
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioinspired modified graphene oxide/polyurethane composites with rapid self-healing performance and excellent mechanical properties†
Yahao Liua,
Jian Zheng*a,
Xiao Zhang *b,
Yongqiang Dua,
Guibo Yua,
Ke Lic,
Yunfei Jiaa and
Yu Zhanga
*b,
Yongqiang Dua,
Guibo Yua,
Ke Lic,
Yunfei Jiaa and
Yu Zhanga
aShijiazhuang Campus, Army Engineering University, Shijiazhuang 050003, China. E-mail: zhengj2020@163.com
bEngineering University of PAP, Xi'an 710086, China. E-mail: zxleo@foxmail.com
cCollege of Naval Architecture and Ocean Engineering, Naval University of Engineering, Wuhan 430033, China
First published on 20th April 2021
Abstract
Self-healing efficiency and mechanical strength are always a pair of mechanical contradictions of a polymer. Herein, a series of novel mussel-inspired modified graphene oxide/polyurethane composites were successfully fabricated via rational molecular design and introducing hyperbranched polymer-modified graphene oxide. The composites exhibit outstanding self-healing performances with a self-healing efficiency of 87.9%. Especially, their self-healing properties possess exceptional water-insensitivity, which presents a high self-healing efficiency of 92.5% under 60 °C water for 2 h and 74.6% under 25 °C water for 6 h. Furthermore, the tensile strength of the composites increased by 107.7% with a high strain of 2170%. In addition, the composites show a remarkable recovery capability of 76.3% and 83.7% under tensile and compression loading, respectively, after 20 cycles. This strategy shows prominent application potential in high-performance solid propellants, protective coating, electronic skin, soft sensors and other water-insensitive devices.
1. Introduction
Polyurethane (PU), a kind of polymeric material with unmatchable properties, has aroused widespread attention in numerous fields, such as insulation, coating and diaphragms.1,2 Hydroxyl-terminated polybutadiene (HTPB)-based PU is one of the promising materials for composite solid propellant binders and protective coating3–5 owing to its outstanding elasticity, toughness and water-resistance, as well as sub-ambient glass transition temperature (Tg).6–10 However, as a kind of conventional thermosetting polymeric material with irreversible crosslinking, it cannot be repaired, once damaged.Over the past decade, self-healing technology has attracted considerable attention, which provides a feasible route to address the above-mentioned problems. Commonly, there are two strategies to achieve self-healing abilities, including extrinsic and intrinsic self-healing, according to the self-healing mechanisms.11–13 The extrinsic self-healing materials generally require repairing agents stored in pre-embedded microcapsules or microvascular networks to achieve the self-healing process, showing a drawback of single/few-time healing with the exhaustion of repairing agents.14–16 In contrast, the intrinsic self-healing materials can achieve repeated repair without external repairing agents by dynamic reversible covalent bonds and non-covalent interactions, such as hydrogen bond,17,18 supramolecular interaction,19,20 metal–ligand interaction,21 π–π interaction,22 Diels–Alder chemistry,23 host–guest interactions,24 and disulfide bond exchange.25–27 Recently, aromatic disulfide bonds have been determined to be a promising reversible bond to achieve self-healing at room temperature owing to their low bond dissociation energy and stable free radicals.28,29 Moreover, reports have also pointed out that hydrogen bonds and disulfide bonds can synergistically promote the self-healing process. To be specific, hydrogen bonds might accelerate initial sticking or interfacial adhesion, promoting the diffusion of molecular chains and accelerating the exchange reactions of disulfide bonds.2,30 Along with this, disulfide bonds will weaken the strength of hydrogen bonds, leading to their easy dissociation under mild conditions.28 Hence, a polymer with rapid room-temperature self-healing capability may be obtained by introducing both hydrogen bonding network and aromatic disulfide bonds into a polymer network simultaneously.
Nature, during a long evolutionary process, has evolved numerous remarkably intelligent biological systems and marine mussel is one of them. Nowadays, marine mussel has aroused considerable attention in various fields, especially in self-healing materials.31–33 The mussel-inspired self-healing materials originate from the fact that the high content of catechol groups in the mussel adhesive proteins can form strong covalent and non-covalent bonds with various surfaces,32,34,35 thereby possessing outstanding adhesion ability. Especially, the dihydroxy of catechol groups can form bidentate hydrogen and metal-mediated reversible coordination, which is important for the self-healing process.32,33 Moreover, mussel adhesive protein exhibits outstanding adhesion properties in an extremely harsh environment of the ocean,36 and the mussel-inspired catechol groups can be well-bonding and maintain high adhesion underwater.37 Inspired by the above-mentioned features, it is expected to design an HTPB-based PU with underwater and room-temperature self-healing ability based on disulfide bonds and mussel-mimetic hydrogen bonds, which is significant and consistent with its long-term working environment.38
Nevertheless, we found that HTPB-based PU based on the above design had a low mechanical strength during our study process, which severely limited its applications in many fields. In fact, mechanical strength and self-healing efficiency are always a pair of mechanical contradictions of the polymer.39–43 It is difficult and necessary to develop materials that possess both adequate mechanical strength and rapid self-healing speed. Recently, graphene oxide (GO), a specific derivative of graphene, has been considered a promising reinforcer for polymer composites to achieve high strength because of its remarkable properties,44–46 and some studies have attempted to add GO to develop novel self-healing materials.47–49 However, it has been reported that GO tends to aggregate in the polymeric matrix and has poor interfacial interaction with the matrix.50,51 Therefore, the current strategy is to attach some active groups onto the surface of GO sheets to react and improve the compatibility between GO and matrix.4,52 Consequently, the above-mentioned information prompted us to believe that integrating modified GO with rational molecule design might generate a novel composite that has not only better self-healing properties but also improved mechanical properties.
Herein, we prepared a series of composites with excellent mechanical properties and rapid room-temperature self-healing performances. In our design, the incorporation of modified GO mainly enables the composite with excellent mechanical strength, while disulfide bonds and mussel-mimetic hydrogen bonds within the polymer chains provide self-healing ability. Effects of modified GO on the mechanical and self-healing properties were investigated carefully.
2. Materials and experiments
2.1. Materials
Monolayer GO with a purity of >99% was obtained from Nanjing XFNANO Material Technology Co., Ltd, China. Hydroxyl-terminated polybutadiene (HTPB, 99.9%, hydroxyl value: 0.75 mmol g−1) was purchased from Liming Research & Design Institute of Chemical Industry Co., Ltd., Luoyang, China. Isophorone diisocyanate (IPDI, 99%), and triphenylbismuthine (TPB) were obtained from Macklin Biochemical Technology Co., Ltd, Shanghai, China. The amino-terminated hyperbranched polyamide (HBPA) was supplied by Wuhan Hyperbranched Polymers Science & Technology Co., Ltd, China. N,N-Dimethylformamide (DMF, ≥99.8%) and tetrahydrofuran (THF, ≥99.5%) were obtained from Damao Chemical Reagent Factory, Tianjin, China and used directly. Dopamine hydrochloride (DA, 98%), O-(7-azabenzotriazole-1-yl)-N,N,N,N′-tetramethyluronium hexafluorophosphate (HATU, 99%) and 2-aminophenyl disulfide (APD, 98%) were purchased from Aladdin Co., Shanghai, China.2.2. Preparation of modified GO (MGO)
In a typical procedure, GO (100 mg) was first suspended in 30 ml DMF followed by stirring and sonication for 1 h to obtain the well-exfoliated GO suspension. Meanwhile, the HBPA (1.0 g) was dissolved in DMF (20 ml) with sonication for 10 min. Subsequently, the GO suspension was added to the HBPA solution and sonicated for 30 min. After that, 10 mg of HATU was dispersed in the obtained mixture and continuously sonicated for 3 h at room temperature. Afterward, the modified GO was separated by centrifugation and washed with excess ethanol at least four times to remove other molecules. Finally, the obtained product was dried under vacuum at 60 °C for 24 h and coded as MGO.2.3. Fabrication of self-healing composites
First, HTPB (3 g, –OH: 2.25 mmol) was placed in a 100 ml three-necked flask and dried at 60 °C overnight under vacuum. After that, HTPB was preheated at 70 °C, then IPDI (2.25 mmol) and TPB (3 mg) in DMF (5 ml) were added to the flask. The mixture was stirred for 3 h at 70 °C under nitrogen protection. Then, 3 mg (0.1 wt%) of MGO was dispersed in DMF by sonicating for 1 h to obtain a uniform MGO suspension that was poured into the flask to react for a further 2 h at 50 °C. Next, APD (0.9 mmol) was added gradually, and the mixture was stirred for another 2 h (PU–S). Thereafter, DA (0.45 mmol) was added and the reaction was continued for another 3 h at 40 °C. Finally, the samples were obtained by pouring the mixture into a Teflon mold and drying at 60 °C for 36 h in a vacuum oven to evaporate the solvent, and coded as SPUM-2. For comparison, samples with different contents of MGO and GO were prepared in the same method as above. The preparation formula of the sample is summarized in Table 1.| Reagent | SPU | SPUM-1 | SPUM-2 | SPUM-3 | SPUM-4 | SPUG-1 | SPUG-2 |
|---|---|---|---|---|---|---|---|
| MGO | 0 | 0.05% | 0.1% | 0.15% | 0.2% | 0 | 0 |
| GO | 0 | 0 | 0 | 0 | 0 | 0.05% | 0.1% |
2.4. Characterization
The Fourier transform infrared spectroscopy (FTIR) measurements performed using an infrared spectrometer (NICOLET iS10) in the wavenumber range of 500–4000 cm−1 with a resolution of 4 cm−1. Ultraviolet-visible (UV-vis) spectra were recorded using a UV-vis spectrophotometer (Shimadzu UV-3600, Japan). X-ray diffraction (XRD) was performed using a Persee XD6 diffractometer with a scanning speed of 10° min−1 from 5° to 80°. Raman spectra were obtained on a Raman spectrometer (inVia, Renishaw) with laser-excited at 532 nm. The microstructure and morphology of the GO and HGO were observed using transmission electron microscopy (TEM, JSM-2100F, Japan), scanning electron microscopy (SEM, JSM-7800F, Japan), and atomic force microscopy (AFM, Nano Man VS, Bruker). Thermogravimetric analysis (TGA, SDT Q600, TA instrument) was performed at a heating rate of 10 °C min−1 from 30 °C to 800 °C under nitrogen atmosphere. Glass transition temperatures of samples were obtained using a differential scanning calorimeter (DSC, Q20, TA Instruments) under nitrogen atmosphere, and the heating rate was set at 10 °C min−1 in the temperature range of −90 °C to 60 °C. The dynamic mechanical analysis of samples was performed on a DMA Q800 (TA Instruments) in a single cantilever beam mode with a frequency range from 0.1 Hz to 100 Hz. Tensile and compression tests were performed on an Instron 5982 at rates of 100 mm min−1 and 5 mm min−1, respectively (testing temperature was 20 °C). Cyclic tensile and compression tests were also conducted using an Instron 5982 instrument with a maximum strain of 100% and 70% in every cycle at a settled stretch rate of 20 mm min−1 and 5 mm min−1, respectively. For the healing experiment, samples were cut into two pieces and immediately put into contact without an exterior force for a certain time and temperature. The self-healing efficiency (η) was defined as:2where σhealed and σoriginal are the tensile strengths of the healed and the original samples, respectively. Dumbbell specimens (gauge length × width × thickness: 35 × 5 × 2 mm) were utilized to measure the mechanical properties and the self-healing performance of the samples.
