
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron-catalyzed dehydrative allylation of 1,3-diketones and β-ketone esters with 1,3-diarylallyl alcohols in water†
Guo-Min Zhangab,
Hua Zhangab,
Bei Wangab and
Ji-Yu Wang *a
*a
aChengdu Institute of Organic Chemistry, Chinese Academy of Sciences, Chengdu 610041, P. R. China. E-mail: JiyuWang@cioc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 10th May 2021
Abstract
A metal-free catalytic allylation with atom economy and green environment friendly was developed. Allylic alcohols could be directly dehydrated in water by B(C6F5)3, without using any base additives. The reaction can afford the corresponding monoallylated product in moderate to high yield and has been performed on a gram-scale, and a quaternary carbon center can be constructed for the active methine compounds of 1,3-diketones or β-ketone esters in this process. The product can be further converted, such as the synthesis of tetra-substituted pyrazole compounds, or 1,4-dienes and functionalized dihydropyrans.
Introduction
The Tsuji–Trost reaction is an important tool for C–C bond construction. According to the different nucleophiles of the substrates, numerous types of reactions have been reported such as allylic alkylation,1 allylic arylation,2 allylic amination,3 or O-allylation.4 The allylation of activated methylene compounds is a typical type of the Tsuji–Trost reaction,5 and the corresponding allylation products are very useful for the synthesis of a variety of valuable products.6 In tradition, the transition metal-catalyzed substitution reaction of activated methylene compounds and allylic alcohols or activated allylic alcohol derivatives with a base was widely reported,7 and the diallylated compounds were often detected.8 In this type of the reaction, the direct Tsuji–Trost reaction of activated methylene compounds with allylic alcohols would be more atom- and step-economical than activated allylic alcohol derivatives.9 In a previous research study, those reactions based on transition metals such as Pd10 or Pt,11 and the key intermediates were π-allyl metal species. Lewis acid12 such as InCl3, FeCl3, Bi(OTf)3, LnOTf (Ln = La, Yb, Sc) or Bronsted acid13 such as p-toluenesulfonic acid have also been reported to catalyze the process efficiently. In addition, iodine,14 hexafluoroisopropanol,15 and graphene oxide CO2H16 as the metal-free catalysts were used in the allylation of 1,3-dicarbonyl compounds with allylic alcohols (Scheme 1a). Recently, a calcium system17-catalyzed allylation of the cyclic β-ketone esters with MBH alcohols in MeCN was developed by Xie17 et al. (Scheme 1b). This process can efficiently construct a quaternary carbon. Despite these developments, some problems such as long reaction time, using metal catalysts or unsafe organic solvents are still remaining. Developing green and economic allylation of activated methylene compounds is of great interest.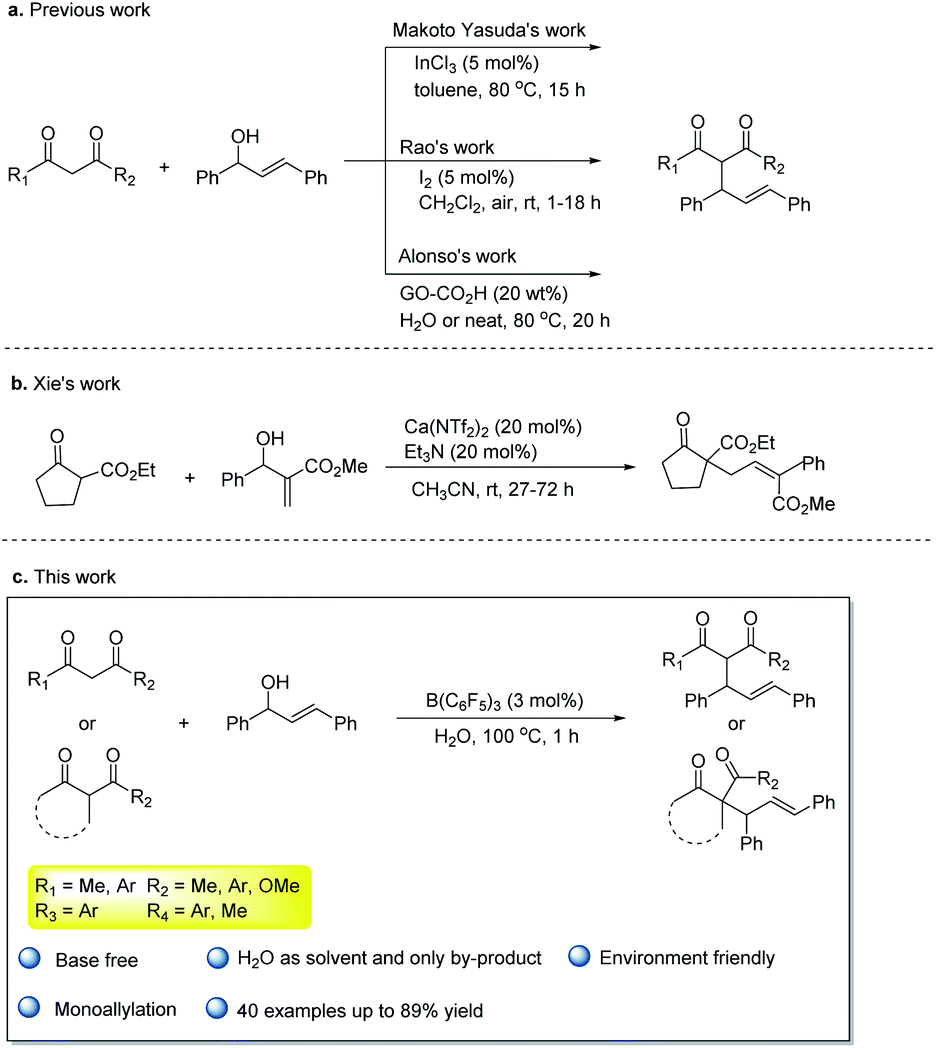 | ||
| Scheme 1 The reaction of 1,3-diketones and β-ketone esters with allylic alcohols. | ||
Water is a solvent that is more environmental-friendly than organic solvents in green chemistry. However, most of the catalysts used in organic solvents are often incompatible with water, and the low solubility of common organic compounds in water makes its practical use highly problematic. Thus, a catalyst that activates the substrate and is stable in air and water needs to be developed.
