wash away the nonconjugated dyes. After the noted pretreatment, we could use the PVA samples for observation under a confocal microscope.Digital image processing method
To obtain the real stress state of the material at each moment during the loading process, we used a camera to take a full picture of the sample through the entire process. Because the hydrogel is opaque and white, we placed a blackboard behind the sample to increase contrast so that we could use edge detection technology to distinguish the boundary of the sample clearly. By calculating the distance of the edges of each photo, we obtained the width and thickness of the sample at each moment, and furthermore, we obtained the true stress–strain relationship of the material.Results and discussion
Characterization of PVA hydrogel
After freezing and thawing, the physical PVA hydrogel obtained had low transparency. During the freezing process, the water in the solution froze and PVA chains were discharged, forming dense regions with high PVA density and sparse regions with low PVA density (Fig. 2(a)). The hydrogen bonding interaction between the polymer chains in the dense regions drove the formation of the microcrystals. As a result, the hydrogels turned opaque.23 After the PVA hydrogel was immersed in the NaOH solution, the hardness increased, but the swelling ratio and transparency decreased further (Fig. 2(b)).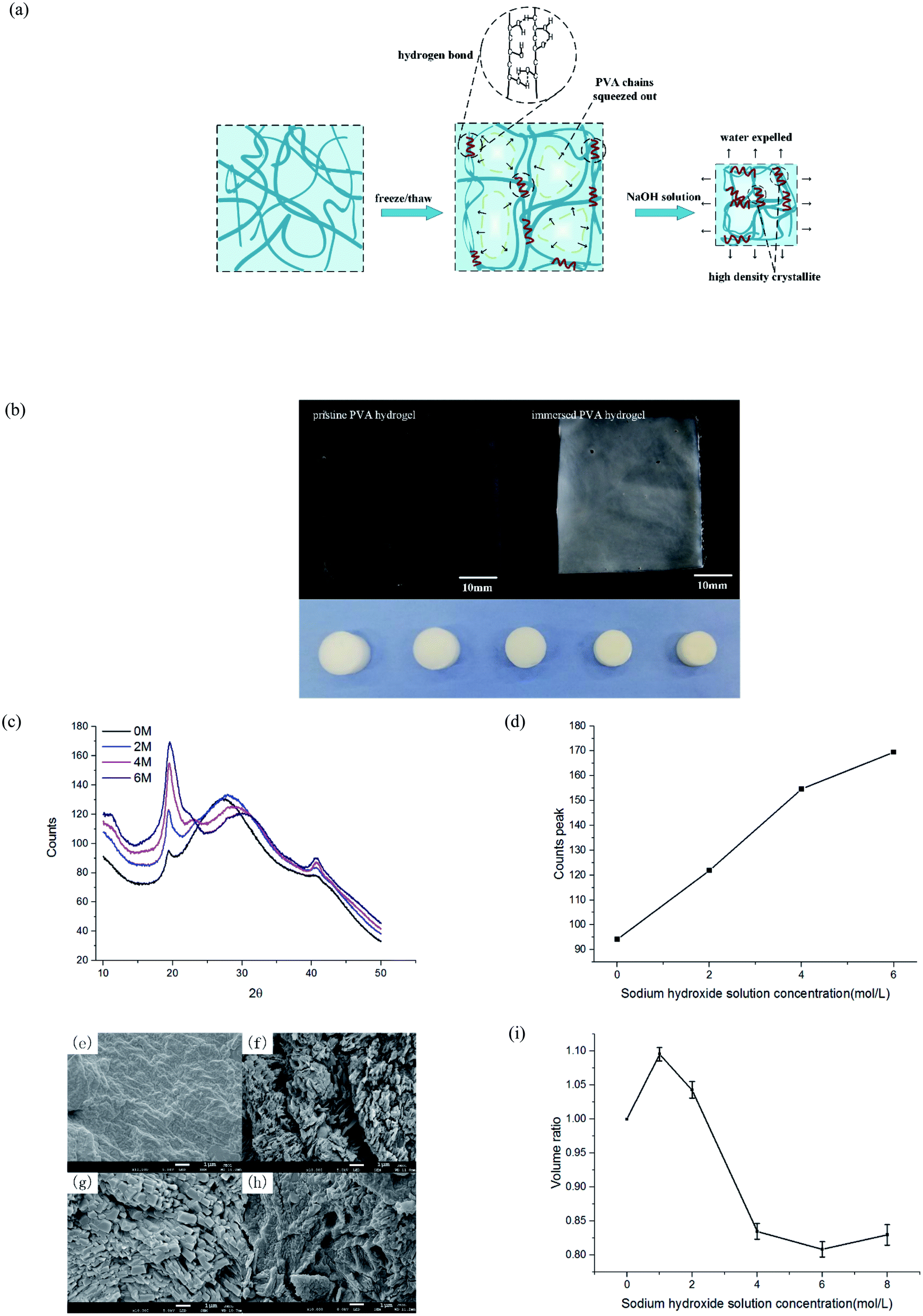 | ||
| Fig. 2 Characterization of PVA hydrogel after immersion. (a) Schematic of phase separation mechanism of freeze–thaw and solution immersion; (b) transparency and volume changes induced by NaOH solution immersion, the photos were taken by a camera; (c) XRD patterns under different concentrations of immersion; (d) statistics of crystallization peaks of XRD patterns at four concentrations; (e)–(h) surface morphology of hydrogels immersed in 0, 2, 4, and 6 M NaOH solution, respectively; and (i) volume ratio of hydrogels immersed in different NaOH concentrations. | ||
To prove that phase separation existed and led to an increase of microcrystalline, we tested the samples by wide-angle XRD and observed their surface morphology under an electron microscope. PVA hydrogels containing 80% water were immersed in 0, 2, 4, and 6 mol L−1 NaOH solution, respectively. The spectrum of the hydrogel contained diffraction peaks at the 2θ angles of 19.5° (Fig. 2(c)). This result was consistent with a previous study24 that also indicated the formation of PVA crystallites. With the increasing of NaOH solution concentration, the diffraction peaks also increased obviously. Fig. 2(d) displays the statistical result of peak intensity. The calculation of crystallinity was
| φ = Ic/(Ic + Ia) | (1) |
Tension and compression behavior of phase-separated PVA hydrogels
To study the mechanical properties of PVA hydrogel immersed in NaOH solution, we conducted uniaxial tensile and compressive tests. NaOH solution concentrations were 0, 1, 2, 4, 6, and 8 M. The experimental results showed that the mechanical properties of PVA hydrogels increased significantly after being immersed in high-concentration solution.The stress–strain curves under uniaxial tensile are shown in Fig. 3(a). The failure strain of hydrogels immersed in 8 M concentration reached more than four times that of pristine PVA hydrogel (Fig. 3(b)). The tensile stress of pristine PVA hydrogel showed nonlinearity: with increases in stretching, the stress increased faster, which was consistent with previous results.26 For hydrogels immersed in NaOH solution, the results showed an opposite trend: in the early stage of stretching, the stress increased rapidly, but in the late stage of stretching, the slope of the stress curve decreased. The shape of the stress curve changed from “lower convex” of the pristine PVA hydrogel to “upward convex” of the PVA hydrogel immersed in NaOH solution. This may have resulted from a gradual destruction inside the polymer network: the decrease in the number of stressed fibers resulted in a decrease in the tangent slope of the stress curve. In terms of compressive behavior (see the stress–strain curves in Fig. 3(c)), the modulus of PVA hydrogels immersed in 8 M NaOH concentration reached seven times that of the pristine PVA hydrogel (Fig. 3(d)).
