
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew aspect of photophysics of 7,7,8,8-tetracyanoquinodimethane and its solvated complexes: intra- vs. inter-molecular charge-transfer†
Satoru Muramatsu a,
Nobumasa Chakia,
Shin-nosuke Kinoshitaa,
Yoshiya Inokuchia,
Manabu Abea,
Toshifumi Iimorib and
Takayuki Ebata*ac
a,
Nobumasa Chakia,
Shin-nosuke Kinoshitaa,
Yoshiya Inokuchia,
Manabu Abea,
Toshifumi Iimorib and
Takayuki Ebata*ac
aDepartment of Chemistry, Graduate School of Advanced Science and Engineering, Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima-shi, Hiroshima, 739-8526, Japan. E-mail: tebata@nctu.edu.tw
bDepartment of Sciences and Informatics, Muroran Institute of Technology, 27-1 Mizumoto-cho, Muroran, Hokkaido 050-8585, Japan
cDepartment of Applied Chemistry, Institute for Molecular Science, National Yang Ming Chiao Tung University, Hsinchu 30010, Taiwan
First published on 24th June 2021
Abstract
We performed laser-induced fluorescence (LIF) spectroscopy of 7,7,8,8-tetracyanoquinodimethane (TCNQ) and its solvated complexes with acetonitrile (ACN) and benzene (Bz), under the jet-cooled gas-phase condition. We also carried out fluorescence and its time profile measurements in TCNQ/Bz/hexane solution to compare with the gas-phase results. The LIF excitation spectrum of the S1 (ππ*)–S0 electronic transition of TCNQ monomer exhibited unusual vibronic structure with the maximum intensity at ∼3000 cm−1 above the band origin. In addition, the fluorescence lifetime is more than 100 times longer than that in hexane solution with most of the bands showing double exponential decay. The unusual feature of the vibronic bands is intrinsic and not due to the presence of other species, as confirmed by UV-UV hole-burning (HB) spectroscopy. These unusual features of S1 are thought to be due to the coupling with the S2 state, where S2 was revealed to have intramolecular charge-transfer (ICT) character. The S1–S0 transition of the TCNQ–ACN complex exhibited sharp vibronic bands which are red-shifted by 120 cm−1 from those of the monomer, indicating van der Waals (vdW) interaction between them; however, the fluorescence lifetime was drastically shortened. In contrast, the TCNQ–Bz complex gave a broad electronic spectrum. The study of the fluorescence and its time profile in TCNQ/Bz/hexane solution clearly shows the formation of the CT complex between TCNQ and Bz. Based on the experimental results and density functional theory (DFT) calculations, we propose that in the TCNQ monomer and TCNQ–ACN complex the S1 (ππ*) state is coupled to the intramolecular CT state, while the S1 state of TCNQ in the TCNQ–Bz complex is more strongly coupled to the intermolecular CT state.
1. Introduction
7,7,8,8-Tetracyanoquinodimethane (TCNQ, Chart 1) is a prototypical electron acceptor which forms charge-transfer (CT) complexes with a variety of electron-donor molecules as well as metal ions.1 Among them, the most famed example is a TCNQ–tetrathiafulvalene (TTF) organic salt, firstly reported in 1973, which exhibits high electric conductivity (∼104 S cm−1 at 66 K).2 Since then, intensive studies have been carried out for TCNQ and its derivative-based complexes,3–5 to focus on practical development for the electron transport materials, as well as for organic magnets,6 Langmuir–Blodgett films,7 field-effect transistors,8,9 organic crystals,10 and nonlinear optical materials.11 Meanwhile, several recent studies have also revealed characteristic electronic structures and photophysics of this molecule. For example, bound excited states were found for the radical anion, TCNQ˙−, by photodetachment spectroscopy,12 which is ascribed by the high electron affinity of TCNQ (3.38 eV);13 time-resolved spectroscopy in the gas phase14 and solutions15 revealed the very fast (fs–ps) decay for these states, typically attributed to internal conversion (IC) to the ground state. For neutral TCNQ molecule, Iimori and coworkers reported that the neutral TCNQ molecule in non-polar solvent (e.g. hexane) emits blue fluorescence (∼460 nm) with fluorescence lifetime as long as 4.5 ns, while the fluorescence lifetime as well as the quantum yield (QY) are drastically shortened with an increase of solvent polarity.16–18 They recently carried out transient absorption and fluorescence spectroscopic studies and described the shorter fluorescence lifetime in higher polar solvent to be due to the faster nonradiative decay to the nonfluorescent state whose energy is lowered at higher polar solvent.19 However, the non-fluorescent state was not clearly assigned and they proposed a possibility of the triplet state. Despite these interesting excited-state characteristics in bulk system, gas-phase spectroscopic study of a neutral TCNQ molecule has not been performed until 2019, when we first reported the S1 (ππ*; B3u)–S0 (Ag) electronic spectrum of TCNQ in a cold free jet by means of laser-induced fluorescence (LIF) spectroscopy.20 In that study, we found several unexpected features. First is the large difference between fluorescence lifetime in solution (4.5 ns)16 and that in free jet (220 ns at origin band).20 Second is the prominent Franck–Condon (FC) active vibrational progressions, and double exponential fluorescence decay profiles at energetically high vibronic bands. These features could not be explained by theoretical calculations of the S1 state, which may indicate coupling with other electronic state(s), although detailed investigations could not be addressed. | ||
| Chart 1 (a) Structure and (b) molecular orbitals of HOMO (π) and LUMO (π*) of TCNQ. | ||
In this study, we revisit LIF spectroscopy of TCNQ by using the excitation laser with higher resolution (∼0.2 cm−1) than the previous report (∼8 cm−1) as well as by quantitative analysis of the fluorescence decay profiles. We also performed UV-UV hole-burning (HB) spectroscopy to discriminate isomers in the jet if they exist. From the experimental results and quantum chemical calculations, we propose that the coupling between S1 and S2 states is responsible to the observed unusual feature of the vibronic bands and fluorescence decay, where S2 state has a longer lifetime (small oscillator strength); DFT calculations revealed that the S2 state has intramolecular charge-transfer (ICT) character. Furthermore, motivated by the acute dependence of the fluorescence properties on solvents, we herein investigated the effects of micro-solvation on the electronic spectra and fluorescence decay of TCNQ by focusing on acetonitrile (ACN) and benzene (Bz) as solvent molecules. We observed sharp vibronic feature in the LIF spectrum for the TCNQ–ACN complex, while only broad feature for the TCNQ–Bz complex. Both bands show faster fluorescence decay. We extend the study in solution for TCNQ–Bz system. We clearly observed the emission attributed to the intermolecular CT complex between TCNQ and Bz. The measurement of the time profile indicates that the transition from the locally excited (LE) state to the CT state occurs at ∼500 ps in hexane solution.
