
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and conversion of primary and secondary 2-aminoestradiols into A-ring-integrated benzoxazolone hybrids and their in vitro anticancer activity†‡
Ferenc Kovács a,
Mohana K. Gopisettyb,
Dóra I. Adameczb,
Mónika Kiricsib,
Éva A. Enyedycd and
Éva Frank*a
a,
Mohana K. Gopisettyb,
Dóra I. Adameczb,
Mónika Kiricsib,
Éva A. Enyedycd and
Éva Frank*a
aDepartment of Organic Chemistry, University of Szeged, Dóm tér 8, H-6720 Szeged, Hungary. E-mail: frank@chem.u-szeged.hu; Tel: +36-62-544-275
bDepartment of Biochemistry and Molecular Biology, Doctoral School of Biology, University of Szeged, Közép fasor 52, H-6726 Szeged, Hungary
cDepartment of Inorganic and Analytical Chemistry, Interdisciplinary Excellence Centre, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary
dMTA-SZTE Lendület Functional Metal Complexes Research Group, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary
First published on 14th April 2021
Abstract
Hybrid systems are often endowed with completely different and improved properties compared to their parent compounds. In order to extend the chemical space toward sterane-based molecular hybrids, a number of estradiol-derived benzoxazol-2-ones with combined aromatic rings were synthesized via the corresponding 2-aminophenol intermediates. 2-Aminoestradiol was first prepared from estrone by a two-step nitration/reduction sequence under mild reaction conditions. Subsequent reductive aminations with different arylaldehydes furnished secondary 2-aminoestradiol derivatives in good yields. The proton dissociation processes of the aminoestradiols were investigated in aqueous solution by UV-visible spectrophotometric titrations to reveal their actual chemical forms at physiological pH. The determined pK1 and pK2 values are attributed to the +NH3 or +NH2R and OH moieties, and both varied by the different R substituents of the amino group. Primary and secondary 2-aminoestradiols were next reacted with carbonyldiimidazole as a phosgene equivalent to introduce a carbonyl group with simultaneous ring-closure to give A-ring-fused oxazolone derivatives in high yields. The novel aminoestradiols and benzoxazolones were subjected to in vitro cytotoxicity analysis and were found to exert cancer cell specific activity.
Introduction
Molecular hybridization is a very promising approach in drug design and can offer therapeutic options for a wide variety of diseases.1,2 The main goal of hybrid development is to enhance or favourably modulate the pharmacokinetic and/or pharmacodynamic properties of a molecule through the synergistic effects of combined structural elements; although combating multidrug resistance and minimizing unwanted side effects are also important considerations. Occasionally, new, unpredictable bioactivities may arise which are different from those of the individual molecules that constitute the hybrid compounds.3,4Thanks to their attractive structural, physicochemical and biological features, natural sex steroids are often used to construct molecular hybrids for anticancer indication,5–7 either by covalent attachment to another pharmacophore directly8 or via a spacer,9 or by integrating key functional elements to form a new chemical entity.10 However, any residual hormonal effect of the steroid moiety is undesirable in these cases, and should be significantly reduced or, preferably, eliminated during hybrid formation.
For estrane-based hybrids, the best way to avoid hormonal side effects is to modify the OH groups at positions C-3 and C-17, which are primarily responsible for the hormone receptor binding via H-bonding interactions.11 Decreased affinity to the target protein, due to electronic and steric reasons, has also been demonstrated for a number of 2-substituted estradiol derivatives (e.g., 2-methoxyestradiol), which, however, are effective in inhibiting the proliferation of various human cancer cells.12–16 Although the cytotoxic effect of 2-nitroestradiol obtained by direct nitration of estradiol was weaker than that of 2-methoxyestradiol, the attachment of a piperidine or morpholine ring to the phenolic-O through a spacer led to particularly potent cytotoxic compounds.17 Interestingly, there are very few examples in the literature for the preparation of A-ring-modified estrane-based derivatives containing a [2,3]-fused heterocyclic moiety.18,19 Elimination of the phenolic OH group in parallel with the substitution at C-2 makes it even more likely that the compound will not be able to bind to hormone receptors, and possibly other major bioactivities can predominate without undesired side effects.
Since 2-aminophenols are valuable building blocks for many biologically active compounds20 and can be used as starting materials for the synthesis of pharmacologically important N,O-heterocycles,21–23 our primary aim was to introduce an amino substituent at the C-2 position of estradiol. In view of the high reactivity of the phenolic A-ring of estrogens toward electrophiles and the two available ortho positions (C-2 and C-4), the regioselective introduction of a substituent into C-2 that can be easily converted to an amino group is a challenging task. Although C-2 attack is slightly favoured, in most of the cases, e.g. during halogenation, nitration or formylation, mixtures of mono- and disubstituted products – which are often difficult to separate – were usually obtained, and the yield of the 2-functionalized derivative was only moderate.24 Accordingly, the first step of the seemingly simplest process involving nitration and subsequent reduction for the synthesis of primary 2-amino estradiol often suffered from the above-mentioned problems.25,26
When forming a 2-aminophenol moiety on estrogens, given the high cost of sex hormones, it is desirable to develop a process that consists of only few reaction steps and provides the product in acceptable yield. Moreover, if the amino derivative is to be used as a starting material for the preparation of N,O-heterocycles, it must be synthesized in a reproducible manner in larger quantities. Although the task does not seem difficult from an organic chemical point of view, it is no coincidence that there are only few literature examples for the preparation of 2-aminoestrone or 2-aminoestradiol, most of which are based on a two-step procedure, i.e. nitration and subsequent reduction after separation of the mono- and dinitro derivatives26–30 (Fig. 1B). All other synthetic methods, e.g. those depicted in Fig. 1A31–36 and Fig. 1C,37,38 involve several steps and require protection/deprotection of the phenolic OH group, expensive reagents or harsh conditions, and suffer from selectivity problems that ultimately lead to the final product in lower yields than route B.
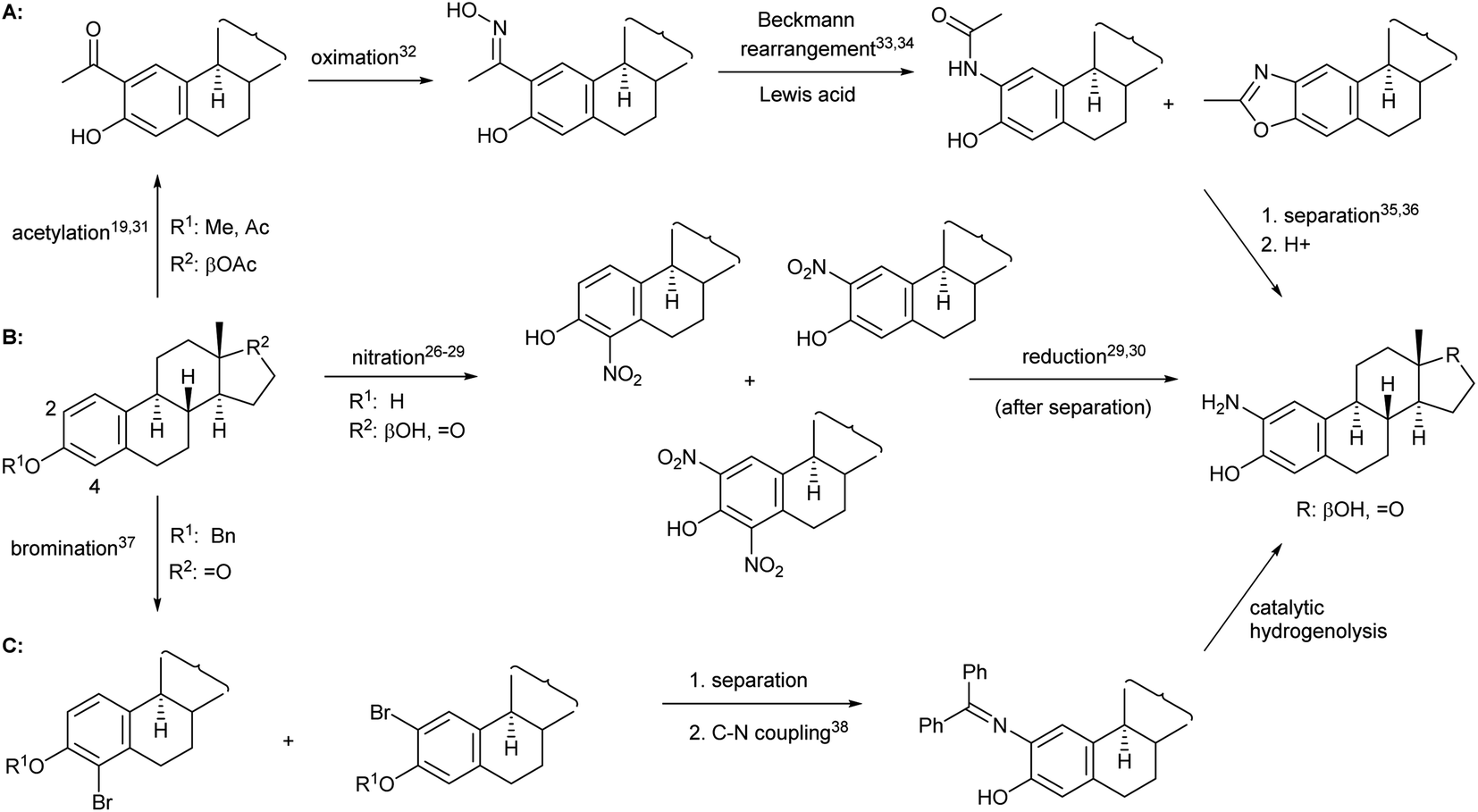 | ||
| Fig. 1 Possible routes for the synthesis of 2-amino estrogens. | ||
In addition to studying a number of methods that may be suitable for the most efficient and scalable access to 2-aminoestradiol, our next goal was to further convert the compound to secondary aminophenols by reductive amination. In order to characterize the actual chemical forms (and charges) of the novel compounds in solution focusing on physiological pH, and to explore the effect of the various substituents on the aromatic ring of the benzylamino moiety in the case of secondary amines, the proton dissociation constants (Ka) of the amphoteric aminophenols were determined by UV-visible spectrophotometric titrations. Finally, bifunctional derivatives were subjected to CDI-induced ring-closure reactions with the simultaneous incorporation of a carbonyl group from the reagent to furnish various novel estradiol–benzoxazolone molecular hybrids. The cytotoxicity of all synthesized derivatives was tested in vitro by the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay using human MCF-7 breast, HeLa cervical as well as DU145 and PC-3 prostate cancer cell lines, and simultaneously non-cancerous MRC-5 fibroblast cells. Based on the primary toxicity screen, five potent compounds were selected and subjected to further experiments to obtain their IC50 values on different cell lines.
