
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic synthesis of novel fructosylated compounds by Ffase from Schwanniomyces occidentalis in green solvents†
David Piedrabuenaad,
Ángel Rumberob,
Elísabet Pires c,
Alejandro Leal-Duasoc,
Concepción Civerad,
María Fernández-Lobato*a and
María J. Hernaiz*d
c,
Alejandro Leal-Duasoc,
Concepción Civerad,
María Fernández-Lobato*a and
María J. Hernaiz*d
aCentro de Biología Molecular Severo Ochoa (CBMSO; UAM-CSIC), Departamento de Biología Molecular, Facultad de Ciencias, Universidad Autónoma de Madrid, Nicolás Cabrera 1, 28049 Madrid, Spain. E-mail: mfernandez@cbm.uam.es
bDepartamento de Química en Ciencias Farmacéuticas, Universidad Complutense de Madrid, Plaza Ramón y Cajal s/n, 28040 Madrid, Spain
cDepartamento de Química Orgánica, Facultad de Ciencias, Universidad Autónoma de Madrid, Francisco Tomás y Valiente 7, 28049 Madrid, Spain
dDepartamento de Química Orgánica, Facultad de Ciencias, Universidad de Zaragoza-Instituto de Síntesis Química y Catálisis Homogénea (ISQCH-CSIC), 50009 Zaragoza, Spain. E-mail: mjhernai@ucm.es
First published on 9th July 2021
Abstract
The β-fructofuranosidase from the yeast Schwanniomyces occidentalis (Ffase) produces potential prebiotic fructooligosaccharides (FOS) by self-transfructosylation of sucrose, being one of the highest known producers of 6-kestose. The use of Green Solvents (GS) in biocatalysis has emerged as a sustainable alternative to conventional organic media for improving product yields and generating new molecules. In this work, the Ffase hydrolytic and transfructosylating activity was analysed using different GS, including biosolvents and ionic liquids. Among them, 11 were compatible for the net synthesis of FOS. Besides, two glycerol derivatives improved the yield of total FOS. Interestingly, polyols ethylene glycol and glycerol were found to be efficient alternative fructosyl-acceptors, both substantially decreasing the sucrose fructosylation. The main transfer product of the reaction with glycerol was a 62 g L−1 isomeric mixture of 1-O and 2-O-β-D-fructofuranosylglycerol, representing 95% of all chemicals generated by transfructosylation. Unexpectedly, the non-terminal 2-O fructo-conjugate was the major molecule catalysed during the process, while the 1-O isomer was the minor one. This fact made Ffase the first known enzyme from yeast showing this catalytic ability. Thus, novel fructosylated compounds with potential applications in food, cosmetics, and pharmaceutical fields have been obtained in this work, increasing the biotechnological interest of Ffase with innocuous GS.
Introduction
Non-conventional yeasts are excellent tools in the discovery of alternative enzymes with new abilities and applications.1 β-fructofuranosidase (EC 3.2.1.26) from Schwanniomyces occidentalis (Ffase) is a robust glycosyl hydrolase (GH) that cleaves glycosidic substrates including β-(2→1) or β-(2→6)-linked fructose units.2,3 In addition, it produces fructooligosaccharides (FOS) by self-transfructosylation of sucrose, specifically the trisaccharides 6-kestose, 1-kestose, and neokestose (with a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio), as well as small traces of the disaccharide blastose by direct fructosylation of glucose.4,5 Indeed, Ffase is one of the highest and more selective known producers of 6-kestose from sucrose.2 FOS, in which one to three fructosyl moieties are linked to a sucrose scaffold, act as prebiotics exerting a beneficial effect on human health. Besides, FOS are attractive substitutes for sugar because they are generally sweet, low in calories, and non-cariogenic.6,7 Ffase also showed high fructosyl-acceptor promiscuity, being able to fructosylate 17 different hydroxylated compounds, including small sugars and alditols, to produce new fructo-conjugates. The best alternatives to sucrose as an acceptor were mannitol and erythritol, that accordingly generated 1-O-β-D-fructofuranosyl-D-mannitol and 1/4-O-β-D-fructofuranosyl-D-erythritol.4 Sugar alcohols are extensively used in food as sweeteners and bulking agents, so their transglycosylation has always been of great interest to improve either their functional properties (particularly the solubility, stability, and flavor) or their bioavailability to reach the large intestine.8–10
:2:1 ratio), as well as small traces of the disaccharide blastose by direct fructosylation of glucose.4,5 Indeed, Ffase is one of the highest and more selective known producers of 6-kestose from sucrose.2 FOS, in which one to three fructosyl moieties are linked to a sucrose scaffold, act as prebiotics exerting a beneficial effect on human health. Besides, FOS are attractive substitutes for sugar because they are generally sweet, low in calories, and non-cariogenic.6,7 Ffase also showed high fructosyl-acceptor promiscuity, being able to fructosylate 17 different hydroxylated compounds, including small sugars and alditols, to produce new fructo-conjugates. The best alternatives to sucrose as an acceptor were mannitol and erythritol, that accordingly generated 1-O-β-D-fructofuranosyl-D-mannitol and 1/4-O-β-D-fructofuranosyl-D-erythritol.4 Sugar alcohols are extensively used in food as sweeteners and bulking agents, so their transglycosylation has always been of great interest to improve either their functional properties (particularly the solubility, stability, and flavor) or their bioavailability to reach the large intestine.8–10
The Ffase 3D structure has been elucidated as a homodimer of 160 kDa, each subunit included in the glycoside hydrolase family 32 (GH32) and with a β-sandwich domain involved in substrate binding.3,11 The protein sequence contains serine instead of leucine at position 196 by non-universal decoding of the leucine codon CUG.12 Substitution of this Ser residue for Leu (ScFfase-Leu196) decreased ∼1000-fold the Ffase apparent sucrose-catalytic efficiency (kcat/Km); but, surprisingly, enhanced its transferase activity by almost 3-fold, as well as its specificity towards the synthesis of especially 6-kestose and, to a lesser extent neokestose, without significant production of 1-kestose.12 Usually, the transferase/hydrolase ratio of those GH showing these two activities depends largely on factors such as the intrinsic nature of the enzyme, acceptor/donor concentrations, and physico-chemical conditions of the reaction media.4,13,14
In this context, the so-called green solvents (GS) are acquiring increasingly relevance compared to classical organic ones to improve the yield and selectivity of enzymatic reactions in an economical, health-safe, and environmentally-friendly manner. This type of solvents encompasses, among others, ionic liquids (IL) mainly composed of imidazole or alkylammonium salts, polyols, carbonyl compounds, organic polymers, and biomass-based chemicals (biosolvents). This latter group chiefly includes different derivatives from carbohydrates, vegetable oils, short-chain carboxylic acids, glycerol, and phenols.15–18 Glycerol ethers were already successfully used, both with or without biocatalysts, to hydrotropically increase the solubility of limiting substrates or to modify the selectivity towards alternative acceptors.16,17,19–21 In general, short-chain polyols are the most common solvents in nature to prevent denaturation and loss of protein activity.22 Effects such as alterations in the spatial arrangement of proteins that improves their active center flexibility, removal of hydrogen-bonded water molecules from strongly hydrated acceptors, or changes in electrostatic interactions could be involved in this process.14,16,22 Until now, the catalytic effect exerted by solvents has been poorly examined in yeast enzymes. For example, β-fructofuranosidases from Saccharomyces cerevisiae and Candida utilis showed satisfactory activity with some alditols, including glycerol and sorbitol,22,23 and that from Cryptococcus laurentii with DMSO, DMF, or acetone.24
In this work, the activity of Ffase from Schwanniomyces occidentalis was analysed over sucrose in reactions including different GS. The implication of selected solvents in the production of fructosylated compounds was also evaluated.