2. Methods
2.1 Experimental section
The experimental apparatus used in this study consists of (1) supersonic jet source with heating nozzle and (2) LIF spectrometer, whose details are described elsewhere.20,21 Briefly, in (1), a sample housing which contained TCNQ powder, attached at the head of a pulse valve, was heated to ∼210 °C. The gaseous mixture of sample vapor and He carrier gas (∼6 atm) was expanded into the vacuum chamber through a 1 mm aperture of the valve, generating a supersonic free jet. For the generation of micro-solvated complexes, the vapor of the solvent (acetonitrile (−25 °C) or benzene (10 °C)) was seeded to the He carrier gas. Then, in (2), the cooled gas sample in the jet was irradiated with the tunable UV laser output at ∼20 mm downstream from the pulse valve. We used two types of tunable lasers. One is a dye laser (Lambda Physik, Scanmate; resolution: 0.2 cm−1) pumped by the second harmonics of a Nd:YAG laser (Continuum, Surelite II). The other is an optical parametric oscillation/amplification (OPO) laser (Ekspla, NT342B; resolution: ∼7 cm−1). The fluorescence was collected by a series of lenses, and detected by a photomultiplier tube (Hamamatsu Photonics, 1P28 or R9880U). A set of long-pass filters was inserted to detect the emission in the >440 nm region, unless otherwise noted. The excitation spectra were recorded by detecting total emission as a function of the UV laser frequency. For the measurement of the UV-UV HB spectra, a tunable UV hole laser (Ekspla, NT342B) was introduced at ∼10 mm upstream of the jet from the crossing point of the probe laser (Lambda Physik, Scanmate) with a timing of ∼3 μs prior to the probe laser. The probe laser frequency was fixed to the vibronic band of a specific species, and we scanned frequency of the hole-creating laser while monitoring the fluorescence intensity. When the hole-creating laser makes a population hole for the same species monitored by the probe laser light, it causes a depletion of the fluorescence signal, which results in the specific species-selective electronic spectrum. We also measured fluorescence spectra in solution by using commercial spectrometer (Hitachi, F-2500) at room temperature. The fluorescence lifetime in solution was measured by using commercial time-correlated single-photon counting system (Hitachi, TemPro 01). The measurements of the fluorescence spectra and fluorescence lifetime in the solutions were performed without degassing or bubbling with inert gas of the solvents.The powder sample of TCNQ (purity: >98.0%) was purchased from Tokyo Chemical Industry and used without purification; we actually compared the obtained LIF excitation spectra of as-purchased samples with those of recrystallized ones,20 where no significant difference was discerned. The purity of TCNQ was examined by 1H NMR measurements (Camcor, Varian Mercury-300) before and after the LIF measurement to confirm no decomposition upon the heating of the sample. Acetonitrile (>99.8%) and benzene (>99.0%) were purchased from Sigma-Aldrich, and used without further purification.
2.2. Computational section
Electronic and geometrical structures of TCNQ and micro-solvated complexes were studied by DFT calculations using the Gaussian 16 program suite.22 The functional and basis set used were M06-2X and 6-311++G(d,p), respectively, unless otherwise noted. Structural optimization was carried out, followed by harmonic frequency calculations to confirm that each optimized structure locates at potential energy minima. Total electronic energy of each conformer is shown after the vibrational zero-point energy (ZPE) corrections. For solvated complexes, we first performed basis set superposition error (BSSE) corrections by means of counterpoise (CP) method, which revealed only the minor effect (see Section 3.2); therefore, the binding energy (solvation energy) is shown without BSSE correction in the present study. Theoretical Sn–S0 excitation energies were calculated by TD-DFT (TD-M06-2X) method. Kohn–Sham orbitals of the calculated structures were depicted with isodensity value of 0.02, by using Gaussview 6.0 program.3. Results and discussion
3.1 TCNQ monomer
Fig. 1a shows an LIF excitation spectrum of jet-cooled TCNQ in the 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–28000 cm−1 region, recorded by a dye laser with resolution of ∼0.2 cm−1. The origin band appears at 24262 cm−1. Many vibronic bands at higher energy region can be assigned by the vibrations of 327, 1098, and 2430 cm−1 and their combination bands. The overall spectral feature well reproduced the previous report in which an OPO laser with low resolution (∼7 cm−1) was used.20 The use of the dye laser resulted in the stronger intensities of low energy vibronic bands (<∼26000 cm−1), compared to those measured by OPO laser, as shown in Fig. S1 (ESI†). The difference is due to that the bandwidth at low energy region, ∼4 cm−1, is narrower than the spectral resolution of the OPO laser, and the width increases with the excess energy: ∼12 cm−1 at the excess energy of ∼3000 cm−1 (Fig. S1†). Thus, only small percentage of the OPO laser is overlapped at lower energy vibronic bands, whereas full of that is overlapped at higher energy vibronic bands. On the other hand, full percentage of the higher resolution dye laser is overlapped with the vibronic bands for all the covered energy region. Thus, the observed LIF spectrum of Fig. 1a exhibits more accurate action spectrum of TCNQ than that reported in our previous study. The increase of the bandwidth of the vibronic bands in higher energy is attributed to the increase of intramolecular vibrational redistribution (IVR) rate at higher energy, since the fluorescence lifetime of several tenth nanosecond, as discussed later, indicates no rapid nonradiative decay which causes the spectral broadening.
000–28000 cm−1 region, recorded by a dye laser with resolution of ∼0.2 cm−1. The origin band appears at 24262 cm−1. Many vibronic bands at higher energy region can be assigned by the vibrations of 327, 1098, and 2430 cm−1 and their combination bands. The overall spectral feature well reproduced the previous report in which an OPO laser with low resolution (∼7 cm−1) was used.20 The use of the dye laser resulted in the stronger intensities of low energy vibronic bands (<∼26000 cm−1), compared to those measured by OPO laser, as shown in Fig. S1 (ESI†). The difference is due to that the bandwidth at low energy region, ∼4 cm−1, is narrower than the spectral resolution of the OPO laser, and the width increases with the excess energy: ∼12 cm−1 at the excess energy of ∼3000 cm−1 (Fig. S1†). Thus, only small percentage of the OPO laser is overlapped at lower energy vibronic bands, whereas full of that is overlapped at higher energy vibronic bands. On the other hand, full percentage of the higher resolution dye laser is overlapped with the vibronic bands for all the covered energy region. Thus, the observed LIF spectrum of Fig. 1a exhibits more accurate action spectrum of TCNQ than that reported in our previous study. The increase of the bandwidth of the vibronic bands in higher energy is attributed to the increase of intramolecular vibrational redistribution (IVR) rate at higher energy, since the fluorescence lifetime of several tenth nanosecond, as discussed later, indicates no rapid nonradiative decay which causes the spectral broadening.
