
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRatchet-like mechanism in a long-life photoproduct of salicylideneaniline enclathrated in a pillared-layer guanidinium disulfonate structure†
Kentaro Nakayama,
Taisuke Manako,
Ryo Koibuchi,
Isao Yoshikawa and
Hirohiko Houjou*
and
Hirohiko Houjou*
Institute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan. E-mail: houjou@iis.u-tokyo.ac.jp
First published on 13th April 2021
Abstract
Salicylideneaniline (SA) was found to exhibit extraordinary long-life photochromism upon being included in a cavity of guanidinium organosulfonate and exhibited a lifetime of ∼30 times that of a pure SA crystal. Crystal structure analysis suggested that the sulfonate molecule in the apo-host provided a flexible cavity space that kinetically trapped SA in its photo-isomerized form, as if it was locked by a ratchet-like mechanism.
Crystal engineering is a promising strategy for the modification of the chemical and physical properties of molecular solids to obtain desirable functions.1 For example, salicylideneanilines (SAs), which are known to exhibit either photochromism or thermochromism depending on their molecular environment,2–4 are often employed to exemplify the effects of polymorphs, cocrystallization, salification, or inclusion in a porous coordination network.5–8 The chromism of SA originates in the interconversion of three tautomers: the enol, cis-keto, and trans-keto forms (Chart 1).9,10 Therefore, the modification of the energy landscape by changing the surroundings of the SA chromophore facilitates chromism. Guanidinium organosulfonate (GS), which is known to spontaneously form a pillared-layer structure,11–14 is a promising candidate for providing a variety of molecular hosts to accommodate SA molecules as guests. In the present study, the use of biphenyl-4,4′-disulfonate as a pillar resulted in the clathrate compound, SA@1, which was a photochromic compound with an extraordinarily long life, whereas the use of naphthalene-2,6-disulfonate afforded a photo-inactive clathrate compound, SA@2.
 | ||
| Chart 1 Three tautomers of salicylideneaniline relevant to its chromic behaviour. | ||
Molecular apo-hosts 1 and 2 were prepared in accordance with studies presented by Ward et al.14,15 After their preparation, 1 and 2, were dissolved in methanol with 3 equivalents (to guanidinium) of SA, respectively, and were left to sit until the open evaporation initiated crystallization.† The products were examined by single-crystal X-ray structural analysis, which revealed that 1 and 2 afforded the molecular clathrates SA@1 and SA@2, respectively. Several crystal structures have been reported for several clathrates and solvates of 1 and 2,14 and their connection topology that features hydrogen-bonding networks is known to vary with the type of guest and solvent molecules. Chart 2 illustrates the framework of the putative apo-hosts 1 and 2 in SA@1 and SA@2. The ratio of SA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :disulfonate was 1:2, which implied that each unit cell (with volumes of 1227.95 Å3 for SA@1 and 1098.28 Å3 for SA@2) contained one molecule of SA (Fig. 1). However, the NMR profiles revealed that the ratio (0.44 and 0.41) of SA to sulfonate molecules was lower than that (0.50) derived from stoichiometry (Fig. S1†), implying that the bulk product contained an appreciable amount of the apo-hosts. FTIR spectra16 (Fig. S2†) and powder XRD patterns (Fig. S3†) also implies the contamination of the apo-host.
:disulfonate was 1:2, which implied that each unit cell (with volumes of 1227.95 Å3 for SA@1 and 1098.28 Å3 for SA@2) contained one molecule of SA (Fig. 1). However, the NMR profiles revealed that the ratio (0.44 and 0.41) of SA to sulfonate molecules was lower than that (0.50) derived from stoichiometry (Fig. S1†), implying that the bulk product contained an appreciable amount of the apo-hosts. FTIR spectra16 (Fig. S2†) and powder XRD patterns (Fig. S3†) also implies the contamination of the apo-host.