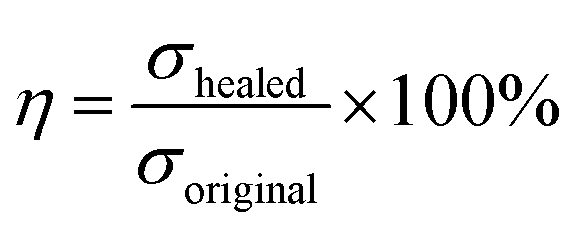
3. Results and discussion
3.1. Design, synthesis and characteristics of materials
First of all, we modified GO with amino-terminated hyperbranched polyamide (HBPA) to improve the compatibility and interfacial interactions of GO with the polymer matrix. We chose HBPA as the modification agent mainly based on the following considerations: HBPA is a kind of hyperbranched polymer with three-dimensional branching structures, nonentangled architectures, low solution viscosity and ultrahigh solubility, which have aroused increasing attention in reinforcing polymer matrix recently.52–54 Meanwhile, the abundant reactive terminal groups on its molecular chains can react with GO and polymer matrix simultaneously, resulting in a stronger interfacial interactions and adhesion between GO and polymer matrix and beneficial to improve the mechanical properties.55–57The modification of GO with HBPA was determined using FTIR spectra. As shown in Fig. 1(a), the characteristic absorption peaks of GO at 1728 cm−1, 1628 cm−1 and 1053 cm−1 correspond to stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC and C–O, respectively, and the wide absorption peak at 3300–3000 cm−1 is ascribed to stretching vibrations of O–H in hydroxyl and carboxylic acid.58,59 In the case of MGO, after modification, the peak at 1728 cm−1 disappeared while some new absorption peaks at 3250 cm−1, 2959–2850 cm−1, 1640 cm−1 and 1555 cm−1 appeared, which are assigned to stretching vibrations of N–H groups, stretching vibrations of –CH3 and –CH2 groups, amide CO stretching (amide I), amide N–H bending and C–N stretching (amide II), respectively, implying that GO was successfully modified with HBPA via the reaction between the terminal amine of HBPA and carboxylic acid groups of GO.51,60 XRD patterns of GO and MGO are shown in Fig. 1(b). Due to the incorporation of HBPA onto the surface of GO, the 2θ peak of MGO was shifted down to 9.05°, indicating a larger interlayer spacing of 0.97 nm. It is known that the peak intensity ratio of D and G bands (ID/IG) on Raman spectra (Fig. 1(c)) is related to disordered structures of carbon, and the lower ID/IG ratio corresponds to fewer defects.61 After functionalization, the ID/IG value of MGO increases slightly from 0.926 to 1.007. Obviously, more defects are formed, reflecting the successful modification with HBPA.
O, CC and C–O, respectively, and the wide absorption peak at 3300–3000 cm−1 is ascribed to stretching vibrations of O–H in hydroxyl and carboxylic acid.58,59 In the case of MGO, after modification, the peak at 1728 cm−1 disappeared while some new absorption peaks at 3250 cm−1, 2959–2850 cm−1, 1640 cm−1 and 1555 cm−1 appeared, which are assigned to stretching vibrations of N–H groups, stretching vibrations of –CH3 and –CH2 groups, amide CO stretching (amide I), amide N–H bending and C–N stretching (amide II), respectively, implying that GO was successfully modified with HBPA via the reaction between the terminal amine of HBPA and carboxylic acid groups of GO.51,60 XRD patterns of GO and MGO are shown in Fig. 1(b). Due to the incorporation of HBPA onto the surface of GO, the 2θ peak of MGO was shifted down to 9.05°, indicating a larger interlayer spacing of 0.97 nm. It is known that the peak intensity ratio of D and G bands (ID/IG) on Raman spectra (Fig. 1(c)) is related to disordered structures of carbon, and the lower ID/IG ratio corresponds to fewer defects.61 After functionalization, the ID/IG value of MGO increases slightly from 0.926 to 1.007. Obviously, more defects are formed, reflecting the successful modification with HBPA.
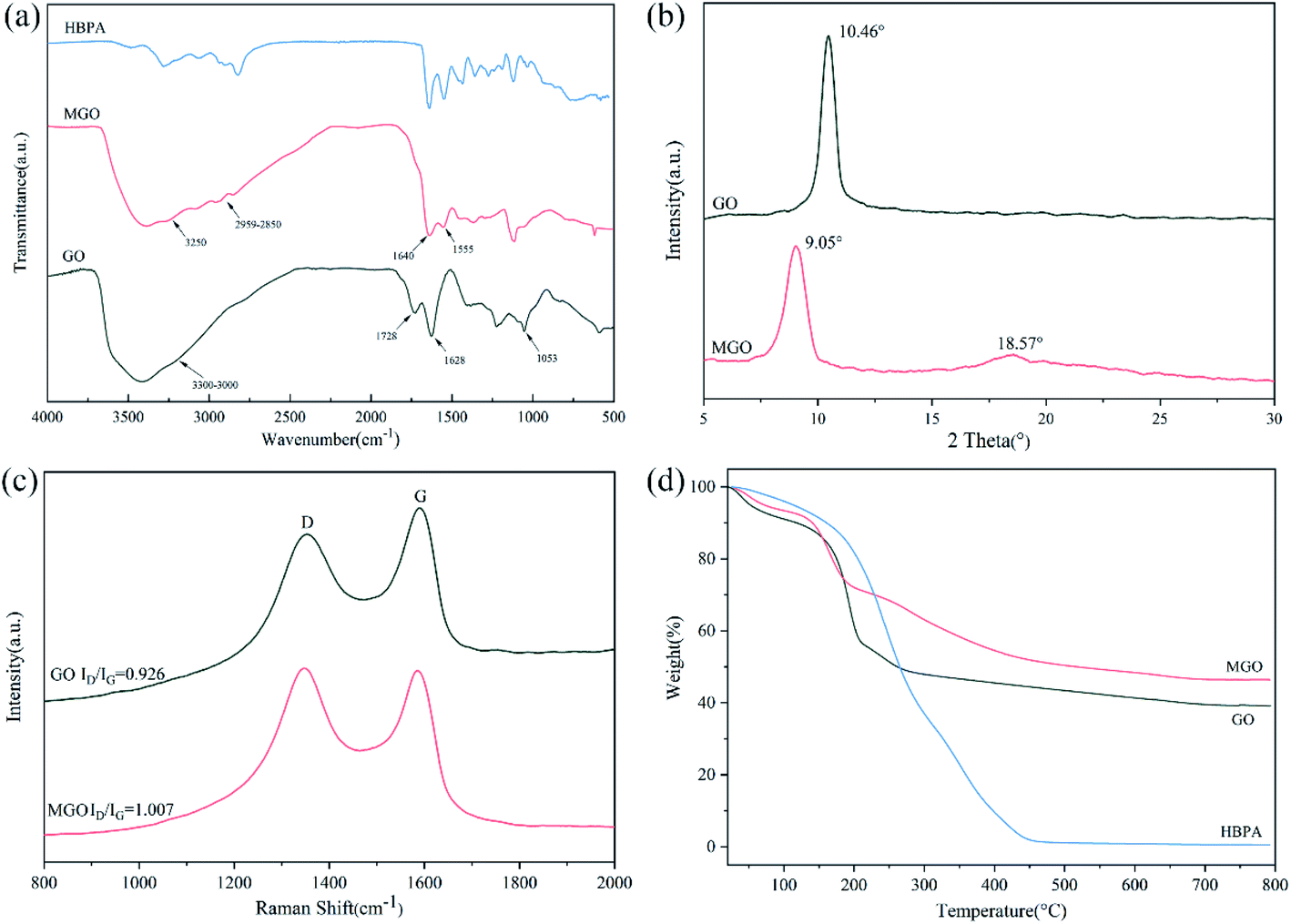 | ||
| Fig. 1 (a) FTIR spectra of GO, MGO and HBPA. (b) XRD spectra of GO and MGO. (c) Raman spectra of GO and MGO. (d) TGA spectra of GO, MGO and HBPA. | ||
TGA was also conducted to confirm the success of functionalization of GO (Fig. 1(d)). It can be seen that there are two main loss stages on the TGA curve of GO, which can be ascribed to the loss of absorbed water and the decomposition of labile oxygen-containing groups on the surface of GO, respectively.62 As for HBPA, there is only a sharp decomposition stage from 200 °C to 460 °C and the residual weight was only 0.54 wt%. In the case of MGO, the weight loss of the second decomposition stage was significantly reduced compared to that of GO. It can be explained in a way that during the modification procedure, HBPA replaced some oxygen-containing groups via the reaction between –COOH of GO and –NH2 of HBPA. Notably, a new decomposition stage of MGO from 200 °C to 450 °C can be observed, which corresponds to the decomposition of grafted HBPA.
