DOI:
10.1039/D1RA00732G
(Paper)
RSC Adv., 2021,
11, 22820-22825
Synthesis of 2-methoxybenzamide derivatives and evaluation of their hedgehog signaling pathway inhibition†
Received
27th January 2021
, Accepted 22nd June 2021
First published on 28th June 2021
Abstract
Aberrant hedgehog (Hh) signaling is implicated in the development of a variety of cancers. Smoothened (Smo) protein is a bottleneck in the Hh signal transduction. The regulation of the Hh signaling pathway to target the Smo receptor is a practical approach for development of anticancer agents. We report herein the design and synthesis of a series of 2-methoxybenzamide derivatives as Hh signaling pathway inhibitors. The pharmacological data demonstrated that compound 21 possessed potent Hh pathway inhibition with a nanomolar IC50 value, and it prevented Shh-induced Smo from entering the primary cilium. Furthermore, mutant Smo was effectively suppressed via compound 21. The in vitro antiproliferative activity of compound 21 against a drug-resistant cell line gave encouraging results.
Introduction
The hedgehog (Hh) signaling pathway is one of the pivotal targets for cancer therapy, and it is fundamental in regulating embryonic development and maintaining homeostasis of adult tissues.1–4 Upon secretion of Hh ligands, the transmembrane protein patched (Ptch) abrogates its inhibition of smoothened (Smo) receptor.5–8 Smo trafficking into the primary cilium (PC) transmits the signal to the downstream cascade.9–11 The glioma (Gli) transcription factors translocate into the nucleus and activate the target genes.12–14 Abnormal activation of the Hh pathway is able to facilitate tumorigenesis and metastasis, inducing multiple solid tumors and non-solid tumors such as basal cell carcinoma (BCC), medulloblastoma (MB), pancreatic carcinoma, colorectal carcinoma and leukaemia.15–20 Hence, the inhibition of the Hh signaling pathway is deemed as a promising option for antitumor therapy.
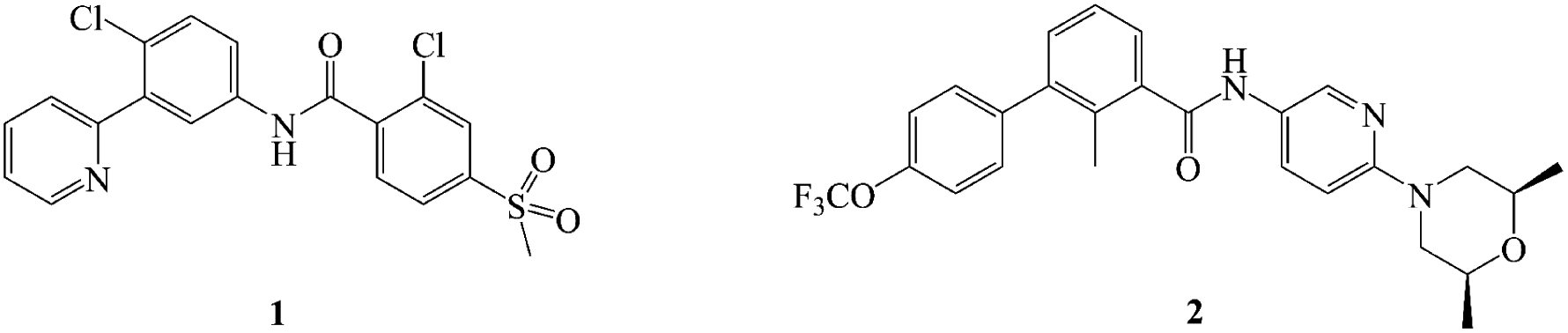
There have been two highly efficacious Hh pathway inhibitors, vismodegib (1) and sonidegib (2) (Fig. 1), launched for treating BCC nowadays.21–23 However, it was reported that 30 percent of patients who had taken 1 suffered from several adverse reactions such as muscle cramps, hair loss, taste disorders, weight loss, and fatigue.24,25 Besides, acquired drug