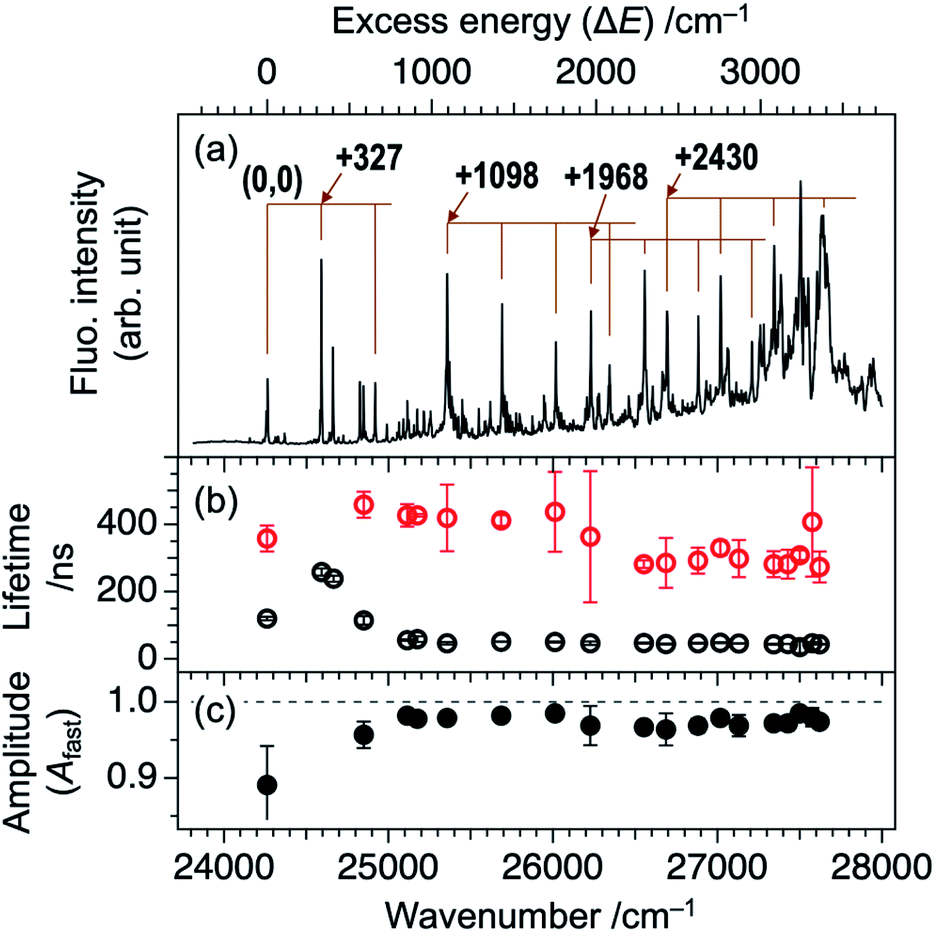 | ||
| Fig. 1 (a) LIF excitation spectrum of jet-cooled TCNQ recorded with high-resolution laser. Bottom axis represents wavelength of the incident laser while top axis is relative wavenumber with respect to the origin band at 24262 cm−1. (b) Plots of lifetimes obtained from fluorescence decay profile of each vibronic band. Black and red indicates fast and slow components, respectively. (c) Plots of amplitude of the fast component for each vibronic band. Error bars indicate estimated standard deviation from several individual measurements. | ||
As described in our previous paper,20 the LIF spectra of Fig. 1a shows unusual intensity pattern: appearance of the sharp vibronic bands in the energy region as wide as 3000 cm−1, congestion at ∼27500 cm−1, and intensity drop at the region higher than 27700 cm−1. One of the possibilities of this unusual intensity pattern is the existence of different isomers which show different vibronic structures. Thus, we performed UV-UV HB spectroscopy. Fig. 2b–d show the HB spectra measured by probing the bands at 24262, 24590, and 27620 cm−1 (refer to Fig. 2a for corresponding bands), respectively. The HB spectra exhibited almost the same spectral pattern regardless of the monitoring vibronic bands, indicating that main bands observed in the LIF spectrum belong to a single species; these bands are certainly assigned to a TCNQ monomer, as supported by vibrationally-resolved dispersed fluorescence spectroscopy with the help of DFT calculations in our previous study.20 It should be noted that the dip intensities at low wavenumber region (<25300 cm−1) are weak or sometimes missing. This is due to insufficient pumping efficiency due to low resolution of OPO laser, as described above (Fig. 2a), as well as long S1 lifetime. We also checked the purity of the sample by NMR measurements before and after the experiment to examine whether there is any decomposition product involved in the spectrum upon heating the sample. We confirmed there is no impurity in the sample (Fig. S2†). Therefore, we conclude that the unusual intensity pattern of the vibronic bands is not due to the existence of other species but is intrinsic to the S1 state of TCNQ monomer.
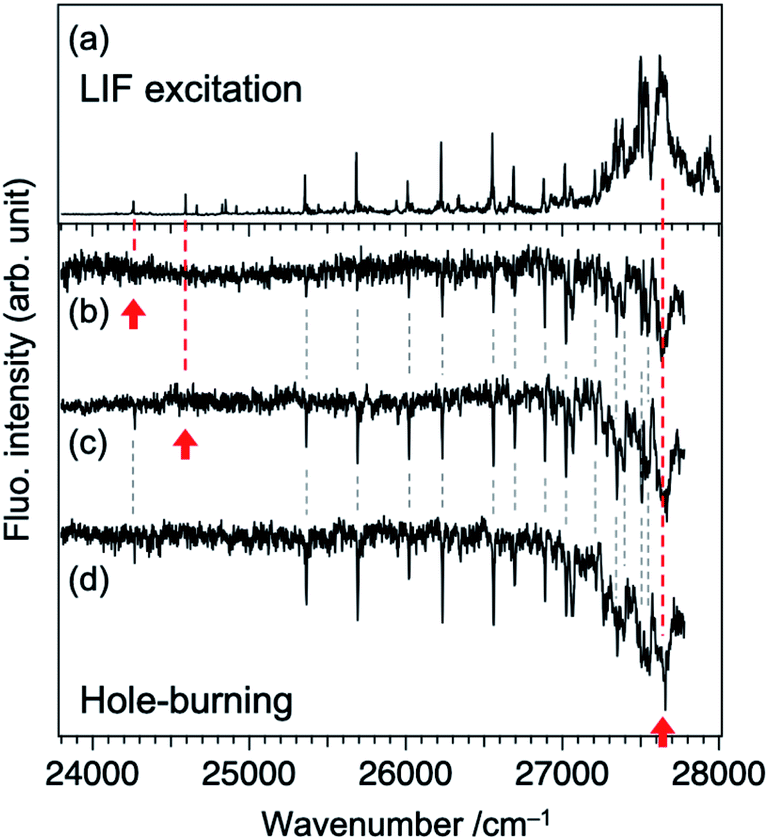 | ||
| Fig. 2 (a) LIF excitation spectrum and (b)–(d) fluorescence-detected UV-UV hole-burning spectra of TCNQ. Spectrum (a) is measured again in this study by using an OPO laser, where experimental condition was adjusted to that for spectra (b)–(d). The monitored bands are marked by red arrows: (b) 24262, (c) 24590, and (d) 27620 cm−1. | ||
In order to gain an insight of the excited state dynamics of TCNQ monomer, we focus on fluorescence decay profiles. We previously mentioned that the profiles of most of the vibronic bands exhibit double exponential decay,20 which is consistent in the present study (Fig. S3†). Herein, we fitted the decay curves with the following formula with fitting parameters of Afast, Aslow, τfast, and τslow (eqn (1)).
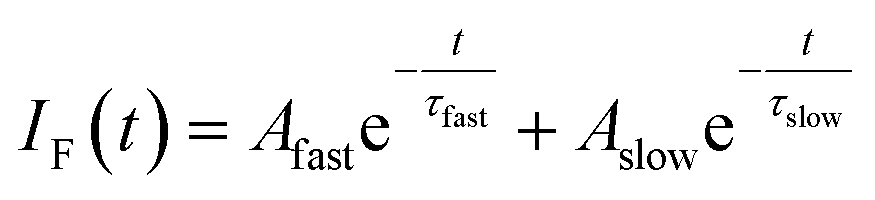 | (1) |
Here, the fluorescence intensity, IF(t), is normalized so that Afast + Aslow = 1 is satisfied. In Fig. 1b and c, we plotted the lifetimes (τfast, τslow) and amplitude of the fast component (Afast) as a function of excess energy (ΔE). First, it should be noted that the decay curve of the origin band (ΔE = 0 cm−1) exhibits two exponential decay with τfast, and τslow are 120 and 358 ns, respectively, and Afast = 0.89. Then, the vibronic bands at low excess energy (ΔE) region (∼300–570 cm−1) exhibit single exponential decay with the lifetime of 240–260 ns. The double exponential decay feature starts to appear at ΔE > 580 cm−1, again. The τfast value, lifetime of the fast component, becomes shorter with ΔE, and reaches to ∼45 ns at ΔE = ∼800 cm−1. Above this energy, the τfast value is almost invariant up to ΔE = ∼3000 cm−1. Also, the change of the τslow value is small within the range of 300–400 ns. In addition, the Afast value is almost constant at ∼0.95 (Aslow ∼ 0.05) in the examined energy region, indicating that the contribution of slow component is very small.