The morphological structure was also studied using TEM. As we can see from TEM images (Fig. 2), GO sheets display better transparency than MGO, which are similar to the images reported in previous related studies.52,63 In addition, elemental mapping of MGO can further demonstrate successful modification with HBPA as presented in its STEM images shown in Fig. 2(c and d). It can be found that the nitrogen element, originated from HBPA, was evenly distributed on its surface. Additionally, AFM results (Fig. 2(e and f)) indicated that the thickness of MGO is ∼4.35 nm, which is obviously thicker than that of GO (∼0.8 nm), owing to the grafting of HBPA. Combined with the results from the above various characterizations, we confirm that the HBPA was successfully grafted onto the surface of GO.
 | ||
| Fig. 2 TEM images of (a) GO and (b) MGO. (c and d) STEM image of MGO and the corresponding elemental mapping of nitrogen. AFM images of (e) GO and (f) MGO. | ||
Subsequently, MGO-reinforced self-healing polyurethane composites were fabricated via a four-step strategy (Fig. 3). First, a terminal –NCO prepolymer was synthesized using HTPB and IPDI as the raw materials. Second, MGO was added to cross-linking with the prepolymer. Then, APD was used as a chain extender to react with the prepolymer, introducing the dynamic reversible disulfide bonds. Finally, DA was added to consume the remaining –NCO groups, yielding the mussel-mimetic adhesion structure, which can provide hydrogen bonds. Just by changing the contents of MGO, a series of self-healing polyurethane were fabricated. As far as we know, high self-healing efficiency is always at the expense of mechanical properties.39–43 In our design, the self-healing properties originating from dynamic covalent bonds and mussel-mimetic structure, and the improved mechanical properties are well-combined in the HTPB-based PU. On the one hand, aromatic disulfide bonds can exhibit dynamic properties at room temperature,29 which is conducive to the self-healing process at room temperature. On the other hand, the catechol groups on DA can form a weak dual hydrogen bond, which plays an initial sticking or interfacial adhesion role to promote the self-healing process, and the presence of the catechol groups is potential to achieve underwater self-healing.37 In addition, well-dispersed MGO sheets greatly improved mechanical properties on the premise of maintaining high self-healing efficiency. Finally, the superior water-resistance of HTPB gives it the innate advantage of underwater self-healing.10
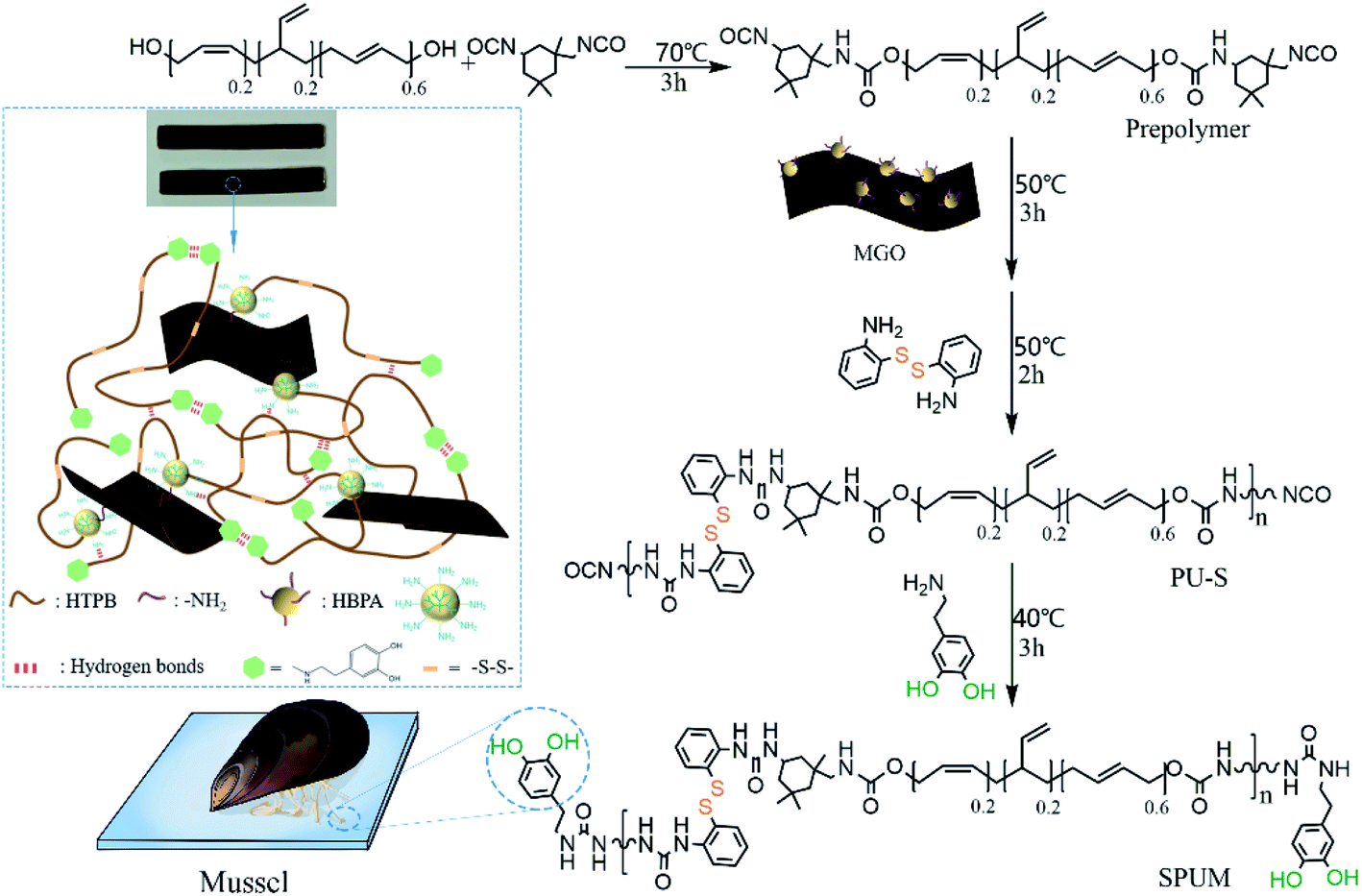 | ||
| Fig. 3 Synthesis of the self-healing polyurethane composites. | ||
The chemical structure of composites can be determined by FTIR. Fig. 4(a) exhibits FTIR spectra of DA, PU–S and SPU. The spectrum of DA shows characteristic peaks of O–H, C–N and N–H.36 For PU–S, there are obvious characteristic peaks at 3331 cm−1 and 1709 cm−1, corresponding to the stretching vibrations of N–H and CO in urethane linkage, and the peaks at 1520 cm−1 and 1350 cm−1 are attributed to urea N–H bending vibration (amide II) and aromatic C–N stretching vibration, respectively.64 Additionally, there is an obvious absorption peak at 2266 cm−1, attributed to the remaining –NCO groups, which disappear absolutely after adding DA in SPU. The presence of DA can be confirmed by utilizing UV-vis spectroscopy as elaborated in Fig. 4(b). The SPU sample shows the absorption peak at ∼280 nm, which is the characteristic peak of DA.33,65 Furthermore, the glass transition temperatures (Tg) of the obtained composites were measured by DSC tests and the curves are displayed in Fig. 4(c). It can be seen that the Tg value increases with the increase of MGO content, which can be assigned to the crosslinking of MGO and polymer chains, limiting the mobility of molecular chains. Notably, the Tg values of all samples are far below room temperature (∼−79 °C), implying the polymer chains hold high mobility at room temperature, which is beneficial to the self-healing process. The dispersion degree and interfacial interaction of the nanofiller have a great influence on the mechanical properties of composites.4 XRD spectra display a sharp peak of GO at the 2θ position of 9.05° (Fig. 1(b)), whereas SPU and SPUM-2 show similar XRD patterns, revealing that GO nanosheets are uniformly dispersed in the PU matrix (Fig. 4(d)).66
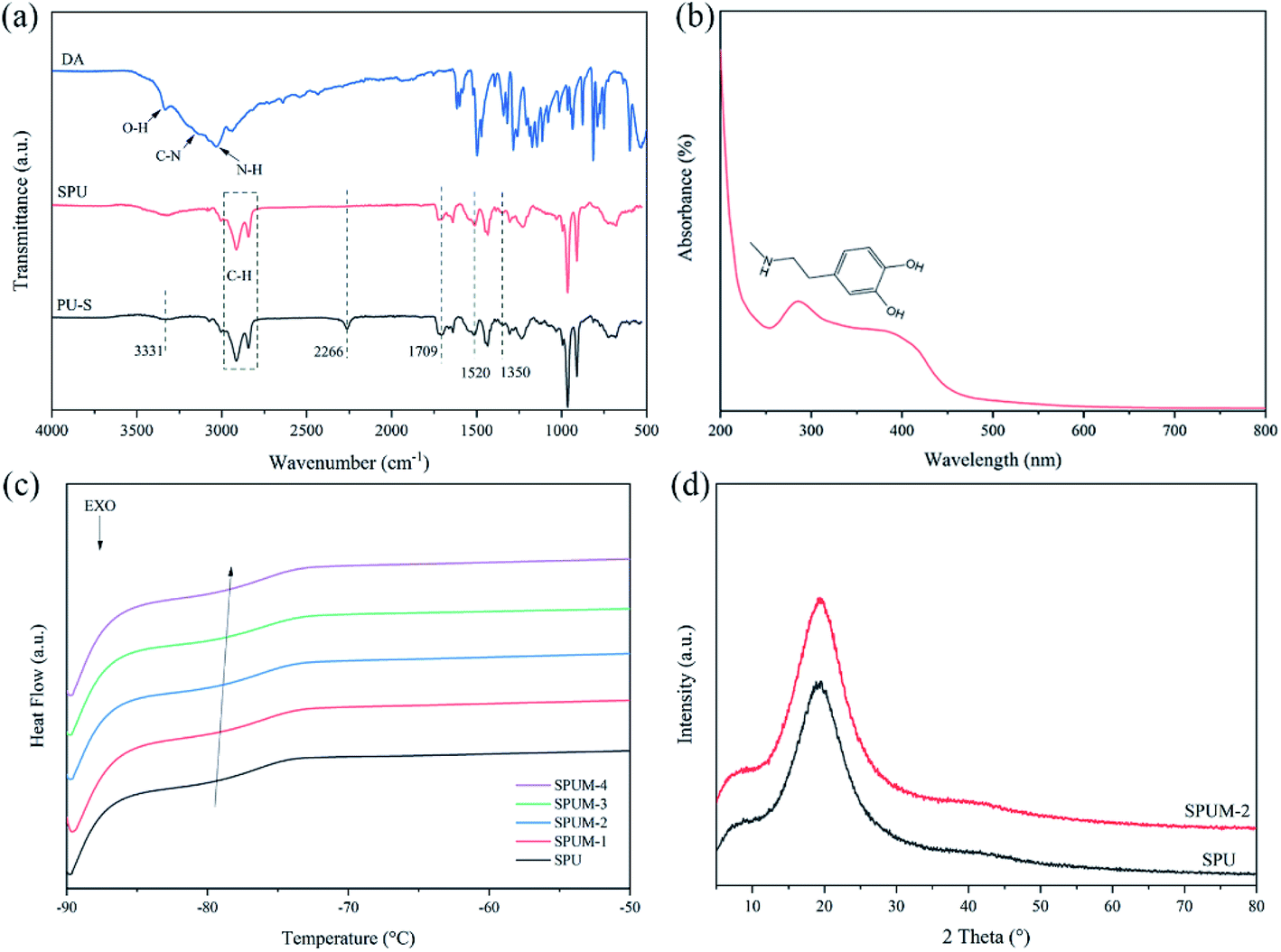 | ||
| Fig. 4 (a) FTIR spectra of DA, PU–S and SPU. (b) UV-vis spectra of SPU. (c) DSC curves of different composites. (d) XRD spectra of SPU and SPUM-2. | ||
The dispersion degree of MGO sheets in the matrix shows significant effect on the mechanical and self-healing properties of the composites,67 as such SEM was used to observe the cryo-fractured surfaces of the obtained composites. As shown in Fig. 5(a), obvious aggregates of GO can be observed, and interfaces of GO are quite bare (Fig. 5(c)), suggesting weak interfaces exist in SPUG matrix. In contrast, uniformly dispersed MGO can be seen in the fracture surface of SPUM-2 (Fig. 5(b and d)). Besides, owing to the crosslinking between MGO and matrix, MGO sheets are covered by the matrix and show indistinct interfaces.