resistance is a major drawback with this class of drugs. The best studied instance was that Smo mutation (D473H) interfered with the binding of the drug to Smo.26,27 Similarly, Smo D477G in the mouse MB allograft model was deemed as a comparable murine Smo mutant.28,29 The bottlenecks of Hh pathway inhibitors have prompted an extensive search for novel chemotype inhibitor.
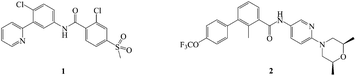 |
| | Fig. 1 Structure of hedgehog pathway inhibitors. | |
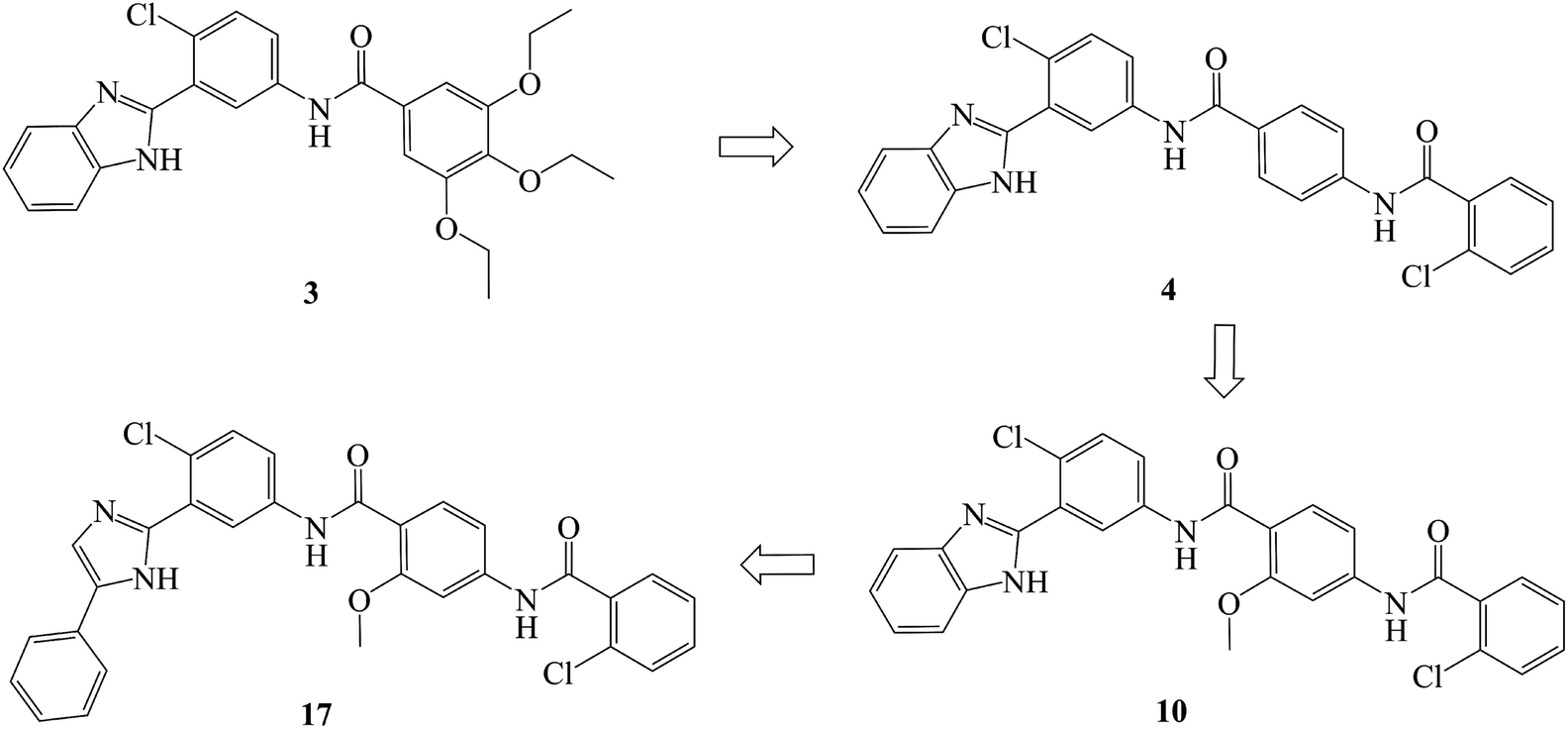
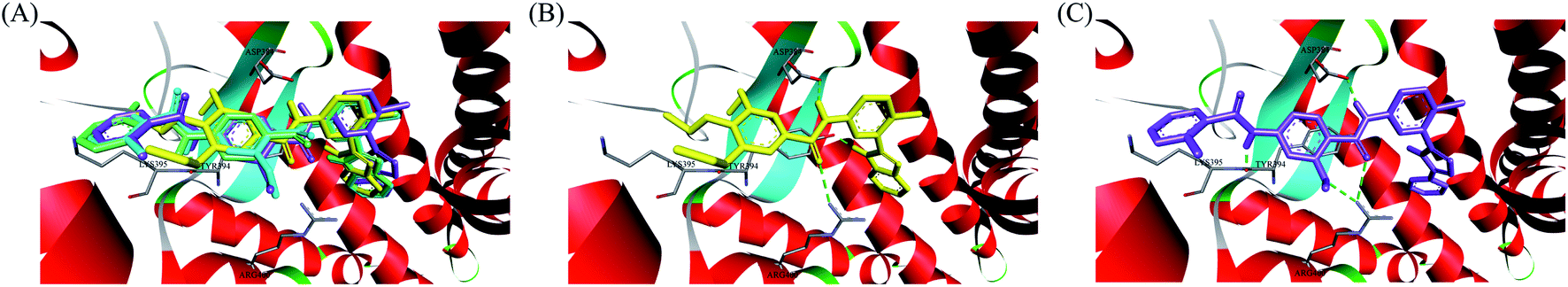
In recent years, several benzimidazole derivatives, such as glasdegib, SANT-2(3), have exhibited potent Hh pathway inhibition, all of which target Smo receptor.30–32 Compound 3 is a known Smo inhibitor with submicromolar IC50 in the Shh light II test.33 Combination of glasdegib and low-dose of arabinoside are currently for the treatment of leukemia.34 Alternatively, aryl amide groups in some Hh antagonists were deemed as the pharmacophores responsible for high affinity with Smo receptor, for instance, taladegib, XL139 and MDB5.35–37 In our effort on the discovery of novel chemotype Hh inhibitors (Fig. 2), aryl amide group was introduced into compound 3 to replace the metabolically labile triethoxy groups. The resulting compound 4 (IC50 of 0.25 μM) displayed similar potency to compound 3 in Gli-luciferase (Gli-luc) reporter assay. With the addition of methoxy, the formation of a new hydrogen bond (H-bond) acceptor was in compound 10 (IC50 of 0.17 μM). To enhance the molecular flexibility, the replacement of benzimidazole with phenyl imidazole fragment offered compound 17 with higher potency (IC50 of 0.12 μM) in the light of the ring-opening strategy. Followed by Smo crystal structures analysis (PDB ID: 5L7I),38 compounds 3, 4, 10 and 17 were docked into the binding pocket of Smo receptor using AutoDock vina.39 The calculated binding energy of their best poses was −9.4, −10.9, −12.5, −12.7 kcal mol−1, respectively, and their binding orientations superimposed well with one another (Fig. 3A). Comparing the interaction patterns between compounds 3 and 17 (Fig. 3B and C), the introduction of aryl amide group and methoxy afforded two extra H-bonds with Tyr394 and Arg400. Herein, the novel compounds with 2-methoxybenzamide skeleton were developed and their Hh pathway inhibition were evaluated.