The double exponential decay feature in fluorescence indicates the coupling or mixing with other electronic states having the long lifetime. One of such candidates is the triplet state. The T1 (ππ*) state of TCNQ was reported to be located at 1.96 eV (15800 cm−1) in Br-containing solvent previously.23 It might be plausible that TCNQ in S1 (ππ*) undergoes intersystem crossing (ISC) to Tn state (n ≥ 2), followed by IC to T1 (ππ*) and phosphorescence with the observed slow component of 300–400 ns. In this case, the intensity ratio between the fast and slow components will be different at different monitoring wavelength, λobs. So, we compared the time profiles of the fluorescence decay measured by monitoring at λobs ≥ 440 nm and λobs ≥ 540 nm. However, we obtained almost the same profile regardless of the conditions, as shown in Fig. 3 for several bands (ΔE = +1098, +2613, and +3238 cm−1). This result suggests that the slow component is not due to the phosphorescence from T1. Actually, a recent theoretical study reported that spin–orbit coupling matrix elements between S1 and Tn (n = 1–3) are negligibly small,24 consistent with this result.
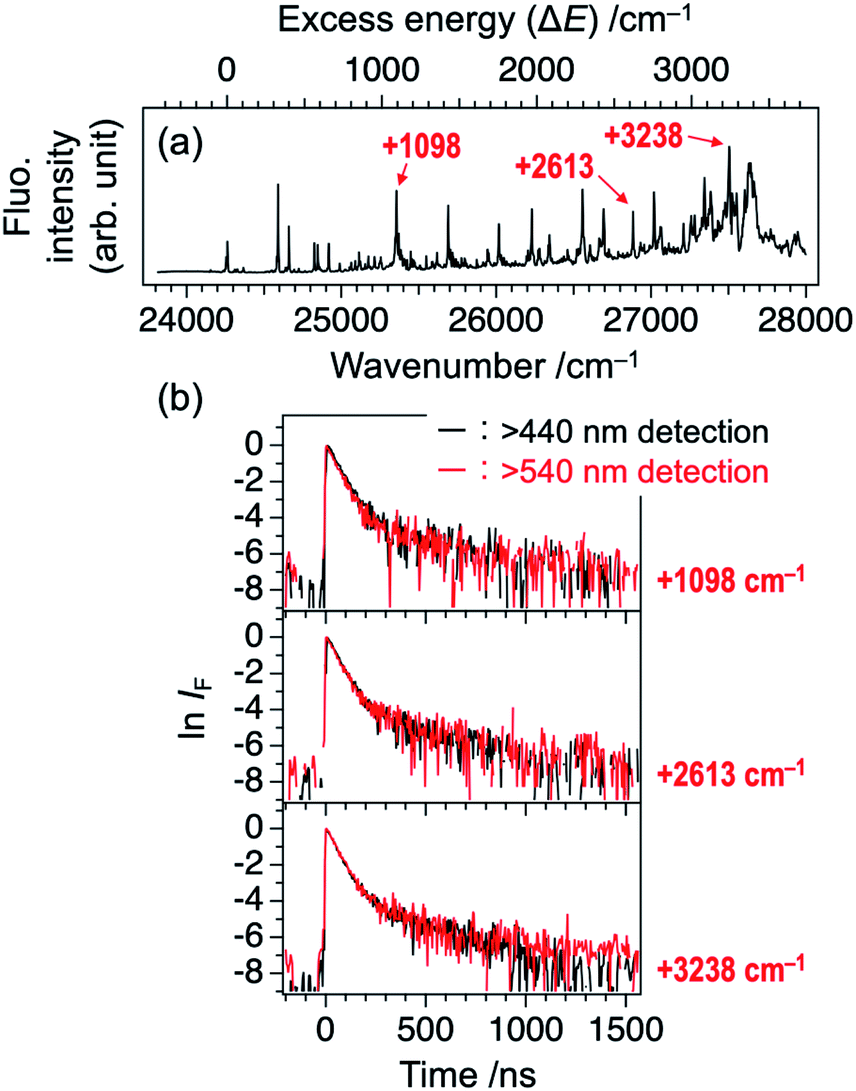 | ||
| Fig. 3 (a) LIF excitation spectrum of TCNQ (identical to Fig. 1a). (b) Logarithmic plots of fluorescence decay curves of the vibronic bands at ΔE = +1098, +2613, and +3238 cm−1 (see Panel (a)) recorded at two different monitoring wavelengths, λobs. Black: λobs ≥ 440 nm and red: λobs ≥ 540 nm. | ||
Then we consider the possibility of the coupling with higher singlet (Sn; n ≥ 2) states. Fig. 4a shows the vertical Sn ← S0 transition energy levels, as well as oscillator strengths (f), calculated by TDDFT method (TD-M06-2X/6-311++G(d,p)). The f value of the S1 ← S0 transition is as large as 1.15, while those for Sn ← S0 transitions with n = 2–7 are less than 0.001. This result agrees well with a calculation by Khvostenko et al. at TD-B3LYP/6-31G level23 as well as faint UV absorption of TCNQ at >∼4 eV region (corresponding to Sn ← S0 (n = 2–5)) in hexane solution.23 Namely, all of the Sn ⇄ S0 transitions (n = 2–7) are forbidden, so that the coupling with these states will elongate the fluorescence lifetime of S1. Especially, the S2 (B1g at geometric structure of the S1) state is the most plausible candidate. Fig. 4b shows the adiabatic energy level and the structure of the S2 state as well the energy of S1 with the same structure. There are two points to be noted for the S2 state calculated at this level. First is the structural change; the C–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N angle is slightly bent, as reflected by C(7)–C(8)–N(9) angle (176.1°; refer to Chart 1b and Fig. 4b for the atom labels) and the central phenyl ring is bent out of the plane, as reflected by C(1)–C(2)–C(3)–C(4) dihedral angle as large as 6.1° (see also the side view). Note that the latter results in lowering the total symmetry of the TCNQ molecule (D2h → C2h) so that the irreducible representation of the S2 state should be described as Bg. Second, the S1–S2 energy gap is as small as 0.09 eV (∼720 cm−1) at the S2 adiabatic energy level. From these considerations, we propose that the S2 state is mainly responsible for the photophysics and unusual vibronic pattern of S1. As shown in Fig. 4c, the S2 state has the (HOMO−1)1(LUMO)1 electronic configuration. The orbital HOMO−1 is distributed only on the phenyl ring, whereas that of LUMO is delocalized over the whole framework of TCNQ. In this sense, we may describe this state as the intramolecular CT (ICT) state with the electron transfer from the phenyl ring to the four CN groups in total,25,26 although the state does not have a dipole moment. The ICT character can be visually indicated by the differential density between the S2 and S0 states as shown in Fig. 4d. The similar energy relationship as well as the ICT character of the S2 state were calculated by the different functional (CAM-B3LYP), as shown in Fig. S4.† The S2 state is dipole-forbidden from S0 so that it does not have an oscillator strength from S0 (f = 0.00). However, the state can be vibronically coupled to the S1 state. At low energy, the S1–S2 mixing mostly affects the long fluorescence lifetime of the S1 state. The S2 state is dipole-forbidden so that the mixing of this state may contribute the forbidden character to the S1 state. In addition, the S2 state is thought to have no nonradiative decay route except the IC to S1. Thus, the mixing may work only to elongate the S1 lifetime. In the energy region at 27500 cm−1, the two states are closely located or may cross with each other, leading the congestion of the vibronic bands.