Tris(pentafluorophenyl)borane18 is widely used in the reduction,19 dehydrogenation,20 addition reaction,21 or other reactions because of non-toxicity, stability and good water solubility. Moreover, Stephen22 came up with the concept of frustrated Lewis pairs in 2006. B(C6F5)3 also became the darling of the frustrated Lewis pairs with its strong electrophilicity and large space resistance. Piers23 (hydrosilylation) and Stephen24 (catalytic hydrogenation) have carried our numerous pioneering work on metal-free catalytic reduction to make B(C6F5)3 a versatile catalyst. However, the strong Lewis acidity of B(C6F5)3 could cause the B–C bond to undergo preconized hydrolysis in water. The interaction between borane with different groups and water has been comprehensively explored.25 The presence of water would affect the catalytic effects of B(C6F5)3, and it limits the development of B(C6F5)3 or frustrated Lewis pairs chemistry involved. Previously, there have been some developments in frustrated Lewis pairs chemistry that B(C6F5)3 could catalyze reactions with a tiny amount of water.26 Recently, some examples were reported about the complex formed by B(C6F5)3 combined with water that could catalyze the reactions. For example, Tang et al.27 reported that B(C6F5)3 catalyzed α-diazo ester insertion into the O–H bond in water to afford numerous α-hydroxy esters in 2018. Our group has reported the coupling reaction of naphthoquinones with indoles catalyzed by B(C6F5)3 in water,28 and the allylation reaction of electron-rich aromatic rings with allyl alcohols catalyzed by B(C6F5)3 and 2,6-lutidine realized the coupling of the sp2 hybrid C–H bond with allyl alcohols in water.29 These successful examples of B(C6F5)3 catalytic reactions in water have greatly aroused our enthusiasm in this field. We have already known that the allyl alcohols can be activated by B(C6F5)3 to form allyl carbocation in water. Also, 1,3-dicarbonyl compounds as the classical nucleophiles, which were activated by B(C6F5)3, have also been reported in previous literature.30 Based on these studies, we proposed the allylation of 1,3-dicarbonyl compouds and allylic alcohols by B(C6F5)3 catalyzed in water, and this approach would be an efficient and environmentally friendly way (Scheme 1c).
Results and discussion
Acetylacetone 1a and (E)-1,3-diphenylallyl alcohol 2a were selected as the model substrates to investigate the allylic reaction under numerous reaction conditions (Table 1). Initially, the reaction was carried out in different catalysts, 100 °C for 1 h in water. First, the reaction was largely impossible without any catalyst (entry 1). Moreover, to our delight, B(C6F5)3 has catalyzed the allylic reaction as well as we thought, 86% yield of product 3aa was obtained (entry 2). B(3,4,5-F3-C6H2)3 or B(2,4,6-F3-C6H2)3 demonstrated activities in this transformation, and the yields of 3aa were just moderate. Compared with B(C6F5)3, the Lewis acidity of B(3,4,5-F3-C6H2)3 or B(2,4,6-F3-C6H2)3 is weaker, which leads to the decrease in the yield of the desired products and the formation of by-products (entries 3 and 4). In addition, CuSO4·5H2O had a partial catalytic effect in the reaction (entry 5), other metal Lewis acid or non-metallic proton acid had a little catalytic effect on the generation of 3aa (entries 6–12). Except for B(C6F5)3, other catalysts with weaker Lewis acidity could produce the bis(1,3-diphenylallyl) ether 4aa and 5aa, 6aa as by-products in this condition. Further screening of solvents showed that the solvent had some effects on the reaction. When the reaction was performed in CH3CN, DCE or cyclohexane, 3aa could be obtained in good yields (entries 13–15). When using EtOH and 1,4-dioxane as solvents, we did not detect any 3aa in the condition (entries 16 and 17). The water did not affect the reaction. Moreover, when the temperature was adjusted to 30 °C, the yield of the expected product was decreased significantly and that of the by-product bis(1,3-diphenylallyl) ether increased (entry 18). With the increase in temperature, the by-product was reduced (entries 19–21). In addition, we also tried to decrease the amount of the catalyst to 3 mol% and the yield could be maintained (entry 22), and when the amount of catalyst to 1 mol% was decreased, the yield of the product was reduced (entry 23). At last, the best conditions for the reaction were determined: 1a (0.3 mmol), 2a (0.3 mmol), B(C6F5)3 (3 mol%), H2O (1.5 mL), 100 °C, 1 h.