 | ||
| Chart 2 Schematic representation of apo-hosts 1 and 2. The connection topology featuring hydrogen-bonding networks can be observed in their SA-clathrates, SA@1 and SA@2. | ||
 | ||
| Fig. 1 Crystal structures of (a) SA@1 and (b) SA@2. Views along other directions are available in Fig. S4.† | ||
Upon irradiation with UV light, the color of the SA@1 crystal changes from pale yellow to orange, whereas that of the SA@2 crystal does not. Absorption spectra were subsequently obtained with a microscope-based UV-vis absorption spectrometer equipped with a temperature controller and a high-pressure Hg lamp (through a 330–380 nm band path filter) to investigate this.17–19 Upon irradiation with the Hg lamp at 15 °C, the SA@1 sample immediately shows an intense absorption band at ∼500 nm with a blunt vibronic structure (Fig. 2a). An unmixed SA molecule is known to exhibit three crystalline polymorphs, i.e. α1-, α2-, and β-forms,20 among which the α1-form is the most common photochromic species. The absorption spectrum of SA@1 in the photo-stationary state is quite similar to those of α1-SA,5 suggesting that the newly appeared band can be attributed to the photo-induced trans-keto form. When the sample is cooled to −60 °C, the spectrum in the photo-stationary state reveal a clearer structure, whereas that in the dark-adapted state is almost unchanged. The spectra of SA@2 without the Hg lamp are noted to be similar to those of SA@1; however, they did not show significant changes upon UV irradiation (Fig. 2b). Both samples were photoluminescent but with different spectral profiles: SA@1 exhibited an orange emission peak at ∼590 nm, and SA@2 exhibited a yellowish-green emission peak at ∼520 nm (Fig. 3). This difference is similar to the behavior observed in the photochromic and thermochromic polymorphs of 3,5-di-tert-butylsalicylideneaniline.17 There are two possible reasons for the spectral shift: (1) the alteration of electronic state in SA molecule due to conformational change and/or intermolecular interactions inside the cavity; (2) reabsorption of a 500–600 nm region of the emission by the photo-product. The first reason is rather unlikely in view of a more planar structure (angle between two phenyl rings is 2.8°) of SA@2 than that (23°) of SA@1. Interestingly, the luminescence spectra of SA@1 appeared similar to those of SA@2 after the SA@1 crystals were pulverized in an agate mortar; however, its photochromism was maintained. This phenomenon implies that the pulverization induced a partial collapse of the pillared-layer structure, but leaves some room for discussion about the coexistence of yellowish-green luminescence and photochromism that seems contradictory to the second reason above. These observations will be a clue to a complete understanding of the current system, and worth further inspecting in the future.
 | ||
| Fig. 2 Single-crystal UV-vis absorption spectra obtained with and without UV irradiation in (a) SA@1 and (b) SA@2. (inset) Microscope images at 25 °C. | ||
 | ||
| Fig. 3 Emission spectrum of SA@1, SA@2, as compared with that of β-SA. Spectra of SA@1 after grinding with a mortar is overlaid. | ||
The duration of photo-coloration for SA@1 was notably longer than that for α1-SA. This was investigated via an examination of the bleaching process by measuring the absorbance decay curves at 480 nm, starting from the photo-stationary state at 15 °C (Fig. 4). As previously noted,18,19 the decay curves do not fit well to a single exponential function; hence, a time constant cannot be extracted. Instead, the values of T50 and T95 (durations at 50% and 95% decay, respectively) were compared, which revealed that the lifetime of SA@1 was 30 times longer than that of α1-SA (Table 1). Although the lifetime is known to vary with conditions such as the temperature or intensity of ambient light, these results clearly showcase the overwhelming kinetic stability of the photoproduct in SA@1. These experiments were repeated under the same conditions with N-3,5-di-tert-butylsalicylidene-3-nitroaniline (SA-NO2), which is known to possess an extremely long lifetime (half-life of ∼40 days and 1200 min as obtained by Kawato et al.21 and Ohashi et al.,22 respectively: these values were measured in the dark); the results suggest that SA@1 possesses a longer lifetime (∼2×) than that of SA-NO2 (Fig. 4).