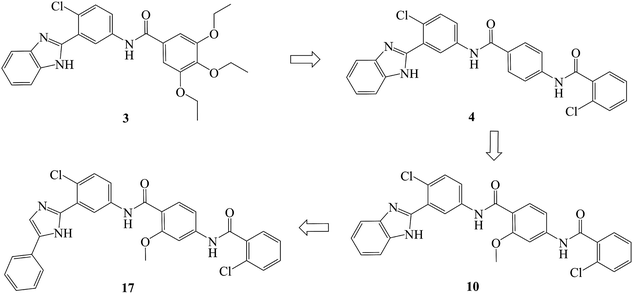 |
| | Fig. 2 Design of 2-methoxybenzamide derivatives. | |
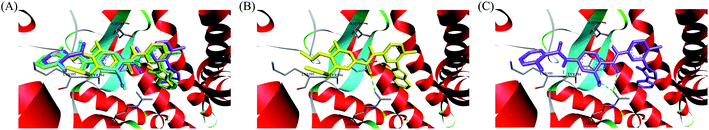 |
| | Fig. 3 (A) Overlay of compounds 3 (yellow), 4 (green), 10 (blue) and 17 (violet) in binding pocket of Smo. (B) Docking conformation of compound 3 at the binding site. (C) Docking conformation of compound 17 at the binding site. Hydrogen bonds are represented by the dashed green lines. Surrounding amino acid are shown in grey stick format and labelled. | |
Result and discussion
Chemistry
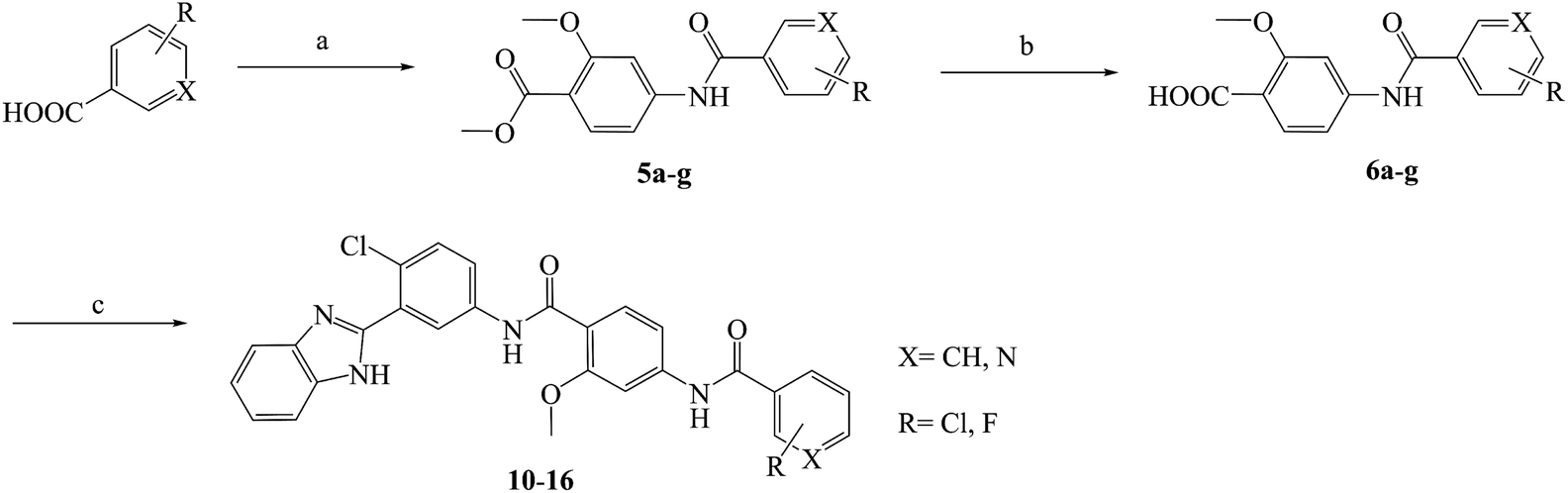
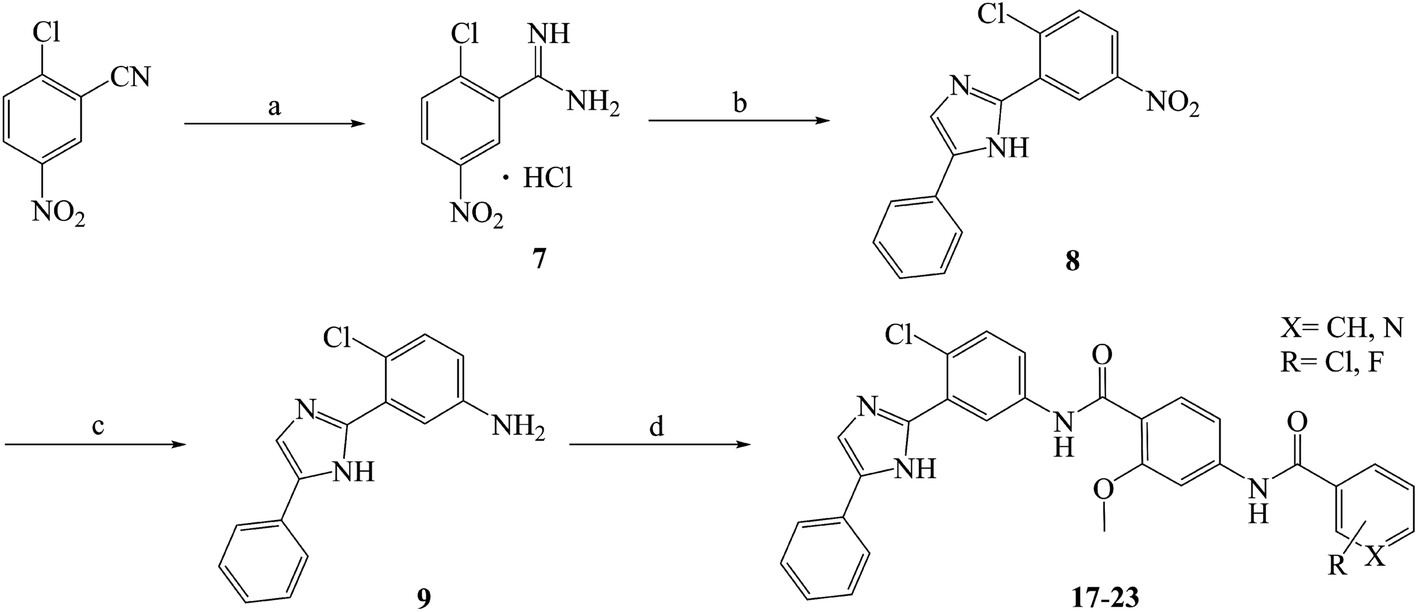
Compounds 10–23 were prepared according to the routes outlined in Schemes 1 and 2. Substituted acids refluxed in SOCl2 for 3 h to generate acyl chlorides, which were condensed with methyl 4-amino-2-methoxybenzoate in the presence of triethylamine in dimethyl formamide at room temperature overnight to obtain the intermediate 5a–g. Then, the esters were hydrolyzed to the carboxylic acid 6a–g, before their amidation with 3-(1H-benzo[d]imidazol-2-yl)-4-chloroaniline33 afforded the target compounds 10–16. In the presence of sodium alkoxide, 2-chloro-5-nitrobenzonitrile and ammonium chloride combined chemically to produce amidine hydrochloride 7 in a yield of 45%. Next, the cyclization of 7 and 2-bromoacetophenone gave imidazole intermediate 8, which converted to amino compound 9 by adding stannous chloride in acidic ethanol solution. The condensation of 9 with 6a–g generated the target compounds 17–23.