N angle is slightly bent, as reflected by C(7)–C(8)–N(9) angle (176.1°; refer to Chart 1b and Fig. 4b for the atom labels) and the central phenyl ring is bent out of the plane, as reflected by C(1)–C(2)–C(3)–C(4) dihedral angle as large as 6.1° (see also the side view). Note that the latter results in lowering the total symmetry of the TCNQ molecule (D2h → C2h) so that the irreducible representation of the S2 state should be described as Bg. Second, the S1–S2 energy gap is as small as 0.09 eV (∼720 cm−1) at the S2 adiabatic energy level. From these considerations, we propose that the S2 state is mainly responsible for the photophysics and unusual vibronic pattern of S1. As shown in Fig. 4c, the S2 state has the (HOMO−1)1(LUMO)1 electronic configuration. The orbital HOMO−1 is distributed only on the phenyl ring, whereas that of LUMO is delocalized over the whole framework of TCNQ. In this sense, we may describe this state as the intramolecular CT (ICT) state with the electron transfer from the phenyl ring to the four CN groups in total,25,26 although the state does not have a dipole moment. The ICT character can be visually indicated by the differential density between the S2 and S0 states as shown in Fig. 4d. The similar energy relationship as well as the ICT character of the S2 state were calculated by the different functional (CAM-B3LYP), as shown in Fig. S4.† The S2 state is dipole-forbidden from S0 so that it does not have an oscillator strength from S0 (f = 0.00). However, the state can be vibronically coupled to the S1 state. At low energy, the S1–S2 mixing mostly affects the long fluorescence lifetime of the S1 state. The S2 state is dipole-forbidden so that the mixing of this state may contribute the forbidden character to the S1 state. In addition, the S2 state is thought to have no nonradiative decay route except the IC to S1. Thus, the mixing may work only to elongate the S1 lifetime. In the energy region at 27500 cm−1, the two states are closely located or may cross with each other, leading the congestion of the vibronic bands.
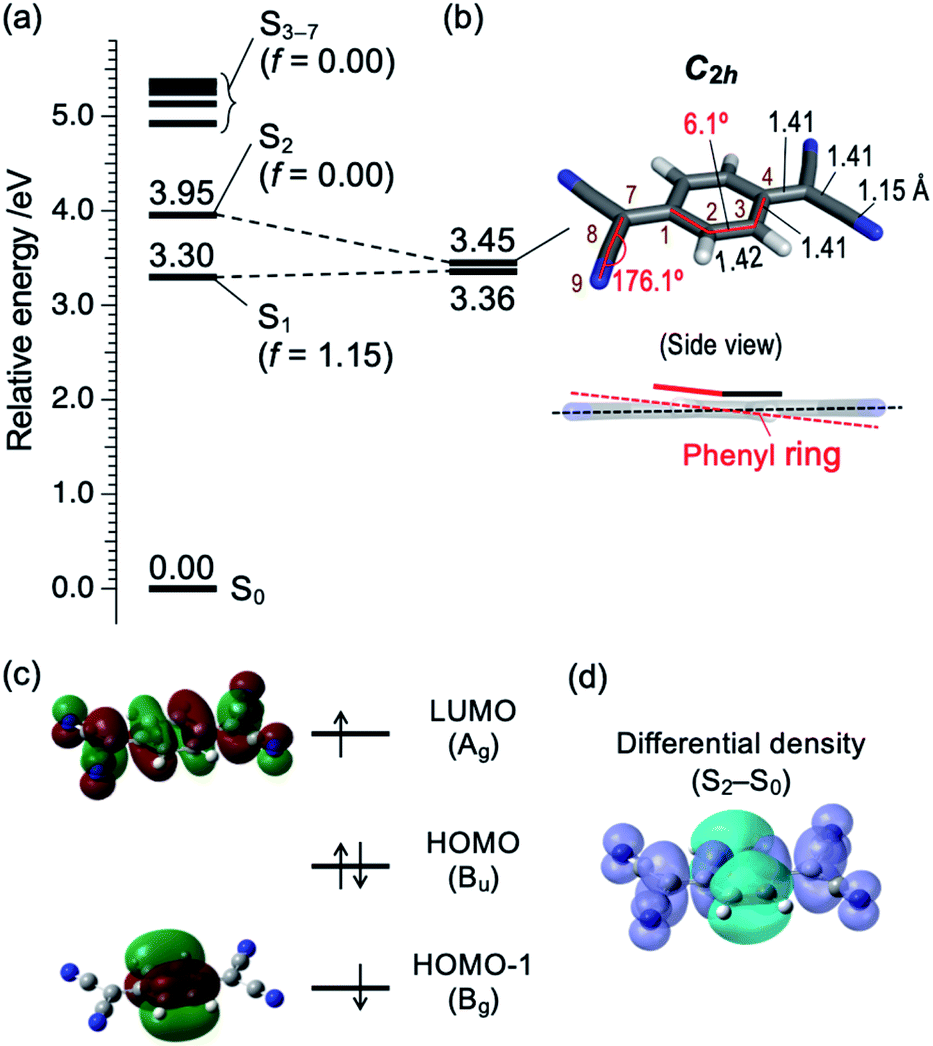 | ||
| Fig. 4 (a) Vertical energy level diagram of Sn (n = 1–7) states with respect to S0 calculated at M06-2X/6-311++G(d,p) level. Calculated oscillator strength (f) is shown in parentheses. (b) Energy level of S1 and S2 at local minimum structure of S2, and its structural details. Bond lengths and angles are shown in angström (Å) and degrees (°), respectively. Atom labels (1–4, 7–9) are identical to those in Chart 1b. Color codes: grey = C, white = H, blue = N. (c) Schematic electronic configuration of S2 state of TCNQ. The irreducible representation of each orbital is based on C2h point group. (d) Differential density surface between S2 and S0 states (isodensity value: 0.0004). Electrons are transferred from the cyan to violet part upon the S0 → S2 transition. | ||
It should be noted that the possibility for presence of the ICT state was previously mentioned by Iimori and coworkers, based on the decrease of the fluorescence QY with increasing the solvent polarity,16,17 although it is not clear whether the S2 state found in this study corresponds to the same state or not. It is also worth mentioning that C(2)–C(3) and C(3)–C(4) bond lengths are almost similar in the S2 state (Fig. 4b) as schematically called “benzenoid” structure.17 It is in contrast to optimized S0 state with “quinoid” structure17 (C(2)–C(3): 1.45 Å, C(3)–C(4): 1.34 Å).20
3.2 TCNQ–acetonitrile complex
Hereafter, we discuss the effects of micro-solvation of the TCNQ molecule on the electronic states and fluorescence features by forming van der Waals (vdW) complexes with other molecules. Fig. 5a shows LIF excitation spectrum obtained by the supersonic expansion of the gaseous mixture of TCNQ and acetonitrile (ACN). Two bands (24142 and 24470 cm−1), which are redshifted by 120 cm−1 from the (0,0) and (0,0) +327 cm−1 bands of TCNQ monomer (Fig. 1a), are clearly observed. We assign the band at 24142 cm−1 to the (0,0) band of the TCNQ–ACN 1:1 complex. The magnitude of the redshift is larger than the benzene-ACN complex (37.6 cm−1),27 indicating a larger stabilization of TCNQ–ACN in the S1 state. Several bands in the 24000–24100 cm−1 region also appeared by adding ACN, which could be attributed to TCNQ–(ACN)n (n ≥ 2) complexes. Fig. 5c shows calculated structure of the TCNQ–ACN 1:1 complex. The binding energy was calculated to be 0.29 eV. Note that the effect of BSSE is not so large in this complex; BSSE correction by CP method still gave the binding energy of 0.27 eV. The binding energy possibly comes from partially polarized C–Hδ+⋯Nδ−C interaction (2.61 Å), as also seen in (ACN)2 dimer oriented in parallel.28,29 Such a relatively strong interaction may account for successful formation of TCNQ–ACN complex in the present study; actually, we attempted to generate vdW complexes of TCNQ with other molecules including Ar, H2O, and n-C6H12, all of which resulted in no spectroscopic evidence for complex formation. In addition to the isomer shown in Fig. 5, we obtained several higher energy isomers with different solvation site as summarized in Fig. S5;† however, the above discussion may not be changed regardless of the isomers.
