
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe guest polymer effect on the dissolution of drug–polymer crystalline inclusion complexes
Lu Chen and
Yanbin Huang *
*
Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: yanbin@tsinghua.edu.cn
First published on 7th April 2021
Abstract
A drug–polymer crystalline inclusion complex (IC) is a novel solid form of drug, in which drug molecules form parallel channels, and linear polymer chains reside in these channels. In this study, we used carbamazepine (CBZ) as a model drug, and directly studied the effect of different types of guest polymers on the dissolution properties of drug–polymer ICs. We successfully prepared ICs formed from CBZ with hydrophilic poly(ethylene glycol) (PEG) and hydrophobic poly(ε-caprolactone) (PCL), respectively, and confirmed that these two drug–polymer ICs both had the same channel-type crystal structure as CBZ form II. During the dissolution test, CBZ–PEG IC showed a faster dissolution rate compared to CBZ form II under both sink and non-sink conditions. CBZ–PCL IC was confirmed to be more stable in aqueous medium, as the guest polymer PCL delayed its transformation to less-soluble crystals during dissolution.
Introduction
Most of the orally-administered drugs are in a solid formulation, and there are multiple choices for the solid state structure of the active pharmaceutical ingredient (API): crystalline or amorphous, pure API or multi-component (e.g., hydrates/solvates, salts, and cocrystals).1,2 In addition to the intrinsic properties of the API itself, its solid state structure significantly influences the drug dissolution profile in the gastrointestinal tract, and consequently the absorption and the efficacy of the drug.Conventionally, multiple-component pharmaceutical solids consist of the API and small molecules such as water/solvent in hydrates/solvates,3–5 acid/base in salts,6–8 and neutral small molecules in cocrystals,9,10 respectively. Recently, crystalline inclusion complexes (IC) between API and linear polymers have been proposed as a relatively un-explored solid form of drugs, where the crystalline structure of the drug molecules possesses continuous and void channels, and the linear polymer chains reside in such channels as guests.11 Though the drug–polymer inclusion complex may not be universally applicable, so far, several drugs have been proven to form such interesting structure, including phenobarbital,12,13 resorcinol,14 mavacoxib,15 griseofulvin,16,17 diflunisal,18–20 nevirapine,21 carbamazepine,22 and dapsone.23 More importantly, outside of the pharmaceutical field, there are many more examples of crystalline inclusion complexes between polymers and small molecules,24–26 suggesting the generality of such structures beyond the abovementioned cases.
In our previous studies, we have shown that the drug–polymer inclusion complexes formed with the hydrophilic guest polymer, poly(ethylene glycol) (PEG), dissolve faster than the API crystal,16 while those with the hydrophobic guest polymer, poly(ε-caprolactone) (PCL), dissolve more slowly.20,21 However, the comparison studies so far were made with pure API crystals whose crystalline structures were completely different from the channel-type structure of the drug–polymer inclusion complexes. Therefore, the effect of the guest polymers on the dissolution profile of drug–polymer inclusion complexes was not conclusive. Fortunately, as shown below, we found that the anticonvulsant drug carbamazepine (CBZ) not only could form inclusion complexes with both PEG and PCL (Scheme 1), but also one of its polymorphs (trigonal form II) possesses the same channel-type structure without guest molecules. Therefore, it would be very interesting to directly compare the dissolution profiles of carbamazepine form II, carbamazepine–PEG inclusion complex, and carbamazepine–PCL inclusion complex crystals.