Results and discussion
Effect of green solvents on the Ffase activity
As mentioned above, the trisaccharides 6-kestose, and to a lesser amount, 1-kestose and neokestose, with small traces of disaccharide blastose, were the main transfructosylation products obtained by Ffase in aqueous sucrose solutions (Fig. 1).4,5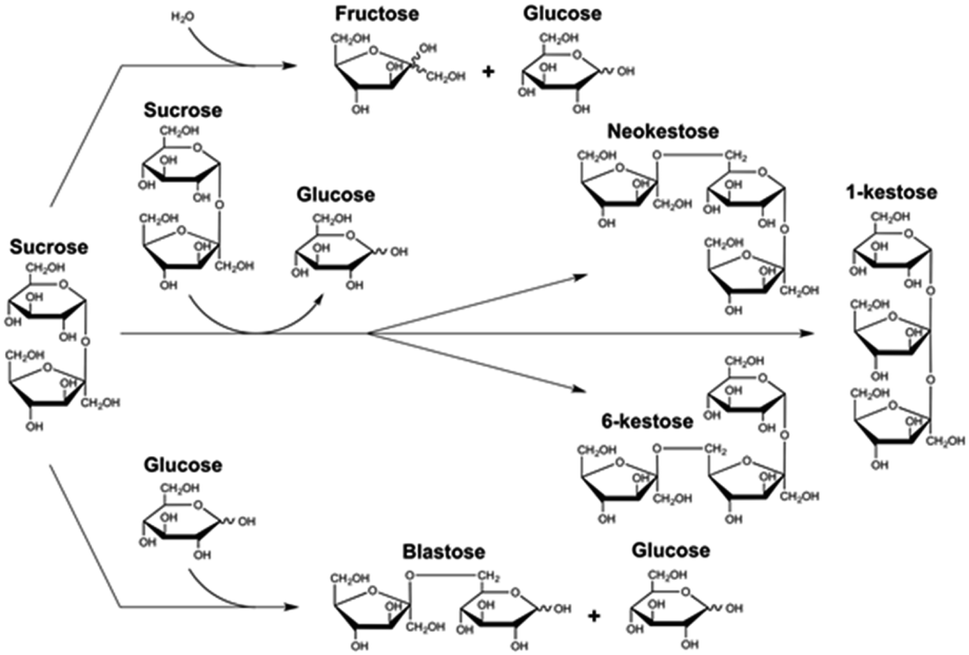 | ||
| Fig. 1 Schematic view of the reactions based on sucrose catalyzed by Ffase. | ||
To analyze this enzymatic activity in the additional presence of GS, a screening was carried out on 56 different solvents.
This selection included some conventional organic compounds, IL, and a set of synthetic derivatives from biomass-origin; all of which were either miscible or immiscible in water. Fig. 2 illustrates the chemical structure of the synthesized biosolvents used in this work and Table S1 in the ESI† some of their physico-chemical characteristics. None of the tested miscible solvents, regardless of their type, were beneficial for sucrose self-transfructosylation (Fig. 3a).
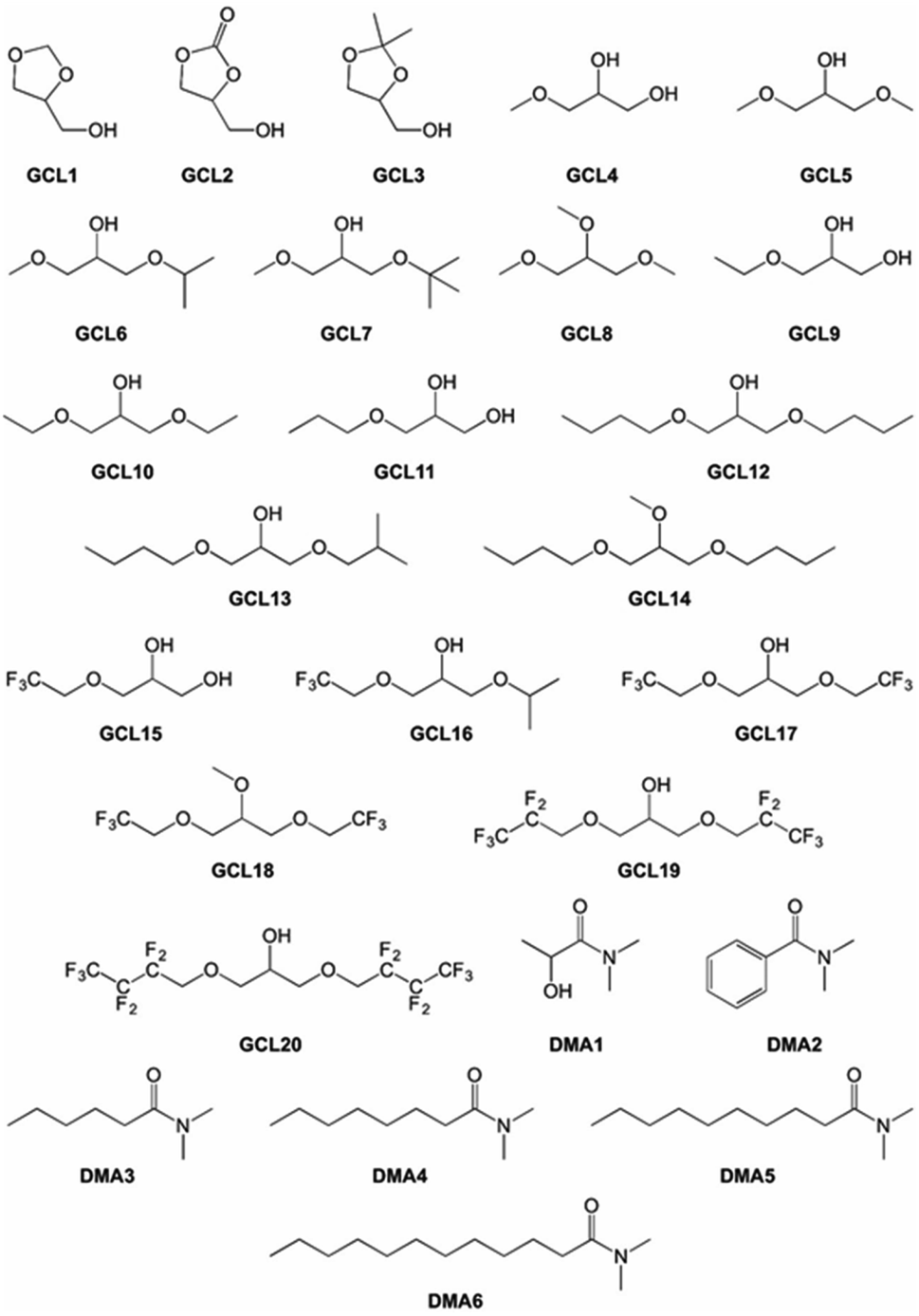 | ||
| Fig. 2 Chemical structure and nomenclature of synthesized biosolvents employed in this study. | ||
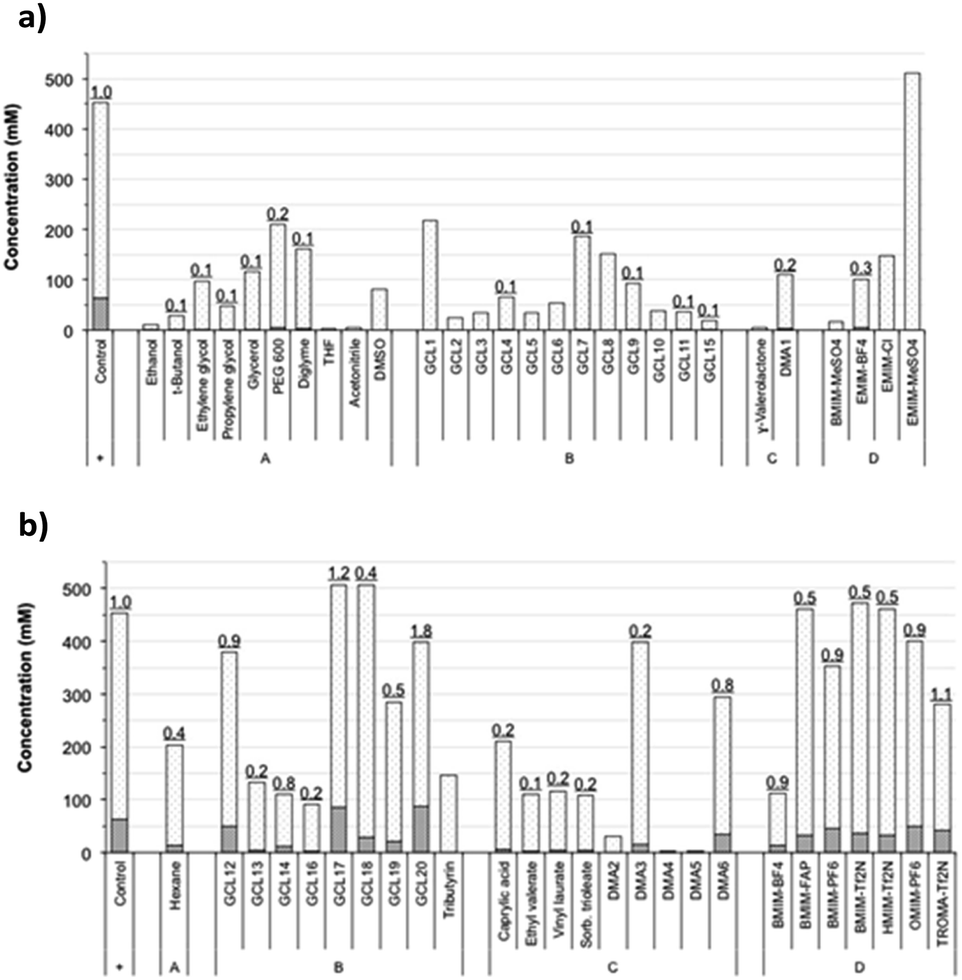 | ||
| Fig. 3 FOS formation and sucrose consumption in transfructosylating reactions using the referred water-miscible (a) or -immiscible (b) solvents. Sucrose consumed and FOS produced (6-kestose, 1-kestose, and neokestose) are presented in white-dotted- and grey-striped-bars, respectively. Underlined numbers at column tops indicate the ratio between the two subsection bars in relation to the value of the control obtained without added solvents (relative transfer efficiency). | ||
The total amount of kestoses obtained in all cases was very small or even negligible when compared to the control, showing relative transfer efficiencies much less than 1.0. Besides, the hydrolytic capacity of Ffase was also negatively affected, excepting for EMIM-MeSO4, which improved it by about 32%. In general, the Ffase activity was shown to be much more compatible with immiscible solvents (Fig. 3b), which seems to be somewhat paradigmatic of some microbial glycosidases.25–27 Indeed, five solvents, namely, GCL17, GCL18, BMIM-FAP, BMIM-Tf2N, and HMIM-Tf2N, enhanced the hydrolytic Ffase activity by about 9–23%, and transfer products were detected in most assays. Furthermore, the yield of total kestoses was almost identical (or somehow similar) to the control in nearly half of the reactions, such as in the case of GCL12, DMA6, BMIM-PF6, OMIM-PF6, or TROMA-Tf2N; and even increased by 34–40% when GCL17 and GCL20 