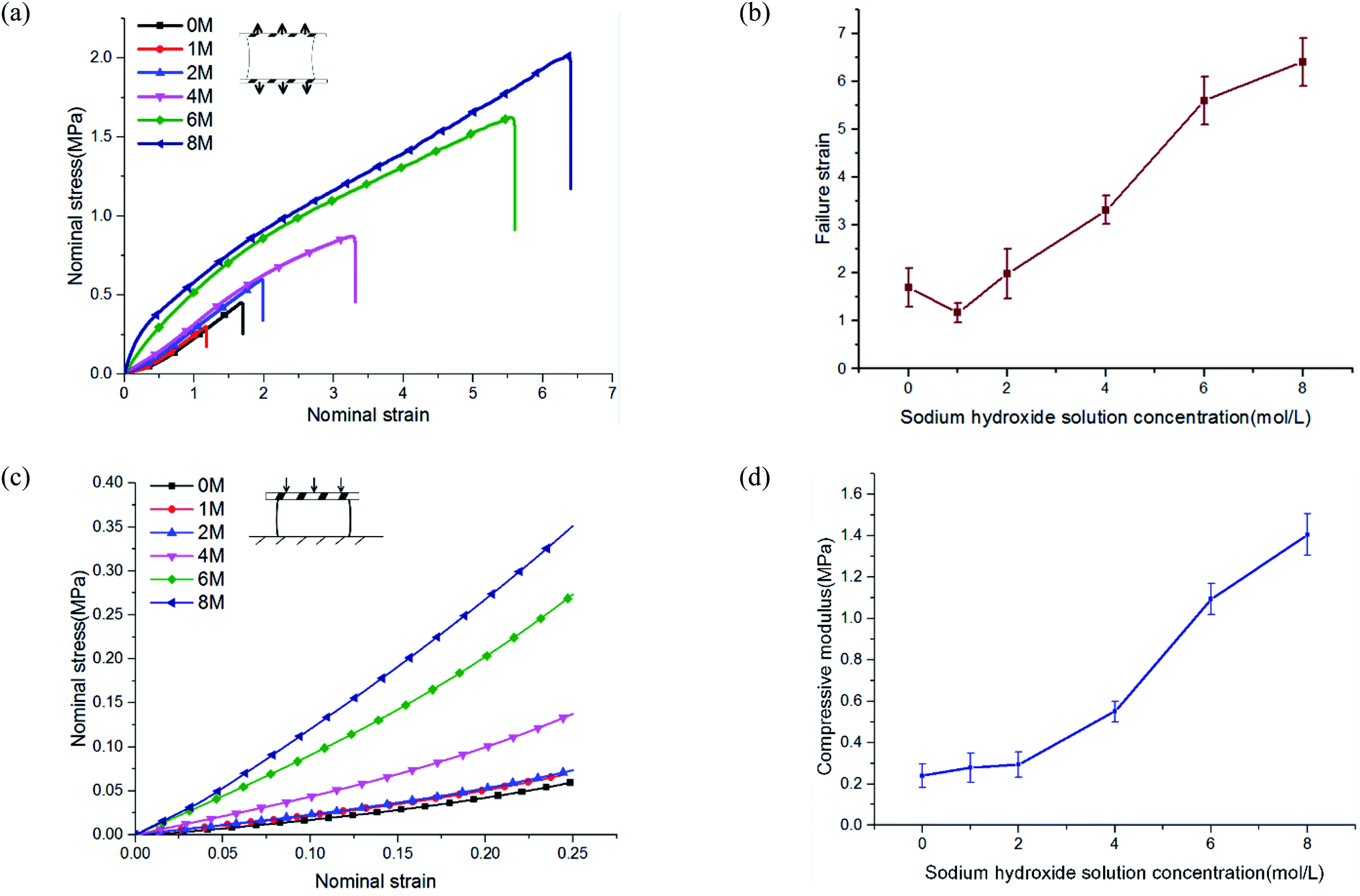 | ||
| Fig. 3 Tension and compression behavior of phase-separated PVA hydrogels: (a) the stress–strain curve under uniaxial tension; (b) failure strain statistics; (c) the stress–strain curve under uniaxial compression; and (d) the compression modulus of PVA hydrogels. | ||
The enhancement of mechanical properties in tension and compression may have been due to two mechanisms. First, the fiber content increased because of a decrease in the swelling ratio. The mechanical properties of the hydrogels were directly related to their fiber content. The mechanical properties of the material could be affected in this way only when the concentration of NaOH solution was lower than 4 M. Previous experiments showed that when the concentration was higher than 4 M, the swelling ratio no longer changed. Second, phase separation changed the polymer structure and further led to the enhancement of mechanical properties. This indicated that multiphase networks with different mechanical properties may have been formed in the polymer network. Moreover, the chemical mechanism of phase separation induced by the interaction of NaOH and PVA has been discussed in detail by Darabi et al.22
Energy dissipation and self-recovery of phase-separated PVA hydrogels
The mechanism of improving the mechanical properties for phase-separated hydrogels was similar to double-network hydrogels. The interactions between polymer chains in dense regions were enhanced and more microcrystals could serve as sacrificial bonds to dissipate energy during stretching to protect the entire polymer network from being destroyed, which has been explained in detail by Gong.27 This mechanism can be proved by the hysteresis phenomenon in loading and unloading tests. The stress curves of six groups immersed in different NaOH concentrations within one cycle are shown in Fig. 4(a). The dissipated energy Uhys is the area Sd enclosed by the loading curve and unloading curve:19
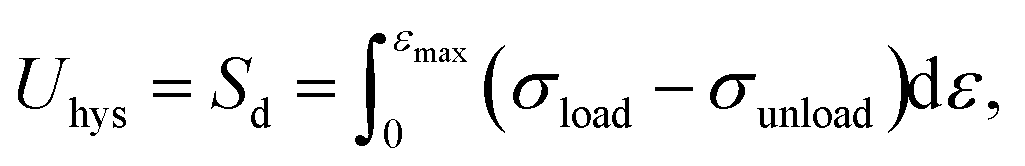 | (2) |
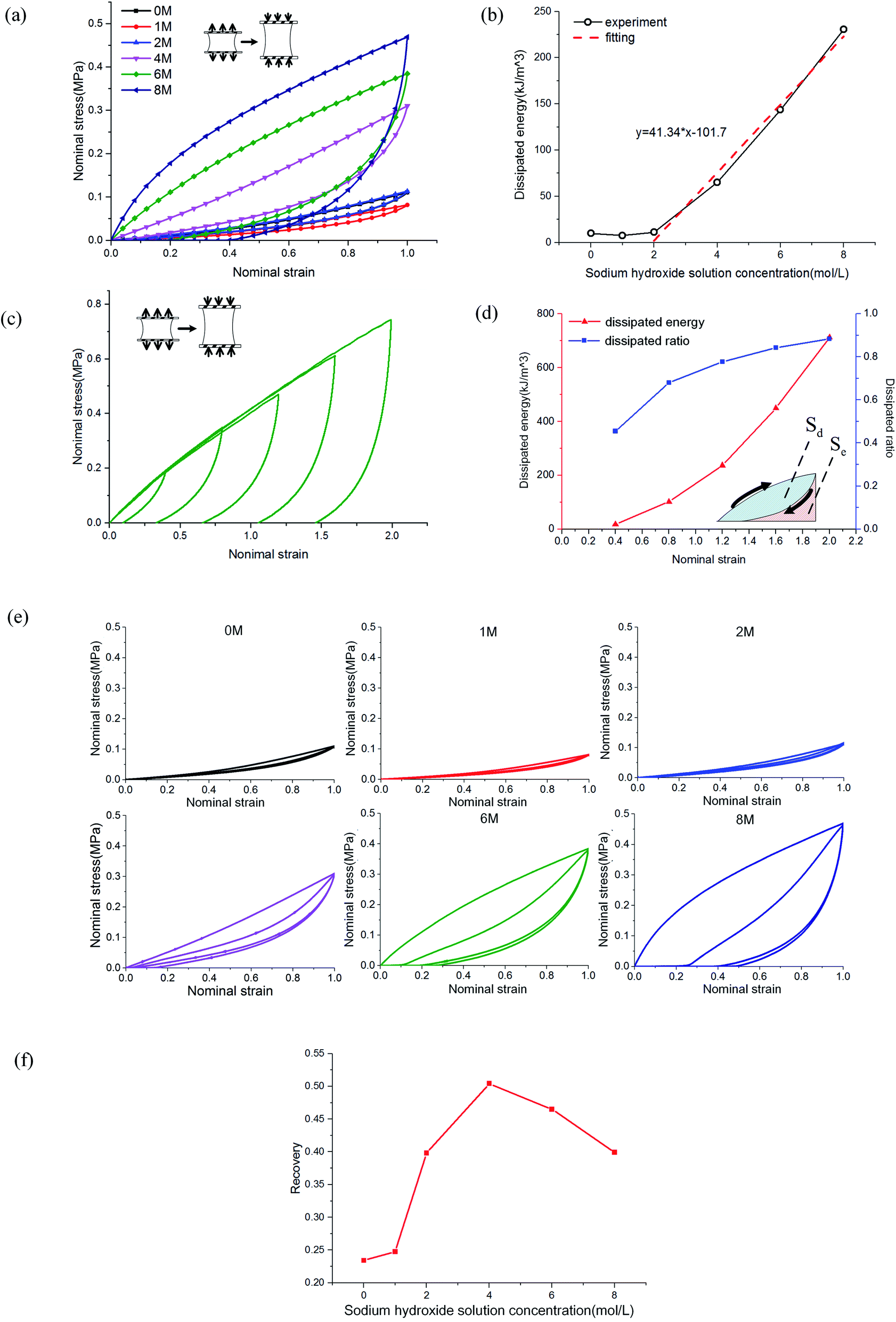 | ||
| Fig. 4 Cyclic load tests: (a) the single loading and unloading curves of PVA samples immersed in different NaOH concentration; (b) energy dissipation statistics of (a); (c) the loading and unloading curves of PVA immersed in 8 M NaOH solution with different tensile strains; (d) statistics of energy dissipation and dissipation ratio of (c); (e) double cycle curves of immersed PVA hydrogel samples; and (f) self-recovery ratio of PVA hydrogels immersed in NaOH solution. | ||