Result and discussion
Synthetic studies
Based on the literature background, we selected the two-step method for the preparation of 2-aminoestradiol; however, both the nitration of estrone and the following reduction were investigated separately. Since mononitration seemed unavoidable at both the C-2 and C-4 positions of the sterane core, we tried to find a proper experimental condition where at least over-nitration and thus the formation of the dinitro product could be eliminated. A number of literature procedures describing the selective mononitration of phenol or estrone either by using 65% (m/m) HNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 or metal nitrates28 were tried to be reproduced in different solvents (such as AcOH, dioxane, EtOH, CH2Cl2 or CHCl3), however, significant amount of 2,4-dinitro derivative reducing the yield of the desired product, was obtained in all cases. Furthermore, unreacted estrone remained in the reaction mixtures when the nitrating agent was not used in excess, which further complicated the separation and purification process.
29 or metal nitrates28 were tried to be reproduced in different solvents (such as AcOH, dioxane, EtOH, CH2Cl2 or CHCl3), however, significant amount of 2,4-dinitro derivative reducing the yield of the desired product, was obtained in all cases. Furthermore, unreacted estrone remained in the reaction mixtures when the nitrating agent was not used in excess, which further complicated the separation and purification process.
Since the selective nitration of phenol and substituted phenols to the corresponding mononitro compounds was reported earlier under mild conditions in a liquid–liquid two-phase system with diluted nitric acid (6% m/m) and in the presence of tetrabutylammonium bromide as phase-transfer catalyst,39 we tried to adapt this method on the substitution reaction of estrone (1). However, given the poor solubility of estrone relative to phenol in water-immiscible organic solvents, a relatively large amount had to be used to prepare a homogeneous solution, so that the volume of water (added with a diluted HNO3 solution) was significantly smaller than that of the organic phase. Therefore, a phase transfer catalyst was not required even during the scale-up synthesis; the reaction proceeded with 1 equiv. of nitrating reagent in boiling CH2Cl2 under vigorous stirring for 1 h to give exclusively a ca. 1:1 mixture of 2- and 4-nitroestrone in 92% overall yield (Scheme 1). The mononitro derivatives were easily separated by column chromatography owing to their large polarity difference. First, 2-nitroestrone (2) eluted from the column; its more apolar nature is due to the intramolecular hydrogen bonding interaction between the phenolic OH and the NO2 group. In contrast, the more polar character of 4-nitroestrone (3) can be attributed to the fact that due to its sterically hindered environment, the nitro group twists out of the plane of the sterane skeleton,40 therefore, intramolecular interaction masking the polarity of the functional groups cannot develop.
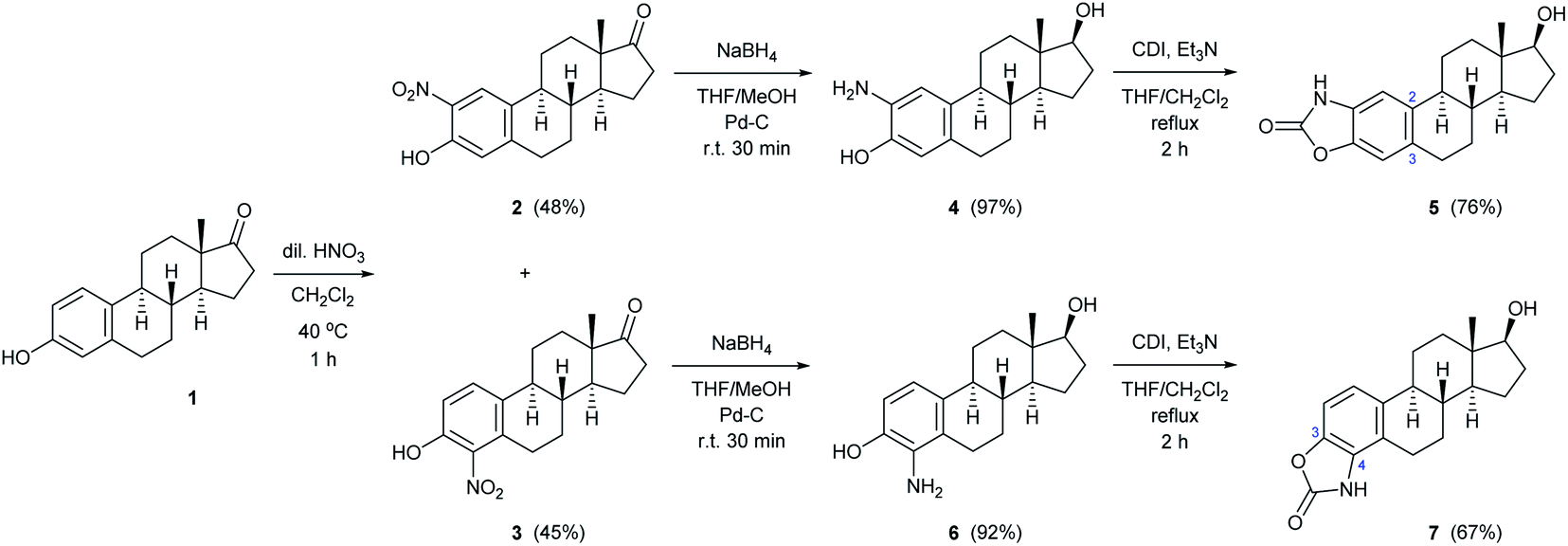 | ||
| Scheme 1 Two-step synthesis of monoamino-estradiol regioisomers and their conversion to A-ring-fused 2-oxazolones. | ||
Although mononitration was not regioselective, both successfully separated nitro compounds (2 and 3) subsequently proved to be suitable, after reduction, for the synthesis of A-ring[2,3]-fused and [3,4]-fused 2-oxazolones.
As a continuation, conversion of 2 and 3 to aminoestradiols was investigated. An insoluble red precipitate was formed as a by-product when 2-nitrophenols (2 or 3) were reduced with iron filings under classical Béchamp conditions, probably due to metal complex formation, and then complex dissociation by adding NaOH was also unsuccessful thanks to the high susceptibility of sodium phenolates to oxidation. Therefore, another hydrogenation method had to be found. In order to avoid introducing flammable hydrogen into the reaction mixture from a gas cylinder, hydrogen generated in situ from NaBH4 in a protic solvent (MeOH) in the presence of Pd–C41,42 was used for catalytic reduction of 2 and 3 (Scheme 1). This method was also suitable to reduce not only the NO2 but also the 17-(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group stereoselectively, thus resulting in 2- (4) or 4-aminoestradiol (6) in a single step with excellent isolated yields. In view of the amphoteric character of the products, adjustment of neutral pH during work-up was of crucial importance.
O) group stereoselectively, thus resulting in 2- (4) or 4-aminoestradiol (6) in a single step with excellent isolated yields. In view of the amphoteric character of the products, adjustment of neutral pH during work-up was of crucial importance.