 | ||
| Fig. 5 SEM images of (a and c) SPUG-1 and (b and d) SPUM-2. Red arrows indicate the MGO sheets. | ||
3.2. Self-healing properties
Through the above molecular structure design, we successfully endow the composites with remarkable self-healing properties. To investigate the self-healing ability, all the samples are cut into two parts by a blade, and the cut surfaces were immediately brought together for a certain time at room temperature. As displayed in Fig. 6(a–e), SPU samples show a high η value after healing for only 6 h. After the addition of MGO, the apparent decline occurred in the η value with increasing content of MGO. Especially, when the MGO content reaches 0.2 wt% (SPUM-4), the η value drastically decreases (62.5% for healing 6 h) even if the healing time was extended to 12 h. By comparison, SPUM-1 and SPUM-2 samples could still maintain a high η value of 87.9% and 87.1%, respectively (Fig. 6(f)). The outstanding self-healing capability of SPU samples could be ascribed to the synergistic effect of disulfide bonds and hydrogen bonds formed by catechol groups. Specifically, the disulfide exchange reactions can occur at a mild temperature due to its low bond dissociation energy and stable free radicals.28,29 Second, the hydrogen bonds formed by catechol groups can accelerate initial sticking or interfacial adhesion, promoting the diffusion of molecular chains and accelerating the exchange reactions of disulfide bonds.2,30 Third, the disulfide bonds will weaken the strength of hydrogen bonds, leading to their easier dissociation under mild conditions.28 In turn, the exchange of hydrogen bonds can provide energy to promote disulfide metathesis. Moreover, APD with a zigzag structure could facilitate the molecular chain motion that readily induces hydrogen bond exchange and generates disulfide metathesis.64 Nevertheless, the crosslinking between MGO and polymer chains will inhibit mobility which is essentially important for the self-healing ability,68 resulting in a sharp decline in the η value when MGO content reaches up to 0.2 wt%. There may be a critical value for the content of MGO. Under this critical value, composites can still possess a high η value. From the results of self-healing experiments, 0.1% may be the critical value, therefore, we choose SPUM-2 for further study.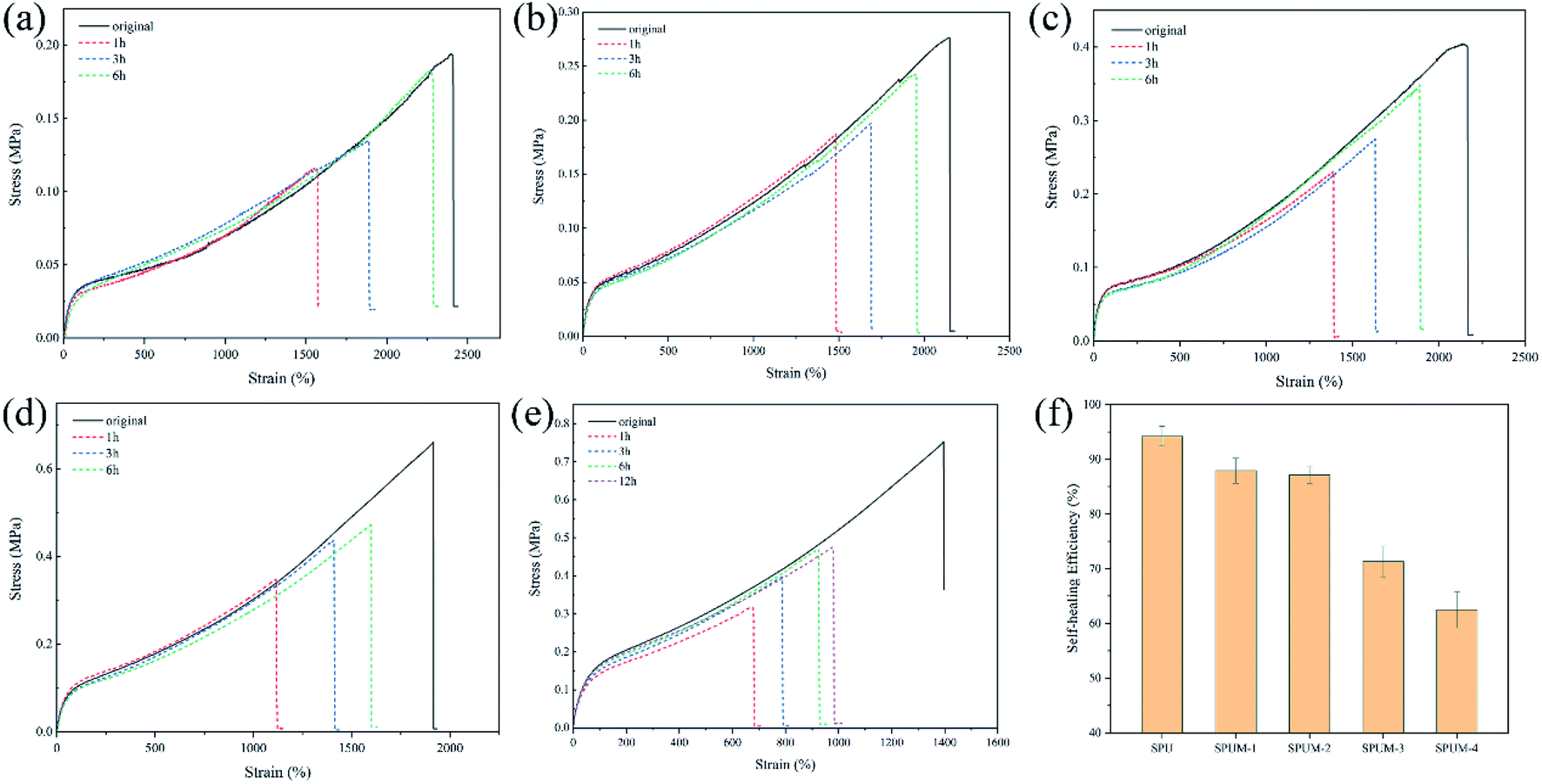 | ||
| Fig. 6 The tensile stress–strain curves of (a) SPU, (b) SPUM-1, (c) SPUM-2, (d) SPUM-3 and (e) SPUM-4 at different healing time under room temperature. (f) Self-healing efficiency (η) of different samples. | ||
To further intuitively study the outstanding self-healing performances, two samples, SPU and SPUM-2, were cut into two pieces and different samples were put together (SPU + SPUM-2) for healing for 6 h at 25 °C. Upon stretching, the healed sample exhibited no breaking (Fig. 7(a)). Subsequently, the tensile test was performed on the SPU + SPUM-2 sample to quantitatively describe the self-healing performance (Fig. 7(b), Movie S1†), and the stress–strain curves are displayed in Fig. 7(c). It can be seen that even after healing with different samples, the stitched sample can still exhibit self-healing capability. Moreover, it is noteworthy that the two different parts show different elongations due to the difference in their moduli.