The area enclosed by the tensile curve and x-axis (Sd + Se) was the work done by external force during the stretching process, and the difference Se was the potential energy stored in the polymer network (Fig. 4(d)). When the NaOH solution concentration was higher than 2 M, the energy dissipated through the network increased obviously, and a linear correlation was evident, as shown in Fig. 4(b). We defined Sd/(Sd + Se) as the external work dissipated ratio. This value reflected the working effect of the mechanism. When nominal strain reached 1.0, the dissipation ratio of pristine PVA hydrogel was 0.23, and that of the samples immersed in 8 M NaOH concentration was 0.7. For pristine PVA hydrogel, most of the external force work was stored as potential energy, which retained the ability to restore the material to its original state. In phase-separated PVA hydrogel, this work left little as stored energy, and most of the energy was consumed through microcrystal expansion. As a result, the sample had obvious residual strain. Fig. 4(c) shows the loading and unloading curves of PVA hydrogel immersed in 8 M NaOH concentration solution under different maximum strains, and the dissipated energy and ratio increased obviously with strain (Fig. 4(d)), which indicated that the dissipation mechanism took more effect as the materials deformed.
Furthermore, we studied the effect of NaOH solution on the self-recovery characteristics of PVA hydrogel. As a feature of hydrogen-bonded hydrogels, self-recovery often has been used to design double-network hydrogels with self-recovery ability.24 This feature is due to the low requirements of PVA chains to interact. Fig. 4(e) shows the stress curves of double cycles of loading and unloading for phase-separated PVA hydrogels. There was no interval between cycles. We explored the recovery degree of polymer networks during the unloading time. The degree of self-recovery was defined as the ratio of the two enclosed areas of stress curve Uhys1 and Uhys2:24
| ϕ = Uhys2/Uhys1, | (3) |
Tearing behavior of phase-separated PVA hydrogels
To study tearing behavior, we conducted uniaxial tensile tests from loading until complete tear for hydrogels. The results showed that their tear modes were quite different. For pristine PVA hydrogel, the crack generated when stress reached a threshold, and then it extended along the transverse direction and directly led to the sample's failure. The subsequent time from crack initiation until the sample completely tore was short, and so the fracture mode was fast fracture.28 For phase-separated PVA hydrogels, as stress increased, multiple cracks appeared and expanded along the loading direction, which was quite different from the pristine PVA hydrogel (Fig. 5(a)). Most of the cracks propagated slowly and suspended at a certain stress, which was not as fast or continuous as in the pristine PVA hydrogel. Thus, the failure mode was ductile fracture.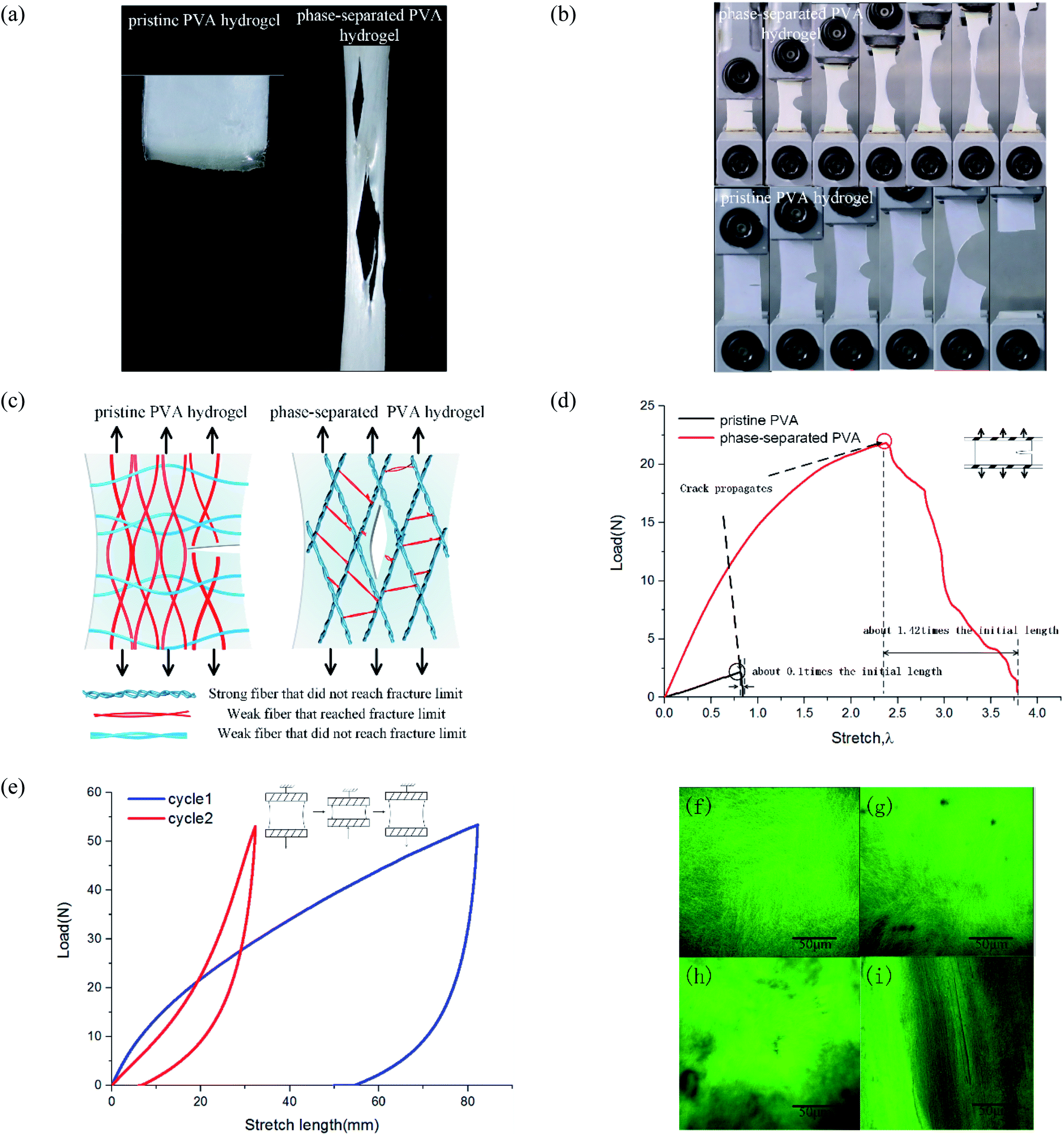 | ||