 | ||
| Scheme 1 Chemical structures of carbamazepine, poly(ethylene glycol), and poly(ε-caprolactone). | ||
Experimental section
Materials
Carbamazepine (raw material, form III) was purchased from Adamas-beta. Poly(ethylene glycol) (PEG) of molecular weight (MW) 6k Da and poly(ε-caprolactone) (PCL) of molecular weight (MW) 10k Da were both obtained from Alfa Aesar. Ethyl acetate (EtOAc) and carbon tetrachloride (CCl4) were purchased from Beijing Chemical Works. All reagents used in this study were of analytical grade and used without further purification.Preparation of CBZ form II27
1.2 g CBZ form III were dissolved in 40 mL EtOAc at 50 °C. The obtained suspension was quickly filtered, and the filtrate was cooled in an ice-water bath for 30 minutes. The precipitated crystals were filtered, and then dried in a vacuum oven at room temperature for 24 hours. The obtained crystals were ground with an agate mortar. Its X-ray diffractogram (Fig. 1) was consistent with that of CBZ form II in the literature27 and confirmed the successful preparation.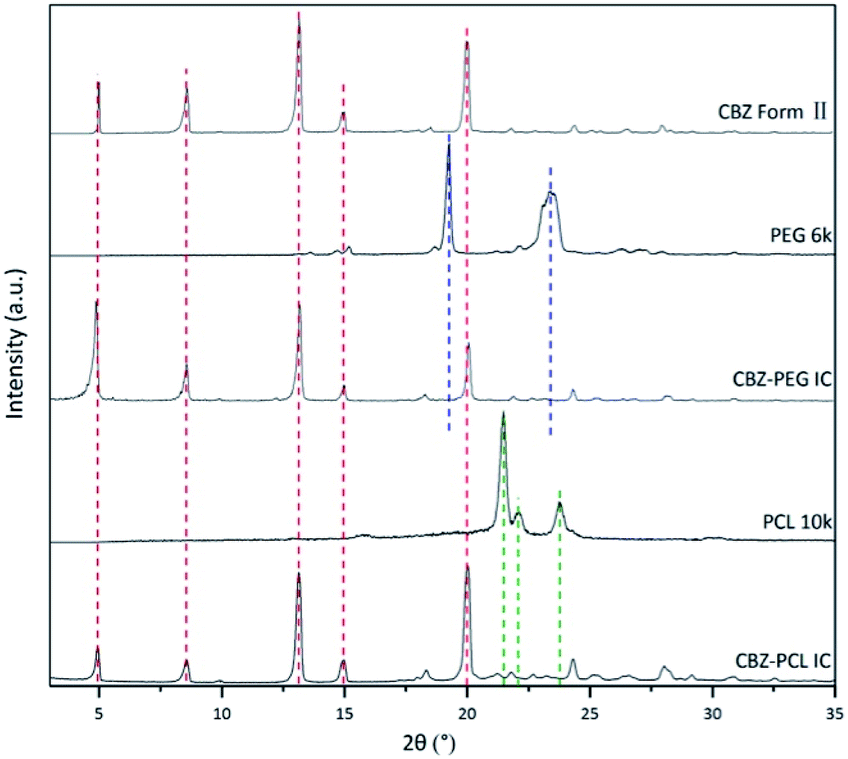 | ||
| Fig. 1 The powder X-ray diffraction patterns of CBZ form II, CBZ–PEG IC, and CBZ–PCL IC crystals. The patterns of pure PEG and PCL were included to show there were no noticeable polymer crystal residues in the two IC samples after the rinsing treatment. | ||
Preparation of the CBZ–PEG inclusion complex
CBZ/PEG 6k mixtures with mass ratios of 80/20, 70/30, 60/40 were ground in an agate mortar for 2 minutes to obtain homogeneous physical mixtures. Approximately 50 mg physical mixtures were placed between two slides and lightly pressed to spread the powder evenly. The slides with the mixture powder were placed on a hot stage, melted at 200 °C for 2 minutes, and then slowly cooled to crystallize at room temperature. To prepare pure CBZ–PEG ICs, the obtained crystals were rinsed with deionized water at 40 °C for 2 minutes to remove excess PEG.Preparation of the CBZ–PCL inclusion complex
The preparation of CBZ–PCL ICs was similar to that of CBZ–PEG ICs. The mass ratio of CBZ/PCL 10k mixtures was 80/20. After the mixture was melted and co-crystallized, the excess PCL was washed several times by CCl4 at 40 °C.Powder X-ray diffraction (PXRD)
PXRD was used to confirm the crystal structures of ICs. PXRD tests were carried out at room temperature. The samples were scanned by Cu Kα ray using D/max-2500 diffractometer from Rigaku, Japan, with a ray wavelength of 1.54184 Å, a scan range of 2θ = 3–35°, and a scan speed of 8° 2θ/min. The step size was 0.02°/2θ. Powder samples were spread over the sample stage to form a complete plane before testing. Data analysis was conducted by Jade software.Thermogravimetric analysis (TGA)
TGA was used to check whether solvates were produced during crystal preparation. TGA tests were performed using a Shimadzu DTG-60 apparatus. Powder samples (approximately 5 mg) were added into aluminium pans, and heated within the temperature range of 25–500 °C (the heating rate was 10 °C min−1). And N2 purge with the flow rate of 50 mL min−1 was maintained in the sample chamber.Differential scanning calorimetry (DSC)
To analyze the stability of ICs, DSC tests were performed using a DSC-60 calorimeter from Shimadzu Corporation, Japan. Approximately 3–5 mg powder samples were added into an aluminium pan and analyzed from 25 to 250 °C with a heating rate of 10 °C min−1. A similar empty pan was used as the reference sample. And N2 purge with the flow rate of 50 mL min−1 was maintained in the calorimeter.Hot stage polarized optical microscope (HS-POM)
Thermodynamic stability analysis of ICs was also conducted by HS-POM. The apparatus was the BX41P model of Olympus Corporation, Japan, coupled with Linkam's LTS420 hot stage for temperature control, and Motic's Moticam Pro 282A camera for image acquisition. A small amount of sample was placed between two slides, and the crystal morphology at different temperatures was observed. The temperature range was 30–250 °C.Nuclear magnetic resonance (1H-NMR)