 | ||
| Fig. 4 Comparison of relaxation times in the photo bleaching of SA@1, SA-NO2, and α1-SA. | ||
| Compound | T50 | T95 |
|---|---|---|
| SA@1 | 329 | 910 |
| α1-SA | 11 | 31 |
| SA-NO2 | 223 | 361 |
The crystal structure of SA@1 was solved under the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, which means that the SA molecule was assumed to have a disorder ratio of 1:1 for opposite orientations, with respect to the pseudo-inversion point at the center of the C
space group, which means that the SA molecule was assumed to have a disorder ratio of 1:1 for opposite orientations, with respect to the pseudo-inversion point at the center of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. In addition, the phenyl rings of biphenyl-4,4′-disulfonate were assumed to have disorders for rotations of 17–40° with respect to the axis along S⋯S (Fig. 5). These observations suggest that the cavity inside 1 can be flexibly shaped in accordance with the structure of the guest molecule(s). For reference, the crystal structure of the o-dichlorobenzene clathrate of 1 (CCDC refcode XAQWUH),23 which has a unit cell and packing structure similar to those of SA@1, was solved without assuming any disorders, and revealed an almost parallel arrangement of the biphenyl moieties in a planar conformation. It is worth mentioning that this arrangement overlays decently with one of the disordered structures (shown in blue and magenta in Fig. 5) in SA@1 (Fig. S5†).
N bond. In addition, the phenyl rings of biphenyl-4,4′-disulfonate were assumed to have disorders for rotations of 17–40° with respect to the axis along S⋯S (Fig. 5). These observations suggest that the cavity inside 1 can be flexibly shaped in accordance with the structure of the guest molecule(s). For reference, the crystal structure of the o-dichlorobenzene clathrate of 1 (CCDC refcode XAQWUH),23 which has a unit cell and packing structure similar to those of SA@1, was solved without assuming any disorders, and revealed an almost parallel arrangement of the biphenyl moieties in a planar conformation. It is worth mentioning that this arrangement overlays decently with one of the disordered structures (shown in blue and magenta in Fig. 5) in SA@1 (Fig. S5†).
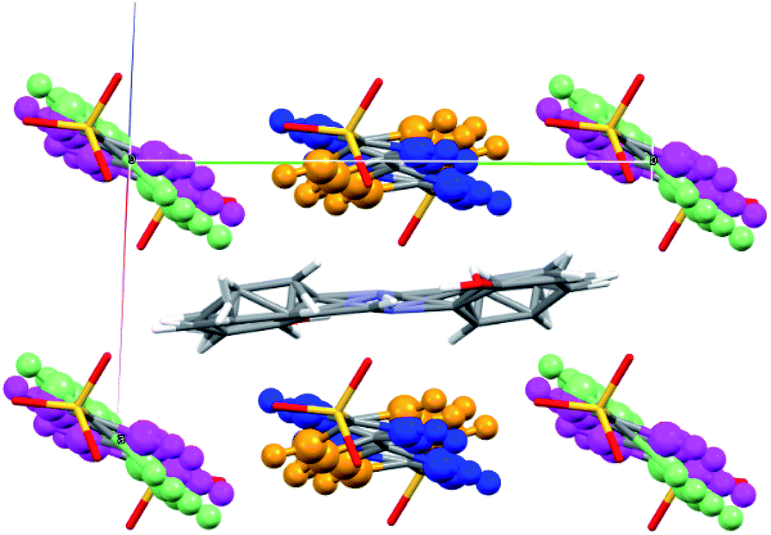 | ||
| Fig. 5 Crystal structure of the biphenyl moieties surrounding the SA molecule in SA@1 (view along 101 direction). The structures are shown using an overlay of two different conformations, in blue and orange, and in magenta and pale green. | ||
The torsion angle (θ) between the two phenyl rings in the SA molecule has been noted to indicate the possibility of photochromism. Previous studies have suggested that SA becomes thermochromic if the molecule is nearly planar (θ < ∼30°), whereas it appears photochromic if the molecule is twisted (θ > ∼30°).2–4 However, SA@1 (θ = 23°) narrowly fails at being categorized as a photochromic crystal in accordance with this criterion, whereas SA@2 (θ = 2.8°) is appropriately predicted as non-photochromic. Another requisite for photochromism involves a loosely packed (open) structure that allows cis-to-trans photoisomerization.1–3 However, there is a lack of definitive discussions on the quantitative relationship between the degree of the open structure and photochromic activity. In addition to the difficulty in quantifying