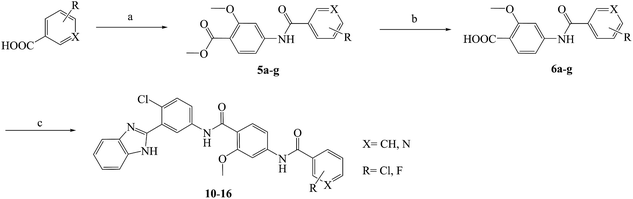 |
| | Scheme 1 Reagents and conditions: (a) (1) SOCl2, toluene, 80 °C, 3 h; (2) methyl 4-amino-2-methoxybenzoate, TEA, DMF, rt, overnight, yield 85%; (b) NaOH, aqueous alcohol, 40 °C, 5 h, yield 83–92%; (c) 3-(1H-benzo[d]imidazol-2-yl)-4-chloroaniline, HATU, DIPEA, DCM, rt, 24 h, yield 71–86%; the specific position of R group directed at Table 1. | |
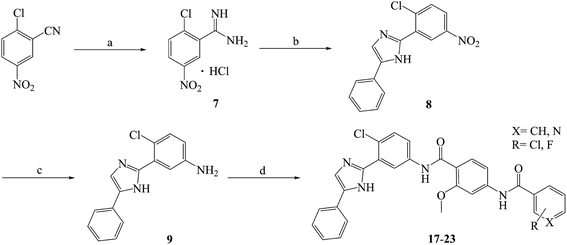 |
| | Scheme 2 Reagents and conditions: (a) CH3ONa, CH3OH, −20 °C, 2 h; NH4Cl, 40 °C, 3 h, yield 46%; (b) 2-bromoacetophenone, NaHCO3, THF, reflux, 40 °C, 24 h, yield 65%; (c) SnCl2·2H2O, ethanol, HCl, 80 °C, reflux, 8 h, yield 86%; (d) 5a–g, HATU, DIPEA, DCM, rt, 24 h, yield 76–84%; the specific position of R group directed at Table 1. | |
Biological evaluation
As shown in Table 1, the targeted compounds contained the following three parts: A, B and a connector. Part A involved imidazole group, and part B contained aryl amide. 2-Methoxybenzamide scaffold was the connector.
Table 1 Hh signaling pathway inhibition of designed compounds
|
| Compd. |
R |
X |
Yield (%) |
Gli-luc reporter IC50a (μM) |
| Results expressed as the mean ± standard deviation of three separate IC50 determinations. For each determination, concentration-inhibition curves were acquired in triplicate and then averaged to afford a single IC50 curve with a 95% confidence interval. Used as a lead compound. Used as a positive control. |
| 10 |
2-Cl |
CH |
75 |
0.17 ± 0.06 |
| 11 |
2,4-Cl2 |
CH |
71 |
0.53 ± 0.05 |
| 12 |
3-F |
CH |
83 |
0.79 ± 0.14 |
| 13 |
4-F |
CH |
85 |
0.34 ± 0.07 |
| 14 |
2-Cl |
N |
80 |
0.05 ± 0.02 |
| 15 |
6-Cl |
N |
73 |
0.86 ± 0.09 |
| 16 |
— |
N |
86 |
0.08 ± 0.02 |
| 17 |
2-Cl |
CH |
79 |
0.12 ± 0.06 |
| 18 |
2,4-Cl2 |
CH |
76 |
0.26 ± 0.08 |
| 19 |
3-F |
CH |
84 |
0.31 ± 0.09 |
| 20 |
4-F |
CH |
82 |
0.25 ± 0.04 |
| 21 |
2-Cl |
N |
81 |
0.03 ± 0.01 |
| 22 |
6-Cl |
N |
77 |
0.15 ± 0.08 |
| 23 |
— |
N |
83 |
0.07 ± 0.02 |
| 3b |
|
|
|
0.18 ± 0.03 |
| 1c |
|
|
|
0.02 ± 0.01 |
Using compound 1 as positive control and compound 3 as the lead compound, the Hh pathway inhibition of compounds 10–23 have been evaluated in Gli luciferase reporter assay. The results were expressed as IC50 values. At the outset, the relationship between part A and its Hh inhibitory activity was discussed. Encouragingly, the activity of phenyl imidazole analogues were higher than that of benzimidazole counterparts (11 vs. 18, 12 vs. 19 and 13 vs. 20), suggesting that ring-opening structural modification was effective. Further analysis revealed that different Hh pathway inhibition was observed as various R groups were introduced into the aromatic ring (part B). Monochlorine substituent 17 exhibited decent activity (IC50 = 0.12 μM), however, dichloride 18 lost potency by 2.2-fold (IC50 = 0.26 μM). Although fluoro analogs such as 19 and 20 (IC50 = 0.31 μM and 0.25 μM) were effective in suppressing Hh signaling pathway, their IC50 values were larger than that of compound 17. We then enhanced the hydrophilicity of part B by replacing phenyl with pyridine, and it was satisfying that the potency of pyridyl derivative 21 (IC50 = 0.03 μM) was higher as compared to that of phenyl derivative 17. When chlorine located at 2′-position of nicotinamide group, its anti-Hh activity improved (21 vs. 23 and 14 vs. 16). Diminished activity was observed as the chlorine transferred to the 6′-position of the nicotinamide group (22 vs. 23). Overall, compound 21 yielded the most potent activity in this class, which was even 6-fold higher as compared to compound 3. Also, the Hh pathway inhibition of compound 21 was comparable to that of compound 1.