were employed. The use of these two fluorinated glycerol ethers involved a relative transfer efficiency of 1.2 and 1.8, respectively, which means an improvement of fructosylation rate over sucrose hydrolysis in relation to control. These results were consistent with those previously obtained with other glycosylases. Thus the β-galactosidase-3 from Bacillus circulans showed excellent catalytic activity in the presence of fluorinated glycerol ether derivatives,28 and a mutated variant of the α-galactosynthase TmGalA from Thermotoga maritima with the solvent GCL17.29 This fact shows the possible benefit of this type of compounds in promoting or optimizing the occurrence of certain transglycosylation reaction.
As seen, Ffase activity was clearly affected by the medium in which the enzymatic reaction takes place, and therefore the conformational state of the enzyme must be somehow altered. This assumption was in accordance with data obtained from a preliminary protein fluorimetric study at 295 nm excitation wavelength (tryptophan maximum absorption), in which three different nonsinkable solvents causing a highly detrimental, GCL5; almost neutral, GCL12; or slightly beneficial, TROMA-Tf2N, effect on Ffase catalytic activity were analyzed (Fig. S1 in the ESI†). This set of solvents was previously proven to be representative while not interfering with the appropriate process itself of protein fluorescence recording. The obtained emission spectra correlated with certain changes in the disposition of tryptophan protein groups, which were linked to conformational alterations with varying impact on the enzyme activity. GCL5 at increasing concentrations causes the enzyme to change its maximum intensity emission peak from 326 nm to 332 nm, a red shift, which is interpreted as greater exposure of tryptophan residues to the surrounding polar environment and therefore more extensive protein denaturation or enzymatic inactivity. On the contrary, GCL12 and TROMA-Tf2N caused a shift of the maximum emission wavelength towards the blue, from 326 nm to 320 nm, suggesting an increased packing of the tryptophan groups into the internal non-polar environment of the protein and thus higher integrity and maintenance of enzyme structure and activity.28,30 This circumstance may be particularly relevant for Ffase as it has two tryptophan residues (Trp-76 and Trp-314) in its active center involved in the recognition of fructose moiety.3 In addition, Ffase contains almost 47% of hydrophobic amino acids (with ∼83% aliphatic index),3 which could be a determining factor for explaining its better stability in non-polar environments.