Solution 1H-NMR was conducted by a ECS-400 NMR spectrometer from JEOL, Japan. Samples were dissolved in deuterated chloroform (CDCl3) before the test. And the spectrum analysis was performed using MestReNova software. The composition of ICs was calculated by comparing the integral area of different compounds.Particle dissolution experiment (under sink and non-sink conditions)
The particle dissolution experiment was conducted in the phosphate buffer (pH 7.4). All the samples used in the test were ground by an agate mortar and sieved to select powders with the particle size of 150–180 μm. For sink condition, the CBZ form II, CBZ–PEG IC and CBZ–PCL IC samples containing equivalent 12.0 mg CBZ were dissolved in 500 mL buffer, respectively. And a small amount of Span 20 (10 μg mL−1, less than the CMC, which is 21 μg mL−1 (ref. 28)) was added to improve the wettability of crystals in aqueous condition. During the test, the stirring rate was 400 rpm. At pre-set time intervals, 3 mL of the suspension was removed and 3 mL buffer was replenished. The removed suspension was filtered through a polyethersulfone membrane (pore size 0.22 μm), and the first 1 mL of the filtrate was discarded. The UV-vis absorption spectra of the remaining filtrate were collected at an optical length of 1 cm. The absorbance at the wavelength of 285 nm was used to determine the amount of dissolved CBZ.For non-sink condition, the CBZ form II, CBZ–PEG IC and CBZ–PCL IC samples containing equivalent 40 mg CBZ were dissolved in 200 mL buffer, respectively. The filtrate collected at pre-set time intervals was diluted 10 times before the UV-vis analysis.
Results and discussion
Confirmation of the CBZ–PEG IC and CBZ–PCL IC structures
With many polymorphs, solvates, and cocrystals, carbamazepine is a popular model drug in the pharmaceutical solid field. Its form II crystal structure possesses continuous and void channels, which are known to accommodate many small molecular solvents and even long aliphatic chain (i.e., tridecane).29–31 In our previous study, we showed that the linear polymer, poly(tetrahydrofuran) (PTHF), could also occupy these channels as guest and form a crystalline inclusion complex.22 The CBZ framework structures of form II and drug–polymer inclusion complex crystals are the same with each other, as evidenced by their X-ray diffraction patterns. Unlike the other drugs in our previous studies on drug–polymer inclusion complexes, the channel-type structure of CBZ form II is apparently stable even without guest occupation, thus providing the rare opportunity to directly study the guest effect. In the current study, we used two pharmaceutically relevant polymers, PEG and PCL, as the guest polymer in the inclusion complex. In addition, PEG is a hydrophilic, water-soluble polymer, while PCL is a hydrophobic, water-insoluble but degradable polymer.CBZ form II, CBZ–PEG IC, and CBZ–PCL IC crystals were successfully prepared. Their X-ray diffraction patterns (Fig. 1) were similar to each other and all consistent with that of the form II crystal in the literature.27 As in other drug–polymer inclusion complex and channel-type solvate crystals,12–23 the guest molecules were in the disordered state and did not contribute to diffraction peaks in diffractograms. The two inclusion complexes were crystallized in the presence of excess polymers, which were removed by rinsing the samples with suitable solvents (water for the PEG and CCl4 for the PCL systems, respectively). There were no noticeable XRD peaks of the corresponding polymer crystals in the patterns of the CBZ–PEG and the CBZ–PCL ICs, suggesting that the removal of excess polymers was successful.