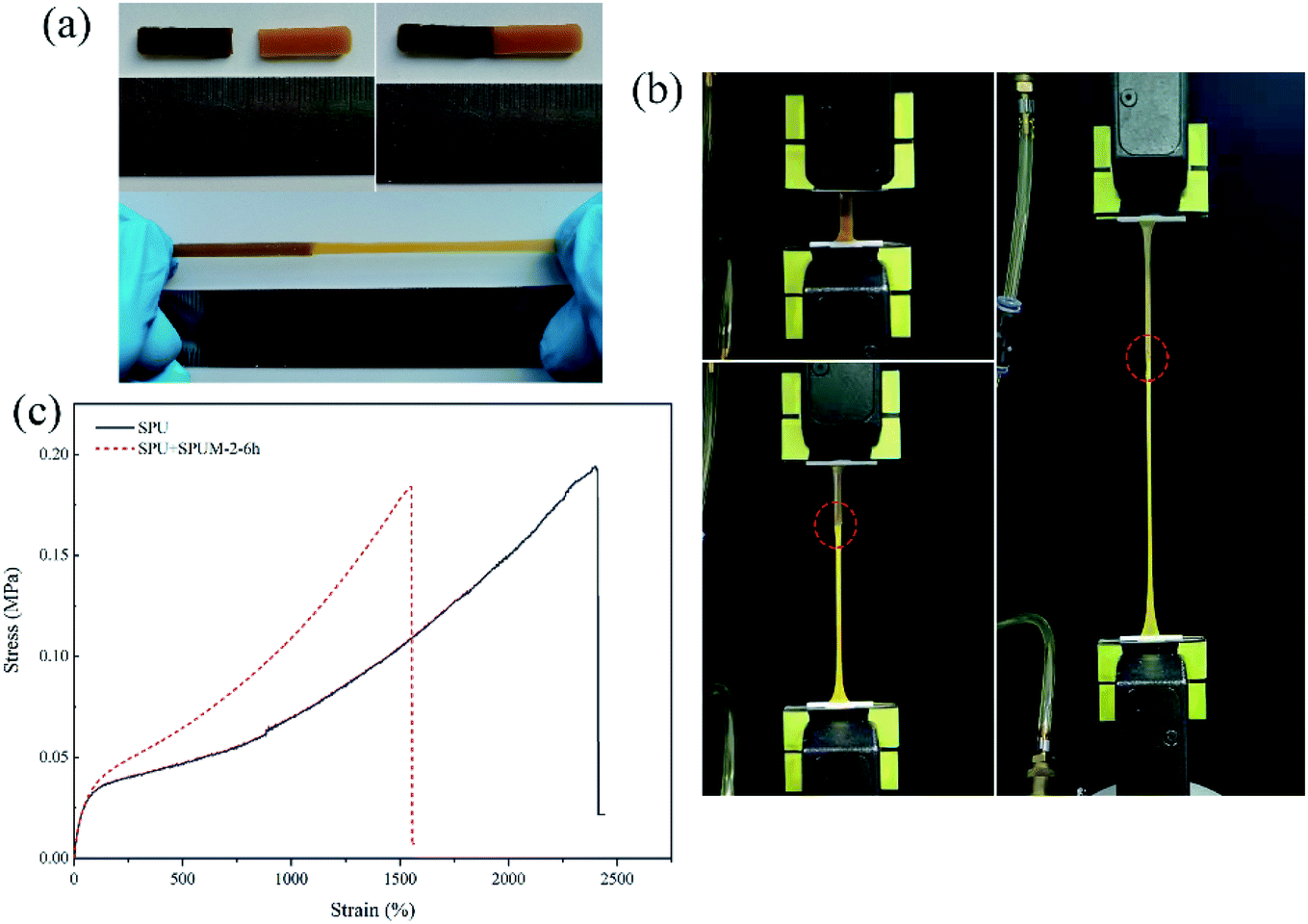 | ||
| Fig. 7 (a) The cut SPU (yellow) and SPUM-2 (brown) samples were put together for healing for 6 h at 25 °C (SPU + SPUM-2) and stretched again. (b) Images of stretching healed SPU + SPUM-2 sample after healing for 6 h at 25 °C. (the red dotted line represents the stitching position) (c) tensile stress–strain curves of the original SPU sample and SPU + SPUM-2 sample. | ||
Various devices based on PU will be inevitably affected by the complex environment during their service life. Therefore, the self-healing experiments were also conducted under different conditions to confirm the environmental adaptability of the self-healing properties. As shown in Fig. 8(a), SPUM-2 samples were healed at different temperatures for 1 h, and the η value shows an upward trend with the increase of temperature. This phenomenon can be attributed to faster exchange reactions of disulfide bonds and hydrogen bonds at higher temperatures. Furthermore, the underwater self-healing property of the composites will significantly promote its applications in prospective coating, electrical skin and wearable devices.69,70 To this end, the SPUM-2 sample was cut into two separate pieces by a blade and soaked in water to wet the fractured surfaces, and then put into a contact. After healing in 60 °C water for 1 h, the healed sample could be stretched to a high strain and without breaking (Movie S2†). Then, the tensile tests were conducted on the samples healed under 25 °C and 60 °C water, and the stress–strain curves were measured as displayed in Fig. 8(b). We can see that the healed samples can be stretched up to 1870% strain with the η value of 92.5% under 60 °C water for 2 h. As for the sample healed under 25 °C water for 6 h, it still shows a high strain of 1750% and the η value of 74.6%, which just shows a slight decline compared with that in air atmosphere for 6 h. These results demonstrated that the obtained composites possess outstanding water-insensitivity and underwater self-healing property, which is meaningful for its applications.
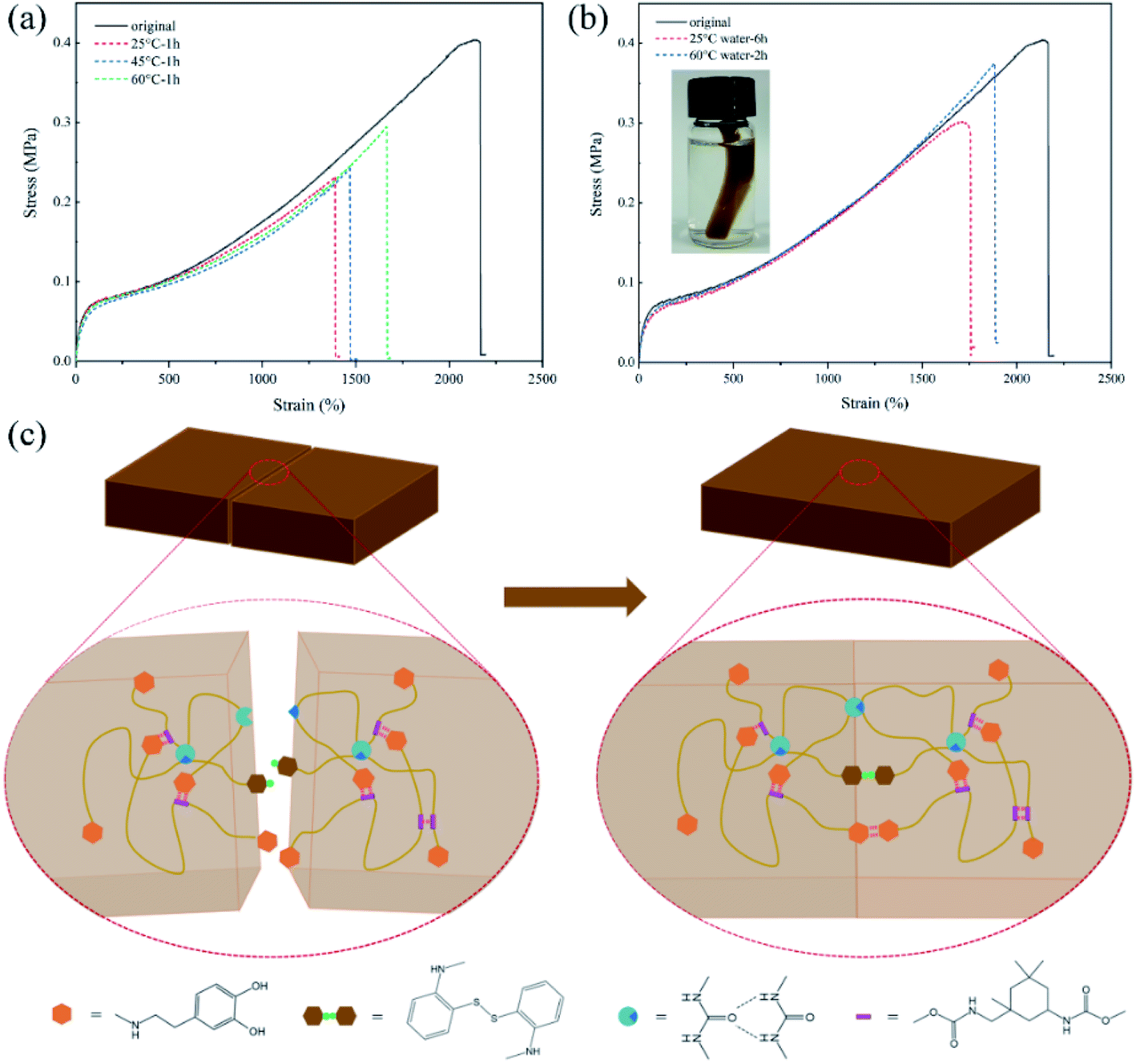 | ||
| Fig. 8 (a) Tensile stress–strain curves of SPUM-2 sample after healing at different temperatures for 1 h. (b) Tensile stress–strain curves of OSPUM-2 sample after healing under 60 °C water for 2 h and 25 °C water for 6 h. (c) Schematic illustration of the self-healing mechanism of the composites. | ||
In order to intuitively elaborate the self-healing process, a schematic diagram is illustrated in Fig. 8(c). Once the damaged surfaces are contacted together, the catechol groups can form hydrogen bonds to achieve initial sticking and interfacial adhesion. Then, the disulfide exchange reactions occurred at surfaces, which will promote the interdiffusion of polymer chains.2 Finally, the continuous randomization of polymer chains gradually completes the self-healing process.71 Moreover, a comparison of the η value, self-healing time and self-healing temperature between SPUM elastomers and other self-healing materials in recent studies is summarized in Fig. 9. It can be found that some self-healing materials exhibit considerable η value but require relatively high temperature, and some can heal at medium temperature but a long time is necessary. On the contrary, SPUM samples not only exhibit a high η value but also show a rapid room-temperature self-healing property.