| Fig. 5 Characterization of tear behavior of phase-separated PVA hydrogels: (a) tensile-fractured pristine PVA hydrogel and phase-separated PVA hydrogels; (b) schematic of different failure modes; (c) uniaxial tensile failure of pre-notched samples; (d) the load–stretch curve of pre-notched samples; (e) the load–displacement curve of phase-separated PVA hydrogels of double cyclic loading test; and (f)–(i) microstructure of materials observed by confocal microscope. | ||
To calculate the toughness of materials, we conducted pure-shear tests for pre-notched samples (Fig. 5(b)). Loading curves are shown in Fig. 5(d). Toughness is equal to29
| Γ = hW(λ) | (4) |
These results showed that the phase separation induced by NaOH solution immersion affected both the toughness and failure mode for PVA hydrogel. To explain this change, we proposed a multiphase network theory. When phase separation occurred, the homogeneous polymer network was separated into dense regions and sparse regions.19 Suppose that in fiber dense regions, the interaction reinforced the screw fibers as strong fibers, and those strong fibers with the same mechanical properties then formed a strong phase network. Thus, the polymer network of phase-separated PVA hydrogels could be characterized by multiphase networks. The modulus and fracture stress varied from different phases. The pristine PVA hydrogel could be regarded as a single-phase network, which meant that the modulus and strengths of all the fibers were close. In the polymer network, those fibers along the loading direction were stressed. When part of these fibers surpassed their fracture limit and were about to break, the surrounding fibers also reached a critical value, and thus, the sample tore apart quickly and the expanding direction was perpendicular to the loading direction (Fig. 5(c)). Strictly speaking, the pristine PVA hydrogel was not a single-phase network because the freeze–thaw process resulted in phase separation, but because of its brittleness and fast fracture behavior, we could simplify it as a single-phase network. For phase-separated PVA hydrogels, fibers could withstand a very high stress, and the strong fibers changed their orientation into loading direction under a high stress, whereas the weak fibers broke early. After those weak fibers that connected the strong fibers broke, the strong fibers separated from each other and the shape of the cracks became the contour formed by the strong fibers (Fig. 5(c)). The crack growth process could be interpreted as the continuous fracture of weak fibers between the strong fibers. Because stress needed for the weak fiber fracture to the strong fiber fracture was discontinuous, the crack propagation was also discontinuous.