The thermo-gravimetric analysis results of the CBZ form II sample (Fig. 2) showed no noticeable mass loss below 200 °C, indicating that the solvent used in its preparation, ethyl acetate, was not included in the channels. Above 320 °C, when the CBZ molecules were completely degraded, there were about 7 wt% mass remained in the CBZ–polymer inclusion complex samples, supposedly the PEG and PCL components in the complex.18,21 The existence of polymers in the two inclusion complexes was further evidenced by the solution NMR characterizations (Fig. 3). Calculated from the area ratios of the corresponding CBZ and polymer NMR peaks, the weight percentage of PEG and PCL was around 5% and 9% in the CBZ–PEG and the CBZ–PCL ICs, which were close to the estimation from the TGA results and to the channel void volume percentage of the CBZ form II crystals.29
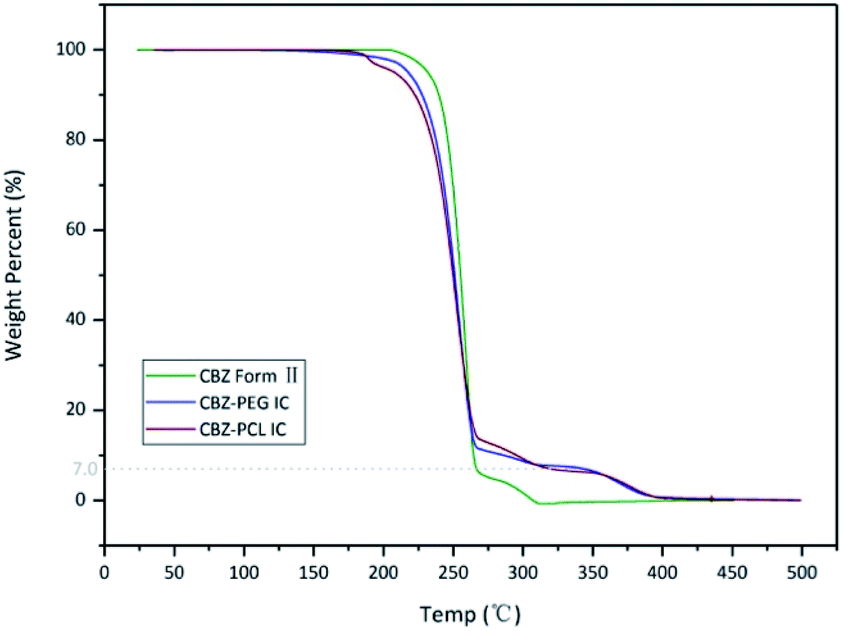 | ||
| Fig. 2 TGA thermograms of CBZ form II, CBZ–PEG IC and CBZ–PCL IC samples. | ||
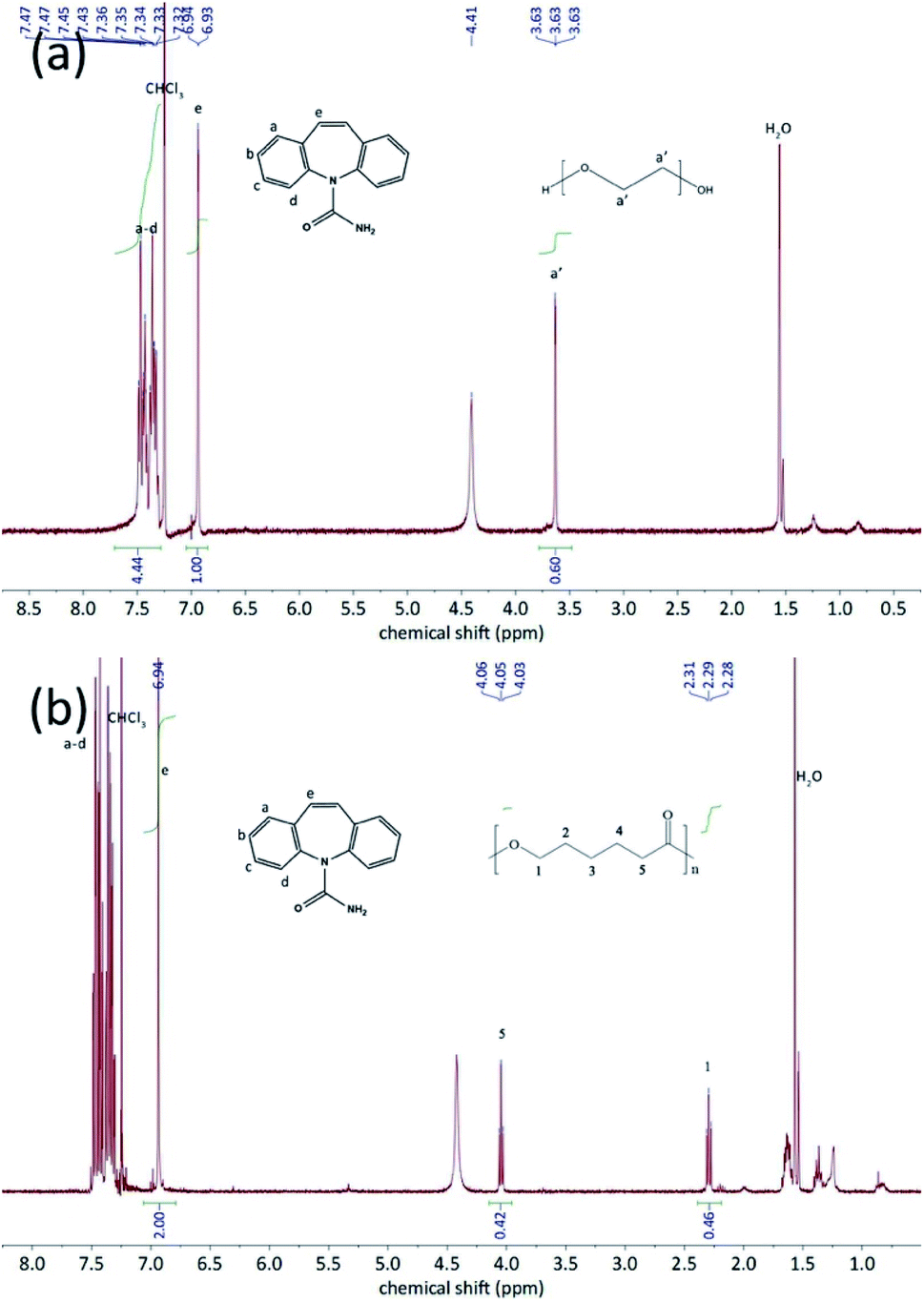 | ||
| Fig. 3 1H-NMR spectra of (a) CBZ–PEG IC an (b) CBZ–PCL IC in CDCl3. The peaks were correspondingly assigned to CBZ and polymers. | ||
The DSC characterization further proved that the polymers in the inclusion complex samples were indeed exist in the complex form and not in a separate phase (Fig. 4). Similar to our previous studies on drug–polymer inclusion complexes, the first heating scan did not show thermal transition signals corresponding to the polymer crystal melting, which would be around 50–60 °C for both PEG and PCL, again supporting that there was no separate polymer phase in these samples. Further heating in the DSC thermograms showed endothermic transition, around 150 and 170 °C for the CBZ–PEG and the CBZ–PCL ICs, respectively. Observed under polarized optical microscope, formation of amorphous droplets and crystallization of new forms of crystals appeared at these thermal transition temperatures, corresponding the dissociation of the original inclusion complexes, the formation of polymer melt droplets, and the re-crystallization of the pure CBZ crystals.17,18 After one heating–cooling cycle, the second heating of the samples did show the melting signals of the polymer crystals around 50 °C (Fig. 4), which formed during the cooling steps from the previous cycle. The absence and appearance of the polymer crystal melting in the first and the second heating scans, respectively, were consistent with our previous studies, and proved the polymers were originally in the inclusion complex state.