In the following, unsubstituted (8a) and para-substituted benzylamino estradiols (8b–h) were prepared from 4 with benzaldehyde and its derivatives by reductive amination (Table 1). Imine formation was performed in the presence of a catalytic amount of AcOH and molecular sieves by boiling the reactants in MeOH for 1 h. Secondary aminophenols (8a–h) were obtained in good to excellent yields by reducing the unstable imines in situ with NaBH4. With the only exception of the p-nitro-substituted compound (8f), all derivatives were found to be stable both in the solid state and in organic solvents (Table 1).
|
|||||
|---|---|---|---|---|---|
| Entry | R | Aminoestradiol | Yielda (%) | Benzoxazolone | Yielda (%) |
| a After purification by column chromatography.b Proved to be sensitive to oxidation even in the solid state. | |||||
| 1 | H | 8a | 76 | 9a | 69 |
| 2 | F | 8b | 76 | 9b | 76 |
| 3 | Cl | 8c | 91 | 9c | 94 |
| 4 | Br | 8d | 75 | 9d | 77 |
| 5 | CN | 8e | 96 | 9e | 92 |
| 6 | NO2 | 8f | 82b | 9f | 92 |
| 7 | CH3 | 8g | 72 | 9g | 78 |
| 8 | OMe | 8h | 94 | 9h | 90 |
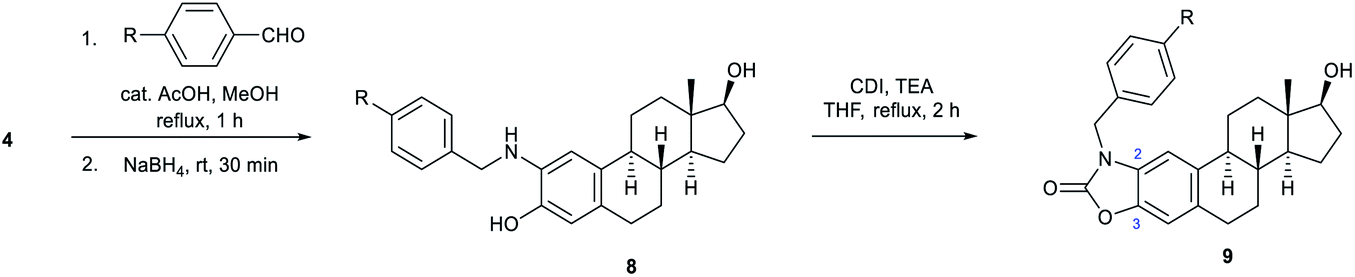
Both the prepared primary (4 and 6) and secondary amines (8a–h) were further converted to estradiol–benzoxazolone hybrids by cyclization and incorporation of a carbonyl group from the applied 1,1-carbonyldiimidazole (CDI) reagent in the presence of triethylamine (TEA) as base by boiling in THF (Scheme 1 and Table 1). The reactions provided the products (5, 7 and 9a–h) in good yield (67–94%) within 2 h.
The structures of all compounds prepared in our experimental work were confirmed by 1H and 13C NMR spectroscopic and high-resolution electrospray ionization mass spectrometry (ESI-HRMS) methods. Due to the nature of the transformations performed, the range of 1H NMR spectra above chemical shift values of 3 ppm was informative; below 3 ppm, the signals of the skeletal protons and the C-13 angular methyl group could be observed. The most important difference between 2- and 4-nitroestrone (2, 3) and 2- and 4-aminoestradiol (4, 6) derivatives can be noticed in the aromatic range of the 1H NMR spectra. For the 2,3-disubstituted compounds (2, 4), the C-1 and C-4 protons give singlet signals, while the adjacent C-1 and C-2 protons of the 3,4-disubstituted analogs (3, 6) appear as doublets with the same coupling constant. In the spectra of the secondary aminophenols (8), the appearance of the extra signals between 7 and 8 ppm as well as the presence of the benzyl-CH2 at 4.3 ppm are indicative for the incorporation of the aromatic ring from the arylaldehyde reagents and the success of the reductive amination. As for the oxazolone (5, 7, 9) derivatives, the negative carbon signal of the introduced CO group can be seen at around 154 ppm in the 13C NMR spectra (J-MOD), which indicated the heterocyclization of the aminophenols upon treatment with CDI.
Proton dissociation processes of primary and secondary aminoestradiols
The methods (NMR, ESI-HRMS) used for the characterization of 2-aminoestradiols (4 and 8a–h) provide information about their purity and chemical structure in the solid phase and in solution of organic solvents. However, knowledge on their behaviour in aqueous solution and proton dissociation is essential for a deeper understanding of their pharmacological properties and structure–activity relationships. Therefore, we aimed to determine the proton dissociation constants (Ka) of the compounds to predict their actual chemical forms at pH 7.4 in solution, and to reveal the influence of the various substituents. The studied sterane-based derivatives (4 and 8a–h) have limited solubility in pure water, thus UV-visible spectrophotometric titrations were performed on samples containing 50 μM compound in a 30% (v/v) DMSO/H2O solvent mixture. However, the halogen and CN substituents in 8b–e resulted in worse solubility, and lower compound concentration (10 μM) had to be applied to avoid the appearance of precipitate. Since 2-aminophenol (2AP) and its derivatives can easily undergo oxidation, especially in their completely deprotonated forms,43,44 a strong argon flow was applied during the titrations to exclude the oxygen. Deprotonation processes of 2AP as a simpler model compound were also investigated for comparison. The studied compounds displayed strong absorption bands in the UV region mostly due to π–π* transitions of the benzene rings. Representative UV-visible spectra recorded for 4 at various pH values are shown in Fig. 2a. Two well-separated processes can be observed upon increasing the pH; namely, the first step is accompanied by an increase of the absorbance and the λmax from 282 nm to 296 nm in the pH range from 2 to 7, while the second deprotonation step, taking place at pH > 9.5 results in a further increase in the λmax (310 nm) with the appearance of a novel band in the wavelength range 350–470 nm. As the deprotonation steps are not overlapping, clear-cut isosbestic points could be found at 284 nm and 302 nm in case of the H2L+ ⇌ HL + H+ and HL ⇌ L− + H+ equilibrium processes, respectively, where L− is the completely deprotonated form of the compound. This finding also indicates that no disturbing processes such as oxidation took place during the titration of 4 under the flow of the inert gas. Proton dissociation constants (Ka) were determined by the deconvolution of the measured spectra and are collected in Table 2.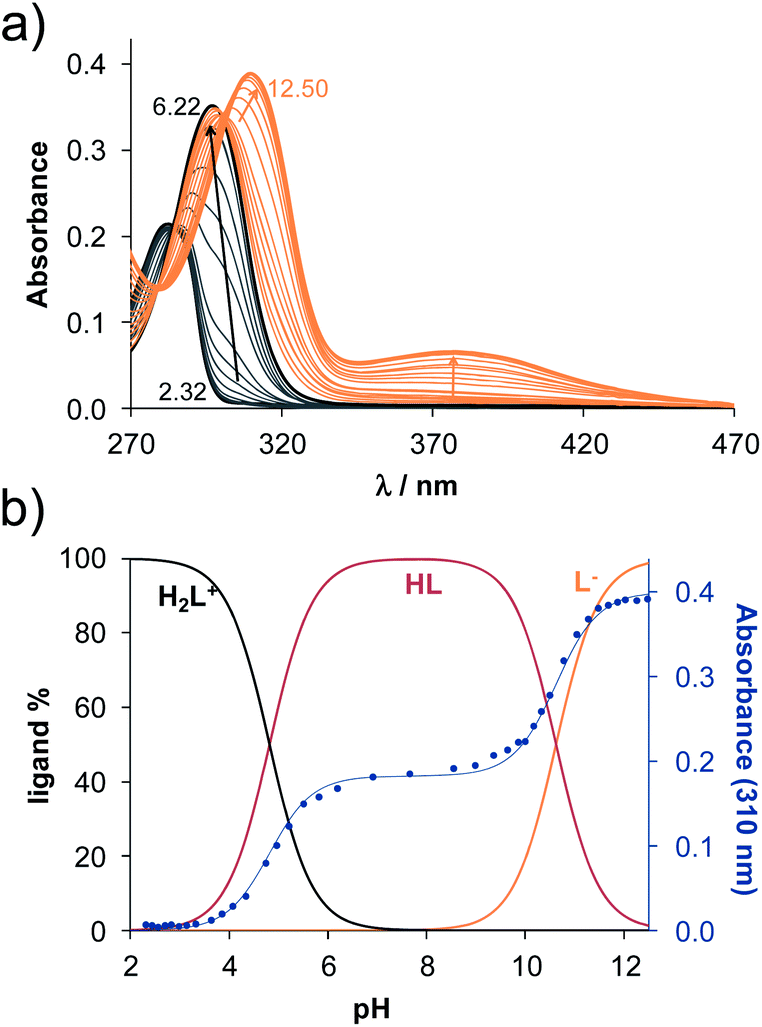 | ||
| Fig. 2 (a) UV-visible spectra recorded for 4 at various pH values. (b) Concentration distribution curves calculated for 4 and the measured absorbance values at 310 nm (●) with the fitted values (blue solid line). {c = 50 μM; l = 2 cm; 30% (v/v) DMSO/H2O; T = 25 °C; I = 0.1 M (KCl)}. | ||
| Compound | R | pKa (NH+amino) | pKa (OHphenolic) | cL | |
|---|---|---|---|---|---|
| a Reference compound. | |||||
| 2AP | 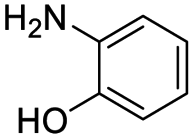 |
—a | 4.41 ± 0.02 | 10.36 ± 0.02 | 50 μM |
| 4 | 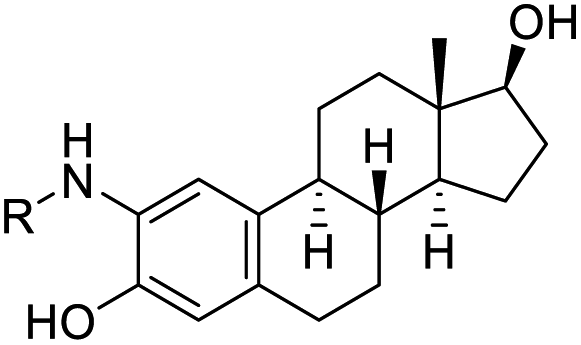 |
H | 4.82 ± 0.01 | 10.63 ± 0.02 | 50 μM |
| 8a | CH2-Ph | 4.58 ± 0.03 | 10.44 ± 0.04 | 50 μM | |
| 8b | CH2-(p-F)C6H4 | 4.23 ± 0.04 | 10.36 ± 0.02 | 10 μM | |
| 8c | CH2-(p-Cl)C6H4 | 4.34 ± 0.05 | ∼10.8 | 10 μM | |
| 8d | CH2-(p-Br)C6H4 | 3.94 ± 0.02 | ∼11.1 | 10 μM | |
| 8e | CH2-(p-CN)C6H4 | 3.85 ± 0.01 | ∼11.0 | 10 μM | |
| 8g | CH2-(p-CH3)C6H4 | 4.75 ± 0.03 | 10.34 ± 0.05 | 50 μM | |
| 8h | CH2-(p-MeO)C6H4 | 4.76 ± 0.03 | ∼10.6 | 50 μM | |
Based on the determined pKa values, concentration distribution curves were computed (Fig. 2b) showing the predominant formation of the HL species in the pH range ca. 6–9.5. The pKa values were also determined for 2AP and sterane-based compounds based on the recorded spectra (Table 2, see representative spectra and concentration distribution curves for 8a in Fig. 3). Notably, the spectra recorded for the compounds with the methoxy (8h), nitrile (8e), chlorine (8c) and bromine substituents (8d) showed a certain extent of oxidation in the basic pH range (as no clear isosbestic point was found at pH > 10); therefore, the pK2 values could not be determined accurately.