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | T/°C | 3aa (%) | 4aa (%) | 5aa (%) | 6aa (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: 1a (30.0 mg, 0.3 mmol), 2a (63.0 mg, 0.3 mmol), and catalyst (5 mol%) in solvent (1.5 mL) for 1 h at T °C.b Yield of the isolated product.c The catalyst was 3 mol%.d The catalyst was 1 mol%. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | — | H2O | 100 | — | Trace | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | B(C6F5)3 | H2O | 100 | 86 | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | B(3,4,5,-F3-C6H2)3 | H2O | 100 | 56 | 22 | 8 | 6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | B(2,4,6-F3-C6H2)3 | H2O | 100 | 49 | 19 | 10 | 8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | CuSO4·5H2O | H2O | 100 | 43 | 27 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | FeCl3 | H2O | 100 | Trace | 22 | 8 | 5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Fe(OTf)3 | H2O | 100 | Trace | 62 | 11 | 12 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | CoCl2·6H2O | H2O | 100 | Trace | 16 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | SnCl2·2H2O | H2O | 100 | Trace | Trace | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | Cu(OTf)2 | H2O | 100 | Trace | 52 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | TsOH | H2O | 100 | — | 64 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | H3BO3 | H2O | 100 | — | 15 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | B(C6F5)3 | MeCN | 100 | 82 | Trace | Trace | Trace | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | B(C6F5)3 | DCE | 100 | 83 | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | B(C6F5)3 | Cyclohexane | 100 | 79 | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 | B(C6F5)3 | EtOH | 100 | Trace | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 17 | B(C6F5)3 | 1,4-Dioxane | 100 | — | 13 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 18 | B(C6F5)3 | H2O | 30 | 39 | 26 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 19 | B(C6F5)3 | H2O | 50 | 57 | 12 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 20 | B(C6F5)3 | H2O | 70 | 73 | Trace | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 21 | B(C6F5)3 | H2O | 90 | 78 | Trace | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 22c | B(C6F5)3 | H2O | 100 | 89 | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 23d | B(C6F5)3 | H2O | 100 | 76 | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Next, the applicability of the protocol for a broad range of substrates was explored. As shown in Table 2, for almost active methyl compounds of 1,3-diketones and β-ketone esters, they could perform the allylic reaction with corresponding product yields of more than 80% (3aa–3ca, 3ja–3oa). The yield of the ring of active methylene compounds such as 1,3-cyclohexanedione was 75% (3da), and the yield of the product was not significantly affected by the ring tension. When the substrates were ring active methine compounds, the yield of the allylic product was moderate to high (3ea–3ha, 3pa–3sa). The steric hindrance of the active methine compounds did not affect the yield of the allylic reaction in these substrates interestingly. In contrast, (E)-3-cinnamylpentane-2,4-dione was reacted with 2a to give the allylic product in moderate (3ia), but the yield of methyl-(4E)-2-acetyl-5-phenyl-
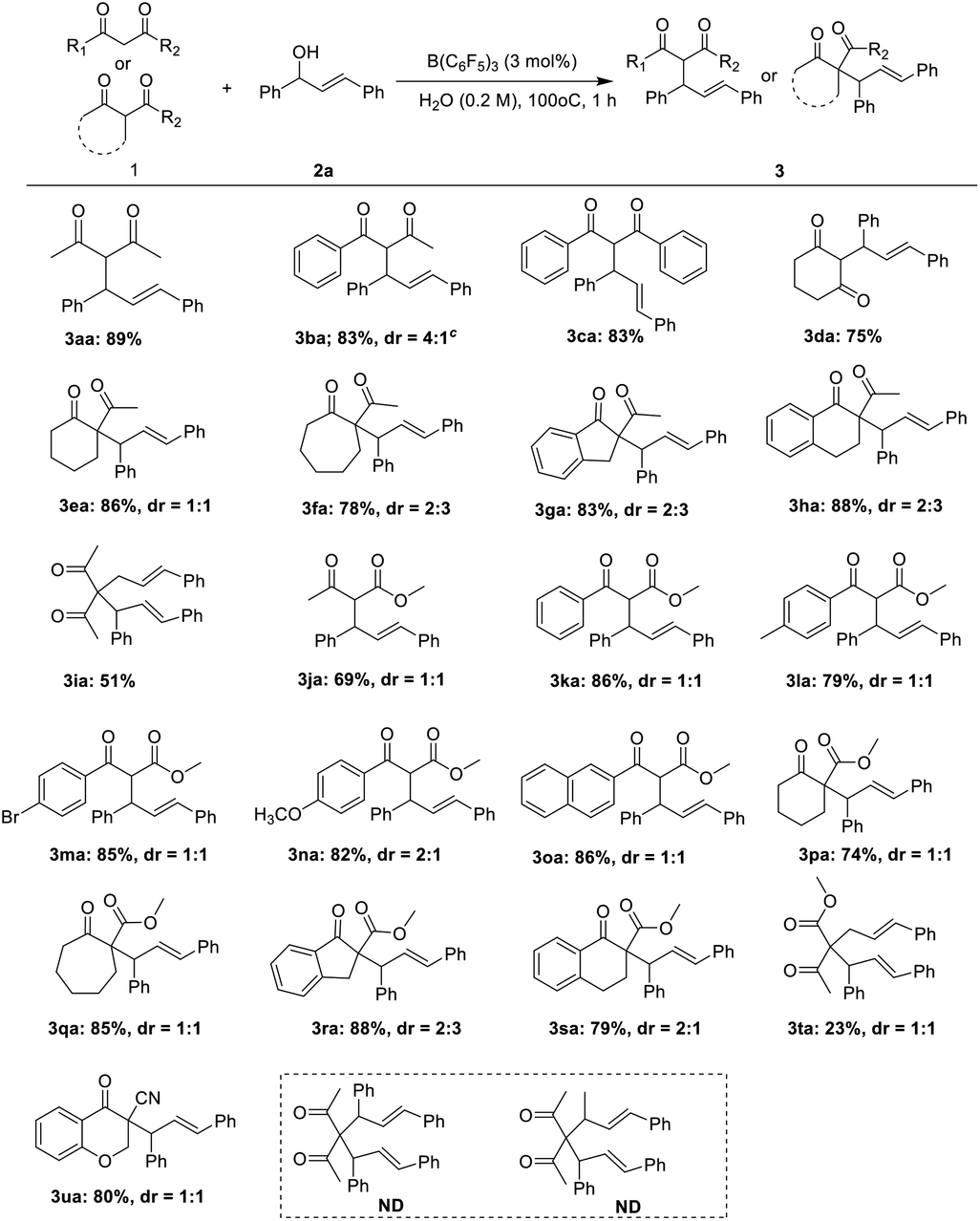
4-Pentenoate was only 23% (3ta). It may be that the presence of two ketone carbonyl in (E)-3-cinnamylpentane-2,4-dione increases the reaction activity. Interestingly, 3-cyano-4-chromanone 3ua afforded the corresponding product in good yield. When the compound 3aa was employed, the corresponding diallylated product was not obtained. Moreover, the compound 3aj also failed. It may be caused by the steric hindrance. Most of the products have two chiral centers, and we obtained a mixture of diastereoisomer. The dr value was obtained by NMR. However, unfortunately, the dr value was not high.