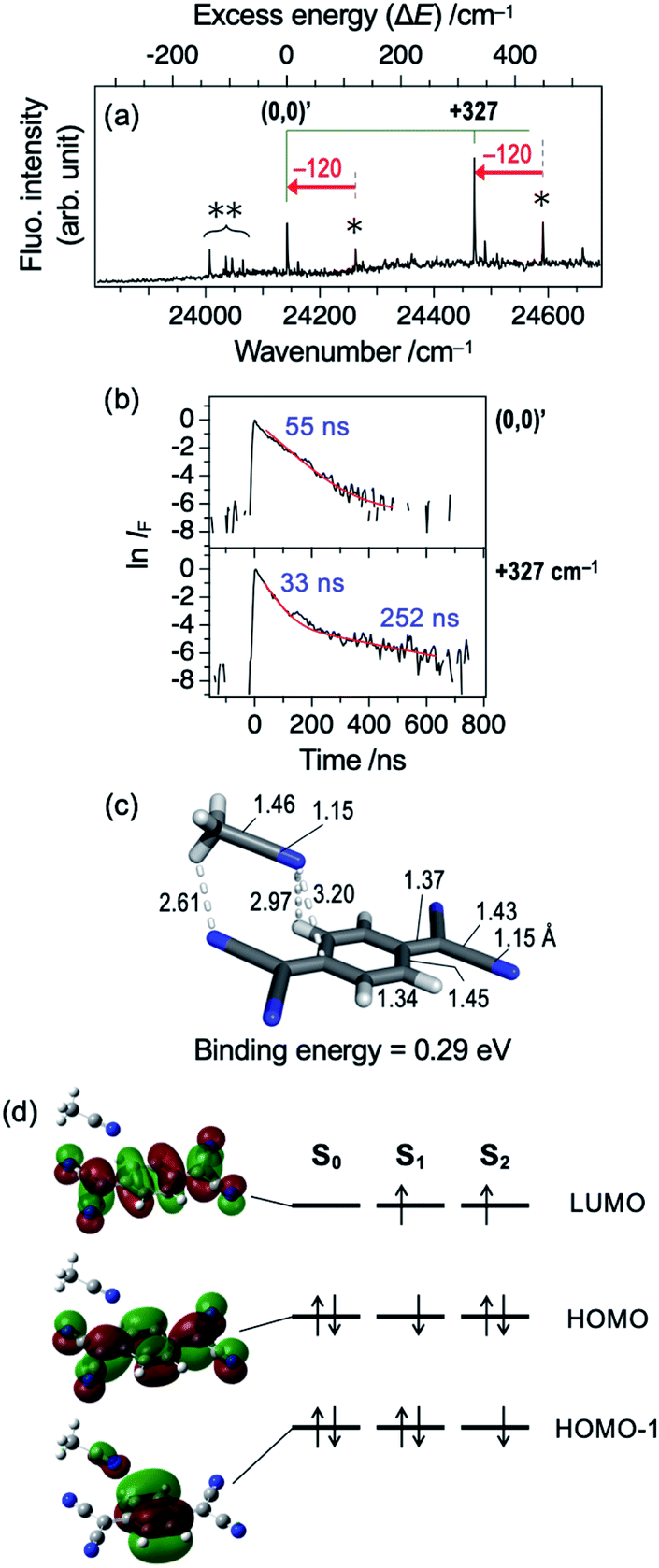 | ||
| Fig. 5 (a) LIF excitation spectrum of jet-cooled TCNQ with presence of acetonitrile vapor in the carrier gas. Bottom axis represents wavelength of the incident laser, while top axis is relative wavenumber with respect to the origin band of TCNQ–ACN complex at 24142 cm−1. Single asterisk indicates the band originated from TCNQ monomer. Double asterisks indicate the bands assigned to TCNQ–(ACN)n (n ≥ 2) complexes. (b) Fluorescence decay curve of TCNQ–ACN complex at the vibronic bands with excess energy of +0 ((0,0)′) and +327 cm−1. The curves are shown in logarithmic scale after normalization at t = 0, to clearly indicate the double decay feature at “(0,0) +327 cm−1” band. (c) Calculated structural details of TCNQ–ACN complex. Bond lengths and angles are shown in angström (Å) and degrees (°), respectively. Color codes: gray = C, white = H, blue = N. (d) Kohn–Sham orbitals of the TCNQ–ACN complex and schematic electron configuration of Sn states (n = 0–2) on the basis of TDDFT results. | ||
Fig. 5b shows fluorescence decay profiles of TCNQ–ACN 1:1 complex. The (0,0) band shows the single decay with the lifetime of 55 ns, and the (0,0) +327 cm−1 band shows double exponential decay with τfast = 33 ns and τslow = 252 ns. This result indicates that the complex formation with ACN shortens the S1 lifetime of TCNQ. Thus, even a single molecule drastically shortens the S1 lifetime. It is in accordance with the results of solution by Tamaya et al.; they reported that the fluorescence QY of TCNQ in hexane was as large as 0.11 whereas that in acetonitrile became 2 × 10−3 times smaller.16 They ascribed the drastic difference in QY to the difference in nonradiative decay rates. It is consistent with another report that the fluorescence lifetime of S1 state of TCNQ in acetonitrile was as short as 800 fs due to the ultrafast IC (S1 → S0).15 However, the lifetime value observed in this study (tens of nanosecond) is still much longer than that in acetonitrile solution. This difference indicates that one acetonitrile molecule is not enough to cause such the short S1 lifetime observed in solution. Table 1 shows vertical energy levels of S1 and S2 states of the TCNQ–ACN complex calculated by TDDFT method. As seen in Fig. 5d, the S1 (ππ*) state is essentially the same with monomer, and the S2 state has slightly the intermolecular CT character, although it is not reflected in the oscillator strength. The energy level of S1 of the complex is only 0.01 eV stabilized with respect to that of the monomer, while S2 state is stabilized by 0.10 eV in the complex; the large stabilization of the S2 state is consistent with its ICT nature, given that acetonitrile is polar molecule.