We provide the longitudinal arrangement of fibers by a double cyclic loading test and observation under a confocal microscope. When the loading direction was consistent with the direction of the fiber orientation, the material showed better mechanical properties. As shown in Fig. 5(e), the material in the second loading stage showed better mechanical property. Fig. 5(f)–(i) show the network microstructure results. We used the fluorescence dye to label the PVA side groups. Fig. 5(f) and (g) show the observation of pristine PVA hydrogels before and after tensile fracture. Stretching to the fracture did not change the uniformity and isotropy of the network. In Fig. 5(h), the high bright green area represented the dense fiber area, and the dark area represented the sparse area. Fig. 5(i) shows that after stretching, the orientation of fibers changed toward the tensile direction, and thus the hydrogels were no longer isotropic.
Probabilistic multiphase network model
In the previous discussion, we explained that NaOH solution immersion caused phase separation in the polymer network, and the interactions in the dense region were enhanced. Thus, we regarded the dense regions as a strong phase network. We next used a multiphase parallel model to explain the tension behavior. Two mechanical parameters representing the mechanical properties of the network were tensile modulus and fracture strain. In the previous experiments, the tangent modulus of the tensile curve for phase-separated PVA hydrogels decreased gradually as stretching, which was quite different from pristine PVA hydrogels (Fig. 6(a)). To explain this difference, we adopted a continuous distribution model. The modulus and fracture strain of the different phases conformed to this continuous probability distribution. During stretching, fibers of different phases broke continuously, and the accumulation of damage led to the appearance of macro cracks, which caused the material to fail completely (Fig. 6(b)).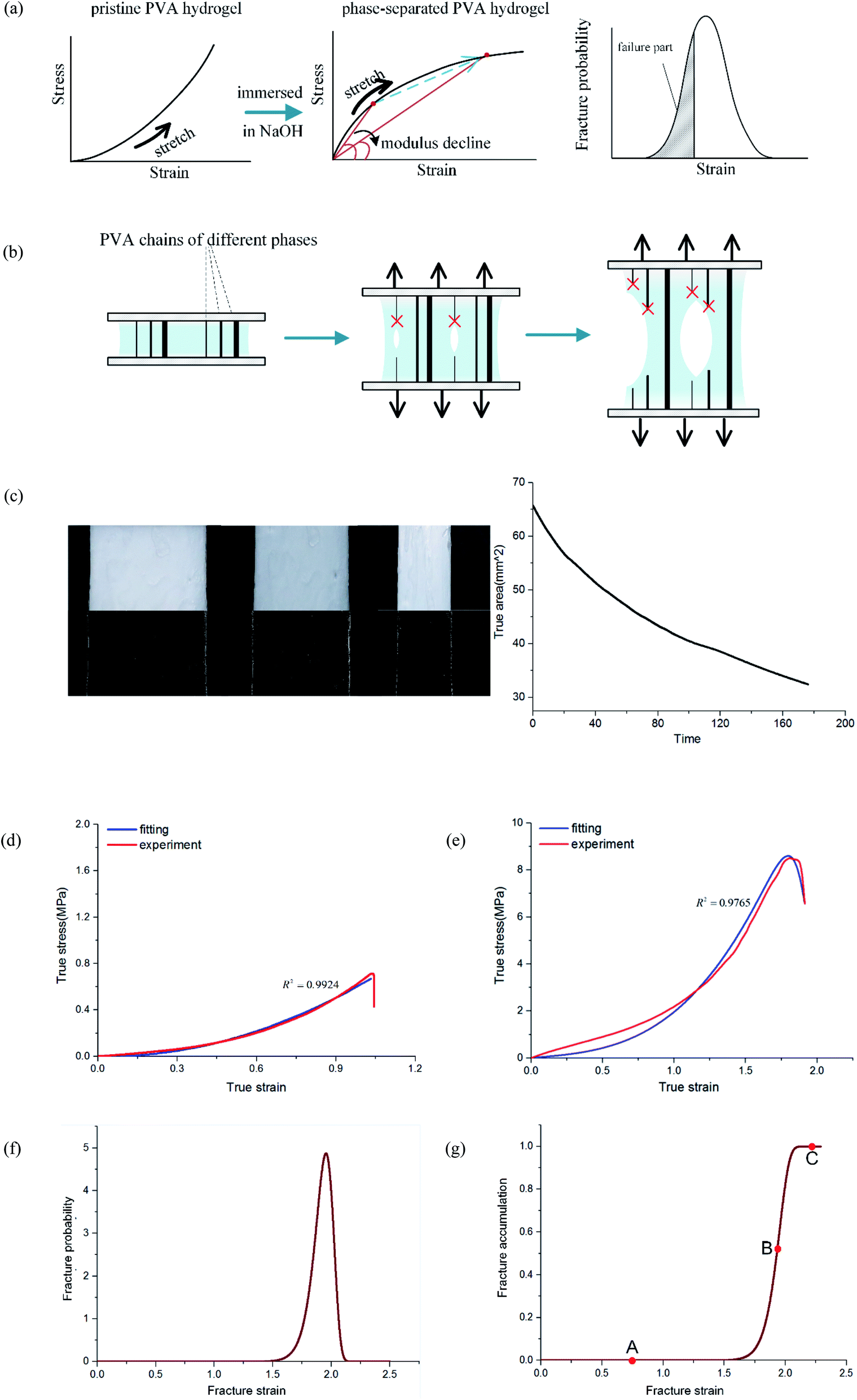 | ||
| Fig. 6 Continuous multiphase network theory: (a) the stress curve change of PVA hydrogel before and after being immersed in NaOH solution; (b) schematic of ductile fracture; (c) edge detection technology method used to measure the true area in real-time; (d) the fitting and experiment data of the pristine PVA hydrogel; (e) the fitting and experiment data of the phase-separated PVA hydrogel; (f) the probability distribution of fracture; and (g) the cumulative distribution of fracture. | ||