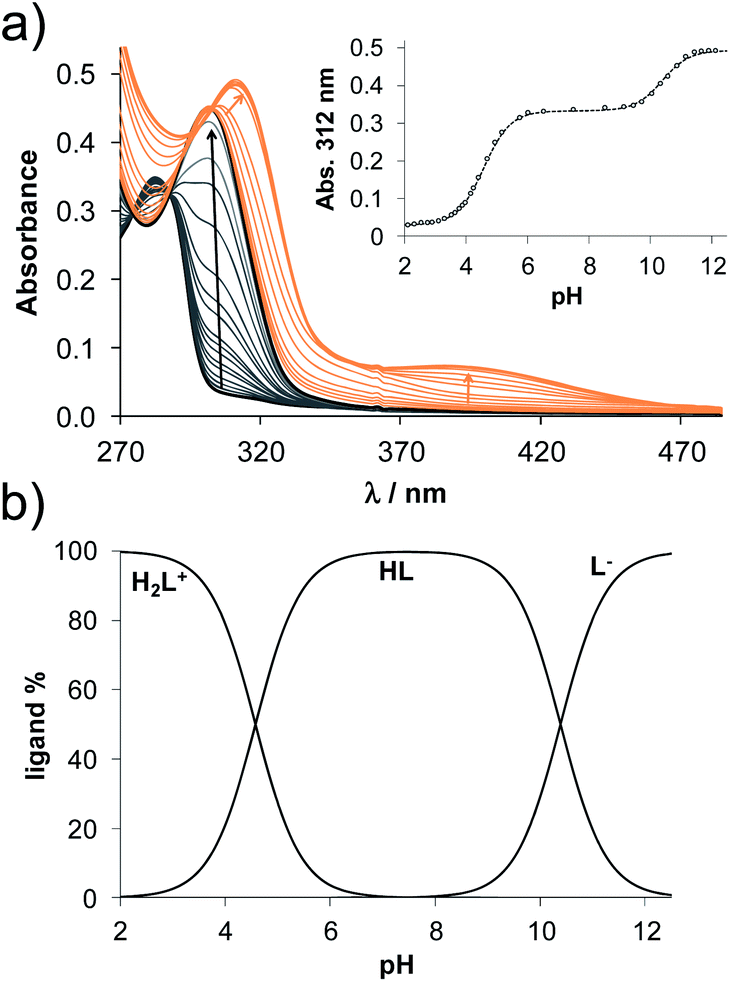 | ||
| Fig. 3 (a) UV-visible spectra recorded for 8a at various pH values. Inserted figure shows the measured absorbance values at 312 nm (●) with the fitted values (solid line) (b) concentration distribution curves calculated for 8a. {c = 50 μM; l = 2 cm; 30% (v/v) DMSO/H2O; T = 25 °C; I = 0.1 M (KCl)}. | ||
Similarly to the case of 2AP,45 the pK1 values are attributed to the amino moiety (NH3+ or NH2+) in all cases, while the pK2 values belong to the phenolic OH group. Since the pK1 and pK2 values are in the range 3.85–4.82 and 10.34–11.1, respectively, it can be concluded that all the studied compounds are in their neutral HL form at the physiological pH, and become air sensitive only in the basic pH range (pH > 9) when the deprotonation of the phenolic OH group starts.
As it is expected, the pKa values determined for 2AP in pure water (pK1 = 4.91 and pK2 = 9.8745) differ somewhat from those obtained in this work using 30% (v/v) DMSO/H2O solvent mixture. Namely, the pKa of the NH3+ group is lower, while that of the phenolic-OH is higher in the presence of the DMSO than in pure water. DMSO has a lower charge neutralization deprotonation/protonation processes are more favourable in the more apolar medium. Comparing the pKa values to each other (Table 2), it can be concluded that the incorporation of 2AP into the sterane core resulted in an increase in both of the pKa values due to the electron-donating effect of the neighbouring B-ring of the sterane core (c.f. 2AP and 4). Derivatization on the amino group resulted in secondary amines, and the introduction of the benzyl functional group led to lower pKa values (c.f. 4 and 8a). The pKa values of all the other derivatives (8b–e, 8g and 8h) are compared to those of 8a as a reference compound. The electron-donating methyl (8g) and methoxy substituents (8h) slightly increased the pKa of the amino group, while the electron-withdrawing CN (8e) and halo substituents (8b–d) decreased it according to the expectations. Notably, the largest effect was observed in the case of the nitrile and bromo substituents. Differences were also observed in the case of the pKa values of the phenolic OH group. This group is located further away from the benzyl ring, and the influence of substituents on pK2 needs some explanations. Effect of the CH3, MeO and F groups was negligible, while significantly higher pK2 values were obtained in the case of the other substituents, although these values are more questionable due to the recognized oxidation process. Most probably, the substituent effect can be realized via the possible N⋯OH hydrogen bond between the deprotonated amino group and the phenolic OH, and the electron-withdrawing groups might result in a stronger hydrogen bond leading to the increase of the pK2 values.
Pharmacological studies of the synthesized compounds
Cytotoxicity of all the synthesized derivatives was tested in vitro on cancerous cell lines MCF-7, PC-3, DU145, and HeLa and also on non-cancerous MRC-5 fibroblasts. For the preliminary screen, compounds were applied in 1.5 μM concentration for 72 h, and a heat map (Fig. 4, ESI, Table S1‡) was constructed with the mean cytotoxicity values obtained from three independent experiments. The results of the screen indicated that at 1.5 μM concentration, all the compounds, except for 8a, 8e, 9a and 9h induced cytotoxicity in at least one or in more cancerous cells (PC-3, DU145, and HeLa), but not in non-cancerous MRC-5 fibroblasts, suggesting that most of the synthesized compounds exhibit a cancer cell-selective toxic feature. Interestingly, none of the compounds showed cytotoxicity on MCF-7 breast cancer cells. These cancer cells lack caspase-3,46 a crucial cellular enzyme required to orchestrate programmed cell death.47 The observed findings imply that the toxicity induced by these compounds might depend on functional caspase-3, however, further studies are required to prove this notion.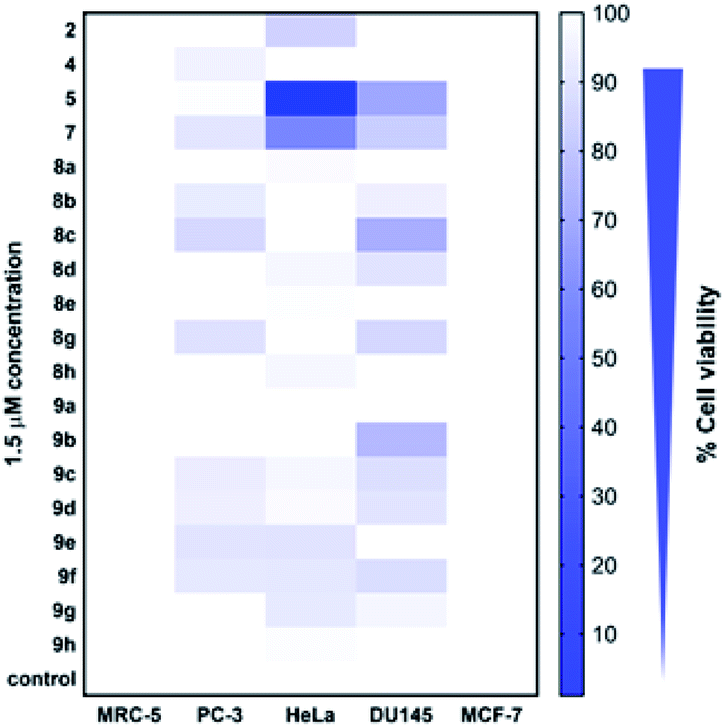 | ||
| Fig. 4 The primary cytotoxic effect of the synthesized aminoestradiols and benzoxazolones on various human cancerous cell lines and on non-cancerous MRC-5 fibroblasts shown on a heat map (c = 1.5 μM; 72 h incubation time). Control represents the viability of untreated cells. | ||
Based on the data of the overall toxicity screen, we selected some derivatives that induced the strongest toxic effects in one or more cancer cell lines. Therefore, we chose compounds 5, 8c and 9b to be tested further on DU145, compounds 2, 5, and 7 for a more profound examination on HeLa, and finally, all the aforementioned molecules (namely 2, 5, 7, 8c and 9b) were examined on MRC-5 cells to assess their IC50 values. Since none of these molecules showed cytotoxicity in MCF-7 breast cancer cells, and only two of them were slightly effective on PC-3 prostate cancer cells, these cell lines were omitted from further analyses.