Solvents used as fructosyl-acceptor by Ffase
As might be expected from the already referred substrate-acceptor promiscuity of Ffase with polyols erythritol and mannitol being efficiently fructosylated,4 new single peaks, which were absent in control assays, were detected in reactions including glycerol or ethylene glycol (Fig. 4).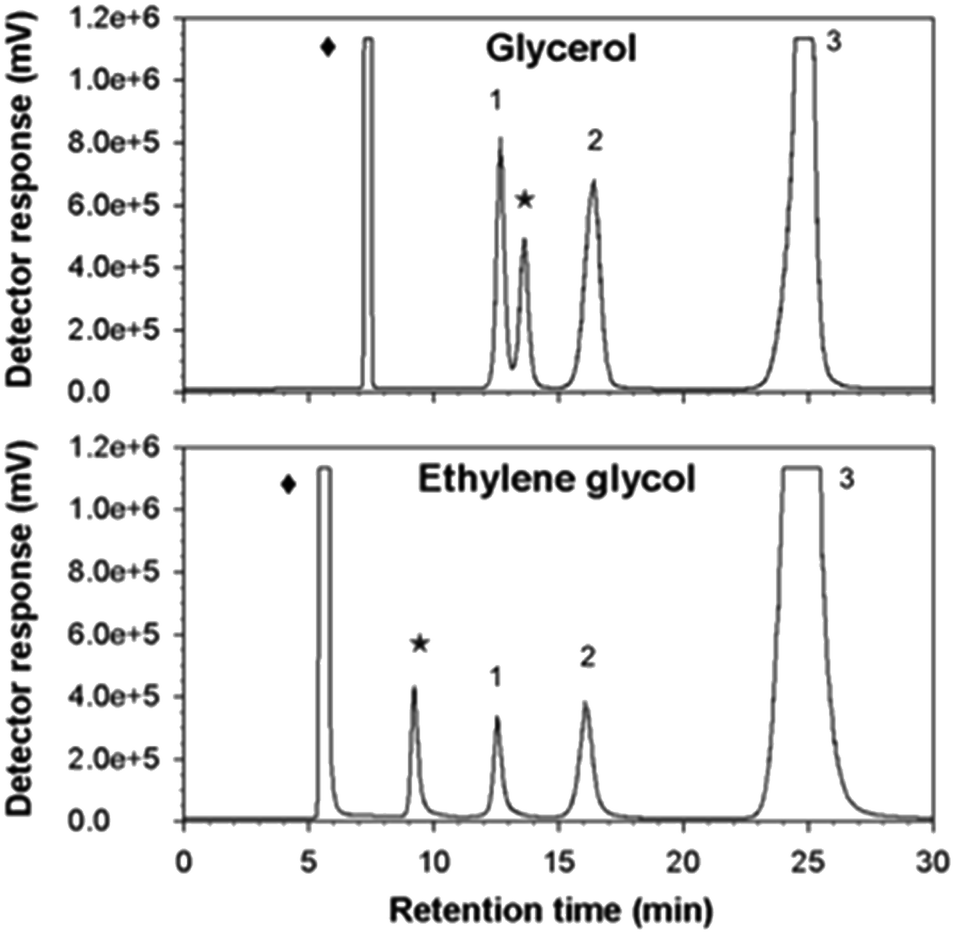 | ||
| Fig. 4 HPLC chromatograms of transfructosylating reactions containing the referred solvents. Mixtures composed of pure Ffase, sucrose, and solvents were incubated for 1 h. Peak assignations: (1) fructose, (2) glucose, (3) sucrose, (star) new fructosylated products obtained in reactions including the indicated solvents (diamond). | ||
Indeed, the size of the peaks corresponding to kestoses was greatly reduced in favor of the new ones. This fact suggested that Ffase could also use these two hydroxylated compounds as alternative fructosyl-acceptors, therefore generating novel fructo-conjugates.
The relative acceptor efficiencies for glycerol and ethylene glycol measured as the molar ratio between the concentrations of new fructosylated products derived from these solvents (accordingly, 53.17 and 53.32 mM) and of FOS from sucrose (1.84 and 1.01 mM) were widely higher than 1.0 in both cases.
The possible fructosylation of glycerol is of enormous relevance and biotechnological interest. This compound is a sweet-tasting, hygroscopic, and viscous polyol with more than 2000 described applications.20,31 It is extensively used in the obtention of numerous pharmaceutical, food, and domestic products usually as humectant, solvent, thickening, or plasticizer agent.31–35 Glycerol is also employed as a conventional sugar substitute additive (E422), mild laxative, ocular hypotensive, or cryoprotectant.31–33,35 Besides, multiple microbial biotransformations and chemical synthesis pathways, most notably, nitroglycerine or epichlorohydrin, are based on this substrate.31,34,36 Interestingly, this polyol might even be useful for cancer therapy as an activator of negative mutant p53, and for energy production as a precursor of diverse biofuels and additives thereof.37,38 In that sense, the glycosylation of glycerol could improve certain functional properties that make it valuable, especially, its solubility, stability, and sweetness; as well as its bioavailability.4,39
In addition, glycerol is generated on a large scale (about 900000 metric tonnes per year) as a common by-product from soap manufacturing, petroleum refining, and molasses fermentation.31,33,34,36,40 Consequently, finding novel and clean processes to convert this compound into new molecules with a higher added value may allow the optimization of its integral use and the achievement of greater profitability to guarantee the sustainability of the industry and the environment.34,36,40 Indeed, this type of glyco-conjugates has been projected as moisturizers, protein stabilizers, oral hypoglycemic drugs, Epstein-Barr virus inhibitors, or even anti-tumor agents.41–43 Certain glycerol glycosides have already been purified from osmotolerant species of higher plants, algae, and microbial extracts;41,44–46 however, until now very little is known concerning their in vitro production. Unlike the invertase SftA from Aspergillus sydowii that did not show to transfer fructose to glycerol,47 Ffase was able to do so. Thereby, and as far as we know, Ffase is the first reported mycotic β-fructofuranosidase with the ability to synthesize a fructosylated derivative from glycerol.