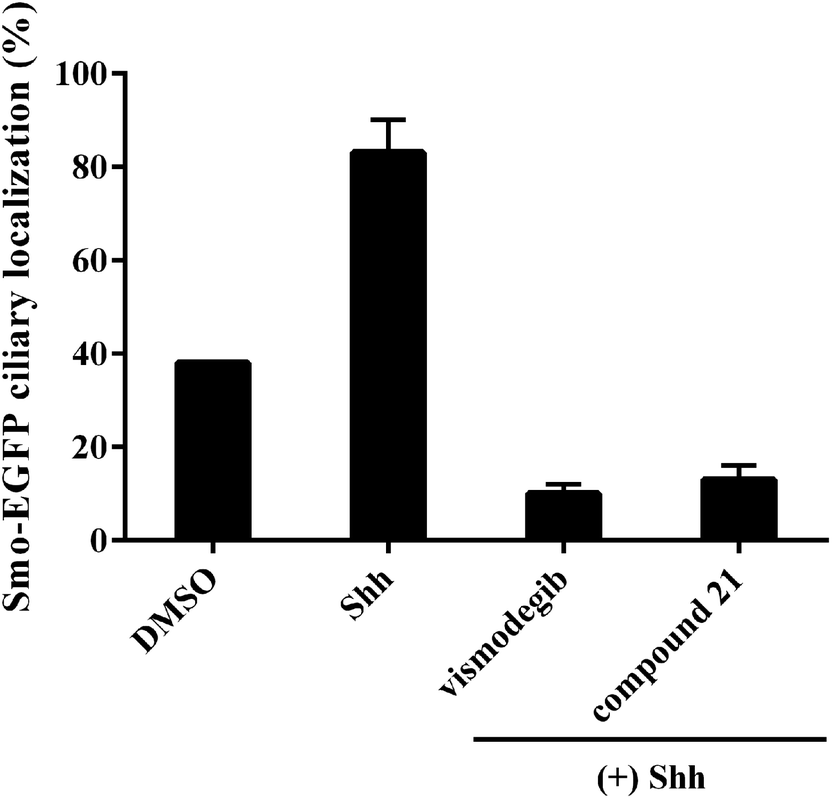
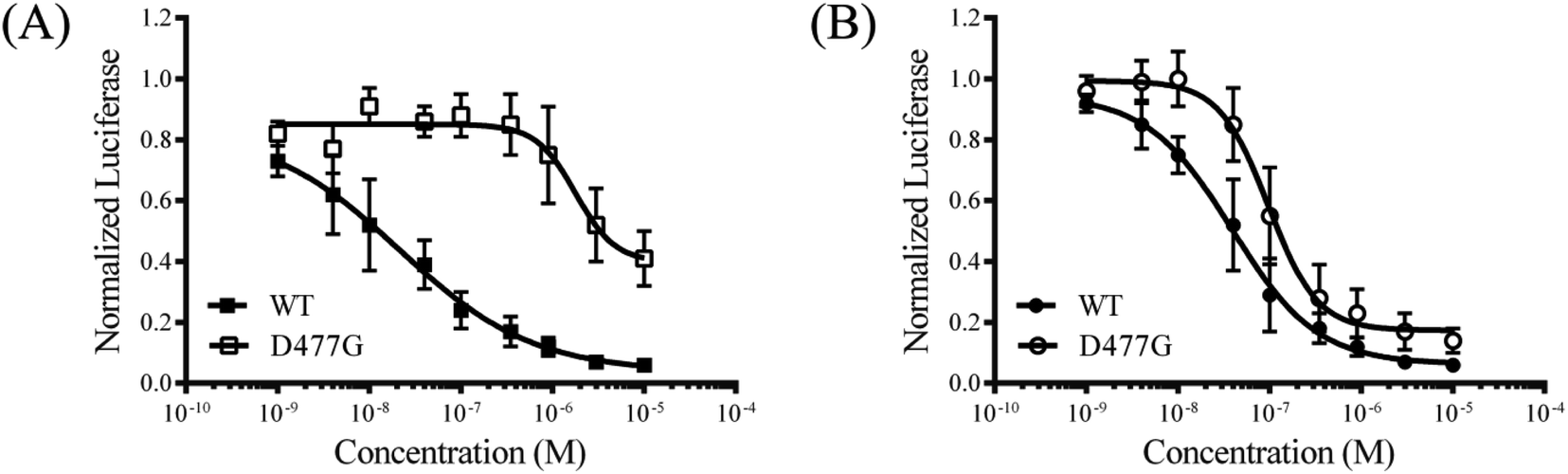
To explore the mode of action of the representative compound 21 in Hh signaling pathway, its effect on Smo ciliary translocation was measured. As presented in Fig. 4, Smo localization in the primary cilia enhanced significantly as the NIH3T3 cells expressing Smo were treated with Shh, an agonist of Hh pathway. In compound 21 group, there was a sharp drop in Smo ciliary translocation, and this result was similar to that of vismodegib group, indicating that the target of compound 21 was Smo receptor. It was reported that drug-resistant mutation in Smo reduced the Hh pathway inhibition of vismodegib by a large of margin.40 To further test the potency of compound 21 on Smo mutation, wild-type (WT) or mutant D477G was over-expressed in the NIH3T3-Gli-Luc reporter cell line. Compliant with previous reports, mutant D477G was refractory to vismodegib (Fig. 5A). In contrast, the Hh pathway inhibitory activity of compound 21 slightly decreased in mutant D477G (Fig. 5B). The 2.4-fold shift in IC50 suggested that mutant D477G did not markedly influence its interaction with Smo (IC50 = 39 nM for WT, IC50 = 96 nM for mutant).