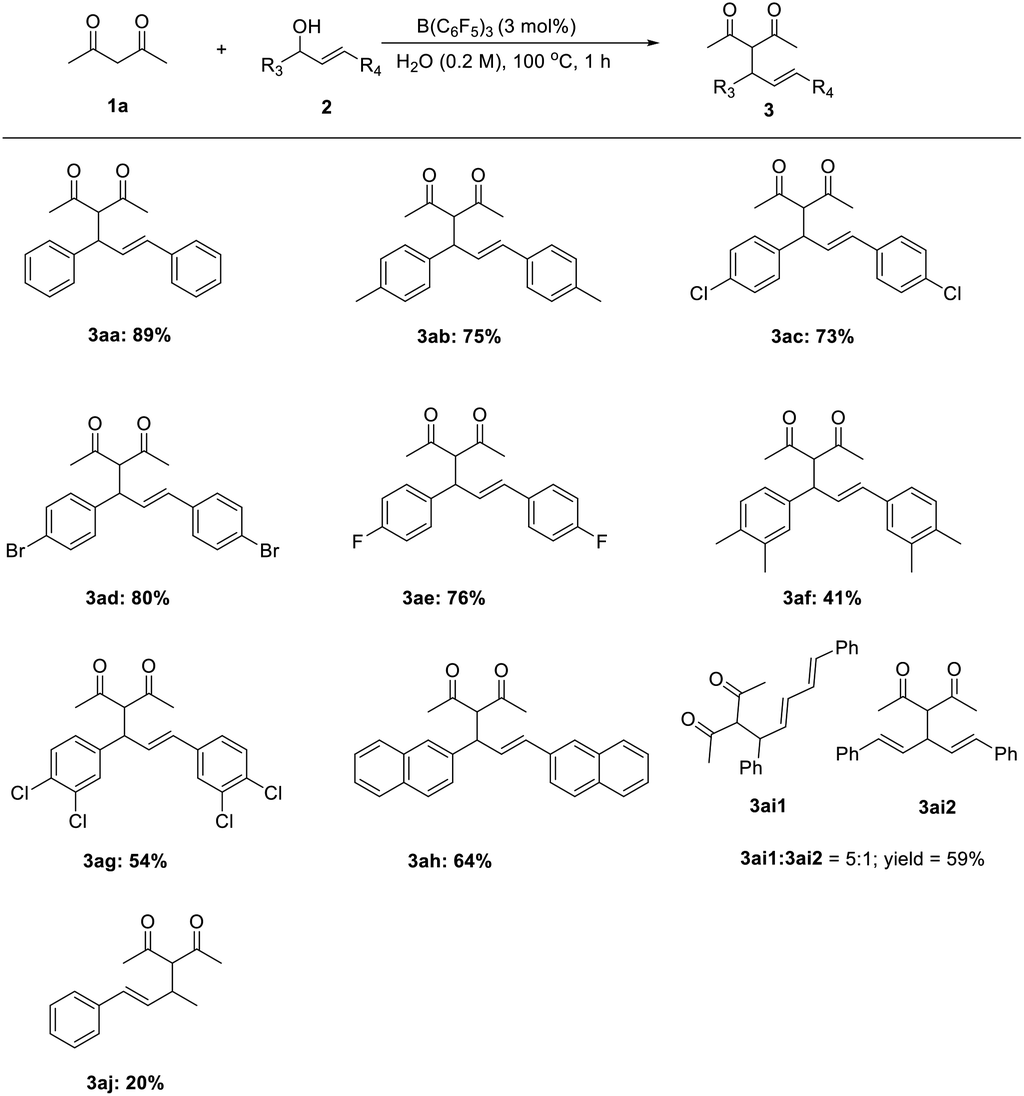
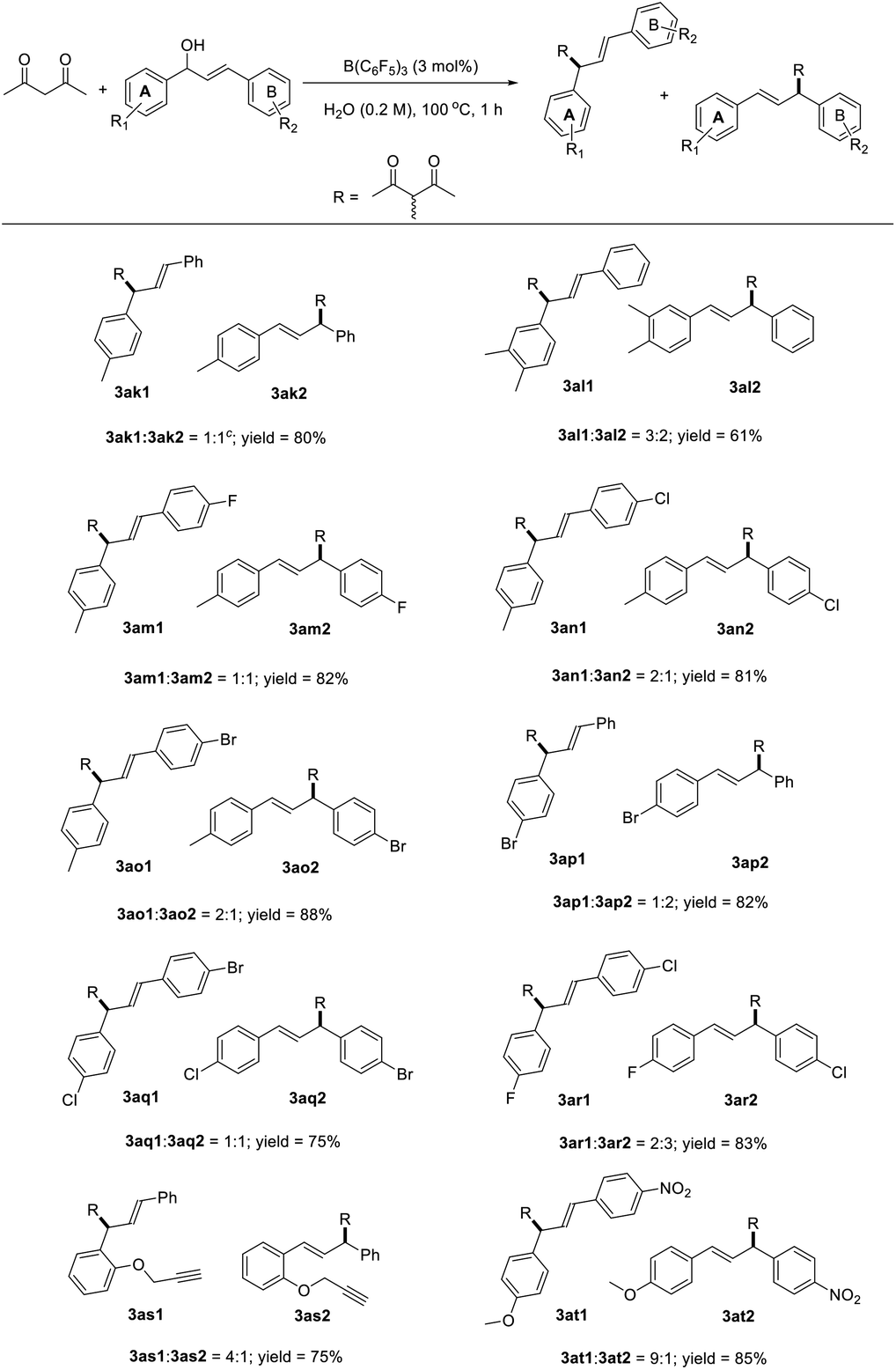
The reaction between numerous substituted allylic alcohols and acetylacetone was then examined. As depicted in Table 3, the reactions proceeded smoothly, affording the desired products in moderate to good yields when the substrate was the Chalone-type allylic alcohols. When allylic alcohols were simple substitutions on the phenyl group, the corresponding products were obtained in good yields (3ab–3ae). Also, when the substituents on the benzene ring increases, the yields of products were decreased (3af–3ag). Besides, the expansion of the conjugate structure or the aromatic ring for the Chalone-type allylic alcohols was also detrimental to this reaction (3ah–3ai). The alkyl allyl alcohol did not perform the reaction smoothly under standard conditions. The yield of product 3aj was just 20%, and the allylic alcohols (E)-4-phenylbut-3-en-2-ol or (E)-1-phenylbut-2-en-1-ol was obtained from the compound 3aj due to the allyl cations that would be stabilized on the side with methyl groups. When asymmetrically substituted Chalone-type allyl alcohol was used in this reaction, the corresponding products can be obtained with good yield, but the allyl alcohol double bond was shifted and a mixture of double bond migrations was obtained (Table 4). The mixture showed a point that could not be separated on the TLC. The ratio of two compounds can be determined by NMR, and the ratio was influenced by different substituents on the benzene ring. When the para position of the allyl alcohol A ring was replaced by a methyl group, the ratio of the double bond migration product to the non-migrated product was about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by NMR (3ak); when a methyl group was added at the meta position of the allyl alcohol A ring, the yield of the product was slightly lower and the ratio increased to 3:2 (3al); and when numerous halogen groups were introduced at the para position of the allyl alcohol B ring, the ratio changed (3am–3ao). In the allyl carbanions formed during the reaction, due to the electron-donating effect of the methyl group on the A ring, the proportion of