For IC50 determination, the selected steroids or cisplatin (positive control) were applied at different concentrations on the cell lines, and at the end of the treatments cell viability was measured via MTT assay. Dose-response curves were fitted on the obtained viability data (ESI, Fig. S1‡), and subsequently the corresponding IC50 values were calculated (Table 3).
| Compound | IC50 (μM)±SD | ||
|---|---|---|---|
| MRC-5 | HeLa | DU145 | |
| 2 | >2.5 ± 1.35 | 1.1 ± 1.01 | — |
| 5 | >2.5 ± 1.41 | 0.09 ± 1.39 | 0.7 ± 1.01 |
| 7 | >2.5 ± 1.02 | 1.2 ± 1.01 | — |
| 8c | >2.5 ± 1.02 | — | 1.1 ± 1.01 |
| 9b | >2.5 ± 1.09 | — | 1.2 ± 1.01 |
| Cisplatin | 31.8 ± 1.09 | 347.3 ± 1.08 | 117.8 ± 1.02 |
In agreement with the preliminary screen, all the tested compounds, notably, 2-nitroestrone (2), the A-ring-fused 2-oxazolones (5, 7), the secondary aminoestradiol 8c and also the A-ring-fused-N-substituted 2-oxazolone 9b were effective on cancerous cells at several fold lower concentrations (Table 3, Fig. S1 and S2‡) compared to positive control cisplatin.
Unlike the tested steroid derivatives, cisplatin did not show any cancer cell specific cytotoxicity, which implies a potential advantage of these compounds as candidates for future therapeutic developments. The most potent molecule on HeLa cells proved to be the A-ring-fused 2-oxazolone (5), whereas the secondary aminoestradiol 8c and the benzoxazolone 9b were the most effective agents on DU145 prostate cancer cells despite being ineffective on HeLa cells. It is noteworthy that compound 8c exerted the strongest effect also on the other prostate cancer cell line PC-3.
Conclusions
In summary, we developed an efficient methodology for the mononitration of estrone at C-2 and C-4 positions. Reduction of nitroestrones by in situ generated hydrogen under catalytic conditions resulted in 2- and 4-aminoestradiol derivatives in a single step. Primary and secondary aminoestradiols, the latter obtained by reductive amination of benzaldehyde and p-substituted arylaldehydes with 2-aminoestradiol, were subjected to cyclization with CDI to afford A-ring-integrated estradiol–benzoxazolone hybrids in good to excellent yields.Compounds 4, 8a–h were characterized by two pKa values that belong to the amino (pK1) and the phenolic-OH (pK2) moieties, respectively. Based on the determined values (pK1 = 3.9–4.8; pK2 = 10.3–11.1), all the studied compounds are present in their neutral form in a wide pH range, including the physiological pH. These aminophenol derivatives become air-sensitive as the deprotonation of the OH functional group starts, thus in the basic pH range (pH > 9). The substituents on the benzyl function of the secondary amines (8) have an impact on the proton dissociation processes mainly of the amino group, namely, the electron-donating CH3 and MeO groups increase pK1, while CN and the halogens decrease it.
Based on a preliminary cytotoxicity screen indicating a possible cancer cell-selective pharmacological effect of the synthesized compounds, the 5 most potent agents – A-ring-fused 2-oxazolones (5, 7), the 2-nitroestrone (2), the secondary aminoestradiol 8c and also the A-ring-fused-N-substituted 2-oxazolone 9b – were selected for more thorough testing. The chosen derivatives were effective at lower concentrations on HeLa and DU145 cancerous cells than cisplatin, highlighting their potential as lead compounds for anticancer drug development.
Experimental
Materials and methods
Chemicals, reagents, and solvents were purchased from commercial suppliers (Sigma-Aldrich and Alfa Aesar) and used without further purification. Melting points (Mp) were determined on an SRS Optimelt digital apparatus and are uncorrected. The transformations were monitored by TLC using 0.25 mm thick Kieselgel-G plates (Si 254 F, Merck). The compound spots were detected by spraying with 5% phosphomolybdic acid in 50% aqueous phosphoric acid. Flash chromatographic purifications were carried out on silica gel 60, 40–63 μm (Merck). NMR spectra were recorded with a Bruker DRX 500 instrument at room temperature in CDCl3 or DMSO-d6 using residual solvent signals as an internal reference. Chemical shifts are reported in ppm (δ scale), and coupling constants (J) are given in Hz. Multiplicities of the 1H signals are indicated as a singlet (s), a doublet (d), doublet of doublets (dd), doublet of triplets (dt) a triplet (t), triplet of doublets (td), or a multiplet (m). 13C NMR spectra are 1H-decoupled and the J-MOD pulse sequence was used for multiplicity editing. In this spin-echo type experiment, the signal intensity is modulated by the different coupling constants J of carbons depending on the number of attached protons. Both protonated and unprotonated carbons can be detected (CH3 and CH carbons appear as positive signals, while CH2 and C carbons as negative signals). ESI-HRMS spectra were recorded on a Q Exactive Plus hybrid quadrupole-orbitrap mass spectrometer (Thermo Scientific) equipped with a heated electrospray ionization (HESI-II) probe that was used in positive and negative ion mode. Samples introduced with FIA (flow injection analysis) method, eluent stream (water, acetonitrile in 1:1 volume ratio with 0.1% formic acid) was provided by a Waters Acquity I-class liquid chromatograph (Waters).
Synthetic procedures
:98).:1 (100 mL) and cooled to 0 °C, followed by the addition of 10% Pd–C (200 mg). To this suspension, NaBH4 (0.91 g, 24 mmol, 4 equiv.) was added in small portions. After 30 min, TLC indicated complete conversion, so the solution was neutralized with diluted HCl, filtered through a Cellite® pad for the removal of the Pd–C catalyst, and the solvent was evaporated under reduced pressure. To the crude product MeOH (20 mL) was added, and the solution was poured onto ice-cold water. The forming white precipitate was filtered off, washed with water, and dried.General procedure for the synthesis of secondary aminoestradiols (8a–h)
To a solution of 2-aminoestradiol (4, 287 mg, 1 mmol) in MeOH (5 mL), catalytic amount of AcOH and molecular sieves were added, followed by the addition of (p-substituted) benzaldehyde (1.1 equiv.). The mixture was kept at reflux temperature for 1 h, cooled to room temperature and reduced with NaBH 4 (76 mg, 2 equiv.). After complete conversion of the Schiff-base (30 min, TLC), the reaction was quenched with water, MeOH was removed, and the remaining residue was extracted with EtOAc (3 × 5 mL). The combined organic phase was washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography using EtOAc/CH2Cl2 (5:95) as eluent.
General procedure for the synthesis of oxazolone hybrids (5, 7 and 9a–h)
The primary (4 or 6) or secondary amine (8a–h) (0.5 mmol) was dissolved in THF/CH2Cl2 = 1:4 (5 mL), CDI (89 mg, 1.1 equiv.) and TEA (140 μL, 1 mmol, 2 eq.) were added and the mixture was kept at reflux temperature for 2 h. The solvent was removed under reduced pressure, the crude product was suspended in water (5 mL), acidified with diluted HCl and extracted with EtOAc (3 × 5 mL). The combined organic phase was washed with water and brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The product was purified by column chromatography.