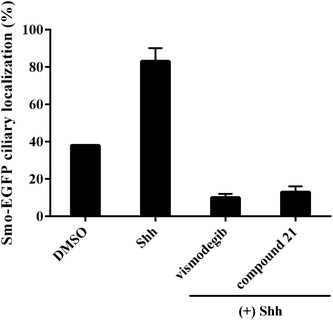 |
| | Fig. 4 Benzamide analogs inhibit Hh pathway through regulating Smo. Error bars represent standard deviation of three parallel groups. | |
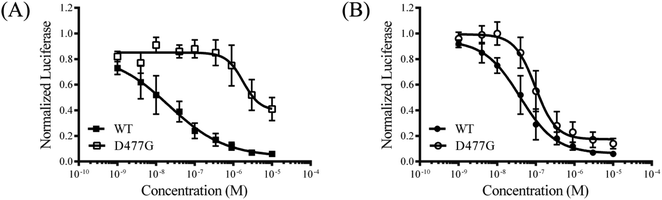 |
| | Fig. 5 The inhibition of drug-resistant Smo mutant for compound 21. The inhibition of Gli-Luc reporter activity by vismodegib (A) and compound 21 (B) in NIH3T3-Gli-Luc cells overexpressing wild-type Smo or Smo D477G. Error data point represents the mean ± SD of three determinations. | |
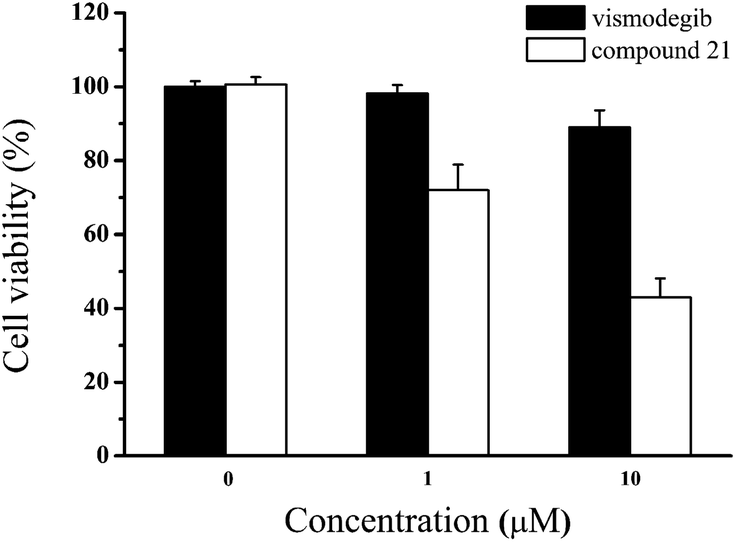
Due to the failure of vismodegib to kill MB cells in patient,41 it was anticipated that compound 21 obtained a positive respond in vitro. Then, we carried out the antiproliferative assay on Daoy cell line which was a proper MB model with consecutive Hh signaling activation. To our delight, compared with vismodegib, compound 21 demonstrated stronger antiproliferation against Daoy cells (Fig. 6). And the cell viabilities of compound 21 were 72% and 43% at concentrations of 1 μM and 10 μM, respectively.
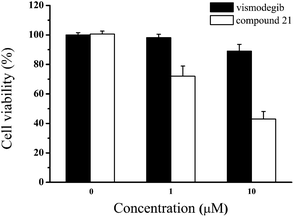 |
| | Fig. 6 Cell viability of Daoy cells incubated with the indicated doses of vismodegib and compound 21 for 48 h. Data represent mean ± SD of experiments performed in triplicate. | |
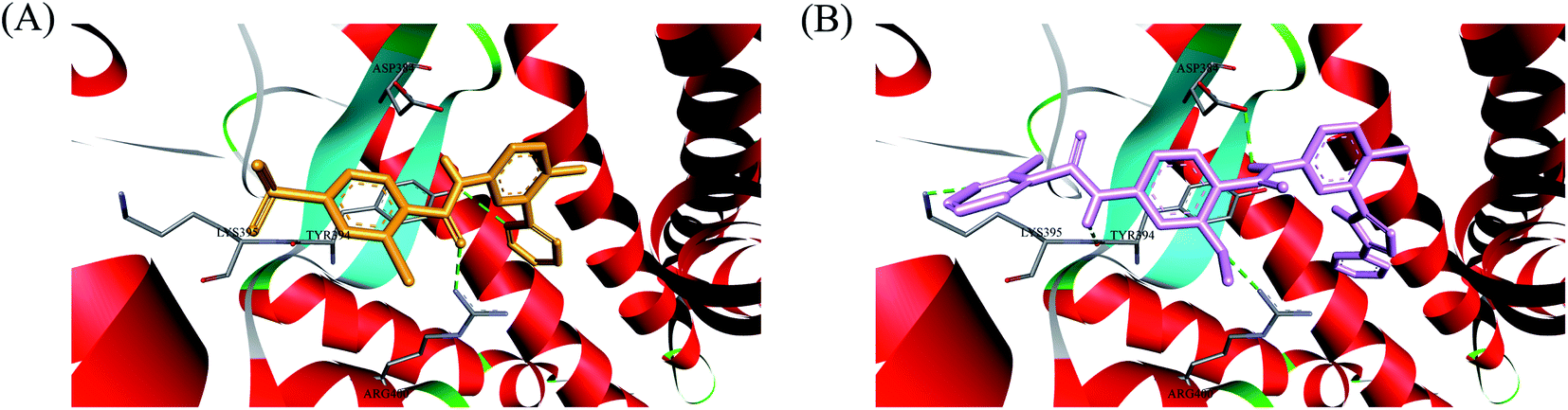
In order to investigate the binding mode of this series of compounds with Smo receptor, a complete molecular docking study was carried out. As presented in Fig. 7A, compound 1 bound with Smo by H-bonds that contained the carbonyl moiety interacting with residue Arg400 and the nitrogen atom of pyridine interacting with residue Tyr394. Additionally, compound 21 kept tight interaction with residues Asp384, Tyr394, Lys395 and Arg400 (Fig. 7B), which showed two more binding sites in comparison with compound 1. The computational modeling study interpreted the better Hh pathway inhibition of compound 21 at the molecular level.