Structural characterization of main acceptor product from glycerol
The new fructo-conjugate generated in reaction mixtures containing Ffase with sucrose and glycerol was successfully isolated by preparative exclusion chromatography after scaling up the corresponding catalytic process, and its degree of purity was corroborated by TLC and HPLC analysis. As shown in Fig. S2a of the ESI,† only one clear spot was observed on the developed plate compared to the multiple spots initially visualized. In conjunction with this, Fig. S2b† reveals the detection of a single signal in the chromatogram in contrast to what was previously seen with the starting sample (Fig. 4a). The purified glycoside was then qualitatively characterized using a combination of 1H and 13C NMR techniques (Table S2, Fig. S3a and b in the ESI†). Analysis of 13C NMR, DEPT and HSQC-HMQC data of 1 and 2 compounds, showed the presence of nine sp3 carbon signals (four CH2, four CH and one C). The 1H NMR and DEPT spectra of 1 revealed three oxygenated methine protons at δH 4.06 (1H, t, J = 10.0 Hz, H-4), which showed correlations COSY H–H with δH 4.13 (1H, d, J = 10.0 Hz, H-3) and H-5 (δH 3.85–3.75). Two methylene protons at δH 3.65–3.60 (2H, m, H-6) and δH 3.70 (1H, d, J = 10.0 Hz, H-1A) and δH 3.63 (1H, d, J = 10.0 Hz, H-1B). The assignment of the signals of the 13C NMR spectrum, corresponding at δ 76.84 (C-3), δ 74.59 (C-4) and δ 81.08 (C-5) was carried out by using HSQC-HMQC spectrum, which showed a correlation via 1δC–H. These signals and the quaternary carbon at δC 103.53 (C-2) were correlated with a fructose ring. HMBC connection of H-2′ (δH 3.85–3.75) of glycerol with C-2 suggested that fructose are linked from anomeric carbon C-2 to C-2′ of glycerol. The compound 2 exhibited a close similarity of the NMR signals to the one of compound 1, with exception of the methylene group C-1′ (δ 70.82) and the HMBC connectivity between C-2 (δ 103.47) and H-1′ (δ 3.70–3.60). This change indicated that fructose are linked from C-2 to C-1′ of glycerol.Based on this analysis two novel regioisomeric structures with an estimated molecular weight of 254.24 g mol−1 each (C9H18O8) were detected, specifically, 1-O-β-D-fructofuranosylglycerol and 2-O-β-D-fructofuranosylglycerol in a proportion of 40% and 60%, respectively. This finding confirmed the biotechnological utility of Ffase as the first known enzyme from yeasts able to synthesize such glycerol derivatives. A mixture of 1/2-O-β-D-fructosyl glycerol was also previously obtained by using levansucrases from Bacillus circulans and B. subtilis (SacB) in reactions containing sucrose and glycerol.48,49 Moreover, the β-fructofuranosidase I (MsFfase) from Microbacterium saccharophilum K-1 (formerly known as Arthrobacter sp. K-1) was shown to fructosylate glycerol and other polyols, but the structure of the products so generated were not characterized.50 Peculiarly, Ffase exhibited a marked bias for glycosylating the secondary hydroxyl group of glycerol, unlike other microbial enzymes, including the aforementioned Bacillus ones. Thus, the α-glucosidase HaG from Halomonas sp. H11 as well as the β-glucosydase CelB from Pyrococcus furiosus and SSG from Sulfolobus shibatae predominantly bound a glucose unit to the terminal OH groups of glycerol.42,44,45 Similarly, only the primary hydroxyl groups of different polyols (other than glycerol) were fructosylated when using the β-fructofuranosidase from the yeast Cryptococcus laurentii.24 The same was also observed for Ffase in reactions including mannitol or erythritol.4 Perhaps, in this case, the secondary hydroxyl group of glycerol is more accessible than the primary one to be preferentially fructosylated by Ffase because of the high flexibility of this small tri-alcohol in aqueous phase and a combination of both intramolecular hydrogen bonds and intermolecular hydroxyl groups solvation.31
Effect of donor/acceptor concentration on the synthesis of fructosyl-glycerol
Production of fructosyl-glycerol was evaluated using different concentrations of sucrose and glycerol. The progression of these reactions was initially monitored by TLC analysis (a representative plate is shown in Fig. S3 in the ESI†) and their products were quantified by HPLC when the maximum level of synthesis was detected. As can be seen in Table 1, the highest amount of fructosyl-glycerol (∼62 g L−1) was obtained by using 400 g L−1 of both sucrose and glycerol, doubling the value obtained with half of these substrates (∼31 g L−1), and without apparent improvement by lowering the sucrose/glycerol ratio through the rise of the acceptor concentration.| Suc (g L−1) | Gcl (g L−1) | Suc/Gcl ratio | Kes (g L−1) | Fru-Gcl (g L−1) | Kes/Fru-Gcl ratio |
|---|---|---|---|---|---|
| a Suc: sucrose; Gcl: glycerol; Kes: total kestoses; and Fru-Gcl: 1/2-O-β-D-fructofuranosyl-glycerol. Each data represents the average of two independent measurements at the reaction time in which the amount of transfer products reach their maxima: 2, 4, 5.5, 20, 3.5, 5, 10 and 336 h for (a), (b), (c), (d), (e), (f), (g) and (h); respectively. Standard deviations were lower than 5%. | |||||
| Ffase | |||||
| 200 | 200 | 1/1(a) | 3.08 | 31.17 | 1/10 |
| 200 | 400 | 1/2(b) | 1.19 | 36.44 | 1/31 |
| 200 | 600 | 1/3(c) | 0.45 | 18.49 | 1/41 |
| 200 | 800 | 1/4(d) | 0.06 | 3.27 | 1/55 |
| 300 | 300 | 1/1(e) | 2.77 | 45.48 | 1/16 |
| 300 | 600 | 1/2(f) | 0.55 | 19.12 | 1/35 |
| 400 | 400 | 1/1(g) | 2.52 | 61.74 | 1/25 |
|
|||||
| ScFfase-Leu196 | |||||
| 400 | 400 | 1/1(h) | 3.13 | 71.97 | 1/23 |
Presumably, the high competence exerted by sucrose and glycerol for gaining access to the catalytic site of Ffase somehow limited the highest yield of transfructosylation since the fructosyl-glycerol increase was correlated with a total FOS decrease. However, when using the protein variant Leu196 of Ffase (ScFfase-Leu196), which a priori showed enhanced transferase activity by almost 3-fold,12 the concentration of both fructosyl-glycerol and kestoses increased by about 1.2 times each (Table 1), but at the expense of a significant increase in the reaction time (336 h vs. 10 h).
In this context, 78 g L−1 of fructosyl-glycerol were previously obtained by using levansucrases from Bacillus circulans or B. subtilis in solutions containing 280 g L−1 sucrose and 500 g L−1 glycerol,48,49 and up to 13 g L−1 of this compound when using the β-fructofuranosidase I from Microbacterium saccharophilum K-1 in 103 g L−1 sucrose and 92 g L−1 glycerol reactions.50 Clearly glycerol is a good fructosylable substrate for Ffase considering that the obtained amount of fructosyl-glycerol (∼62 g L−1) exceeded the already published 44 g L−1 of fructosyl-mannitol and the 35 g L−1 of fructosyl-erythritol generated from mannitol and erythritol, respectively,4 as well as the yield of other yeast β-fructofuranosidases able to use alternative glycosidic acceptors, such as the Xd-INV from Xanthophyllomyces dendrohous, which produced in the best case analysed about 38 g L−1 of neo-erlose from maltose.51
Kinetics of fructosyl-glycerol production
The composition of substrates and products in the reaction catalyzed by Ffase with 400 g L−1 of both glycerol and sucrose was analyzed during 14 h (Fig. 5). As expected for a fructosyltransferase activity, the amount of fructose was always slightly lower than that of glucose.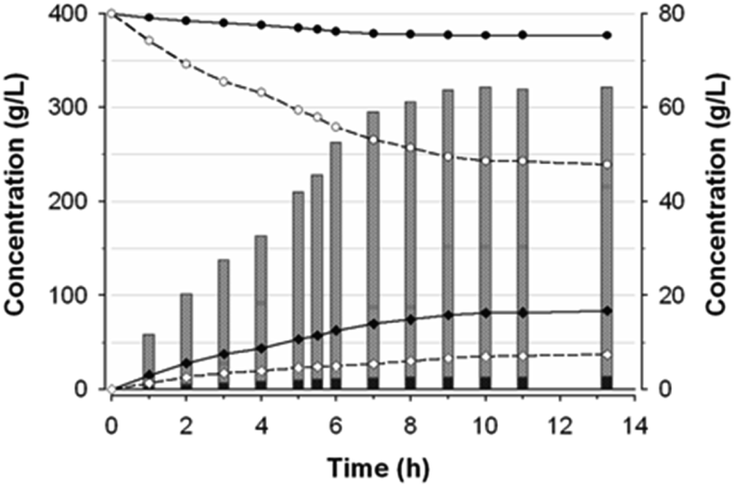 | ||
| Fig. 5 Time-course of fructosyl-glycerol production. Glycerol and sucrose concentrations are indicated with closed and open circles, and those of glucose and fructose with black and white diamonds, respectively. Bars data are associated with the right axis scale. Grey-striped-pattern subsections show the fructosyl-glycerol produced and the black-striped ones the total FOS at the indicated times. | ||
The maximum production of fructosyl-glycerol and kestoses was reached after 10 h. At this point, sucrose and glycerol conversions were, respectively, of about 39% and 6% (w/w), referred to the total amount of substrates and products of the mixture. Furthermore, the reaction mixture contained 34.8 g L−1 fructose, 81.4 g L−1 glucose, 243.1 g L−1 sucrose, 376.7 g L−1 glycerol, 61.7 g L−1 fructosyl-glycerol, 2.5 g L−1 total FOS (of which 1.74 g L−1 6-kestose, 0.44 g L−1 1-kestose, and 0.37 g L−1 neokestose), together with 0.5 g L−1 blastose and minimal traces of some unidentified products. This outcome means fructosyl-glycerol was by far the major transfer product present in this reaction mixture, accounting for around 7.7% (w/w) of all compounds thereof (including polyols), and 95.3% (w/w) of all generated fructo-conjugates. Besides, the ratio between kestoses and fructosyl-glycerol was found to be 1:25. Interestingly, the glycosidic linkage formed between the fructosyl unit and glycerol seemed to be hydrolysis-resistant and stable under the assay conditions. Thereby, the concentration of fructosyl-glycerol continuously increased until it reached a plateau beyond 10 h of reaction. This behavior contrasts with the typical FOS formation dynamic observed in previous works with this enzyme, whose concentration reached a maximum peak followed by a rapid hydrolysis process.2,4 Hence, Ffase has proven to be an excellent tool for catalysing in a non-pollutant way a novel fructosylated derivative from glycerol with biotechnological potential. The applicability of this reaction would be even enhanced by trying to carry out the whole process directly in yeast cultures supplemented with sucrose and glycerol, a future goal that we could address. Further research into the isolation and characterization of the major transfer product from ethylene glycol will also help us to understand the peculiar activity of Ffase and to find additional utilities for this enzyme. Likewise, a prospective structural study of the underlying interaction between Ffase and these new alternative acceptors would contribute to shed light on the determinants responsible for its transferase activity.