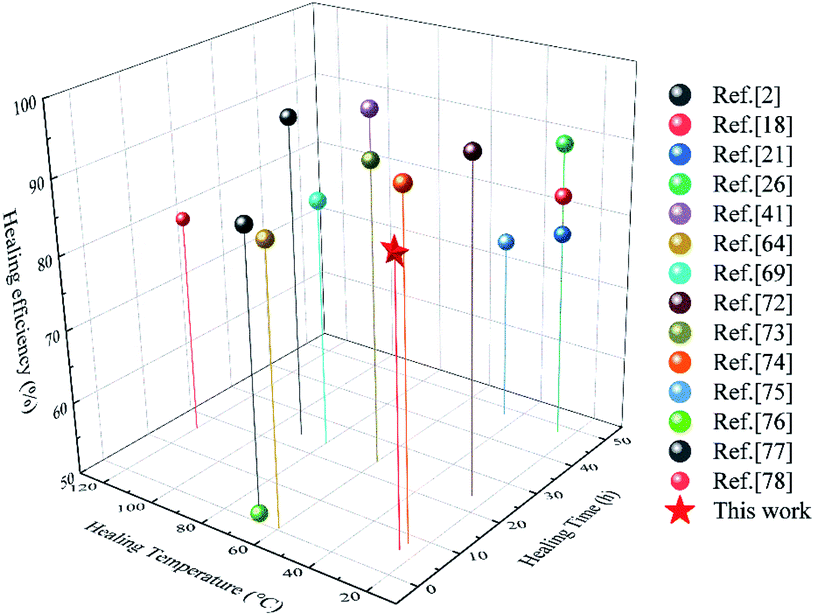 | ||
| Fig. 9 Comparison of the η value, self-healing time and self-healing temperature of the elastomers reported in this work and those reported in recent studies.2,18,21,26,41,64,69,72–78 | ||
3.3. Mechanical properties
Reports have indicated that the self-healing properties are always achieved by sacrificing mechanical properties.39–43 The aim of our design was to fabricate PU with both rapid room-temperature self-healing properties and excellent mechanical properties by molecular structure design and controlling the content of MGO. It was expected that the mechanical properties of SPUM samples can be significantly improved by MGO. Consequently, the mechanical properties were carefully evaluated as follows. As shown in Fig. 10(a), the original SPU samples show the elongation at break of nearly 2400% but low tensile strength and Young's modulus. After the incorporation of MGO, the tensile strength of SPUM samples increases with increase in MGO loading, while the elongation at break decreases except for SPUM-2 samples. To be specific, by increasing the content of MGO, the tensile strength of SPUM samples increases by 37.7%, 107.7%, 240.2% and 287.1%, relative to SPU. It is notable that the SPUM-2 samples exhibit no decline in elongation at break (2170%) when their tensile strength was improved, compared to SPUM-1 samples. It is different from the regular phenomenon reported in previous works, which are reporting that the addition of nanofiller into polymer will continuously decrease the elongation at break.4,79 There is an optimal value of MGO loading, under which the tensile strength of the samples can be significantly improved while maintaining high elongation at break (SPUM-2). Instead, SPUG samples exhibit more apparent decline in the elongation at break and only slight improvement in tensile strength. Combining the SEM images, we infer that these phenomena can be attributed to the following factors: (1) The relative strong interfacial interactions promote stress transfer from matrix to MGO sheets. (2) Well-dispersion of MGO in matrix reduces the stress concentration from the agglomeration of MGO sheets. Subsequently, dynamic mechanical analysis was also conducted on these samples. As shown in Fig. 10(b), the storage modulus (G′) shows an increasing trend with the increase in MGO loading over the entire frequency range, which is in good agreement with tensile tests.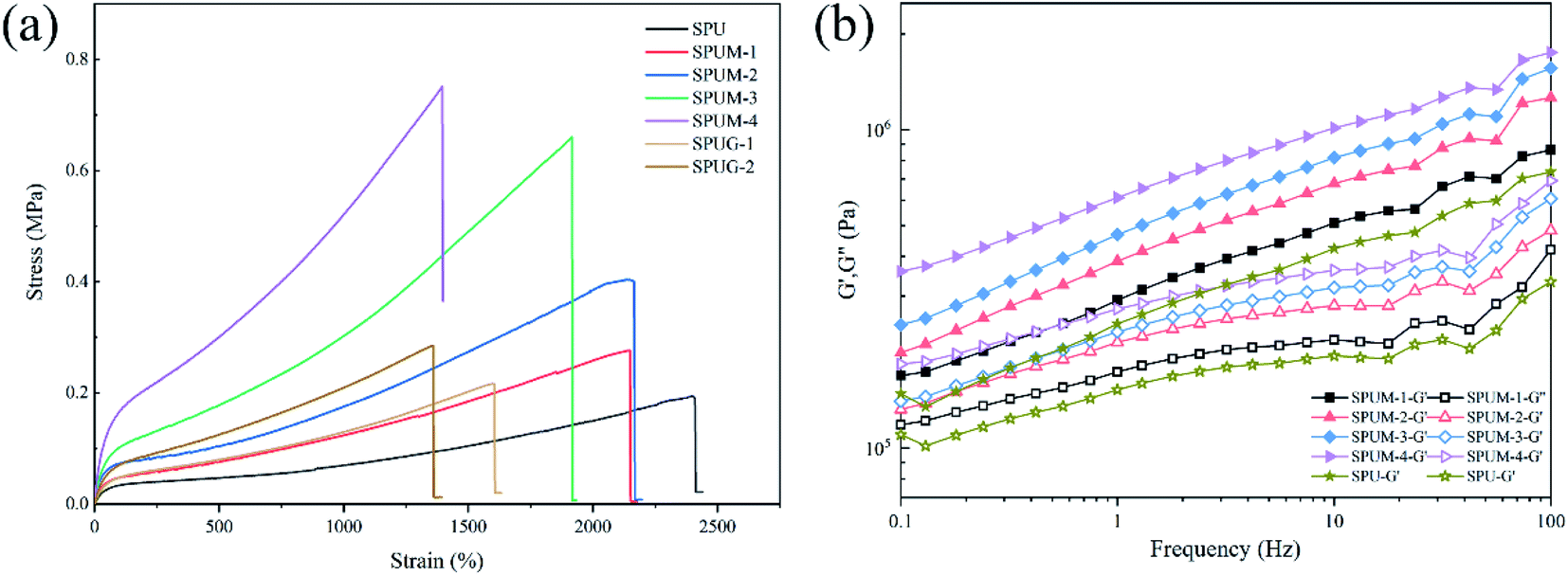 | ||
| Fig. 10 (a) Stress–strain curves and (b) DMA results of various samples. | ||
As is known, PU has been widely used in numerous fields, and it always suffers from various exterior loading during applications.1,2 Therefore, the deformation recovery capability of the composites is important for its long-term working. Given the outstanding self-healing properties and excellent tensile properties of SPUM-2 samples, we chose it for further research.
As presented in Fig. 11(a and b), cycle tensile tests were carried out on SPU and SPUM-2 samples. Both the stress–strain curves of SPU and SPUM-2 show significant and pronounced hysteresis in consecutive cycles without any relaxation time, implying a tremendous energy dissipation from bond breakage of disulfide bonds and hydrogen bonds during the tensile process. But the tensile stress gradually decreases with cycles because the polymer networks cannot be reformed in a short time.80 Subsequently, they were allowed to relax for 10 min and stretched again. It is obvious that SPUM-2 samples show a similar cycle curve to that of the first cycle, while the SPU samples require more time even for 30 min, indicating the outstanding recovery capability of SPUM-2 samples. To further study the tensile recovery capability, 20 successive cycle tensile tests with 10 min of relaxation time between each cycle was carried out on SPUM-2 samples (Fig. 11(c)). To be more quantitative, we define both the recovery degree and dissipated energy by calculating the ratios of stress at the strain of 100% and the hysteresis loop area to those values at the first one. It is noticeable in Fig. 11(d) that, after 5 cycles, the recovery degree and dissipated energy of SPUM-2 recovered by 97.6% and 92.6%, respectively. Even after 20 cycles, they were still maintained at 93.5% and 76.3%, respectively. All the above results confirm that SPUM-2 possesses excellent recovery ability. Due to the outstanding mechanical properties of MGO and crosslinking between MGO and polymer chains, there have formatted a stronger network in polymer matrix.
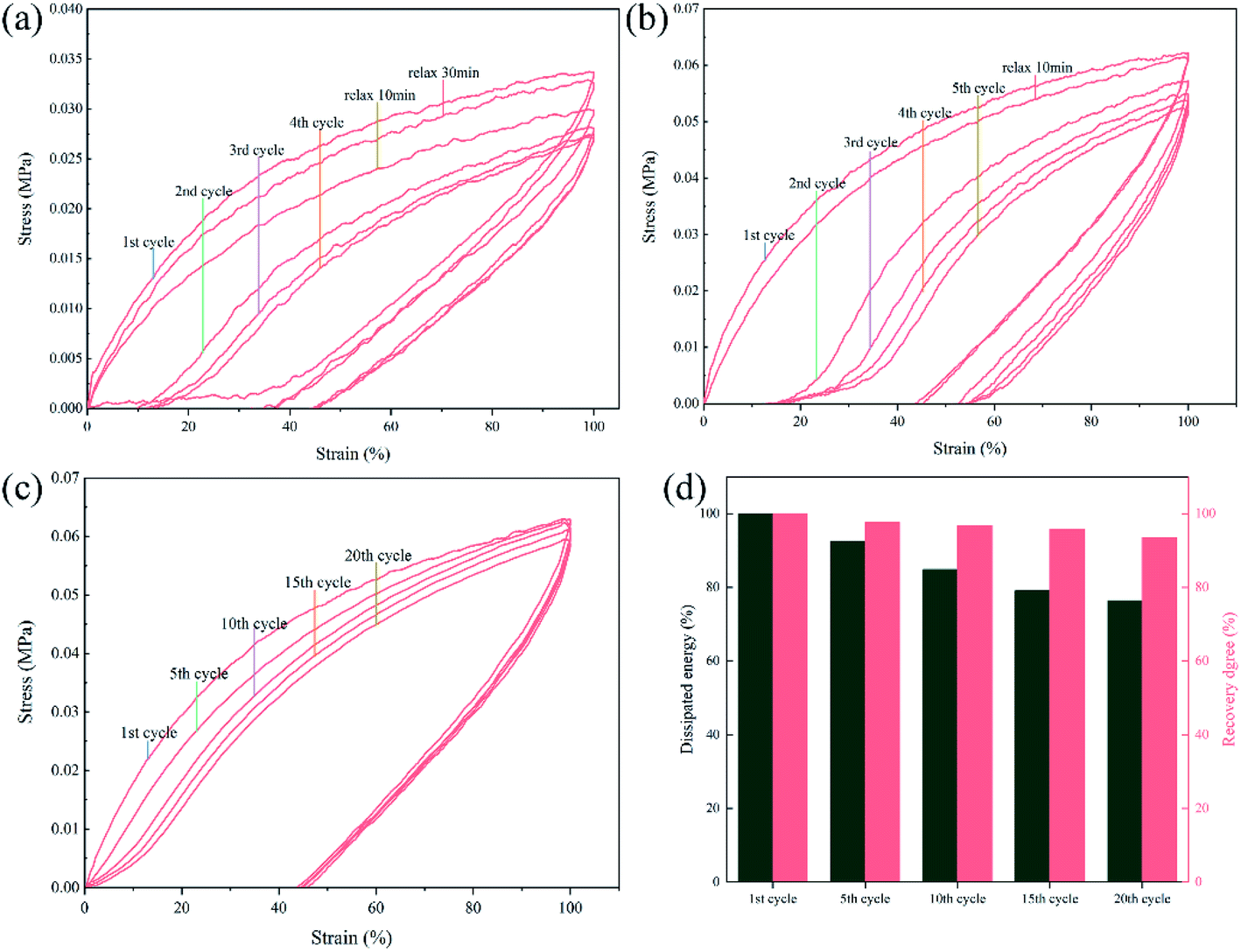 | ||
| Fig. 11 Cyclic tensile curves of (a) SPU and (b) SPUM-2 with a strain of 100%. The sample was stretched and released 5 cycles continuously, then relaxed for another cycle. (c) Twenty successive cycle tests of SPUM-2 with 10 min of relaxing between each cycle. (d) Dissipated energy and recovery degree of SPUM-2 with 20 cycles. | ||
The compressive load is also a typical external load that PU will suffer from during the entire service life. Therefore, the compression cycle tests were also performed to reflect the deformation recovery capability. As shown in Fig. 12(a) and Movie S3,† SPUM-2 samples can recover their initial shape after manual compression in a few seconds. Then, different strains were applied on SPUM-2 (Fig. 12(b and c)). Its compressive stress increased with an increase in the strain and it can recover under such a high strain of 70%. Subsequently, the compression recovery of SPUM-2 was further investigated via a successive loading-unloading cycle test at the strain of 60% for 20 cycles with the recovery time of 1 min between each cycle (Fig. 12(d and e)). Obviously, the hysteresis loop area and compression stress exhibit a slight decline. Here, we also define both the recovery degree and dissipated energy by calculating the ratios of compression stress to the strain of 60% and the hysteresis loop area to those values at the first one, and the results are summarized in Fig. 12(f). After 20 cycles, the value of recovery degree still remains at 83.7% and the dissipated energy shows a slight decline but still stayed above 71%. These results further demonstrated the excellent recovery ability of SPUM-2 samples under different external loads. The crosslinking between MGO and polymer chains reforms an elastic network where well stress transfer can effectively disperse the stress during loading–unloading procedures.81,82