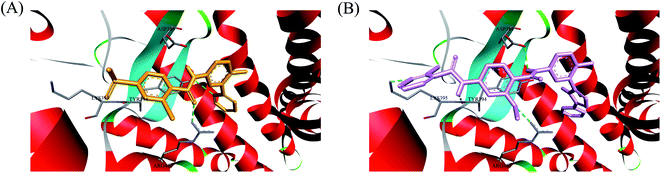 |
| | Fig. 7 (A) Docking conformation of compound 1 (orange) at the binding site. (B) Docking conformation of compound 21 (pink) at the binding site. Hydrogen bonds are represented by the dashed green lines. Surrounding amino acid are shown in grey stick format and labelled. | |
Conclusions
In this report, novel series of Hh signaling pathway antagonists with 2-methoxybenzamide skeleton were explored by structural modification based on SANT-2. In Gli-luc reporter assay, this class of inhibitors entirely showed submicromolar range of IC50 value, signifying that the 2-methoxybenzamide scaffold as connector was advantageous to their Hh pathway inhibition. Thereinto, the IC50 value of compound 21 was 0.03 μM, and the molecular basis of its inhibition was ascribed to the blockade of Smo. Especially, compound 21 retained the suppressive potency against mutant and WT Smo, moreover, it displayed better antiproliferative activity against vismodegib-resistant Daoy cells. Further structural optimization is currently in progress and will be reported in due course.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported financially by the National Natural Science Foundation of China (82003656), Liaoning Science and Technology Project Management (2020-MS-313), the Science and Technology Fund Project of Shenyang Medical College (20201004 and 20205041).
Notes and references
- H. J. Sharpe, W. R. Wang, R. N. Hannoush and F. J. de Sauvage, Nat. Chem. Biol., 2015, 11, 246–255 CrossRef CAS PubMed.
- I. Galperin, L. Dempwolff, W. E. Diederich and M. Lauth, J. Med. Chem., 2019, 62, 8392–8411 CrossRef CAS PubMed.
- A. E. Owens, I. D. Paola, W. A. Hansen, Y. W. Liu, S. D. Khare and R. Fasan, J. Am. Chem. Soc., 2017, 139, 12559–12568 CrossRef CAS PubMed.
- M. H. Xin, X. Y. Ji, L. K. D. L. Cruz, S. Thareja and B. H. Wang, Med. Res. Rev., 2018, 38, 870–913 CrossRef PubMed.
- A. Salaritabar, I. Berindan-Neagoe, B. Darvish, F. Hadjiakhoondi, A. Manayi, K. P. Devi, D. Barreca, I. E. Orhan, I. Suntar, A. A. Farooqi, D. Gulei, S. F. Nabavi, A. Sureda, M. Daglia, A. R. Dehpour, S. M. Nabavi and S. Shirooie, Pharmacol. Res., 2019, 141, 466–480 CrossRef CAS PubMed.
- I. Deshpande, J. H. Liang, D. Hedeen, K. J. Roberts, Y. X. Zhang, B. Ha, N. R. Latorraca, B. Faust, R. O. Dror, P. A. Beachy, B. R. Myers and A. Manglik, Nature, 2019, 571, 284–288 CrossRef CAS PubMed.
- J. E. Cortes, R. Gutzmer, M. W. Kieran and J. A. Solomon, Cancer Treat. Rev., 2019, 76, 41–50 CrossRef CAS PubMed.
- H. Farrokhpour, V. Pakatchian, A. Hajipour, F. Abyar, A. N. Chermahini and F. Fakhari, RSC Adv., 2015, 5, 68829–68838 RSC.
- J. Kim, E. Y. C. Hsia, A. Brigui, A. Plessis, P. A. Beachy and X. Y. Zheng, Sci. Signaling, 2015, 8, ra55 CrossRef PubMed.
- D. Bhattarai, J. H. Jung, S. Han, H. Lee, S. J. Oh, H. W. Ko and K. Lee, Eur. J. Med. Chem., 2017, 125, 1036–1050 CrossRef CAS PubMed.
- A. M. Arensdorf, M. E. Dillard, J. M. Menke, M. W. Frank, C. O. Rock and S. K. Ogden, Cell Rep., 2017, 19, 2074–2087 CrossRef CAS PubMed.
- D. Z. Ji, W. W. Zhang, Y. G. Xu and J. J. Zhang, Bioorg. Med. Chem., 2020, 28, 115354 CrossRef PubMed.
- T. N. Trinh, E. A. McLaughlin, C. P. Gordon, I. R. Bernstein, V. J. Pye, K. A. Redgrove and A. McCluskey, Org. Biomol. Chem., 2017, 15, 3046–3059 RSC.
- Y. Wang, A. C. Arvanites, L. Davidow, J. Blanchard, K. Lam, J. W. Yoo, S. Coy, L. L. Rubin and A. P. McMahon, ACS Chem. Biol., 2012, 7, 1040–1048 CrossRef CAS PubMed.
- E. Y. Lee, H. K. Ji, Z. Q. Ouyang, B. Y. Zhou, W. X. Ma, S. A. Vokes, A. P. McMahon, W. H. Wong and M. P. Scott, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9736–9741 CrossRef CAS PubMed.
- B. Biehs, G. J. P. Dijkgraaf, R. Piskol, B. Alicke, S. Boumahdi, F. Peale, S. E. Gould and F. J. de Sauvage, Nature, 2018, 562, 429–433 CrossRef CAS PubMed.
- S. M. Waszak, G. W. Robinson, B. L. Gudenas, K. S. Smith, A. Forget, M. Kojic, J. Garcia-Lopez, J. Hadley, K. V. Hamilton, E. Indersie, I. Buchhalter, J. Kerssemakers, N. Jäger, T. Sharma, T. Rausch, M. Kool, D. Sturm, D. T. W. Jones, A. Vasilyeva, R. G. Tatevossian, G. Neale, B. Lombard, D. Loew, J. Nakitandwe, M. Rusch, D. C. Bowers, A. Bendel, S. Partap, M. Chintagumpala, J. Crawford, N. G. Gottardo, A. Smith, C. Dufour, S. Rutkowski, T. Eggen, F. Wesenberg, K. Kjaerheim, M. Feychting, B. Lannering, J. Schüz, C. Johansen, T. V. Andersen, M. Röösli, C. E. Kuehni, M. Grotzer, M. Remke, S. Puget, K. W. Pajtler, T. Milde, O. Witt, M. Ryzhova, A. Korshunov, B. A. Orr, D. W. Ellison, L. Brugieres, P. Lichter, K. E. Nichols, A. Gajjar, B. J. Wainwright, O. Ayrault, J. O. Korbel, P. A. Northcott and S. M. Pfister, Nature, 2020, 580, 396–401 CrossRef CAS PubMed.