:50; yield: 120 mg (76%); white powder; Mp 280–282 °C; ESI-HRMS: m/z 314.1761 [M + H]+, 314.1751 calcd for C19H24NO3+; 1H NMR (500 MHz, DMSO-d6): δ 0.67 (3H, s), 1.06–1.45 (7H, m), 1.58 (1H, m), 1.79 (1H, m), 1.88 (2H, m), 2.16 (1H, td, J 10.9, 4.2), 2.23 (1H, dt, J 12.9, 3.6), 2.80 (2H, dt, J 8.1, 4.1, 6-H2), 3.52 (1H, t, J 8.5, 17α-H), 4.45 (1H, s, 17-OH), 6.92 (2H, d, J 7.4, 1-H and 4 H), 11.32 (1H, s, NH).13C NMR (125 MHz, DMSO-d6): δ 11.1 (18-CH3), 22.7 (CH2), 26.1 (CH2), 26.6 (CH2), 29.0 (CH2), 29.8 (CH2), 36.5 (CH2), 38.2 (8-CH), 42.7 (13-C), 43.8 (9-CH), 49.6 (14-CH), 79.9 (17-CH), 106.2 (CH), 109.1 (CH), 128.2, 130.0, 135.6, 141.5, 154.6 (2′-C).:50; yield: 105 mg (67%); white powder; Mp 258–260 °C; ESI-HRMS: m/z 314.1757 [M + H]+, 314.1751 calcd for C19H24NO3+; 1H NMR (500 MHz, DMSO-d6): δ 0.67 (3H, s, 18-CH3), 1.07–1.44 (7H, m), 1.60 (1H, m), 1.88 (3H, m), 2.17 (1H, td, J 11.1, 4.3), 2.29 (1H, m), 2.59–2.70 (1H, m), 2.73–2.81 (1H, m), 3.53 (1H, td, J 8.5, 4.3, 17α-H), 4.43 (1H, d, J 4.8, 17-OH), 7.00 (2H, s, 1-H and 4-H), 11.44 (1H, s, NH); 13C NMR (125 MHz, DMSO-d6): δ 11.1 (18-CH3), 22.7 (CH2), 24.1 (CH2), 25.8 (CH2), 26.2 (CH2), 29.8 (CH2), 36.5 (CH2), 38.0 (8-CH), 42.6 (13-C), 43.6 (9-CH), 49.4 (14-CH), 79.9 (17-CH), 106.3 (2-CH), 118.3 (1-CH), 119.1 (5-C), 128.4 (4-C), 135.7 (3-C), 140.8 (10-C), 154.9 (2′-C).:95; yield: 139 mg (69%); white powder; Mp 148–150 °C; ESI-HRMS: m/z 404.2228 [M + H]+, 404.2220 calcd for C26H30NO3+; 1H NMR (500 MHz, CDCl3): δ 0.77 (3H, s), 1.17 (1H, m), 1.23–1.54 (7H, m), 1.70 (1H, m), 1.88 (1H, m), 1.94 (1H, m), 2.06–2.23 (3H, m), 2.79–2.94 (2H, m), 3.72 (1H, t, J 8.5, 17α-H), 4.90–5.03 (2H, m, N–CH2), 6.75 (1H, s, 1-H), 6.91 (1H, s, 4-H), 7.31 (1H, dt, 4′′-CH), 7,31–7.39 (4H, m, 2′′-H, 3′′-H, 5′′-H and 6′′-H); 13C NMR (125 MHz, CDCl3): δ 11.2 (18-CH3), 23.3 (CH2), 26.7 (CH2), 27.2 (CH2), 29.85 (CH2), 30.8 (CH2), 36.8 (CH2), 38.7 (8-CH), 43.3 (13-C), 44.4 (9-CH), 46.2 (N–CH2), 50.3 (14-CH), 81.9 (17-CH), 105.9 (4-CH), 110.1 (1-CH), 127.8 (3′′-CH and 5′′-CH), 128.3 (4′′-CH), 129.1 (1′′-C), 129.1 (2′′-CH and 6′′-CH), 131.6 (2-C), 135.2 (5-C), 136.4 (10-C), 141.2 (3-C), 155.3 (2′-C).:95; yield: 160 mg (76%); white powder; Mp 146–148 °C; ESI-HRMS: m/z 422.2136 [M + H]+, 422.2126 calcd for C26H29FNO3+; 1H NMR (500 MHz, CDCl3): δ 0.77 (3H, s, 18-CH3), 1.13–1.75 (9H, m), 1.88 (1H, m), 1.96 (1H, dt, J 12.2, 2.9), 2.07–2.24 (3H, m), 2.80–2.94 (2H, m), 3.73 (1H, t, J 8.5, 17α-H), 4.88–4.99 (2H, m, N–CH2), 6.74 (1H, s, 1-H), 6.91 (1H, s, 4-H), 7.04 (2H, td, J 8.5, 1.4), 7.29–7.38 (2H, m); 13C NMR (125 MHz, CDCl3): δ 11.2 (18-CH3), 23.3 (CH2), 26.7 (CH2), 27.2 (CH2), 29.8 (CH2), 30.8 (CH2), 36.8 (CH2), 38.7 (8-CH), 43.3 (13-C), 44.4 (9-CH), 45.4 (N–CH2), 50.3 (14-CH), 81.9 (17-CH), 105.7 (1-CH), 110.3 (4-CH), 116.0, 116.2, 128.8 (2-C), 129.5, 129.6, 131.0, 131.00, 131.8 (5-C), 136.4, 141.1, 155.2 (2′-C), 161.7, 163.7.:95; yield: 205 mg (94%); white powder; Mp 128–130 °C; ESI-HRMS: m/z 438.1840 [M + H]+, 438.1830 calcd for C26H29ClNO3+; 1H NMR (500 MHz, DMSO-d6): δ 0.65 (3H, s, 18-CH3), 1.05–1.43 (7H, m), 1.57 (1H, m), 1.78 (1H, m), 1.87 (2H, m), 2.14 (1H, td, J 11.0, 4.2), 2.28 (1H, m), 2.77–2.83 (2H, m), 3.52 (1H, td, J 8.5, 4.9, 17α-H), 4.47 (1H, d, J 4.8, 17-OH), 5.02 (2H, s, N–CH2), 7.01 (1H, s, 4-H), 7.13 (1H, s, 1-H), 7.36–7.45 (4H, m, 2′′-H, 3′′-H, 5′′-H and 6′′-H); 13C NMR (125 MHz, DMSO-d6): δ 11.2 (18-CH3), 22.7 (CH2), 26.1 (CH2), 26.6 (CH2), 29.1 (CH2), 29.9 (CH2), 36.5 (CH2), 38.2 (8-CH), 42.7 (13-C), 44.0 (overlapping, 9-CH and N–CH2), 49.5 (14-CH), 79.9 (17-CH), 106.2 (1-CH), 109.5 (4-CH), 128.6 (2 C), 128.7 (3′′-CH and 5′′-CH), 129.4 (2′′-CH and 6′′-CH), 130.9 (4′′-C), 132.4 (5-C), 134.9 (1′′-C), 136.0 (10-C), 140.2 (3-C), 154.1 (2′-C).:95; yield: 186 mg (77%); white powder; Mp 159–161 °C; ESI-HRMS: m/z 482.1335 [M + H]+, 482.1325 calcd for C26H29BrNO3+; 1H NMR (500 MHz, CDCl3): δ 0.77 (3H, s, 18-CH3), 1.13–1.54 (8H, m), 1.70 (1H, m), 1.88 (1H, m), 1.96 (1H, m), 2.07–2.24 (3H, m), 2.80–2.94 (2H, m), 3.73 (1H, t, J 8.5, 17α-H), 4.85–4.98 (2H, m), 6.72 (1H, s, 4 H), 6.92 (1H, s, 1-H), 7.22 (2H, s, J 8.4, 2′′-H and 6′′-H), 7.48 (2H, d, J 8.4, 3′′-H and 5′′-H); 13C NMR (125 MHz, CDCl3): δ 11.2 (18-CH3), 23.3 (CH2), 26.7 (CH2), 27.2 (CH2), 29.9 (CH2), 30.8 (CH2), 36.8 (CH2), 38.6 (8-CH), 43.3 (13-C), 44.4 (9-CH), 45.5 (N–CH2), 50.3 (14-CH), 81.9 (17-CH), 105.6 (1-CH), 110.3 (4-CH), 122.4 (4′′-C), 128.8 (2-C), 129.4 (2′′-CH and 6′′-CH), 131.9 (5-C), 132.3 (3′′-CH and 5′′-CH), 134.2 (1′′-C), 136.5 (10-C), 141.1 (3-C), 155.2 (2′-C).:90; yield: 197 mg (92%); white powder; Mp 134–137 °C; ESI-HRMS: m/z 429.2181 [M + H]+, 429.2173 calcd for C27H29N2O3+; 1H NMR (500 MHz, DMSO-d6): δ 0.65 (3H, s, 18-CH3), 1.04–1.43 (7H, m), 1.57 (1H, m), 1.74–1.93 (3H, m), 2.15 (1H, td, J 10.9, 4.0), 2.23–2.31 (1H, m), 2.81 (2H, dd, J 8.0, 3.9), 3.52 (1H, td, J 8.5, 4.8 17α-H), 4.47 (1H, d, J 4.8, 17-OH), 5.14 (2H, s, N–CH2), 7.04 (1H, s, 4-H), 7.15 (1H, s, 1-H), 7.53 (2H, d, J 8.3, 2′′-H and 6′′-H), 7.83 (2H, d, J 8.3, 3′′-H and 5′′-H); 13C NMR (125 MHz, DMSO-d6): δ 11.2 (18-CH3), 22.7 (CH2), 26.2 (CH2), 26.6 (CH2), 29.1 (CH2), 29.9 (CH2), 36.4 (CH2), 38.2 (8-CH), 42.7 (13-C), 44.0 (9-CH), 44.3 (N–CH2), 49.5 (14-CH), 79.9 (17-CH), 106.1 (1-CH), 109.5 (4-CH), 110.5 (4′′-CN), 118.5 (4′′-C), 128.2 (2′′-CH and 6′′-CH), 128.6 (2-C), 131.0 (5-C), 132.6 (3′′-CH and 5′′-CH), 136.1 (10-C), 140.2 (3-C), 141.5 (1′′-C), 154.1 (2′-C).:95; yield: 206 mg (92%); yellowish white powder; Mp 168–170 °C; ESI-HRMS: m/z 449.2082 [M + H]+, 449.2071 calcd for C26H29N2O5+; 1H NMR (500 MHz, DMSO-d6): δ 0.65 (3H, s, 18-CH3), 1.06–1.43 (7H, m), 1.58 (1H, m), 1.74–1.93 (3H, m), 2.15 (1H, td, J 10.9, 3.9), 2.24–2.32 (1H, m), 2.81 (2H, dd, J 8.1, 3.9), 3.51 (1H, td, J 8.5, 4.9, 17α-H), 4.47 (1H, d, J 4.8, 17-OH), 5.20 (2H, s, N–CH2), 7.05 (1H, s, 4-H), 7.17 (1H, s, 1-H), 7.60 (2H, d, J 8.8, 2′′-H and 6′′-H), 8.22 (2H, d, J 8.8, 3′′-H and 5′′-H); 13C NMR (125 