| Excitation character | TCNQ | TCNQ–ACN | TCNQ–Bz | |||
|---|---|---|---|---|---|---|
| Energy/eV | Oscillator strength (f) | Energy/eV | Oscillator strength (f) | Energy/eV | Oscillator strength (f) | |
| a Calculated at TD-M06-2X/6-311++G(d,p) level. | ||||||
| Intermolecular CT | — | — | 3.07 (S1) | 0.03 | ||
| — | — | 3.07 (S2) | 0.02 | |||
| ππ* (LE) | 3.30 (S1) | 1.15 | 3.29 (S1) | 1.13 | 3.25 (S3) | 0.89 |
| Intramolecular CT (ICT) | 3.95 (S2) | 0.00 | 3.85 (S2) | 0.00 | 3.87 (S4) | 0.01 |
3.3. TCNQ–benzene complex
We also carried out the complex formation of TCNQ with benzene (Bz) to examine the effect of π-π interaction on the absorption and emission. The π–π interaction of TCNQ is typically seen in TCNQ-based organic semiconductor including TCNQ–TTF.1 Fig. 6a shows LIF excitation spectrum of the gaseous mixture of TCNQ and benzene vapor expanded with the helium carrier gas. Different from TCNQ–ACN, we only observed broad absorption feature with an onset of ∼24000 cm−1 (shaded in red) for the TCNQ–Bz complex. Fig. S6† directly compares the spectra with and without benzene vapor in the carrier gas, confirming that the broad band originates from TCNQ–Bz complex. Fig. 6b shows fluorescence decay curves obtained by exciting at 24096, 24390, and 24540 cm−1 (marked with A, B, and C, respectively). The curves exhibit almost identical double exponential decay feature regardless of the band position even at low wavenumber region. This behavior is different from TCNQ monomer and TCNQ–ACN complex as shown above. This result raises a possibility that photophysics of TCNQ–Bz complex might be different from TCNQ and TCNQ–ACN.
 | ||
| Fig. 6 (a) LIF excitation spectrum of jet-cooled TCNQ with presence of benzene vapor in the carrier gas. Single asterisks indicate the bands originated from TCNQ monomer. (b) Fluorescence decay curve of TCNQ–benzene complex measured at 24096 (A), 24390 (B), and 24540 cm−1 (C). The curves are shown in logarithmic scale after normalization at t = 0, to clearly indicate the double decay feature. (c) Calculated structural details of TCNQ–benzene complex. Bond lengths are shown in angström (Å). To illustrate distance between TCNQ and benzene, the closest C–C distance (3.39 Å) is explicitly shown. Color codes: gray = C, white = H, blue = N. (d) Kohn–Sham orbitals of the TCNQ–Bz complex and schematic electron configuration of Sn states (n = 0–4) on the basis of TDDFT results. | ||
Then we theoretically investigated the electronic states of TCNQ–Bz complex by assuming that they form 1:1 complex. Fig. 6c shows most stable structure of TCNQ–Bz complex. Less stable isomers are summarized in Fig. S7.† In the most stable complex, bond lengths of TCNQ moiety is almost identical to isolated (non-solvated) TCNQ,20 and benzene molecule is attached to TCNQ with binding energy of 0.40 eV. Thus, it is clear that TCNQ–Bz complex forms π–π stacked structure in S0. Table 1 summarizes vertical transition energy levels of Sn states (n = 1–4) of the TCNQ–Bz 1:1 complex calculated by TDDFT method. Different from TCNQ monomer or TCNQ–ACN complex, the ππ* state is calculated to be S3 state (oscillator strength: 0.89), and lower-lying other excited states (S1 and S2) with small oscillator strength (≤0.03) were predicted. Kohn–Sham orbitals related to these excitations in Fig. 6d clearly show that both S1 and S2 states are intermolecular charge-transfer (CT) states. Based on the calculation results, it is quite possible that the emission of TCNQ–Bz complex reflects the effects of the intermolecular CT state. Previous report by Gorishnyi mentioned the formation of CT complex of TCNQ in benzene solvent,30 although no theoretical calculations nor fluorescence measurements were conducted.
We extended this study to fluorescence measurement in benzene/hexane solution, as shown in Fig. 7a. The spectra were measured at different benzene/hexane ratio (0–10 vol%), while keeping the TCNQ concentration same at 2.0 × 10−5 M. The spectral profile of pure hexane solution (benzene ratio: 0%) is consistent with a report by Tamaya et al.; the intensity maximum appears at 463 nm.16 An addition of benzene by 0.1 vol% (∼0.1 M) drastically decreases the fluorescence intensity peaked at 463 nm by ∼40%, while the intensity at ≥530 nm remains the same. Further addition of benzene until 1.0 vol% mostly diminishes the blue part emission and clarifies the appearance of largely Stokes-shifted CT emission peaked at 530 nm (see also Fig. 7b). That is, local excitation (LE) of TCNQ to its ππ* (S3) state is followed by the energy transfer to the low-lying CT states (S1 and S2; see Table 1) and emission from the CT states. One may anticipate the contribution of Bz–O2 complexes to the fluorescence, since the solution sample was used without degassed condition in the present study. The free energy of the complex formation of Bz–O2 is reported to be as small as −1.2 kcal mol−1 (420 cm−1) in the gas phase.31 Although this weak binding energy will produce small amount of Bz–O2 complexes, the effect on the TCNQ–Bz CT complex will be very small based on the high stability of the CT complex. Then, further addition of benzene (1.0–10.0 vol%) resulted in the emission of the CT state. Fig. 7c and d shows fluorescence decay profiles of TCNQ in pure hexane and benzene (10 vol%)/hexane solvents, respectively. The lifetime in pure hexane solvent was 3.8 ns (detection wavelength: 463 nm), which roughly agrees with previously reported one (4.5 ns) by Tamaya et al.16 It is not clear that the difference is within the experimental error limit or due to the quenching by O2 in our experimental condition. Since the difference is very small, we think the effect of O2 to the present finding is negligibly small. In contrast to the pure hexane solution, the lifetime in the mixed solvent (detection wavelengths: 576, 590, 610, and 630 nm) was clearly elongated to 5.8 ns. There is small contribution of LE emission at this concentration of benzene as seen in Fig. 7a. Note that this value (5.8 ns) still reflects the contribution of both LE and CT emissions; however, the lifetimes of these states are not largely different to each other, which results in apparently single decay profiles. Actually, the decay lifetime in pure benzene solvent at 630 nm detection, possibly corresponding to lifetime of the CT state, was determined to be 6.7 ns (Fig. S8†). The inset of Fig. 7d shows the enlarged portion of the rise of the emission profiles. We see that the peak position of the rise systematically shifts by ∼500 ps from the 576 nm-monitored one to 630 nm-monitored one. Given that the emission at longer wavelength part includes larger contribution of the CT state, this timescale corresponds to the energy transfer from the LE (S3) to CT (S1 and/or S2) states. Although the analysis of the ultrafast time-resolved fluorescence spectroscopic measurement is necessary, we can conclude that the energy transfer from LE to CT will occur in the timescale of ∼500 ps.
 | ||
| Fig. 7 (a) Fluorescence spectra of TCNQ dissolved in benzene/hexane mixed solvent (TCNQ concentration: 2.0 × 10−5 M; benzene ratio: 0–10 vol%). The excitation wavelength is 394 nm. (b) Plot of fluorescence intensity at 463 and 529 nm as a function of benzene ratio. The intensity at 529 nm was shown after subtracting the contribution of fluorescence in pure hexane solvent. (c) and (d) Logarithmic plots of fluorescence decay curves of TCNQ in pure hexane (c) and benzene (10 vol%)/hexane (d) solvents. The excitation wavelength is 390 nm. The detection wavelengths are indicated (also see arrows in Panel (a)). Dotted lines are background (scattered) signals. Inset in Panel (d) shows an expanded view of the selected region of decay curve (shaded in gray). | ||
Finally, we should note that the effect of the coupling between S1 and CT states in the gas-phase free jet spectrum. As discussed above, we did not observe sharp vibronic bands but only observed broad LIF excitation spectra as shown in Fig. 6a. The broad feature may be attributed to the strong coupling between the ππ* and CT states. As to the decay time profile, it is quite possible that the slow component of the decay profile (Fig. 6b) is attributed to the emission from the CT state in the case of TCNQ–Bz complex. The large difference between lifetimes observed in the gas phase and those in solution is consistent with our previous report on TCNQ monomer.20 It can be partly explained by quenching by solvent molecules (see Section 3.2), although it cannot totally be clarified at the current stage.