products near to the A ring in the mixtures might be larger. The para position of the allyl alcohol A ring was replaced by bromine, and the ratio of the double bond migration product to the non-migrated product was about 1:2 by NMR (3ap) due to the electronegativity of bromine, which is stronger than that of hydrogen, and the changes of the proportion about 3aq and 3ar also conformed to the law of the electronegative size of the atom, and the reaction is also suitable for the reduction of terminal alkenes (3as). When A ring was attached to a strong electron donating substituent and the B ring was connected to the strong electron withdrawing group, the uneven distribution of the allyl cation charge occurred, resulting in a product ratio of 9:1 (3at). Unfortunately, the huge limitation was shown in this reaction concerning the allylic alcohol substrates (Fig. 1).
:1 by NMR (3ak); when a methyl group was added at the meta position of the allyl alcohol A ring, the yield of the product was slightly lower and the ratio increased to 3:2 (3al); and when numerous halogen groups were introduced at the para position of the allyl alcohol B ring, the ratio changed (3am–3ao). In the allyl carbanions formed during the reaction, due to the electron-donating effect of the methyl group on the A ring, the proportion of products near to the A ring in the mixtures might be larger. The para position of the allyl alcohol A ring was replaced by bromine, and the ratio of the double bond migration product to the non-migrated product was about 1:2 by NMR (3ap) due to the electronegativity of bromine, which is stronger than that of hydrogen, and the changes of the proportion about 3aq and 3ar also conformed to the law of the electronegative size of the atom, and the reaction is also suitable for the reduction of terminal alkenes (3as). When A ring was attached to a strong electron donating substituent and the B ring was connected to the strong electron withdrawing group, the uneven distribution of the allyl cation charge occurred, resulting in a product ratio of 9:1 (3at). Unfortunately, the huge limitation was shown in this reaction concerning the allylic alcohol substrates (Fig. 1).