The constitutive equation of pristine PVA hydrogel during uniaxial tension follows:30
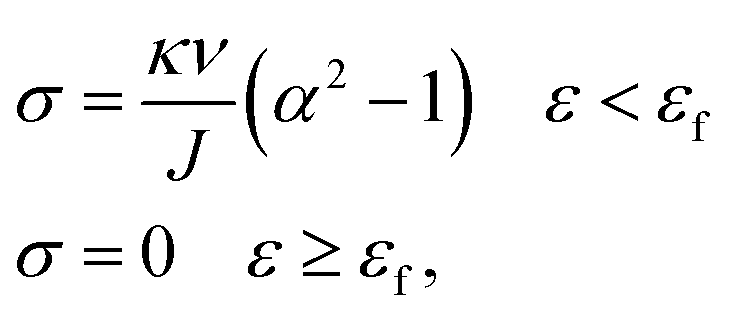 | (5) |
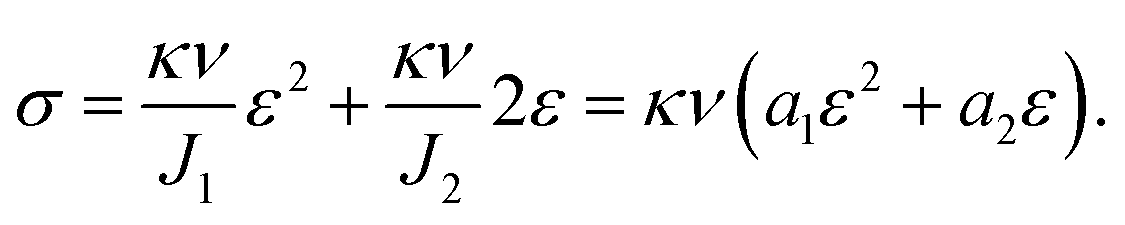 | (6) |
We used this formula to obtain an equation that described the tensile behavior of the pristine PVA hydrogel, where σ and ε are true stress and true strain, respectively. Usually, we can directly measure only the nominal stress–strain in the experiment, and thus we need to transfer the nominal stress–strain into true stress–strain. The relationship between nominal stress–strain and true stress–strain is as follows:
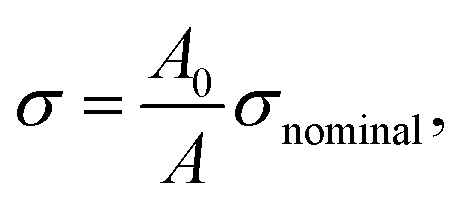 | (7) |
| ε = ln(1 + εnominal), | (8) |
Because Weibull distribution often has been used to describe the strength and life-related parameters of materials,31 we assumed that modulus and fracture strain of different phase networks also satisfy this distribution:
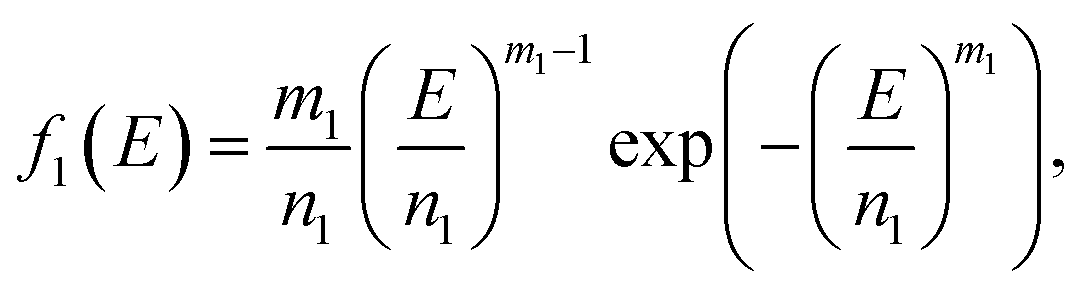 | (9) |
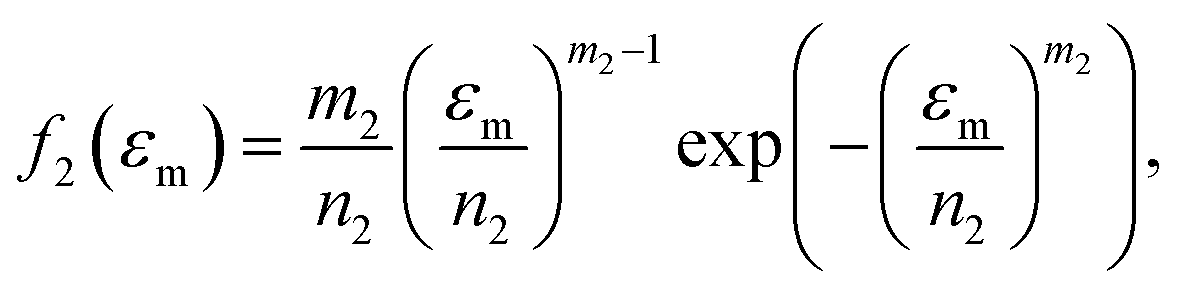 | (10) |
We assumed that different phase networks were in parallel, and every phase had the same strain ε when stretching. Considering the continuous distribution of modulus, the number of networks with modulus E was f(E)dE. Thus, the constitutive equation of the whole network is as follows:
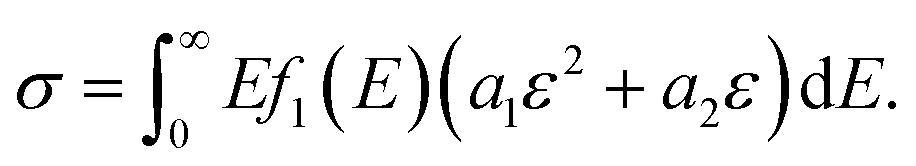 | (11) |
Furthermore, when fracture is considered, the amount that breaks 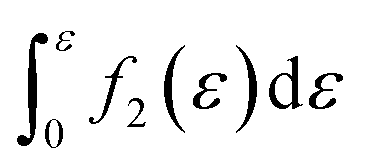 is a cumulative area function. This part of the network loses its loading capacity, and thus the constitutive equation becomes
is a cumulative area function. This part of the network loses its loading capacity, and thus the constitutive equation becomes
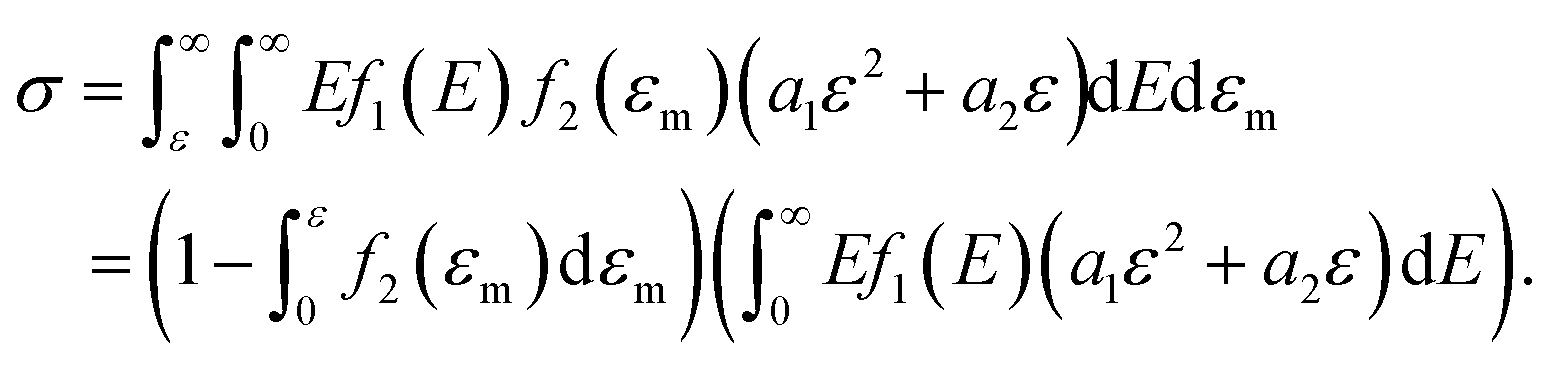 | (12) |
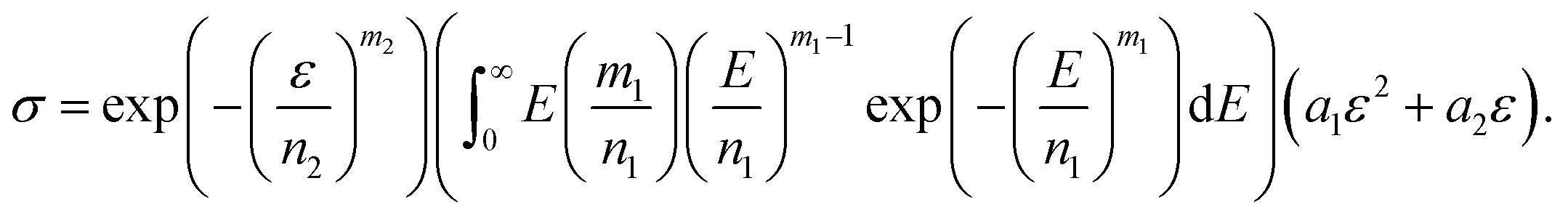 | (13) |
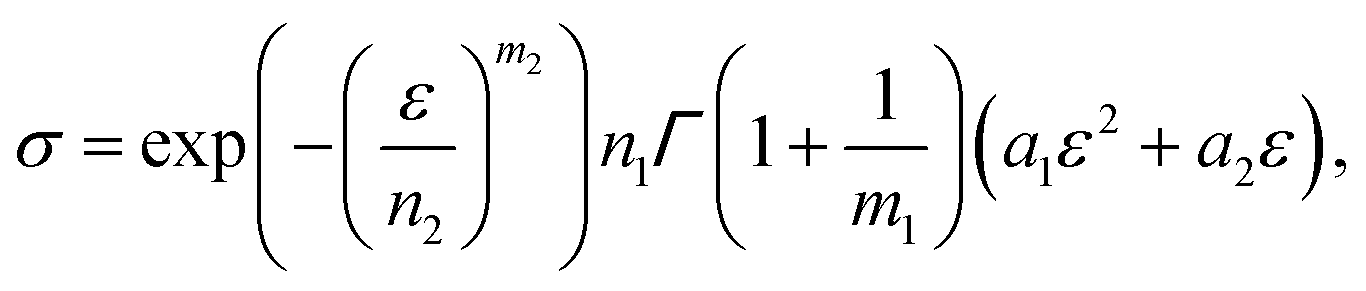 | (14) |
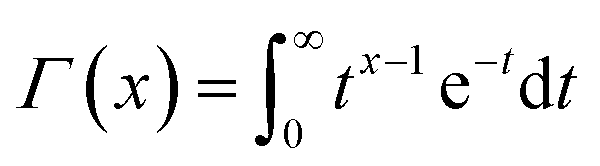 is a gamma function, where t = (E/n1)m1.
is a gamma function, where t = (E/n1)m1.
We used the uniaxial tensile test data of the phase-separated PVA hydrogel with 20% fiber content immersed in 6 M NaOH solution for fitting. First, we fitted the constitutive equation of the pristine PVA hydrogel (2) and obtained
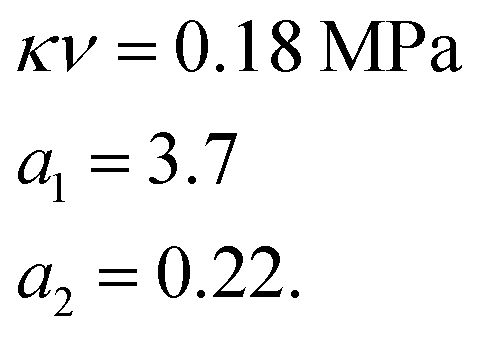
The comparison between fitting and experimental was shown in Fig. 6(d). Then we fitted the experimental data of the phase-separated PVA hydrogels to calculate the four parameters m1, n1, m2, n2 in the probability distribution.