4. Conclusions
Electronic states and photophysics of TCNQ and its solvated complexes (with acetonitrile and benzene) were investigated by means of LIF spectroscopy in the supersonic free jet and fluorescence spectroscopy in solution. The S1 (ππ*; B3u)–S0 (Ag) LIF spectrum of TCNQ monomer in the free jet exhibited unusual intensity pattern: weak origin band, intensity increase with the excess energy (ΔE), and congestion at ΔE ∼ 3000 cm−1. Furthermore, the lifetime is much longer than that in solution, with double exponential decay feature. We proposed that these characteristic features of the S1 state are attributed to the mixing with the S2 state, which has the ICT character. The S1–S2 mixing is still kept in the TCNQ–ACN 1:1 complex; the LIF spectrum of the complex showed the similar vibronic profile with TCNQ monomer, with the redshift of ∼120 cm−1 and the fluorescence lifetime was shortened less than half of the monomer. TDDFT calculations also showed the similarity of the electronic state characters between monomer and TCNQ–ACN complex. In sharp contrast, the TCNQ–Bz complex exhibited only broad absorption, even under the jet-cooled condition. DFT calculations predicted a strong coupling to the intermolecular CT states, which is consistent with the broad absorption spectrum in the jet as well as the appearance of largely Stokes-shifted emission in benzene/hexane solution. As discussed above, we showed that TCNQ has both intra- and inter-molecular CT states, and they appear in a different manner in the emission depending on the nature of solvent molecules. We believe that all these observations will shed new light on the photophysics of TCNQ, which has been thought to be the well understood molecule.
Author contributions
Satoru Muramatsu: methodology, investigation, formal analysis, visualization, writing – original draft. Nobumasa Chaki: methodology, investigation, formal analysis. Shin-nosuke Kinoshita: investigation, writing – review & editing. Yoshiya Inokuchi: writing – review & editing, supervision. Manabu Abe: methodology, writing – review & editing. Toshifumi Iimori: conceptualization, resources, writing – review & editing. Takayuki Ebata: conceptualization, methodology, investigation, writing – original draft, supervision, project administration, funding acquisition.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank Prof. Dr Takeharu Haino (Hiroshima Univ.) for providing access to the NMR spectrometer. This work was financially supported by JSPS KAKENHI (Grant Nos JP19K15507 (S. M.) and JP20H00374 (Y. I.)). T. E. acknowledges the support from Ministry of Science and Technology, Taiwan (Grant Nos MOST106-2113-M-009-016-MY3 and MOST108-2811-M-009-508). Calculations were partly performed using computing systems at the Research Center for Computational Science, Okazaki, and the Research Institute for Information Technology, Kyushu University, Japan.Notes and references
- J. B. Torrance, Acc. Chem. Res., 1979, 12, 79–86 CrossRef CAS.
- J. Ferraris, D. O. Cowan, V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef CAS.
- K. I. Pokhodnya, N. Petersen and J. S. Miller, Inorg. Chem., 2002, 41, 1996–1997 CrossRef CAS PubMed.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS PubMed.
- T. Shimada, Y. Takahashi, J. Harada, H. Hasegawa and T. Inabe, J. Phys. Chem. Lett., 2018, 9, 420–424 CrossRef CAS PubMed.
- H. Miyasaka, Acc. Chem. Res., 2013, 46, 248–257 CrossRef CAS PubMed.
- T. Akutagawa, M. Uchigata, T. Hasegawa, T. Nakamura, K. A. Nielsen, J. O. Jeppesen, T. Brimert and J. Becher, J. Phys. Chem. B, 2003, 107, 13929–13938 CrossRef CAS.
- C. Reese and Z. Bao, Mater. Today, 2007, 10, 20–27 CrossRef CAS.
- A. R. Brown, D. M. de Leeuw, E. J. Lous and E. E. Havinga, Synth. Met., 1994, 66, 257–261 CrossRef CAS.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- M. Szablewski, P. R. Thomas, A. Thornton, D. Bloor, G. H. Cross, J. M. Cole, J. A. K. Howard, M. Malagoli, F. Meyers, J.-L. Brédas, W. Wenseleers and E. Goovaerts, J. Am. Chem. Soc., 1997, 119, 3144–3154 CrossRef CAS.
- E. A. Brinkman, E. Gunther, O. Schafer and J. I. Brauman, J. Chem. Phys., 1994, 100, 1840 CrossRef CAS.
- G. Z. Zhu and L. S. Wang, J. Chem. Phys., 2015, 143, 221102 CrossRef PubMed.
- G. M. Roberts, J. Lecointre, D. A. Horke and J. R. R. Verlet, Phys. Chem. Chem. Phys., 2010, 12, 6226–6232 RSC.
- L. Ma, P. Hu, C. Kloc, H. Sun, M. E. Michel-Beyerle and G. G. Gurzadyan, Chem. Phys. Lett., 2014, 609, 11–14 CrossRef CAS.
- H. Tamaya, H. Nakano and T. Iimori, J. Lumin., 2017, 192, 203–207 CrossRef CAS.
- H. Tamaya, Y. Torii, T. Ishikawa, H. Nakano and T. Iimori, ChemPhysChem, 2019, 20, 2531–2538 CrossRef CAS.
- T. Iimori, T. Ishikawa, Y. Torii, H. Tamaya, H. Nakano and M. Kanno, Chem. Phys. Lett., 2020, 738, 136912 CrossRef CAS.
- T. Iimori, Y. Torii, T. Ishikawa and N. Tamai, J. Phys. Chem. B, 2020, 124, 7918–7928 CrossRef CAS PubMed.
- N. Chaki, S. Muramatsu, Y. Iida, S. Kenjo, Y. Inokuchi, T. Iimori and T. Ebata, ChemPhysChem, 2019, 20, 1–6 CrossRef.
- T. Ebata and Y. Inokuchi, Chem. Rec., 2016, 16, 1034–1053 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresmanand and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- O. G. Khvostenko, R. R. Kinzyabulatov, L. Z. Khatymova and E. E. Tseplin, J. Phys. Chem. A, 2017, 121, 7349–7355 CrossRef CAS PubMed.
- T. Iimori, Comput. Theor. Chem., 2021, 1199, 113211 CrossRef CAS.
- Such intramolecular charge-transfer states formed by electronic transition from a localized orbital to a delocalized one can be found in literatures. For example, ref. 26.
- H. K. Sinha and K. Yates, J. Chem. Phys., 1990, 93, 7085–7093 CrossRef CAS.
- D. M. Daly, D. Wright and M. S. El-Shall, Chem. Phys. Lett., 2000, 331, 47–56 CrossRef.
- H. J. Böhm, I. R. McDonald and P. A. Madden, Mol. Phys., 1983, 49, 347–360 CrossRef.
- E. M. Cabaleiro-Lago and M. A. A. Ríos, J. Phys. Chem. A, 1997, 101, 8327–8334 CrossRef CAS.
- M. P. Gorishnyi, Ukr. J. Phys., 2004, 49, 1158–1162 CAS.
- E. A. Gooding, K. R. Serak and P. R. Ogilby, J. Phys. Chem., 1991, 95, 7868–7871 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison of LIF excitation spectra recorded at different resolution, NMR charts, fluorescence decay curves of TCNQ monomer, structural isomers of TCNQ–ACN and TCNQ–Bz complexes, and fluorescence decay curve of TCNQ/benzene (100%) solution. See DOI: 10.1039/d1ra01430g |
| This journal is © The Royal Society of Chemistry 2021 |