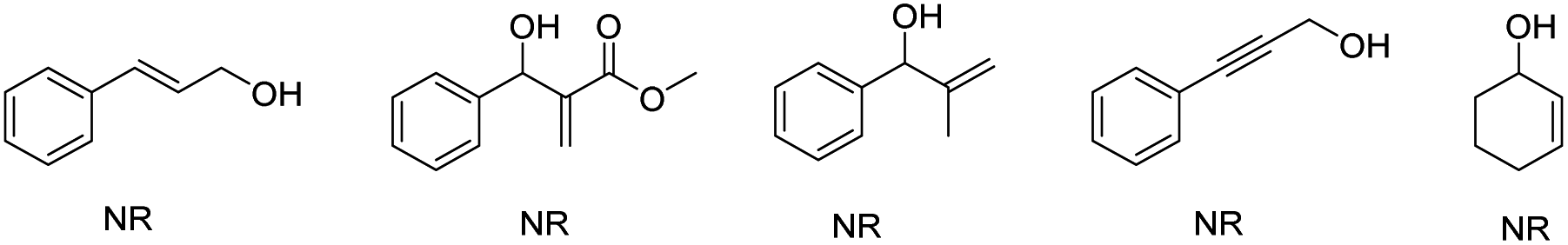 | ||
| Fig. 1 The allyl alcohols of the corresponding allylated products cannot be obtained in this reaction. | ||
To illustrate the synthetic utility of the developed process, an amplification scale reaction was implemented. A 1 g scale reaction was carried out for acetylacetone 1a and (E)-1,3-diphenylallyl alcohol 2a, and the product 3aa was isolated in 85% yield. The result showed that the product yield was not affected after the reaction was scaled up, further indicating that the reaction has great potential for application. We derived the product of the allylic reaction obtained, and the reaction of 3ca and phenylhydrazine in acetic acid at 100 °C for one hour could produce tetrasubstituted pyrazole compound 4 in 69% yield (Fig. 2A). The compound 3ja could be converted to a methyl (E)-2-(1-hydroxyethyl)-3,5-diphenylpent-4-enoate with NaBH4 (Fig. 2B). In addition, the ester group could be removed with NaOH to form α,δ-unsaturated ketone (Fig. 2C). The compound 3sa could be converted to compound 7 or compound 9 by DIBAL-H under different reaction conditions. Also, compound 7 reacted with PTSA in toluene and DMSO at 80 °C for 12 h could be generated from compound 8 (Fig. 2D). Compound 7 might have formed from a four-membered cyclic ether31 by p-toluenesulfonic acid catalyzed in the reaction, and then the ring was cleaved32 to produce compound 8. The skipped dienes are common structural motifs in numerous biologically active molecules due to the stereochemical identity of the diene unit.33 However, the direct dehydrogenative cycloetherification of compound 9 promoted by DDQ afforded compound 10 in moderate yield (Fig. 2E). The functionalized dihydropyrans are also an important class of heterocyclic target molecules, serving as the common core of the structure in natural products or precursors to diverse C-glycosides.34
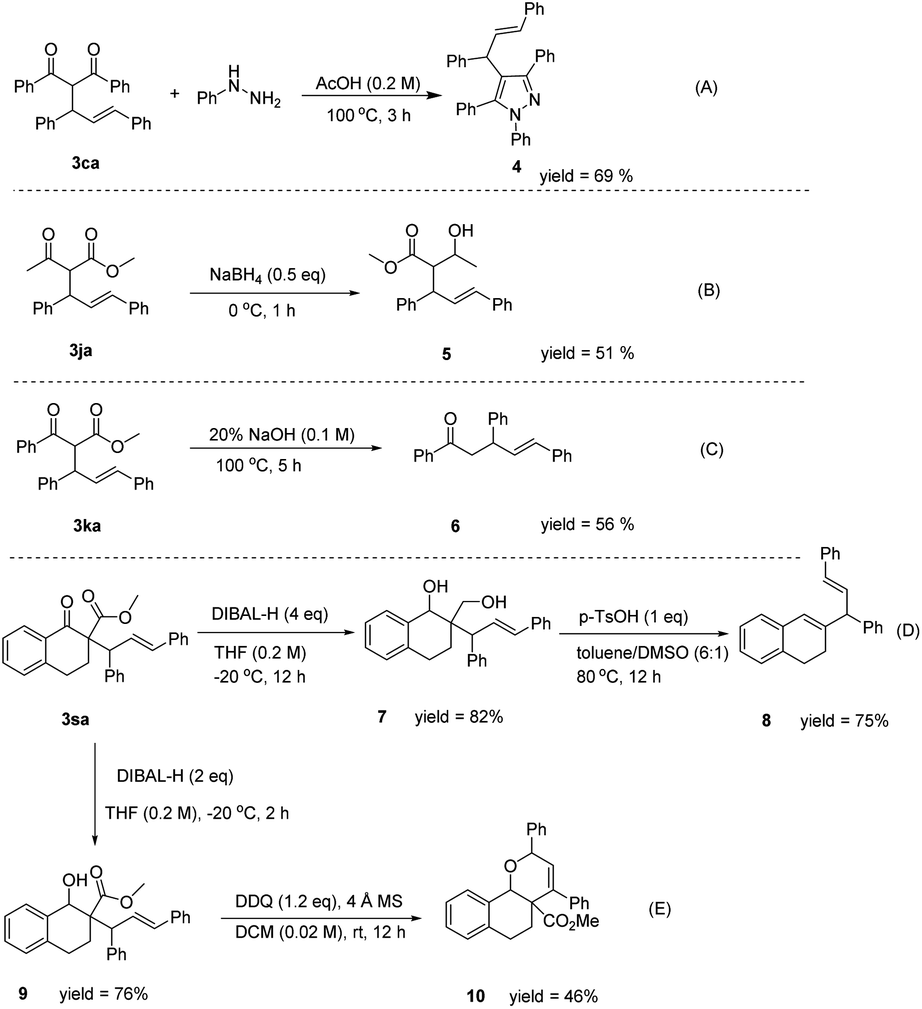 | ||
| Fig. 2 Preliminary protocol applications. | ||
In the next study, we found that the etherification product (4aa) reacted with acetylacetone could give 3aa in good yield under the standard conditions. Also, the yield of the product could be comparable to the template reaction (Scheme 2). Simultaneously, the by-product 4aa would be decomposed into 5aa and 6aa under the standard conditions (Scheme 3). Based on the reported literature,35 we proposed the mechanism of this reaction (Scheme 4). The reaction is a nucleophilic attack about the enol form of 1,3-dicarbonyl compounds and allyl carbocation. First, the carbonyl group of 1,3-diketone can be extremely activated by B(C6F5)3. Second, there are two possible ways in the formation of allyl carbocation: one is that due to the high temperature and strong acidity for the protonated product produced by the catalyst, and the other is that the allyl carbocation in the reaction attacked by the hydroxyl oxygen of the allylic alcohol produced the by-product bis(1,3-diphenylallyl) ether (4aa), and the allyl carbocation can also be formed when the etherified product is cleaved by a protonated catalyst.