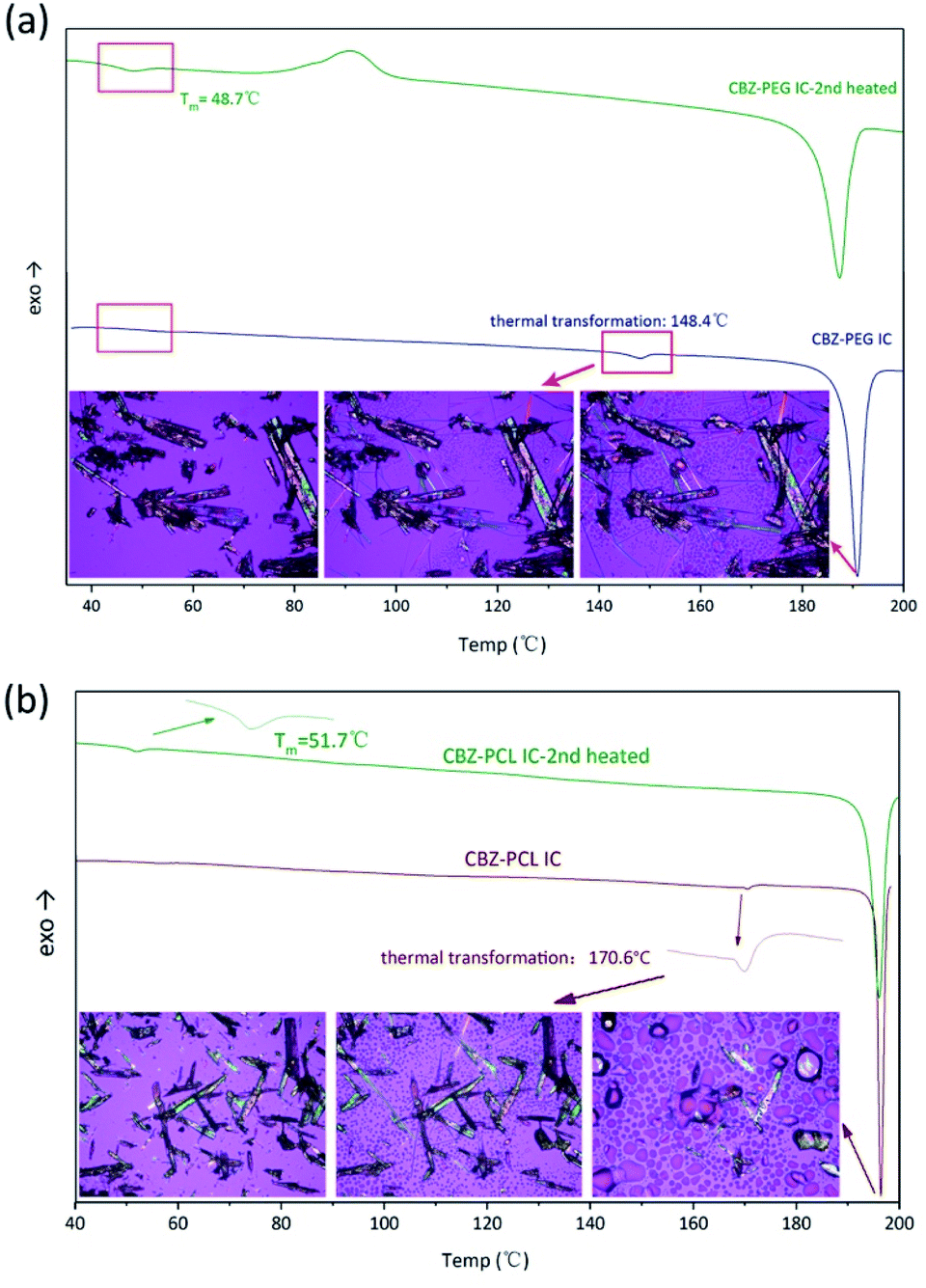 | ||
| Fig. 4 DSC heating scans and in situ HS-POM observation of (a) CBZ–PEG IC; (b) CBZ–PCL IC. | ||
Dissolution profiles
The dissolution profiles of CBZ form II, CBZ–PEG IC, and CBZ–PCL IC particles (all in the size range of 150–180 μm) were directly compared, under both sink and non-sink conditions (Fig. 5a and b). In the sink condition, the dissolution of CBZ–PEG IC was significantly faster than that of CBZ form II, while CBZ–PCL IC and CBZ form II behaved similarly to each other. Unlike our previously studied systems (e.g., griseofulvin,16 diflunisal,20 and nevirapine21), these three crystals all had the same CBZ framework structures, and the only difference was the absence/presence and the nature of the guest polymers. Compared with CBZ, PCL is similarly hydrophobic while PEG is much more hydrophilic and water soluble. Therefore, the hydrophilic guest polymer accelerated the dissolution rate of the drug crystals, probably because PEG improved wetting of the particle surface and/or the dissolved PEG near the particle surface increased the local drug solubility.32–34 In contrast, the insoluble PCL guest polymer did not have such advantages.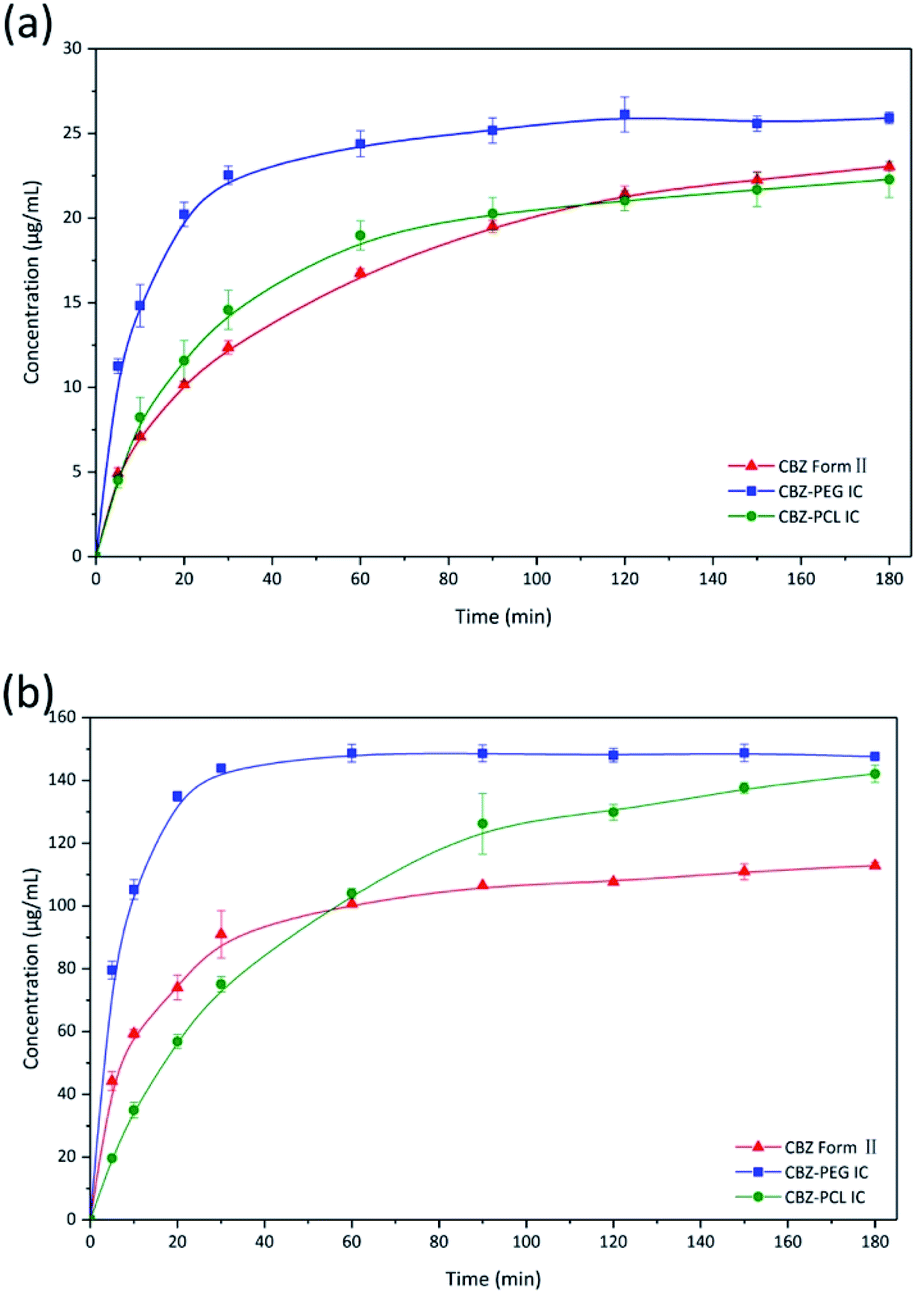 | ||
| Fig. 5 Dissolution profiles of CBZ form II, CBZ–PEG IC, and CBZ–PCL IC particles under (a) sink and (b) non-sink conditions. | ||