Experimental
Solvents, media and reagents
Commercial chemical compounds used throughout this work and their respective suppliers are collected in Table S3 in the ESI.† Glycerol-derived ethers (GCL4–20 in Fig. 2) were synthesized using the procedures previously described.52–54 GCL1–3 and N,N-dimethylamide derivatives of carboxylic acids (DMA1–6 in Fig. 2) were kindly provided by Cognis GmbH (Monheim am Rhein, Germany). Some of their essential physical–chemical properties were listed in Table S1 of the ESI.†Ffase production and purification
Schwanniomyces occidentalis ATCC 26077 was cultivated in YEI medium (20 g L−1 YE, 15 g L−1 inulin) at 28 °C. The density of culture was followed at 660 nm (Abs660) and extracellular Ffase was purified as referred.3 Briefly, culture filtrates showing enzymatic activity (600 mL; Abs660 = 12.5; 110 U mL−1; 9 mg mL−1; 12 U mg−1) were concentrated-fractionated, applied to DEAE-chromatography, and analysed by SDS-PAGE (8%) gels stained with ProtoBlue-Safe colloidal coomassie (National Scientific; Atlanta, USA). Fractions containing apparently pure proteins were pooled, concentrated (760 U mL−1; 1.74 mg mL−1; 437 U mg−1) through Amicon® Ultra-15 (Merck KGaA; Darmstadt, Germany), and stored at −70 °C (Fig. S4a of the ESI†). Protein concentration was determined using Bradford assay (Bio-Rad Lab. Inc.; Hercules, USA).ScFfase-Leu196 production and purification
The strain of invertase-negative Saccharomyces cerevisiae EUROSCARF Y02321 transformed with vector pYES2.0 for expressing recombinant Ffase protein variant containing Leu at position 196 (ScFfase-Leu196) was previously generated.12 ScFfase-Leu196 expression was induced in 1 L of YEPGal (10 g L−1 YE, 10 g L−1 peptone, 20 g L−1 galactose) at 30 °C. At the beginning of the yeast stationary phase (Abs660 = 6–7), culture filtrates (6 U mL−1; 15 mg mL−1; 0.4 U mg−1) were concentrated-fractionated, applied to DEAE-chromatography, and concentrated through Amicon® Ultra-15 (430 U mL−1; 9.4 mg mL−1; 46 U mg−1). Proteins were stored, quantified, and analyzed by SDS-PAGE (8%) as in Section 2.2 (Fig. S4b of the ESI†).Standard β-fructofuranosidase hydrolytic activity assay
Standard β-fructofuranosidase hydrolytic activity was assayed at 50 °C for 20 min by measuring reducing sugars released from 25 g L−1 sucrose in 0.2 M sodium acetate buffer pH 5.5 using the DNS method adapted to 96-well microplates as described.2 Reaction mixtures without enzyme or sucrose were used as negative controls. One unit of activity (U) was defined as that catalysing the formation of 1 μmol of reducing sugar per minute under the described conditions. Reactions were performed at least in triplicate with a standard deviation of less than 5%.Enzyme-catalyzed transfructosylating reactions with solvents
Reactions (500 μL) contained 10 U mL−1-standard β-fructofuranosidase activity, 200 g L−1 sucrose in 0.2 M sodium acetate buffer pH 5.5, and 30% (v/v) final concentration of solvents, except for GCL and DMA at 2 M.28,55,56 Mixtures were maintained at 50 °C with 100 rpm magnetic stirring on a Carouse 6 Plus hotplate (R. B. Radley Co. Ltd; Saffron Walden, UK) during different times, and then boiled for 10 min and stored at −20 °C. Positive controls did not incorporate solvents and negative controls lacked enzyme or sucrose. The absence of cross-reactivity and stability over time of reaction components was preliminarily verified by TLC before adding the enzyme. Analytical determinations of reaction components were carried out by HPLC, after suitable extraction of the aqueous phase (if required). The relative fructosyl-transfer efficiency of the enzyme with each solvent was defined as the ratio between moles of synthesized kestoses and consumed sucrose in relation to the positive control. All assays were carried out at least in duplicate.TLC and HPLC analysis
TLC was applied to monitor the progress of transfructosylating reactions. It was based on a silica gel 60 (Merck; Darmstadt, Germany) with 2-propanol/water/aqueous ammonia 25% (7:2:1) as eluent. UV-lamp light, spraying with 10% H2SO4 in MeOH, and oven heating were used for compounds visualization. Precise analytical determination of products was performed by HPLC. The equipment consisted of a quaternary pump (Jasco Inc.; Easton, USA) with a manual injection valve (Agilent Technologies; Santa Clara, USA) coupled to a Asahipak-NH2P-50-4E amino column (4.6 mm × 250 mm, 5 μm) from Shodex (Showa Denko Europe GmbH; Munich, Germany) followed by an evaporative light-scattering detector (Softa Corp.; Westminster, USA). Acetonitrile/water (8:2) degassed by bath sonication was used as a mobile phase at 0.8 mL min−1 flow. The column was kept at room temperature, while the detector was equilibrated at 54 °C with a constant supply of 50-psi molecular nitrogen as nebulizer gas. Before injection, each sample was diluted with deionized water and passed through a Branchia 0.22 μm nylon syringe filter (Dismadel S.L.; Madrid, Spain). Data obtained after 65 min total analysis time were processed by Borwin software (v. 1.5; Jasco Inc.). All compounds were correspondingly identified and quantified on the basis of the retention time and integrated area of detected chromatographic peaks. Calibration curves in a 0.02–0.75 g L−1 range were first generated using high-purity fructose, glucose, sucrose, 1-kestose, nystose, glycerol, and ethylene glycol. In addition, 6-kestose, neokestose, and blastose, which were previously synthesized from sucrose by β-fructofuranosidases as referred before,4,5 were also employed to ensure appropriate peak assignation. When standards could not be obtained commercially, the same detector response factor of the most related available compound was assumed. Thus, the concentration of blastose and novel fructosylated derivatives from ethylene glycol and glycerol was estimated considering the sucrose calibration, and every FOS type of three sugar units as 1-kestose. Each analysis was conducted at least in duplicate.