the open structure, the conventional dualistic classification into photochromic and thermochromic (non-photochromic) crystals is also an issue; it is possible for a photochromic crystal with an extremely loose packing to have a very high bleaching speed, and get classified as non-photochromic. Therefore, a sufficient long life of the photoproduct can be another requisite for photochromism. For SA-NO2 and polymorphs of its carboxylate analogue, a positive correlation between the lifetime and stability of the trans-keto form has been inferred.22
Combining the above analyses, an explanation for the long-life photochromism of SA@1 can be proposed. The putative apo-host 1 possesses a cavity volume that is sufficient to accommodate the SA molecule, and the cavity shape is flexibly adjusted by the rotation of the phenyl rings of the pillars. When photo-isomerization occurs, the phenyl rings can rotate to follow the structural change. The rings rotate almost freely; however, they do not rotate independently because of the inter-pillar distance of 6.2 Å, which is insufficient for the two phenyl rings to avoid van der Waals contacts. Therefore, the biphenyl moieties serve as meshing gears to prevent the photoproduct from relaxing to the thermodynamically stable enol form; this mechanism can be compared to that of a ratchet gear. By contrast, SA@2, in which the cavity (480 Å3) is slightly smaller than that (546 Å3) of SA@1,24 neither has enough volume to accommodate the twisting of the SA molecule, nor the flexibility to follow the structural change upon isomerization.
In summary, drastic changes in the photochemical properties of SAs were observed via the formation of clathrates of GS compounds. Since the discovery of GS compounds by Etter and Ward,11–13 their pillared-layer structures have been extensively applied for the isolation and stabilization of guest molecules.25 The inclusion of functional dyes such as azobenzene,14 coumarin,15 and ROY26 into GS hosts has been accomplished. It is worth noting that the torsion angle of ROY can be adjusted by changing the type of bifunctional organosulfonic acid employed. The present study demonstrates the possibility of GS clathrates to facilitate new systems with which the various properties of photochromic compounds can be examined in a controlled molecular environment.
Author contributions
The preparation and characterization of the compounds were conducted by K. Nakayama and T. Manako. The microscope absorption spectra and related data were collected by R. Koibuchi. The crystal structures were analyzed by I. Yoshikawa. The whole research project was supervised by H. Houjou. The original draft was written by K. Nakayama, and it was reviewed and edited by H. Houjou.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A part of this work (X-ray single crystallographic analysis) was supported by Nanotechnology Platform project by the Ministry of Education, Culture, Sports, Science and Technology of Japan. The grant number JPMXP09A20UT0081.Notes and references
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- M. D. Cohen and G. M. J. Schmidt, J. Phys. Chem., 1962, 66(12), 2442–2446 CrossRef CAS.
- E. Hadjoudis and I. M. Mavridis, Chem. Soc. Rev., 2004, 33, 579–588 CAS.
- K. Amimoto and T. Kawato, J. Photochem. Photobiol., A, 2005, 6, 207–226 CrossRef CAS.