In the non-sink condition, CBZ–PEG IC again dissolved much faster and reached a higher drug concentration plateau at the end of the 3 hour measurement than CBZ form II (148 vs. 113 μg mL−1). The dissolution profile of CBZ–PCL IC was more complicated: at first its dissolution was a little bit slower than CBZ form II, but the dissolved drug concentration kept increasing, surpassed that of the CBZ form II, and reached almost the same value of CBZ–PEG IC at the end of the measurement. It is known in the literature that various CBZ crystals in the aqueous medium would transform into its dihydrate crystal form with the lowest equilibrium solubility.35 Therefore, we collected the solid residues from the non-sink dissolution tests for powder XRD characterizations (Fig. 6). Indeed, both CBZ form II and CBZ–PEG IC completely converted to CBZ dihydrate crystals after 8 hours in water. However, though the newly-formed CBZ dihydrate structure was obviously observed, CBZ–PCL IC crystals still retained significant amount of its original crystal structure. Since CBZ dihydrate is the most stable crystal form of CBZ with the lowest solubility in the aqueous condition, transformation to the CBZ dihydrate would decrease the apparent dissolution rates of CBZ polymorphs. Compared with CBZ form II, the transformation of CBZ–PCL IC to CBZ dihydrate was delayed and showed apparently faster dissolution after 60 minutes (Fig. 5).