Purification of glycerol-acceptor product and NMR analysis
Transfructosylating reaction including 400 g L−1 sucrose and 400 g L−1 glycerol was scaled up to 20 mL. The produced fructosyl-glycerol derivative was purified through Bio-Gel® P-2 chromatography column (1.5 × 120 cm, 45 μm, Bio-Rad). Samples of 500 μL were loaded onto the column at room temperature and eluted at 0.2 mL min−1 with deionized water previously degassed by bath sonication. After successive rounds, fractions enriched in the glycerol derivative as analysed by TLC were lyophilized, and then their purity evaluated by HPLC. The mass performance of the isolated glycoside after the purification process was around 2% when compared to the original sample content. The structure of the fructosyl derivatives based on glycerol were determined using a combination of 1H and 13C NMR techniques. NMR spectra were recorded in D2O at room temperature using a Bruker WM 500 spectrometer [500 MHz (1H NMR) and 125 (13C NMR)]. Chemical shifts are given on the δ-scale and were referenced to the TMS as internal standard. The pulse programs of the following 2D experiments were taken from the Bruker software library and the parameters were as follows: 500/125 MHz gradient-selected HMQC spectra: relaxation delay D1 = 1.5 s; 500/125 MHz gradient-selected HMBC spectra: relaxation D1 = 1.5 s; evolution delay D2 = 3.33 ms; delay for evolution of long range coupling D6 = 60 ms. 500 MHz gradient selected 1H, 1H COSY spectra: relaxation delay D1 = 1.5 s; 90° pulse for 1H.Conclusions
The hydrolytic and transferase activity of Ffase was largely compatible with the presence of various GS. Among them, two fluorinated glycerol ethers improved the yield of total FOS obtained by this enzyme. This fact shows the possible benefit of these compounds in promoting or optimizing the occurrence of this type of reaction. Most of the hydrophilic solvents assayed here were detrimental to Ffase activity, which seems somewhat paradigmatic of some microbial glycosidases. Despite that, ethylene glycol and glycerol resulted in being suitable alternative acceptors by Ffase, which is in line with previous research using other polyols. The fructosylated derivative from glycerol was purified and characterized, finding 2-O isomer as the major product (60%) and 1-O isomer (40%) as the minor one, thus conferring Ffase a new biotechnological utility as the first known enzyme from yeasts able to carry out this biosynthetic process. This new compound could be applied as a possible sugar substitute, prebiotic, humidifier, stabilizer, or even as a pharmacological agent. After optimizing the process, a yield of about 62 g L−1 of fructosyl-glycerol was obtained, which also increased (∼16%) by using a mutated variant of this enzyme, albeit with an excessively prolonged reaction time. Besides, the glycosidic linkage of this novel fructo-conjugate appeared to be resistant to the hydrolytic action of the enzyme. In that sense, this work opens a window of opportunity to synthesize enzymatically in a non-pollutant way a novel fructosylated derivative from glycerol with biotechnological potential.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Ministry of Science, Innovation, and Universities [Grants: RTI2018-096037-B-I00, RTI2018-093431-B-I00 and PID2019-105838RB-C3-2] and of Economy and Competitiveness [BIO2016-76601-C3-2-R], as well as Fundación Ramón Areces [XIX Call of Research Grants in Life and Material Sciences]. Besides, funding has been received from the Gobierno de Aragón (E37_20R) and the European Regional Development Fund (ERDF). Centro de Biología Molecular Severo Ochoa was benefited from an institutional grant from Fundación Ramón Areces. Mr D. Piedrabuena and Mr A. Leal-Duaso were the recipient of a doctoral fellowship from the Spanish Ministry of Education, Culture, and Sports [FPU014/01004 and FPU014/04338].References
- I. Soares, Z. Távora, R. Patera-Barcelos and S. Baroni, in Scientific, health and social aspects of the food industry, ed. B. Valdez, InTech Europe, Rijeka, 1st edn, 2012, vol. 5, pp. 83–94 Search PubMed.
- M. Álvaro-Benito, M. de Abreu, L. Fernández-Arrojo, F. J. Plou, J. Jiménez-Barbero, A. O. Ballesteros, J. Polaina and M. Fernández-Lobato, J. Biotechnol., 2007, 132, 75–81 CrossRef PubMed.
- M. Álvaro-Benito, A. Polo, B. González, M. Fernández-Lobato and J. Sanz-Aparicio, J. Biol. Chem., 2010, 285, 13930–13941 CrossRef PubMed.
- D. Piedrabuena, N. Míguez, A. Poveda, F. J. Plou and M. Fernández-Lobato, Appl. Microbiol. Biotechnol., 2016, 100, 8769–8778 CrossRef CAS PubMed.
- D. Rodrigo-Frutos, D. Piedrabuena, J. Sanz-Aparicio and M. Fernández-Lobato, Appl. Microbiol. Biotechnol., 2019, 103, 279–289 CrossRef CAS PubMed.
- G. R. Gibson, H. M. Probert, J. V. Loo, R. A. Rastall and M. B. Roberfroid, Nutr. Res. Rev., 2004, 17, 259–275 CrossRef CAS PubMed.
- S. Tadesse, J. Biol. Act. Prod. Nat., 2012, 2, 124–134 Search PubMed.
- J. Ballongue, C. Schumann and P. Quignon, Scand. J. Gastroenterol., Suppl., 1997, 32, 41–44 CrossRef CAS PubMed.
- J. W. Yoon, E. J. Jeon, I. H. Jung, M. J. Min, H. Y. Lee, M. J. Kim, J. S. Baek, H. S. Lee, C. S. Park, S. Oh, K. H. Park and T. W. Moon, Biosci., Biotechnol., Biochem., 2003, 67, 525–531 CrossRef CAS PubMed.
- S. Ghosh and M. L. Sudha, Int. J. Food Sci. Nutr., 2012, 63, 372–379 CrossRef CAS PubMed.
- M. Álvaro-Benito, M. A. Sainz-Polo, D. González-Pérez, B. González, F. J. Plou, M. Fernández-Lobato and J. Sanz-Aparicio, J. Biol. Chem., 2012, 287, 19674–19686 CrossRef PubMed.
- M. Álvaro-Benito, M. de Abreu, F. Portillo, J. Sanz-Aparicio and M. Fernández-Lobato, Appl. Environ. Microbiol., 2010, 76, 7491–7499 CrossRef PubMed.
- J. F. Robyt, Adv. Carbohydr. Chem. Biochem., 1995, 51, 133–168 CrossRef CAS PubMed.
- B. Zeuner, C. Jers, J. D. Mikkelsen and A. S. Meyer, J. Agric. Food Chem., 2014, 62, 9615–9631 CrossRef CAS PubMed.