MHz, DMSO-d6): δ 11.2 (18-CH3), 22.7 (CH2), 26.2 (CH2), 26.6 (CH2), 29.1 (CH2), 29.9 (CH2), 36.4 (CH2), 38.2 (8-CH), 42.7 (13-C), 44.0 (9-CH), 44.1 (N–CH2), 49.5 (14-CH), 79.9 (17-CH), 106.2 (1-CH), 109.6 (4-CH), 123.8 (3′′-CH and 5′′-CH), 128.5 (2′′-CH and 6′′-CH), 128.6 (2-C), 131.1 (5-C), 136.2 (10-C), 140.2 (3-C), 143.6 (1′′-C), 147.0 (4′′-C), 154.1 (2′-C).:95; yield: 163 mg (78%); white powder; Mp 129–131 °C; ESI-HRMS: m/z 418.2385 [M + H]+, 418.2377 calcd for C27H32NO3+; 1H NMR (500 MHz, DMSO-d6): δ 0.66 (3H, s, 18-CH3), 1.04–1.47 (7H, m), 1.58 (1H, m), 1.69–1.82 (1H, m), 1.87 (2H, m), 2.15 (1H, td, J 11.2, 10.7, 4.0), 2.26 (4H, m), 2.80 (2H, dt, J 8.4, 3.6, 6-H2), 3.52 (1H, td, J 8.5, 4.9, 17α-H), 4.42 (1H, d, J 4.8, 17-OH), 4.96 (2H, s, N–CH2), 7.00 (1H, s, 4-H), 7.09 (1H, s, 1-H), 7.16 (2H, d, J 7.8, 3′′-H and 5′′-H), 7.25 (2H, d, J 8.0, 2′′-H and 6′′-H); 13C NMR (125 MHz, DMSO-d6): δ 11.1 (18-CH3), 20.5 (CH2), 22.6 (CH2), 26.1 (CH2), 26.5 (CH2), 29.0 (CH2), 29.8 (CH2), 36.4 (CH2), 38.1 (8-CH), 42.6 (13 C), 43.9 (9-CH), 44.5 (N–CH2), 49.5 (14-CH), 79.9 (17-CH), 106.1 (1-CH), 109.3 (4-CH), 127.4 (2′′-CH and 6′′-CH), 128.6 (2 C), 129.1 (3′′-CH and 5′′-CH), 130.7 (1′′-C), 132.7 (5 C), 135.9 (4′′-C), 136.9 (10-C), 140.1 (3-C), 154.1 (2′-C).:98; yield: 195 mg (90%); white powder; Mp 136–138 °C; ESI-MS: m/z 434.2335 [M + H]+, 434.2326 calcd for C27H32NO4+; 1H NMR (500 MHz, DMSO-d6): δ 0.66 (3H, s, 18-CH3), 1.10–1.43 (7H, m), 1.57 (1H, m), 1.78 (1H, m), 1.87 (2H, m), 2.15 (1H, td, J 10.9, 4.0), 2.25–2.33 (1H, m), 2.79 (2H, dt, J 8.4, 3.4), 3.52 (1H, td, J 8.5, 4.7, 17α-H), 3.72 (3H, s, 4′′-OMe), 4.48 (1H, d, J 4.8, 17-OH), 4.94 (2H, s, N–CH2), 6.91 (2H, d, J 8.6, 3′′-H and 5′′-H), 7.00 (1H, s, 4-H), 7.12 (1H, s, 1-H), 7.32 (2H, d, J 8.6, 2′′-H and 6′′-H); 13C NMR (125 MHz, DMSO-d6): δ 11.2 (18-CH3), 22.7 (CH2), 26.2 (CH2), 26.6 (CH2), 29.1 (CH2), 29.9 (CH2), 36.5 (CH2), 38.2 (8-CH), 42.7 (13-C), 44.0 (9-CH), 44.2 (N–CH2), 49.5 (14-CH), 55.1 (4′′-OMe), 79.9 (17-CH), 106.3 (1-CH), 109.4 (4-CH), 114.1 (3′′-CH and 5′′-CH), 127.8 (1′′-C), 128.6 (2 C), 129.0 (2′′-CH and 6′′-CH), 130.7 (5-C), 135.9 (10-C), 140.1 (3-C), 154.1 (2′-C), 158.9 (4′′-C).UV-visible spectrophotometric titrations
Stock solutions of tested compounds were prepared in DMSO at 5 mM concentration. UV-visible spectra were recorded for compounds in order to determine proton dissociation constants (pKa) in the pH range 2–12 in a 30% (v/v) DMSO/H2O mixture at 25.0 ± 0.1 °C. Ionic strength of 0.10 M (KCl) was used in order to keep the activity coefficients constant. The titrations were performed with carbonate-free KOH solution of known concentration (0.10 M) with 30% (v/v) DMSO content. The electrode system was calibrated to the pH = −log[H+] scale by means of blank titrations (HCl vs. KOH) according to the method suggested by Irving et al.48 The average water ionization constant (pKw) is 14.52 ± 0.05 in the 30% (v/v) DMSO/H2O mixture. Argon was always passed over the solutions during the titrations. An Agilent Cary 8454 diode array spectrophotometer was used to record the UV-visible spectra at an interval of 200–800 nm. The path length was 2 or 5 cm. Spectrophotometric titrations were performed on samples containing the compounds at 10 or 50 μM concentration. Proton dissociation constants (Ka) of the compounds and the UV-visible spectra of the individual species in the differently protonated forms were calculated by the computer program PSEQUAD.49Cell culture
All the cell lines were purchased from ATCC. MCF-7, DU145 and PC-3 cells were maintained in RPMI-1640 (LONZA) complete medium (10% FBS, 2 mM glutamine and penicillin–streptomycin) at 37 °C and 5% CO2. HeLa and MRC-5 cells were maintained in EMEM (LONZA) complete medium at 37 °C and 5% CO2. Treatments were applied in cell specific complete medium described above.MTT cytotoxicity assay
Each synthesized compound was dissolved in cell culture grade DMSO (Sigma) to a final concentration of 10 mM. Cytotoxicity of these compounds on cancerous (MCF-7, HeLa, DU145, PC-3) and non-cancerous (MRC-5) cell lines was tested using MTT cell viability assay. Briefly, in a 96-well plate cells were seeded at 5000 cells per well density, and were left to grow overnight in 5% CO2 atmosphere at 37 °C. Cells were incubated for 72 hours at 37 °C under 5% CO2 with each synthesized compound separately in 1.5 μM concentration upon the primary screen and only with the selected compounds in concentrations 0.5, 1, 1.5, 2 and 2.5 μM for IC50 determination. At the end of treatment, cell viability was measured by MTT assay as described previously.19 The positive control cisplatin was applied for 24 h in 20, 40, 80, 160, and 330 μM concentrations. Final results were obtained by three independent replicates. GraphPad Prism 7 software was used to perform statistics and to construct the heat map.Author contributions
Conceptualization, É. F.; É. A. E. and M. K.; chemical synthesis and optimization experiments, F. K.; pharmacological studies, M. K. G. and D. I. A.; UV-visible spectrophotometric titrations, É. A. E.; structural analysis, F. K.; formal analysis and interpretation of data, M. K. G. and D. I. A.; methodology, resources, supervision, É. F.; M. K. and É. A. E.; writing—original draft preparation, F. K.; M. K. G. and D. I. A.; writing—review and editing, É. F.; M. K. and É. A. E. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Research, Development and Innovation Office-NKFIA (Hungary) through projects FK 124240, GINOP-2.3.2-15-2016-00038, GINOP-2.3.2-15-2016-00035, and Ministry of Human Capacities (Hungary) grant TKP-2020. The New National Excellence Program of the Ministry for Innovation and Technology (ÚNKP-20-5-SZTE-655 for M. K.) and the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/00878/19/8 for M. K.) are gratefully acknowledged.References
- L. F. Tietze, H. P. Bell and S. Chandrasekhar, Angew. Chem., Int. Ed., 2003, 42, 3996 CrossRef CAS PubMed.