The results were
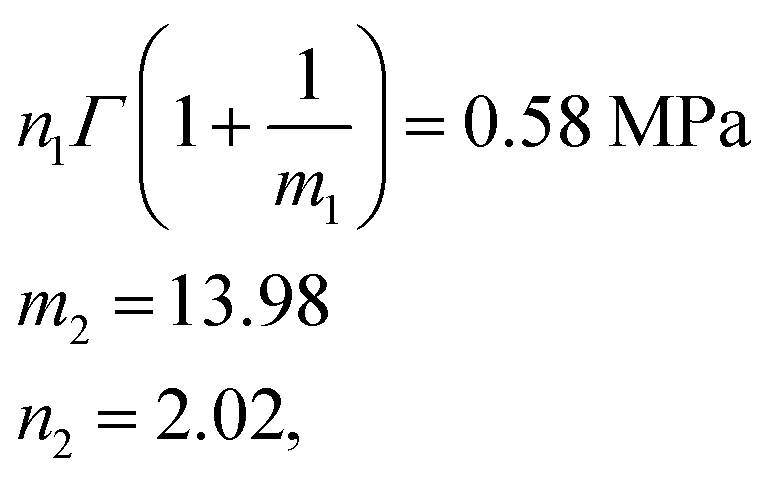
After obtaining the noted parameters, we obtained the probability distribution and cumulative distribution of the fracture function, as shown in Fig. 6(f) and (g):
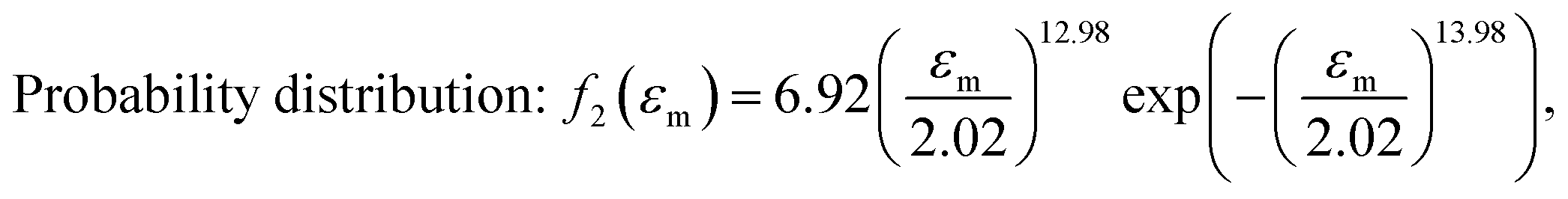 | (15) |
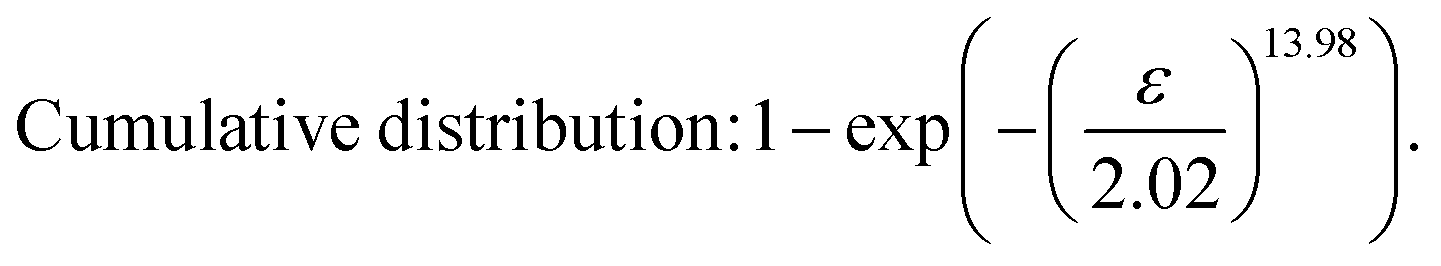 | (16) |
For pristine PVA hydrogel, when the strain reached the fracture limit, the material tore rapidly. It is not meaningful to discuss the mechanical behavior in the failure stage for fast fracture behavior. Therefore, the existing constitutive theories are valuable before fracture happens. For phase-separated PVA hydrogel, however, the failure mode of the material was ductile fracture. Thus, it was necessary to study the fracture behavior during the failure stage. By introducing the continuous probability distribution of failure and combining the constitutive theory of the single-phase network, we well described the mechanical behavior of the material for a considerable period of time after fracture happens. Considering that the relationship between real strain and nominal strain was ε = ln(1 + εnominal), the real stress range seemed to be a short time in the middle and late stages of loading, which actually had quite dense experimental data points, and the loading time was quite long.
Fig. 6(g) shows the fracture of phase-separated hydrogel had three stages. When the strain was relatively low, corresponding to stage A in the graph, fibers were hardly broken, and the stress curve of the phase-separated hydrogels was similar to that of the pristine hydrogel (ε < 1.5). After strain exceeded a critical value, the fracture started to accumulate (stage B), and the stress changed from growth to a gradual decline (1.5 < ε < 2.0). When most fibers fractured, the material was completely destroyed, which corresponded to stage C (ε > 2.0). In comparison, the fracture cumulative distribution function of the pristine PVA hydrogel could be regarded as the following step function:
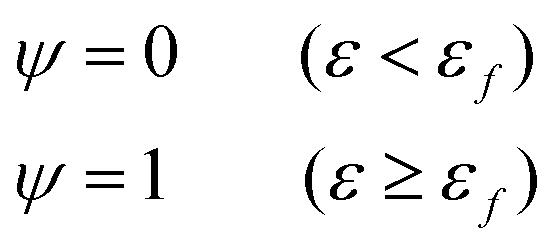 | (17) |
In the middle-late stage, the fibers followed a longitudinal direction. This was similar to the parallel mode that we used in the model; therefore, the fitting result was in better agreement with the experiment. In the fracture probability distribution diagram (Fig. 6(f)), the strain corresponding to the peak was 1.9, which meant that the probability of failure of networks at this point reached the maximum. The strain corresponding to failure was 1.8 in the experiment, which indicated that most of the breaks in the networks led to the failure of the macrostructure. The continuous phase network distribution model not only was in good agreement with the tensile curve but also described the failure behavior of the material. We admit that the theory was not always satisfactory in some aspects. As a simple network model, our hypothesis is still quite different from the real network structure in PVA hydrogels.
Conclusion
Alkaline solution immersion is an important step to prepare magnetic hydrogel in in situ precipitation. Our study indicated that immersion in high-concentration NaOH solution enhanced the mechanical properties of physical PVA hydrogel, including its elastic modulus, failure strength, toughness, and self-recovery ability. The mechanical properties changed obviously with the solution concentration.The enhancement of mechanical properties of the material was mainly due to the change of the structure of hydrogel. Phase separation led the polymer structure to change from a homogeneous network to an inhomogeneous network consisting of multiple phases with different strength and modulus. The sacrifice of weak phases gave the network a high toughness and the existence of strong phases allowed the hydrogel to endure a high stress. The enhancement also changed the failure mode from fast fracture to ductile fracture.
On the basis of the aforementioned experiments, we proposed a model of a continuous multiphase network. The model is in good agreement with the experimental results under uniaxial tension. Additionally, the probability peak corresponding to the macroscopic failure of the materials further explained the failure behavior of the hydrogel. These basic experiments may enhance our understanding of PVA hydrogels and thus improve practical applications.
Author contributions
L. Z. and L. T., conceived the project. Z. Z. and Y. Z. synthesized the samples; performed the mechanical tests, XRD tests, and SEM tests; and processed the data. Z. Z., Y. Z., Z. L., Z. J., and Y. L. discussed and analyzed the results. Z. Z., L. T., and Y. Z. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Natural Science Foundation of China (Grant No. 11932007, 11802062, 11972162, 12072115, 12072116, 11772131, 11772132, and 11772134) and the Science and Technology Program of Guangzhou, China (Grant No. 201903010046). We thank LetPub (http://www.letpub.com) for its linguistic assistance during the preparation of this manuscript.References
- J. Li, L. T. Mo, C. H. Lu, T. Fu, H. H. Yang and W. H. Tan, Functional nucleic acid-based hydrogels for bioanalytical and biomedical applications, Chem. Soc. Rev., 2016, 45, 1410–1431 RSC.
- A. Sionkowska, Current research on the blends of natural and synthetic polymers as new biomaterials: Review, Prog. Polym. Sci., 2011, 36, 1254–1276 CrossRef CAS.