- V. Kumar, G. Mondal, P. Slavik, S. Rachagani, S. K. Batra and R. I. Mahato, Mol. Pharmaceutics, 2015, 12, 1289–1298 CrossRef CAS PubMed.
- L. Vesci, F. M. Milazzo, M. A. Stasi, S. Pace, F. Manera, C. Tallarico, E. Cini, E. Petricci, F. Manetti, R. D. Santis and G. Giannin, Eur. J. Med. Chem., 2018, 157, 368–379 CrossRef CAS PubMed.
- C. Zhao, A. Chen, C. H. Jamieson, M. Fereshteh, A. Abrahamsson, J. Blum, H. Y. Kwon, J. Kim, J. P. Chute, D. Rizzieri, M. Munchhof, T. VanArsdale, P. A. Beachy and T. Reya, Nature, 2009, 458, 776–779 CrossRef CAS PubMed.
- M. Wahid, A. Jawed, R. K. Mandal, S. A. Dar, S. Khan, N. Akhter and S. Haque, Critical Reviews in Oncology/Hematology, 2016, 98, 235–241 CrossRef PubMed.
- A. M. Giannetti, H. Wong, G. J. P. Dijkgraaf, E. C. Dueber, D. F. Ortwine, B. J. Bravo, S. E. Gould, E. G. Plise, B. L. Lum, V. Malhi and R. A. Graham, J. Med. Chem., 2011, 54, 2592–2601 CrossRef CAS PubMed.
- S. Jain, R. L. Song and J. W. Xie, OncoTargets Ther., 2017, 10, 1645–1653 CrossRef CAS PubMed.
- A. Kumari, A. N. Ermilov, M. Grachtchouk, A. A. Dlugosz, B. L. Allen, R. M. Bradley and C. M. Mistretta, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10369–10378 CrossRef PubMed.
- F. Ghirga, M. Mori and P. Infante, Bioorg. Med. Chem. Lett., 2018, 28, 3131–3140 CrossRef CAS PubMed.
- S. X. Atwood, K. Y. Sarin, R. J. Whitson, J. R. Li, G. Kim, M. Rezaee, M. S. Ally, J. Kim, C. Yao, A. L. S. Chang, A. E. Oro and J. Y. Tang, Cancer Cell, 2015, 27, 342–353 CrossRef CAS PubMed.
- X. C. Dong, C. L. Wang, Z. J. Chen and W. L. Zhao, Drug Discovery Today, 2018, 23, 704–710 CrossRef CAS PubMed.
- S. Coni, P. Infante and A. Gulino, Biochem. Pharmacol., 2013, 85, 623–628 CrossRef CAS PubMed.
- H. Y. Tao, Q. H. Jin, D. Koo, X. B. Liao, N. P. Englund, Y. Wang, A. Ramamurthy, P. G. Schultz, M. Dorsch, J. Kelleher and X. Wu, Chem. Biol., 2011, 18, 432–437 CrossRef CAS PubMed.
- P. L. Bedard, D. M. Hyman, M. S. Davids and L. L. Siu, Lancet, 2020, 395, 1078–1088 CrossRef CAS.
- M. J. Munchhof, Q. F. Li, A. Shavnya, G. V. Borzillo, T. L. Boyden, C. S. Jones, S. D. LaGreca, L. Martinez-Alsina, N. Patel, K. Pelletier, L. A. Reiter, M. D. Robbins and G. T. Tkalcevic, ACS Med. Chem. Lett., 2012, 3, 106–111 CrossRef CAS PubMed.
- J. K. Chen, J. Taipale, K. E. Young, T. Maiti and P. A. Beachy, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14071–14076 CrossRef CAS PubMed.
- A. Büttner, K. Seifert, T. Cottin, V. Sarli, L. Tzagkaroulaki, S. Scholz and A. Giannis, Bioorg. Med. Chem., 2009, 17, 4943–4954 CrossRef PubMed.
- J. E. Cortes, F. H. Heidel, A. Hellmann, W. Fiedler, B. D. Smith, T. Robak, P. Montesinos, D. A. Pollyea, P. DesJardins, O. Ottmann, W. W. Ma, M. N. Shaik, A. D. Laird, M. Zeremski, A. O'Connell, G. Chan and M. Heuser, Leukemia, 2019, 33, 379–389 CrossRef CAS PubMed.
- C. Wang, H. X. Wu, V. Katritch, G. W. Han, X. P. Huang, W. Liu, F. Y. Siu, B. L. Roth, V. Cherezov and R. C. Stevens, Nature, 2013, 497, 338–343 CrossRef CAS PubMed.
- M. R. Tremblay, M. Nesler, R. Weatherhead and A. C Castro, Expert Opin. Ther. Pat., 2009, 19, 1039–1056 CrossRef CAS PubMed.
- V. Kumar, A. K. Chaudhary, Y. X. Dong, H. A. Zhong, G. Mondal, F. Lin, V. Kumar and R. I. Mahato, Sci. Rep., 2017, 7, 1665 CrossRef PubMed.
- E. F. Byrne, R. Sircar, P. S. Miller, G. Hedger, G. Luchetti, S. Nachtergaele, M. D. Tully, L. Mydock-McGrane, D. F. Covey, R. P. Rambo, M. S. Sansom, S. Newstead, R. Rohatgi and C. Siebold, Nature, 2016, 535, 517–522 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- S. Ohshima-Hosoyama, M. A. Davare, T. Hosoyama, L. D. Nelon and C. Keller, J. Neuro-Oncol., 2011, 105, 475–483 CrossRef CAS PubMed.
- R. L. Yauch, G. J. Dijkgraaf, B. Alicke, T. Januario, C. P. Ahn, T. Holcomb, K. Pujara, J. Stinson, C. A. Callahan, T. Tang, J. F. Bazan, Z. Kan, S. Seshagiri, C. L. Hann, S. E. Gould, J. A. Low, C. M. Rudin and F. J. de Sauvage, Science, 2009, 326, 572–574 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00732g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Dajun Zhang,
Tian Luan,
Youbing Wang,
Wenhu Zhang,
Lin Lin
*,
Dajun Zhang,
Tian Luan,
Youbing Wang,
Wenhu Zhang,
Lin Lin