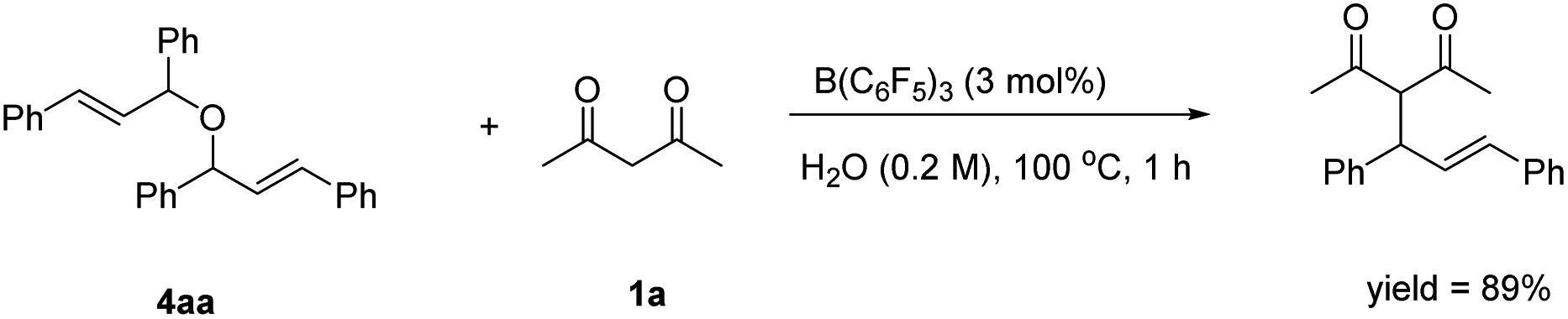 | ||
| Scheme 2 The allylization of 4aa. | ||
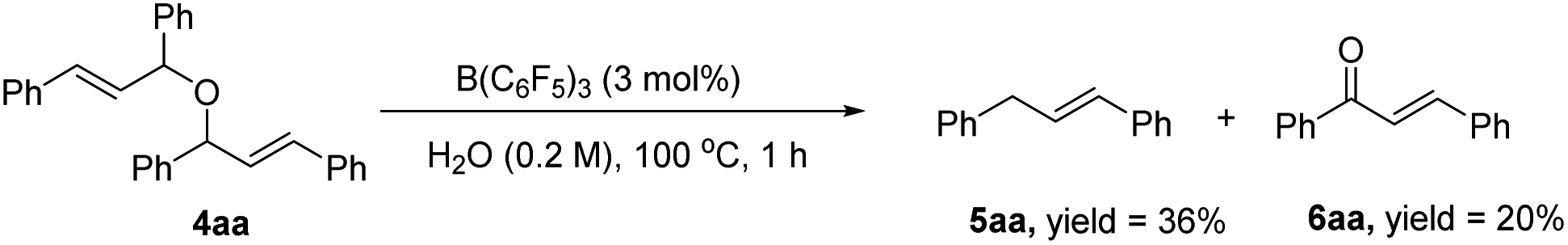 | ||
| Scheme 3 The decomposition of 4aa. | ||
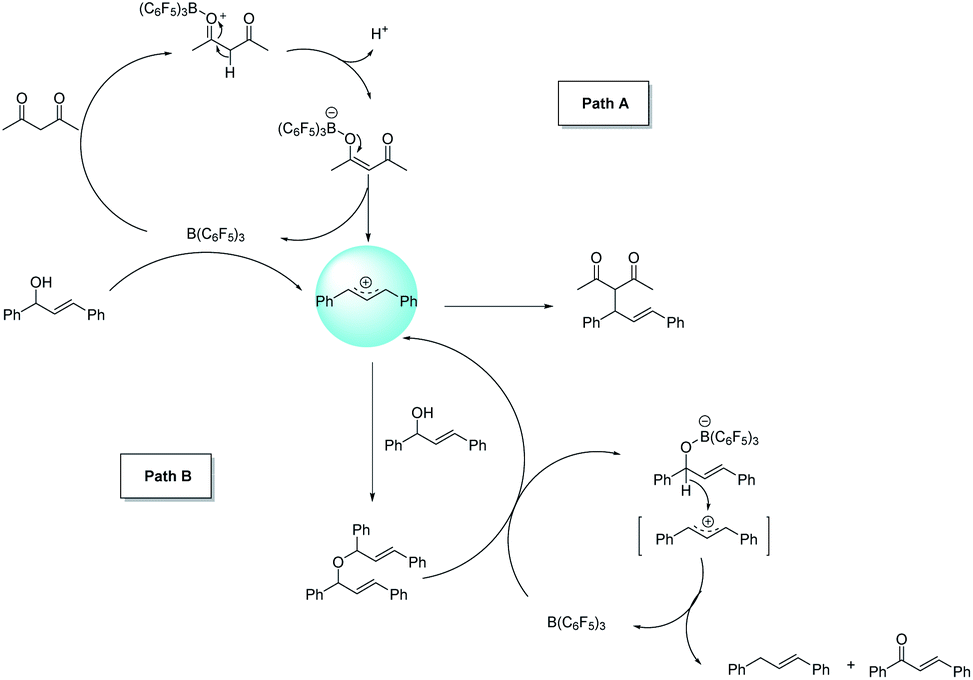 | ||
| Scheme 4 Plausible reaction mechanism. | ||
Conclusions
In summary, a metal-free catalyzed allylization of 1,3-diketones or β-ketone esters with allyl alcohols in water has been reported. The allyl alcohols with 1,3-diketones and β-ketone esters are dehydrated in water, and a quaternary carbon center can be well constructed for active methine of 1,3-dicarbonyl compounds in this method. The products can be further converted into other compounds such as tetrasubstituted pyrazole or 1,4-dienes and functionalized dihydropyrans. This reaction has a very good atom economy and water is only the by-product and a green solvent simultaneously. The protocol would offer new insight into the reaction catalyzed by B(C6F5)3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from CAS “Light of West China” program, the Applied Basic Research Programs of Sichuan province, China (no. 20YYJC0492).Notes and references
-
(a) C. Chevrin, J. L. Bras, F. Hénin, J. Muzart, A. Pla-Quintana, A. Roglans and R. Pleixats, Organometallics, 2004, 23, 4796–4798 CrossRef CAS
; (b) B. M. Trost, Tetrahedron Lett., 2015, 71, 5708–5733 CrossRef CAS PubMed
-
(a) T. Mino, T. Kogure, T. Abe, T. Koizumi, T. Fujita and M. Sakamoto, Eur. J. Org. Chem., 2013, 8, 1501–1505 CrossRef
-
(a) A. M. Johns, Z. Liu and J. F. Hartwig, Angew. Chem., Int. Ed., 2007, 46, 7259–7261 CrossRef CAS PubMed
-
(a) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1998, 120, 815–816 CrossRef CAS
-
(a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–402 CrossRef CAS PubMed
-
(a) Z. Lu and S. M. Ma, Angew. Chem., Int. Ed., 2008, 47, 258–297 CrossRef CAS PubMed
-
(a) H. Kinoshita, H. Shinokubo and K. Oshima, Org. Lett., 2004, 6, 4085–4088 CrossRef CAS PubMed
- B. C. Ranu, K. Chattopadhyay and L. Adak, Org. Lett., 2007, 9, 4595–4598 CrossRef CAS PubMed
-
(a) M. Bandini and M. Tragni, Org. Biomol. Chem., 2009, 7, 1501–1507 RSC
-
(a) F. Ozawa, H. Okamoto, S. Kawagishi, S. Yamamoto, T. Minami and M. Yoshifuji, J. Am. Chem. Soc., 2002, 124, 10968–10969 CrossRef CAS PubMed
- R. Shibuya, L. Lu, Y. Nakahara, K. Z. Mashima and T. Ohshima, Angew. Chem., Int. Ed., 2014, 53, 4377–4381 CrossRef CAS PubMed
-
(a) U. Jana, S. Biswas and S. Maiti, Tetrahedron Lett., 2007, 48, 406–4069 Search PubMed
-