- E. A. Kamoun, X. Chen, M. S. Mohy Eldin and E. S. Kenawy, Crosslinked poly(vinyl alcohol) hydrogels for wound dressing applications: a review of remarkably blended polymers, Arabian J. Chem., 2015, 8, 1–14 CrossRef CAS.
- X. L. Qi, X. Y. Hu, W. Wei, H. Yu, J. J. Li, J. F. Zhang and W. Dong, Investigation of Salecan/poly(vinyl alcohol) hydrogels prepared by freeze/thaw method, Carbohydr. Polym., 2015, 118, 60–69 CrossRef CAS PubMed.
- W. X. Li, D. Wang, W. Yang and Y. Song, Compressive mechanical properties and microstructure of PVA–HA hydrogels for cartilage repair, RSC Adv., 2016, 6, 20166–20172 RSC.
- N. A. Peppas and E. W. Merrill, Differential Scanning Calorimetry of Crystallized PVA Hydrogels, J. Appl. Polym. Sci., 1976, 20, 1457–1465 CrossRef CAS.
- E. G. Fuller, H. Sun, R. D. Dhavalikar, M. Unni, G. M. Scheutz, B. S. Sumerlin and C. Rinaldi, Externally Triggered Heat and Drug Release from Magnetically Controlled Nanocarriers, ACS Appl. Polym. Mater., 2019, 1(2), 211–220 CrossRef CAS.
- T. Y. Liu, S. H. Hu, T. Y. Liu, D. M. Liu and S. Y. Chen, Magnetic-Sensitive Behavior of Intelligent Ferrogels for Controlled Release of Drug, Langmuir, 2016, 22, 5974–5978 CrossRef PubMed.
- R. V. Ramanujan and L. L. Lao, The mechanical behavior of smart magnet-hydrogel composites, Smart Mater. Struct., 2006, 15, 952–956 CrossRef.
- S. A. Meenach, J. Z. Hilt and K. W. Anderson, Poly(ethylene glycol)-based magnetic hydrogel nanocomposites for hyperthermia cancer therapy, Acta Biomater., 2010, 6, 1039–1046 CrossRef CAS PubMed.
- S. Mandal and K. Chaudhuri, Engineered magnetic core shell nanoprobes: Synthesis and applications to cancer imaging and therapeutics, World J. Biol. Chem., 2016, 7, 158–167 CrossRef PubMed.
- M. T. S. Alcantara, A. J. C. Brant, D. R. Giannini, J. O. C. P. Pessoa, A. B. Andrade, H. G. Riella and A. B. Lugao, Influence of dissolution processing of PVA blends on the characteristics of their hydrogels synthesized by radiation—Part I: gel fraction, swelling, and mechanical properties, Radiat. Phys. Chem., 2012, 81, 1465–1470 CrossRef CAS.
- K. Parivar, F. M. Fard, M. Bayat, S. M. Alavian and M. Motavaf, Evaluation of Iron Oxide Nanoparticles Toxicity on Liver Cells of BALB/c Rats, Iran. Red Crescent Med. J., 2016, 18, e28939 Search PubMed.
- L. Zhang, J. Zhao, J. T. Zhu, C. C. He and H. L. Wang, Anisotropic tough poly(vinyl alcohol) hydrogels, Soft Matter, 2012, 8, 10439–10447 RSC.
- W. Cha, S. H. Hyon, M. Oka and Y. Ikada, Mechanical and wear properties of poly(v1nyl alcohol) hydrogels, Macromol. Symp., 1996, 109, 115–126 CrossRef CAS.
- A. Suzuki and S. Sasaki, Swelling and mechanical properties of physically crosslinked poly(vinyl alcohol) hydrogels, J. Eng. Med., 2015, 229, 828–844 CrossRef PubMed.
- S. T. Lin, J. Liu, X. Y. Liu and X. H. Zhao, Muscle-like fatigue-resistant hydrogels by mechanical training, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10244–10249 CrossRef CAS PubMed.
- G. Q. Guo, Y. Z. Chen, X. Y. Liu, D. Y. Zhu, B. Zhang, N. M. Lin and L. Gao, Tough and durable hydrogels with robust skin layers formed via immersing treatment, J. Mater. Chem. B, 2018, 6, 8043–8054 RSC.
- K. Sato, T. Nakajima, T. Hisamatsu, T. Nonoyama, T. Kurokawa and J. P. Gong, Phase-Separation-Induced Anomalous Stiffening, Toughening, and Self-Healing of Polyacrylamide Gels, Adv. Mater., 2015, 27, 6990–6998 CrossRef CAS PubMed.
- J. D. Tang, Z. F. Tong, Y. K. Xia, M. Liu, Z. Y. Lv, Y. Gao, T. Q. Lu, S. J. Xie, Y. M. Pei, D. N. Fang and T. J. Wang, Super tough magnetic hydrogels for remotely triggered shape morphing, J. Mater. Chem. B, 2018, 6, 2713–2722 RSC.
- X. C. Jiang, N. P. Xiang, H. X. Zhang, Y. J. Sun, Z. Lin and L. X. Hou, Preparation and characterization of poly(vinyl alcohol)/sodium alginate hydrogel with high toughness and electric conductivity, Carbohydr. Polym., 2018, 186, 377–383 CrossRef CAS PubMed.
- M. A. Darabi, A. Khosrozadeh, Y. Wang, N. Ashammakhi, H. Alem, A. Erdem, Q. Chang, K. G. Xu, Y. Q. Liu, G. X. Luo, A. Khademhosseini and M. Xing, An Alkaline Based Method for Generating Crystalline, Strong, and Shape Memory Polyvinyl Alcohol Biomaterials, Adv. Sci., 2020, 7, 1902740 CrossRef CAS PubMed.
- J. L. Holloway, A. M. Lowmanb and G. R. Palmese, The role of crystallization and phase separation in the formation of physically cross-linked PVA hydrogels, Soft Matter, 2013, 9, 826–833 RSC.
- J. Y. Li, Z. G. Suo and J. J. Vlassak, Stiff, strong, and tough hydrogels with good chemical stability, J. Mater. Chem. B, 2014, 2, 6708–6713 CAS.
- Y. Hou, C. Chen and K. Liu, Preparation of PVA hydrogel with high-transparence and investigations of its transparent mechanism, RSC Adv., 2015, 5, 24023–24030 RSC.
- K. J. Xu, L. Q. Tang, Y. R. Zhang, R. Ding, X. Y. Wang, D. F. Huang and J. S. Lin, Study on the Microscopic Network Model of PVA Hydrogel Based on the Tensile Behavior, Acta Mech. Solida Sin., 2019, 32, 663–674 CrossRef.
- J. P. Gong, Why are double network hydrogels so tough?, Soft Matter, 2010, 6, 2583–2590 RSC.
- J. D. Tang, J. Y. Li, J. J. Vlassaka and Z. G. Suo, Fatigue fracture of hydrogels, Extreme Mech. Lett., 2017, 10, 24–31 CrossRef.
- R. S. Rivlin and A. G. Thomas, Rupture of Rubber. I. Characteristic Energy for Tearing, J. Polym. Sci., 1953, 10, 291–318 CrossRef CAS.
- Y. R. Zhang, K. J. Xu, Y. L. Bai, L. Q. Tang, Z. Y. Jiang, Y. P. Liu, Z. J. Liu, L. C. Zhou and X. F. Zhou, Features of the volume change and a new constitutive equation of hydrogels under uniaxial compression, J. Mech. Behav. Biomed. Mater., 2018, 85, 181–187 CrossRef CAS PubMed.
- W. Weibull and Stockholm, A statistical distribution function of wide applicability, J. Appl. Mech., 1995, 293–297 Search PubMed.
Footnote |
| † Both authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2021 |