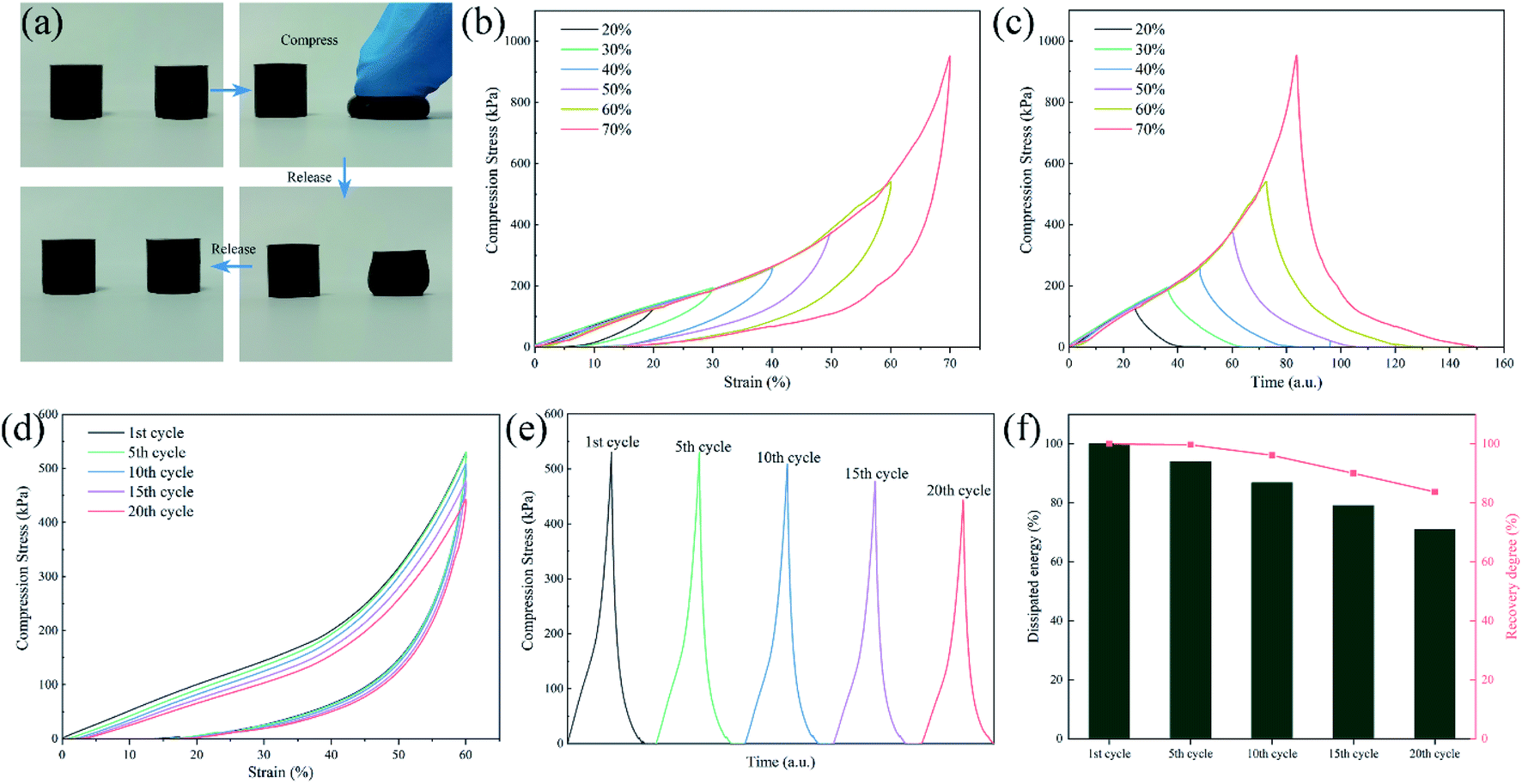 | ||
| Fig. 12 (a) Images of compressed SPUM-2 samples. (b) Compression curves of SPUM-2 at different compression strains. (c) Corresponding changes in compression stress overtime during cycles. (d) Twenty successive compression cycles of SPUM-2 with 1 min recovery between each cycle. (e) Corresponding compression stress overtime during cycles. (f) Dissipated energy and recovery degree of SPUM-2 with 20 cycles. | ||
According to the study in the last section, composites still possessed excellent self-healing properties under the appropriate content of MGO. Hence, we could obtain a PU with both outstanding self-healing properties and superb mechanical properties from molecular structure design and controlling the content of MGO. From the above results, we believe that the SPUM-2 is an ideal PU.
In summary, the superb self-healing and mechanical properties of our composites are attributed to the following factors: (1) the low Tg (∼−79 °C) value of composites endows high mobility to the molecule chains, which enable self-healing process even at room temperature. (2) The aromatic disulfide exchange reactions can occur at mild temperature due to its low bond dissociation energy and stable free radicals.28,29 (3) The mussel-inspired catechol groups can form weak dual hydrogen bonds to accelerate initial sticking or interfacial adhesion, which will promote the diffusion of molecular chains and accelerate the exchange reactions of disulfide bonds.2,30 (4) The water-resistant adhesive characteristics of catechol groups promote initial sticking and interfacial adhesion under a water environment.83 (5) Owing to the superior water-resistance of HTPB chains,10,84 composites are not significantly affected by water molecules during the self-healing process. (6) The crosslinking between MGO and polymer chains can promote the stress transfer from the matrix to MGO, resulting in improved mechanical properties.
4. Conclusions
In this work, a series of novel mussel-inspired modified graphene oxide/polyurethane composites were designed and successfully synthesized. Owing to the successful graft of hyperbranched polymer onto GO sheets, MGO can crosslink with polymer chains. As a result, MGO could dramatically improve the mechanical properties of composites. The tensile strength of SPUM-2 increased by 107.7% with a high strain of 2170%. Notably, SPUM-2 also exhibits outstanding deformation recovery capability under cycle tensile and compression loading with recovery degree of 76.3% and 83.7%, respectively, after 20 cycles. Furthermore, owing to the synergistic effect of aromatic disulfide bonds and catechol groups, SPUM-2 shows a high self-healing efficiency of 87.1% at room temperature for only 6 h. More importantly, the self-healing performance of SPUM-2 exhibits excellent water-insensitivity with high self-healing efficiency of 92.5% under 60 °C water for 2 h and 74.6% under 25 °C water for 6 h. In summary, the synthesized HTPB-based PU composites possess excellent mechanical and self-healing properties, simultaneously, which may be potentially applied in solid propellants, protective coating, electronic skin, soft sensors and other water-insensitive devices.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.References
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- M. Liu, J. Zhong, Z. Li, J. Rong, K. Yang, J. Zhou, L. Shen, F. Gao, X. Huang and H. He, Eur. Polym. J., 2020, 124, 109475 CrossRef CAS.
- H. Xu, J. Tu, G. Xiang, Y. Zhang and X. Guo, Macromol. Chem. Phys., 2020, 221, 2000273 CrossRef CAS.
- X. Zhang, J. Zheng, H. Fang, Y. Zhang and S. Bai, Composites, Part A, 2017, 103, 208–218 CrossRef CAS.
- K. Ghosh, L. V. Gaikwad, R. K. Kalal, P. A. Kulkarni, A. Kumar, S. Banerjee and M. Gupta, Def. Technol., 2020, 17(2), 559–570 CrossRef.
- B. N. Rao, P. J. P. Yadav, K. Malkappa, T. Jana and P. U. Sastry, Polymer, 2015, 77, 323–333 CrossRef CAS.
- Q. Zhou, S. Jie and B.-G. Li, Polymer, 2015, 67, 208–215 CrossRef CAS.
- K. Ganesh, S. Sundarrajan, K. Kishore, K. N. Ninan, B. George and M. Surianarayanan, Macromolecules, 2000, 33, 326–330 CrossRef CAS.
- A. Nazmi, M. Yarmohammadi, M. Zamani Pedram and M. Shahidzadeh, Int. J. Chem. Kinet., 2019, 51, 28–36 CrossRef CAS.
- B. K. Sikder and T. Jana, ACS Omega, 2018, 3, 3004–3013 CrossRef CAS.
- N. Zhong and W. Post, Composites, Part A, 2015, 69, 226–239 CrossRef CAS.
- K. Urdl, A. Kandelbauer, W. Kern, U. Müller, M. Thebault and E. Zikulnig-Rusch, Prog. Org. Coat., 2017, 104, 232–249 CrossRef CAS.
- V. K. Thakur and M. R. Kessler, Polymer, 2015, 69, 369–383 CrossRef CAS.
- Y. Zhao, J. Fickert, K. Landfester and D. Crespy, Small, 2012, 8, 2954–2958 CrossRef CAS PubMed.
- K. S. Toohey, N. R. Sottos, J. A. Lewis, J. S. Moore and S. R. White, Nat. Mater., 2007, 6, 581–585 CrossRef CAS.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- L. Zhang, D. Wang, L. Xu, X. Zhang, A. Zhang and Y. Xu, J. Mater. Chem. C, 2020, 8, 2043–2053 RSC.
- J. Kang, D. Son, G.-J. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. B. H. Tok and Z. Bao, Adv. Mater., 2018, 30, 1706846 CrossRef PubMed.
- J. Cao, C. Lu, J. Zhuang, M. Liu, X. Zhang, Y. Yu and Q. Tao, Angew. Chem., Int. Ed., 2017, 56, 8795–8800 CrossRef CAS PubMed.
- Y. Wang, Q. Guo, G. Su, J. Cao, J. Liu and X. Zhang, Adv. Funct. Mater., 2019, 29, 1906198 CrossRef CAS.
- C.-H. Li, C. Wang, C. Keplinger, J.-L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X.-Z. You and Z. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- S. Yang, X. Du, S. Deng, J. Qiu, Z. Du, X. Cheng and H. Wang, Chem. Eng. J., 2020, 398, 125654 CrossRef CAS.
- K. Miyamae, M. Nakahata, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2015, 54, 8984–8987 CrossRef CAS PubMed.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- Y. Yang, X. Lu and W. Wang, Mater. Des., 2017, 127, 30–36 CrossRef CAS.
- S. Nevejans, N. Ballard, M. Fernández, B. Reck and J. M. Asua, Polymer, 2019, 166, 229–238 CrossRef CAS.