(a) R. Sanz, A. Martínez, D. Miguel, J. M. Álvarea-Gutiérrez and F. Rodríguez, Adv. Synth. Catal., 2006, 348, 1841–1845 CrossRef CAS
- W.-D. Rao, L.-P. Wai, H. L. T. Adeline, P.-J. Goh, J. M. L. Choy, J. Kaijie-Ke and P. W. H. Chan, Tetrahedron Lett., 2008, 49, 122–126 CrossRef CAS
- T. Paz, B. Alejandro and N. Carmen, J. Org. Chem., 2012, 77, 7344–7354 CrossRef PubMed
- G. M. Melania, B. Alejandro and D. A. Alonso, ChemCatChem, 2017, 9, 1032–1039 CrossRef
- P.-Z. Xie, S.-S. Li, Y.-A. Liu, X.-Y. Cai, J.-Y. Wang, X.-B. Yang and T.-P. Loh, Org. Lett., 2020, 22, 31–35 CrossRef CAS PubMed
- A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245–250 CrossRef CAS
-
(a) E. Gábor, K. Nagy, H. Mehdi, I. Pápai, N. Péter, P. Király, G. Tárkányi and T. Sooś, Chem.–Eur. J., 2012, 18, 574–585 CrossRef
-
(a) F. G. Alexander, S. Grimme, S. Tobias, U. Florke, Z.-W. Qu, S. Grimme and J. Paradies, Angew. Chem., Int. Ed., 2016, 55, 12219–12223 CrossRef PubMed
-
(a) S. Mahesh, K. Guddi and R. V. Anand, RSC Adv., 2016, 6, 80718–80722 RSC
- G. C. Welch, R. R. San-Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1128 CrossRef CAS PubMed
- D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed
- M. Tayseer and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef
- C. Bergquist, B. M. Bridgewater, C. J. Harlan, J. R. Norton, R. A. Friesner and G. Parkin, J. Am. Chem. Soc., 2000, 122, 10581–10590 CrossRef CAS
-
(a) G. Ádám, B. Mária, F. Tamás, P. Imre, D. Attila and T. Soós, ACS Catal., 2015, 5, 5366–5372 CrossRef
- H.-H. San, S.-J. Wang, M. Jiang and X.-Y. Tang, Org. Lett., 2018, 20, 4672–4676 CrossRef CAS PubMed
- Y. Dong, H. Zhang, J. Yang, S. He, Z.-C. Shi, X.-M. Zhang and J.-Y. Wang, ACS Omega, 2019, 4, 21567–21577 CrossRef CAS PubMed
- H. Zhang, X.-Y. Zhan, Y. Dong, J. Yang, S. He, Z.-C. Shi, X.-M. Zhang and J.-Y. Wang, RSC Adv., 2020, 10, 16942–16948 RSC
- D. J. Parks and W. E. T. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS
- M. G. Bolster, B. J. M. Jansen and A. D. Groot, Tetrahedron Lett., 2001, 57, 5657–5662 CrossRef CAS
- Z. Grobelny, A. Stolarzewicz and A. Maercker, J. Organomet. Chem., 2000, 604, 283–286 CrossRef CAS
-
(a) M. Jaime, R. C. Eva and F. M. Martín, ACS Catal., 2017, 7, 5340–5344 CrossRef
-
(a) S. V. Ganapathy, J. Yang and C.-J. Li, Org. Lett., 1999, 7, 993–995 Search PubMed
- W. Huang, Z.-X. Zhang, X. Xiang, R.-T. Liu, X.-P. Zhou and J.-L. Wang, J. Org. Chem., 2009, 74, 3299–3304 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01922h |
| This journal is © The Royal Society of Chemistry 2021 |