- G. Bérubé, Expert Opin. Drug Discovery, 2016, 11, 281 CrossRef PubMed.
- K. Nepali, S. Sharma, M. Sharma, P. M. S. Bedi and K. L. Dhar, Eur. J. Med. Chem., 2014, 77, 422 CrossRef CAS PubMed.
- G. Mehta and V. Singh, Chem. Soc. Rev., 2002, 31, 324 RSC.
- P. Borah and P. Chowdhury, Curr. Drug Ther., 2017, 12, 3 CrossRef CAS.
- P. Saha, C. Debnath and G. Bérubé, J. Steroid Biochem. Mol. Biol., 2013, 137, 271 CrossRef CAS PubMed.
- A. Gupta, B. Sathish Kumar and A. S. Negi, J. Steroid Biochem. Mol. Biol., 2013, 137, 242 CrossRef CAS PubMed.
- I. Scherlitz-Hofmann, M. Dubs, R. Krieg, B. Schönecker, M. Kluge and D. Sicker, Helv. Chim. Acta, 1997, 80, 2345 CrossRef CAS.
- S. D. Kuduk, F. F. Zheng, L. Sepp-Lorenzino, N. Rosen and S. J. Danishefsky, Bioorg. Med. Chem. Lett., 1999, 9, 1233 CrossRef CAS PubMed.
- L. F. Tietze, G. Schneider, J. Wölfling, T. Nöbel, C. Wulff, I. Schubert and A. Rübeling, Angew. Chem., Int. Ed., 1998, 37, 2469 CrossRef CAS PubMed.
- G. M. Anstead, K. E. Carlson and J. A. Katzenellenbogen, Steroids, 1997, 62, 268 CrossRef CAS PubMed.
- É. Frank and G. Schneider, J. Steroid Biochem. Mol. Biol., 2013, 137, 301 CrossRef PubMed.
- R. Dutour, J. Roy, F. Cortés-Benítez, R. Maltais and D. Poirier, J. Med. Chem., 2018, 61, 9229 CrossRef CAS PubMed.
- G. Panchapakesan, V. Dhayalan, N. Dhatchana Moorthy, N. Saranya and A. K. Mohanakrishnan, Steroids, 2011, 76, 1491 CrossRef CAS PubMed.
- T. V. Petrasheuskaya, M. A. Kiss, O. Dömötör, T. Holczbauer, N. V. May, G. Spengler, A. Kincses, A. Č. Gašparović, É. Frank and É. A. Enyedy, New J. Chem., 2020, 44, 12154 RSC.
- M. Numazawa, M. Ando, Y. Watari, T. Tominaga, Y. Hayata and A. Yoshimura, J. Steroid Biochem. Mol. Biol., 2005, 96, 51 CrossRef CAS PubMed.
- D. Bandyopadhyay, G. Rivera, J. L. Sanchez, J. Rivera, J. C. Granados, A. M. Guerrero, F.-M. Chang, R. K. Dearth, J. D. Short and B. K. Banik, Eur. J. Med. Chem., 2014, 82, 574 CrossRef CAS PubMed.
- B. Xiao, T.-J. Gong, Z.-J. Liu, J.-H. Liu, D.-F. Luo, J. Xu and L. Liu, J. Am. Chem. Soc., 2011, 133, 9250 CrossRef CAS PubMed.
- B. Molnár, M. K. Gopisetty, D. I. Adamecz, M. Kiricsi and É. Frank, Molecules, 2020, 25, 4039 CrossRef PubMed.
- X. Yang, G. Shan and Y. Rao, Org. Lett., 2013, 15, 2334 CrossRef CAS PubMed.
- J. Poupaert, P. Carato and E. Colacino, Curr. Med. Chem., 2005, 12, 877 CrossRef CAS PubMed.
- M. Smist and H. Kwiecien, Curr. Org. Synth., 2014, 11, 676 CrossRef CAS.
- S. Bilginer, H. I. Gul, F. S. Erdal, H. Sakagami, S. Levent, I. Gulcin and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2019, 34, 1722 CrossRef CAS PubMed.
- G. Neef, Estrogens and Antiestrogens I, Handbook of Experimental Pharmacology, Springer-Verlag Berlin Heidelberg, 1999, Vol 135/I, ch. 2, p. 21 Search PubMed.
- A. Cornelis, P. Laszlo and P. Pennetreau, J. Org. Chem., 1983, 48, 4771 CrossRef CAS.
- A. Bose, W. P. Sanjoto, S. Villarreal, H. Aguilar and B. K. Banik, Tetrahedron Lett., 2007, 48, 3945 CrossRef CAS.
- Y. Wang, Z. Zhan, Y. Zhou, M. Lei and L. Hu, Monatsh. Chem., 2018, 149, 527 CrossRef CAS.
- J.-M. Poirier and C. Vottero, Tetrahedron, 1989, 45, 1415 CrossRef CAS.
- L. W. Lawrence Woo, B. Leblond, A. Purohit and B. V. L. Potter, Bioorg. Med. Chem., 2012, 20, 2506 CrossRef CAS PubMed.
- T. M. Lavallee, V. S. Pribluda, J. Simons, N. Mabjeesh, P. Giannakakou, US Pat., WO 2003073985 A2, 2003 Search PubMed.
- P. N. Rao and J. W. Cessac, Steroids, 2002, 67, 1065 CrossRef CAS PubMed.
- P. Crabbé, L. A. Maldonado and I. Sánchez, Tetrahedron, 1971, 27, 711 CrossRef.
- A. Loupy and S. Régnier, Tetrahedron Lett., 1999, 40, 6221 CrossRef CAS.
- N. Ganguly and P. Mondal, Synthesis, 2010, 3705 CrossRef CAS.
- G. Wu, P. N. Ibrahim, Y. Zhou, S. Mamo, S. J. Gillette, Y.-L. Zhu, J. Liu, C. Zhang, K. Zhang, D. R. Artis, US Pat., WO 2010129467, A1, 2010 Search PubMed.
- M. Armitage, G. Bret, B. M. Choudary, M. Kingswood, M. Loft, S. Moore, S. Smith and M. W. J. Urquhart, Org. Process Res. Dev., 2012, 16, 1626 CrossRef CAS.
- I. Bacsa, D. Szemerédi, J. Wölfling, G. Schneider, L. Fekete and E. Mernyák, Beilstein J. Org. Chem., 2018, 14, 998 CrossRef CAS PubMed.
- I. Bacsa, R. Jójárt, G. Schneider, J. Wölfling, P. Maróti, B. E. Herman, M. Szécsi and E. Mernyák, Steroids, 2015, 104, 230–236 CrossRef CAS PubMed.
- A. V. Joshi, M. Baidoosi, S. Mukhopadhyay and Y. Sasson, Org. Process Res. Dev., 2003, 7, 95–97 CrossRef CAS.
- Y. A. Mostafa, B. Kralt, P. P. N. Rao and S. D. Taylor, Bioorg. Med. Chem., 2015, 23, 5681–5692 CrossRef CAS PubMed.
- N. Al Soom and T. Thieman, Int. J. Org Chem., 2016, 6, 1–11 CrossRef CAS.
- S. I. El-Hout, S. M. El-Sheikh, H. M. A. Hassan, F. A. Harraz, I. A. Ibrahim and E. A. El-Sharkawy, Appl. Catal., A, 2015, 503, 176–185 CrossRef CAS.
- S. C. Mitchell, P. Carmichael and R. Waring, Kirk-Othmer Encyclopedia of Chemical Technology, ed. 2003 Search PubMed.
- J. Kaizer, R. Csonka and G. Speier, J. Mol. Catal., 2002, 180, 91–96 CrossRef CAS.
- A. Vaidyan and P. Bhattacharya, Indian J. Chem., 1994, 33, 103–1007 Search PubMed.
- R. U. Janicke, Breast Cancer Res. Treat., 2009, 117, 219–221 CrossRef PubMed.
- A. G. Porter and R. U. Jänicke, Cell Death Differ., 1999, 6, 99–104 CrossRef CAS PubMed.
- H. M. Irving, M. G. Miles and L. D. Petit, Anal. Chim. Acta, 1967, 38, 475–488 CrossRef CAS.
- L. Zékány and I. Nagypál, Computational Methods for the Determination of Stability Constants, Plenum Press, New York, 1985 Search PubMed.
Footnotes |
| † Dedicated to Prof. Gyula Schneider on his upcoming 90th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01889b |
| This journal is © The Royal Society of Chemistry 2021 |