- T. Li, Z. Xie, J. Xu, Y. Weng and B.-H. Guo, Eur. Polym. J., 2018, 107, 249–257 CrossRef CAS.
- J. M. Matxain, J. M. Asua and F. Ruipérez, Phys. Chem. Chem. Phys., 2016, 18, 1758–1770 RSC.
- A. M. Grande, J. C. Bijleveld, S. J. Garcia and S. van der Zwaag, Polymer, 2016, 96, 26–34 CrossRef CAS.
- L. Li, B. Yan, J. Yang, L. Chen and H. Zeng, Adv. Mater., 2015, 27, 1294–1299 CrossRef CAS PubMed.
- Z. Xu, L. Chen, L. Lu, R. Du, W. Ma, Y. Cai, X. An, H. Wu, Q. Luo, Q. Xu, Q. Zhang and X. Jia, Adv. Funct. Mater., 2020, 2006432 Search PubMed.
- H. Ceylan, M. Urel, T. S. Erkal, A. B. Tekinay, A. Dana and M. O. Guler, Adv. Funct. Mater., 2013, 23, 2081–2090 CrossRef CAS.
- W. Wei, Y. Tan, N. R. Martinez Rodriguez, J. Yu, J. N. Israelachvili and J. H. Waite, Acta Biomater., 2014, 10, 1663–1670 CrossRef CAS PubMed.
- Z. Jia, Y. Zeng, P. Tang, D. Gan, W. Xing, Y. Hou, K. Wang, C. Xie and X. Lu, Chem. Mater., 2019, 31, 5625–5632 CrossRef CAS.
- A. V. Menon, G. Madras and S. Bose, ACS Appl. Polym. Mater., 2019, 1, 2417–2429 CrossRef CAS.
- Z. Shafiq, J. Cui, L. Pastor-Pérez, V. S. Miguel, R. A. Gropeanu, C. Serrano and A. d. Campo, Angew. Chem., 2011, 124, 4408–4411 CrossRef.
- Y. Cao, H. Wu, S. I. Allec, B. M. Wong, D.-S. Nguyen and C. Wang, Adv. Mater., 2018, 30, 1804602 CrossRef PubMed.
- C. Wang, N. Liu, R. Allen, J. B. H. Tok, Y. Wu, F. Zhang, Y. Chen and Z. Bao, Adv. Mater., 2013, 25, 5785–5790 CrossRef CAS PubMed.
- J. Mao, C. Zhao, Y. Li, D. Xiang and Z. Wang, Compos. Commun., 2020, 17, 22–27 CrossRef.
- T. Wan and D. Chen, Prog. Org. Coat., 2018, 121, 73–79 CrossRef CAS.
- S. Nevejans, N. Ballard, M. Fernández, B. Reck, S. J. García and J. M. Asua, Polymer, 2019, 179, 121670 CrossRef CAS.
- J. Yang, Z. Zhang, Y. Yan, S. Liu, Z. Li, Y. Wang and H. Li, ACS Appl. Mater. Interfaces, 2020, 12, 13239–13247 CrossRef PubMed.
- R. Cao, Z. Chen, Y. Wu, Y. Tu, G. Wu and X. Yang, Composites, Part A, 2017, 93, 100–106 CrossRef CAS.
- Y. Zhang, Q. n. Zhang, D. Hou and J. Zhang, Appl. Surf. Sci., 2020, 504, 144152 CrossRef.
- A. Surnova, D. Balkaev, D. Musin, R. Amirov and A. M. Dimiev, Composites, Part B, 2019, 162, 685–691 CrossRef CAS.
- S. Thakur and N. Karak, J. Mater. Chem. A, 2015, 3, 12334–12342 RSC.
- L. Huang, N. Yi, Y. Wu, Y. Zhang, Q. Zhang, Y. Huang, Y. Ma and Y. Chen, Adv. Mater., 2013, 25, 2224–2228 CrossRef CAS PubMed.
- Y.-G. Luan, X.-A. Zhang, S.-L. Jiang, J.-H. Chen and Y.-F. Lyu, Chin. J. Polym. Sci., 2018, 36, 584–591 CrossRef CAS.
- L. Guo, H. Yan, Z. Chen, Q. Lv, T. Bai and Y. Zhang, SN Appl. Sci., 2020, 2, 473 CrossRef CAS.
- J. Li, W. Zhu, S. Zhang, Q. Gao, J. Li and W. Zhang, Polym. Test., 2019, 76, 232–244 CrossRef CAS.
- X. Wang, N. Li, J. Wang, G. Li, L. Zong, C. Liu and X. Jian, Compos. Sci. Technol., 2018, 155, 11–21 CrossRef CAS.
- H. Feng, W. Ma, Z.-K. Cui, X. Liu, J. Gu, S. Lin and Q. Zhuang, J. Mater. Chem. A, 2017, 5, 8705–8713 RSC.
- S. O. Ilyin, V. V. Makarova, M. Y. Polyakova and V. G. Kulichikhin, Mater. Today Commun., 2020, 22, 100833 CrossRef CAS.
- S. Chen, J. Zhang, J. Zhou, D. Zhang and A. Zhang, Chem. Eng. J., 2018, 334, 1371–1382 CrossRef CAS.
- L. Shi, L. Ma, P. Li, M. Wang, S. Guo, P. Han and G. Song, Appl. Surf. Sci., 2019, 479, 334–343 CrossRef CAS.
- L. Wang, Y. Tan, X. Wang, T. Xu, C. Xiao and Z. Qi, Chem. Phys. Lett., 2018, 706, 31–39 CrossRef CAS.
- B. Ramezanzadeh, G. Bahlakeh, M. H. Mohamadzadeh Moghadam and R. Miraftab, Chem. Eng. J., 2018, 335, 737–755 CrossRef CAS.
- X. Zhang, J. Zheng, H. Fang, Y. Zhang, S. Bai and G. He, Compos. Sci. Technol., 2018, 161, 124–134 CrossRef CAS.
- Y. Li, M. Zhou, Z. Xia, Q. Gong, X. Liu, Y. Yang and Q. Gao, Colloids Surf., A, 2020, 602, 125172 CrossRef CAS.
- Z. Cao, J. Zhang, Y. Ding, Y. Li, M. Shi, H. Yue, Y. Qiao, Y. Yin and S. Yang, J. Mater. Chem. A, 2016, 4, 8636–8644 RSC.
- Y. Tang, H. Liu, X. Wang, S. Cheng, Z. Jin, T. Zhuang, S. Guan and L. Li, J. Mol. Struct., 2021, 1224, 129196 CrossRef CAS.
- F. Zhang, W. Liu, S. Wang, C. Jiang, Y. Xie, M. Yang and H. Shi, Appl. Surf. Sci., 2019, 491, 301–312 CrossRef CAS.
- H. Huang, W. Zhou, Z. Zhong, S. Peng and X. Peng, Prog. Org. Coat., 2020, 146, 105717 CrossRef CAS.
- Z. Li, Y. Shan, X. Wang, H. Li, K. Yang and Y. Cui, Chem. Eng. J., 2020, 394, 124932 CrossRef CAS.
- C. Pan, L. Liu, Q. Chen, Q. Zhang and G. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 38052–38061 CrossRef CAS PubMed.
- S. Wu, J. Li, G. Zhang, Y. Yao, G. Li, R. Sun and C. Wong, ACS Appl. Mater. Interfaces, 2017, 9, 3040–3049 CrossRef CAS PubMed.
- E. Zhang, T. Wang, L. Zhao, W. Sun, X. Liu and Z. Tong, ACS Appl. Mater. Interfaces, 2014, 6, 22855–22861 CrossRef CAS PubMed.
- Z. Yang, H. Li, L. Zhang, X. Lai and X. Zeng, J. Colloid Interface Sci., 2020, 570, 1–10 CrossRef CAS PubMed.
- S.-M. Jeong, Y. Kang, T. Lim and S. Ju, Adv. Mater. Interfaces, 2018, 5, 1801376 CrossRef.
- M. Zhang, F. Zhao and Y. Luo, ACS Omega, 2019, 4, 1703–1714 CrossRef CAS PubMed.
- L. Zhao, Y. Yin, B. Jiang, Z. Guo, C. Qu and Y. Huang, J. Colloid Interface Sci., 2020, 573, 105–114 CrossRef CAS PubMed.
- X. Wu, J. Wang, J. Huang and S. Yang, J. Colloid Interface Sci., 2020, 559, 152–161 CrossRef CAS PubMed.
- Y. Li, Z. Yang, L. Ding, L. Pan, J. Zhang, X. Zheng and C. Lin, React. Funct. Polym., 2019, 144, 104342 CrossRef CAS.
- M. Zhong, Y.-T. Liu and X.-M. Xie, J. Mater. Chem. B, 2015, 3, 4001–4008 RSC.
- M. Shahidzadeh, Z. Khansari Varkaneh, B. Ramezanzadeh, M. Zamani Pedram and M. Yarmohammadi, Prog. Org. Coat., 2020, 140, 105503 CrossRef CAS.
- J.-F. Mei, X.-Y. Jia, J.-C. Lai, Y. Sun, C.-H. Li, J.-H. Wu, Y. Cao, X.-Z. You and Z. Bao, Macromol. Rapid Commun., 2016, 37, 1667–1675 CrossRef CAS PubMed.
- Z.-J. Li, J. Zhong, M.-C. Liu, J.-C. Rong, K. Yang, J.-Y. Zhou, L. Shen, F. Gao and H.-F. He, Chin. J. Polym. Sci., 2020, 38, 932–940 CrossRef CAS.
- L. Hu, P. Jiang, P. Zhang, G. Bian, S. Sheng, M. Huang, Y. Bao and J. Xia, J. Mater. Sci., 2016, 51, 8296–8309 CrossRef CAS.
- A. Dutta, S. Maity and R. K. Das, Macromol. Mater. Eng., 2018, 303, 1800322 CrossRef.
- C. Shao, M. Wang, H. Chang, F. Xu and J. Yang, ACS Sustainable Chem. Eng., 2017, 5, 6167–6174 CrossRef CAS.
- M. Hu, X. Gu, Y. Hu, T. Wang, J. Huang and C. Wang, Macromolecules, 2016, 49, 3174–3183 CrossRef CAS.
- N. N. Xia, X. M. Xiong, J. Wang, M. Z. Rong and M. Q. Zhang, Chem. Sci., 2016, 7, 2736–2742 RSC.
- M. Khatib, O. Zohar, W. Saliba, S. Srebnik and H. Haick, Adv. Funct. Mater., 2020, 30, 1910196 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00944c |
| This journal is © The Royal Society of Chemistry 2021 |