- M. J. Hernaiz, A. R. Alcántara, J. I. García and J. V. Sinisterra, Chem.–Eur. J., 2010, 16, 9422–9437 CrossRef CAS PubMed.
- M. Pérez-Sánchez, M. Sandoval, M. J. Hernaiz and P. Domínguez de María, Curr. Org. Chem., 2013, 17, 1188–1199 CrossRef.
- A. Farrán, C. Cai, M. Sandoval, Y. Xu, J. Liu, M. J. Hernaiz and R. J. Linhardt, Chem. Rev., 2015, 115, 6811–6853 CrossRef PubMed.
- C. J. Clarke, W. C. Tu, O. Levers, A. Bröhl and J. P. Hallet, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- A. Wolfson, A. Snezhko, T. Meyouhas and D. Tavor, Green Chem. Lett. Rev., 2012, 5, 7–12 CrossRef CAS.
- J. I. García, H. García-Marín and E. Pires, Green Chem., 2014, 16, 1007–1033 RSC.
- B. P. Soares, D. O. Abranches, T. E. Sintra, A. Leal-Duaso, J. I. García, E. Pires, S. Shimizu, S. P. Pinho and J. A. P. Coutinho, ACS Sustainable Chem. Eng., 2020, 8, 5742–5749 CrossRef CAS.
- Gangadhara, P. R. Kumar and V. Prakash, Protein J., 2008, 27, 440–449 CrossRef CAS PubMed.
- J. Sunitha and P. K. Saiprakash, Indian J. Chem., 1994, 33, 995–998 Search PubMed.
- J. Dudíková, M. Mastihubová, V. Mastihuba and N. Kolarova, J. Mol. Catal. B: Enzym., 2007, 45, 27–33 CrossRef.
- C. Laane, S. Boeren, K. Vos and C. Veeger, Biotechnol. Bioeng., 1987, 30, 81–87 CrossRef CAS PubMed.
- C. Bucke, J. Chem. Technol. Biotechnol., 1996, 217–220 CrossRef CAS.
- S. Bielecki and R. I. Somiari, Biotechnol. Prog., 1998, 15, 423–428 CAS.
- C. Bayón, A. Cortés, A. Aires-Trapote, C. Civera and M. J. Hernaiz, RSC Adv., 2013, 3, 12155–12163 RSC.
- C. Bayón, M. Moracci and M. J. Hernaiz, RSC Adv., 2015, 5, 55313–55320 RSC.
- J. T. Vivian and P. R. Callis, Biophys. J., 2001, 80, 2093–2109 CrossRef CAS PubMed.
- M. Pagliaro and M. Rossi, The future of glycerol: new usages for a versatile raw material, Royal Society of Chemistry, Cambridge, 1st edn, 2008, 1, pp. 1–17 Search PubMed.
- D. Brisson, M. C. Vohl, J. St-Pierre, T. J. Hudson and D. Gaudet, BioEssays, 2001, 23, 534–542 CrossRef CAS PubMed.
- P. M. Collins, Dictionary of carbohydrates, Chapman and Hall/CRC, Boca Raton, 2nd edn, 2005, pp. 584–585 Search PubMed.
- X. Luo, X. Ge, S. Cui and Y. Li, Bioresour. Technol., 2016, 215, 114–154 CrossRef PubMed.
- R. Hudgens, R. Hercamp, J. Francis and D. Nyman, SAE [Tech. Pap.], 2007, 1, 1–18 Search PubMed.
- P. B. Smith, Plast. Eng., 2013, 69, 44–48 CrossRef.
- J. Q. Albarelli, D. T. Santos and M. R. Holanda, Braz. J. Chem. Eng., 2011, 28, 691–698 CrossRef CAS.
- R. L. Arechederra and S. D. Minteer, Fuel Cells, 2009, 9, 63–69 CrossRef CAS.
- H. Chen, X. Jin, L. Zhu, Y. Lu, Z. Ma, S. Liu and X. Chen, Appl. Microbiol. Biotechnol., 2020, 104, 9523–9534 CrossRef CAS.
- M. Anitha, S. K. Kamarudin and N. T. Kofli, Chem. Eng. Sci., 2016, 295, 119–130 CrossRef CAS.
- C. Goedl, T. Sawangwan, M. Mueller, A. Schwarz and B. A. Nidetzky, Angew. Chem., Int. Ed., 2008, 47, 10086–10089 CrossRef CAS.
- A. Schwarz, M. S. Thomsen and B. Nidetzky, Biotechnol. Bioeng., 2009, 103, 865–872 CrossRef CAS.
- F. Takenaka and H. Uchiyama, Biosci., Biotechnol., Biochem., 2001, 65, 1458–1463 CrossRef CAS PubMed.
- D. H. Jung, D. S. Seo, J. H. Park, M. J. Kim, N. I. Baek and C. S. Park, J. Microbiol. Biotechnol., 2019, 29, 562–570 CrossRef CAS PubMed.
- T. Ojima, W. Saburi, T. Yamamoto and T. Kudo, Appl. Environ. Microbiol., 2012, 78, 1836–1845 CrossRef CAS PubMed.
- P. M. Collins, Dictionary of carbohydrates, Chapman and Hall/CRC, Boca Raton, 2nd edn, 2005, pp. 474–483 Search PubMed.
- M. Muramatsu and T. Nakakuki, Biosci., Biotechnol., Biochem., 1995, 59, 208–212 CrossRef CAS PubMed.
- F. González-Muñoz, J. Pérez-Oseguera, M. Cassani, M. Jiménez-Estrada, R. Vázquez-Duhalt and A. López-Munguía, J. Carbohydr. Chem., 1999, 18, 275–283 CrossRef.
- N. Galonde, N. Dyubankova, Q. Dongyan, J. P. Boutique, E. Lescrinier and W. Van den Ende, Biocatal. Biotransform., 2009, 27, 328–339 CrossRef CAS.
- K. Fujita, K. Hara, H. Hashimoto and S. Kitahata, Agric. Biol. Chem., 1990, 54, 2655–2661 CrossRef CAS.
- M. Gimeno-Pérez, P. Santos-Moriano, L. Fernández-Arrojo, A. Poveda, J. Jiménez-Barbero, A. O. Ballesteros, M. Fernández-Lobato and F. J. Plou, Process Biochem., 2014, 49, 423–429 CrossRef.
- A. Leal-Duaso, M. Caballero, A. Urriolabeitia, J. A. Mayoral, J. I. García and E. Pires, Green Chem., 2017, 19, 4176–4185 RSC.
- A. Leal-Duaso, P. Pérez, J. A. Mayoral, J. I. García and E. Pires, ACS Sustainable Chem. Eng., 2019, 7, 13004–13014 CrossRef CAS.
- A. Leal-Duaso, S. Gracia-Barberán, J. A. Mayoral, J. I. García and E. Pires, Org. Process Res. Dev., 2020, 24, 154–162 CrossRef CAS.
- M. Pérez-Sánchez, M. Sandoval, A. Cortés-Cabrera, H. García-Marín, J. V. Sinisterra, J. I. García and M. J. Hernaiz, Green Chem., 2011, 13, 2810–2817 RSC.
- M. Pérez-Sánchez, A. Cortés-Cabrera, H. García-Martín, J. V. Sinisterra, J. I. García and M. J. Hernaiz, Tetrahedron, 2011, 67, 7708–7712 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01391b |
| This journal is © The Royal Society of Chemistry 2021 |