- D. S. Lo, Appl. Opt., 1974, 13(4), 861–865 CrossRef CAS PubMed.
- A. Carletta, X. Buol and T. Leyssens, J. Phys. Chem. C, 2016, 120, 10001–10008 CrossRef CAS.
- P.-L. Jacquemin, K. Robeyns, M. Devillers and Y. Garcia, Chem.–Eur. J., 2015, 21, 6832–6845 CrossRef CAS PubMed.
- T. Haneda, M. Kawano, T. Kojima and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 6643–6645 CrossRef CAS PubMed.
- K. Ogawa, Y. Kasahara, Y. Ohtani and J. Harada, J. Am. Chem. Soc., 1998, 120, 7107–7108 CrossRef CAS.
- J. Harada, H. Uekusa and Y. Ohashi, J. Am. Chem. Soc., 1999, 121, 5809–5810 CrossRef CAS.
- V. A. Russell, M. C. Etter and M. D. Ward, J. Am. Chem. Soc., 1994, 116, 1941–1952 CrossRef CAS.
- V. A. Russell, M. C. Etter and M. D. Ward, Chem. Mater., 1994, 6, 1206–1217 CrossRef CAS.
- V. A. Russell, C. C. Evans, W. Li and M. D. Ward, Science, 1997, 276, 575–579 CrossRef CAS PubMed.
- A. C. Soegiarto, A. Comotti and M. D. Ward, J. Am. Chem. Soc., 2010, 132, 14603–14616 CrossRef CAS PubMed.
- A. C. Soegiarto and M. D. Ward, Cryst. Growth Des., 2009, 9, 3803–3815 CrossRef CAS.
- A. Karmakar, R. Illathvalappilz, B. Anothumakkool, A. Sen, P. Samanta, A. V. Desai, S. Kurungot and S. K. Ghosh, Angew. Chem., Int. Ed., 2016, 55, 10667–10671 CrossRef CAS PubMed.
- H. Houjou, H. Ikedo and I. Yoshikawa, Chem. Commun., 2017, 53, 10898–10901 RSC.
- H. Houjou, T. Kato, H. Huang, Y. Suzuki, I. Yoshikawa and T. Mutai, Cryst. Growth Des., 2019, 19, 1384–1390 CrossRef CAS.
- Y. Suzuki, T. Kato, H. Huang, I. Yoshikawa, T. Mutai and H. Houjou, J. Photochem. Photobiol., A, 2019, 385, 112096 CrossRef CAS.
- F. Arod, P. Pattison, K. Schenk and G. Chapuis, Cryst. Growth Des., 2007, 7, 1679–1685 CrossRef CAS.
- T. Kawato, H. Koyama, H. Kanatomi and M. Isshiki, J. Photochem., 1985, 28, 103–110 CrossRef CAS.
- K. Johmoto, A. Sekine, H. Uekusa and Y. Ohashi, Bull. Chem. Soc. Jpn., 2009, 82, 50–57 CrossRef CAS.
- J. A. Swift, A. M. Reynolds and M. D. Ward, Chem. Mater., 1998, 10, 4159–4168 CrossRef CAS.
- The values were calculated by subtracting the approximate vdW volume of the constituent molecules in the apo-host from the unit cell volume: Y. H. Zhao, M. H. Abraham and A. M. Zissimos, J. Org. Chem., 2003, 68, 7368–7373 CrossRef CAS PubMed.
- W. Xiao, C. Hu and M. D. Ward, Cryst. Growth Des., 2013, 13(7), 3197–3200 CrossRef CAS.
- S. Tang, A. Yusov, Y. Li, M. Tan, Y. Hao, Z. Li, Y. Chen, C. T. Hu, B. Kahr and M. D. Ward, Mater. Chem. Front., 2020, 4, 2378–2383 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2062419 and 2062420. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra01823j |
| This journal is © The Royal Society of Chemistry 2021 |