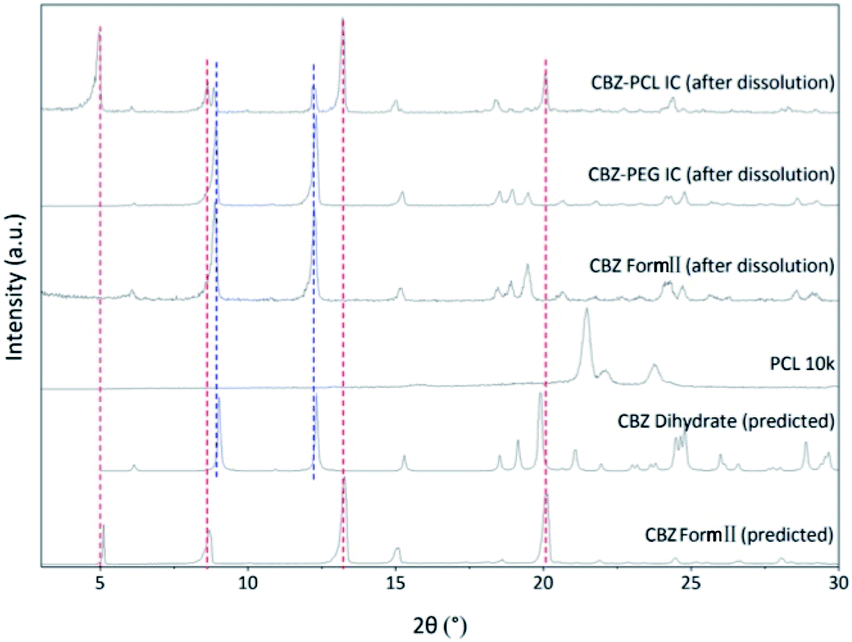 | ||
| Fig. 6 The powder X-ray diffraction patterns of solid residues from the non-sink dissolution tests after 8 h. | ||
The behavior of the insoluble guest polymer PCL during the IC dissolution deserved more discussion. In our previous studies on diflunisal–PCL and nevirapine–PCL ICs, we proposed the following mechanism: as the drug molecules dissolved away from the IC crystals, the insoluble PCL polymer chains, which initially resided in separate channels, coalesced together, crystallized, and formed porous shell structures; in addition, during the dissolution process, the particles retained its original size and shape.20,21
The morphology of the residue particles was observed under SEM (Fig. 7), and porous structure was observed, which was similar to other drug–PCL IC systems. However, when the solid residue of CBZ–PCL IC from the non-sink dissolution study was characterized with powder XRD (Fig. 6), we noticed that no characteristic diffraction peaks of PCL crystals were observed, i.e., the remained PCL apparently did not crystallize (different from other drug–PCL IC systems). Interestingly, after annealed at 40 °C for 60 minutes, the same PCL residual particles showed clear PCL crystal diffraction peaks (Fig. 8). Therefore, after the dissolution of CBZ from the CBZ–PCL IC particles at the room temperature, the remained PCL formed a porous structure, whose crystallinity was much lower than those of the diflunisal–PCL and nevirapine–PCL IC systems. Though the detailed mechanism of guest polymer crystallization in these polymer inclusion complex systems is not clear, one possible contribution to the difference may be that the PCL content in CBZ–PCL IC was lower than the other two systems (9 wt% vs. 10 wt% and 31 wt%).
 | ||
| Fig. 7 SEM images of the surface morphology of CBZ–PCL IC particles as prepared and dissolved under non-sink conditions after 4 h. Scale bars: 1 μm. | ||
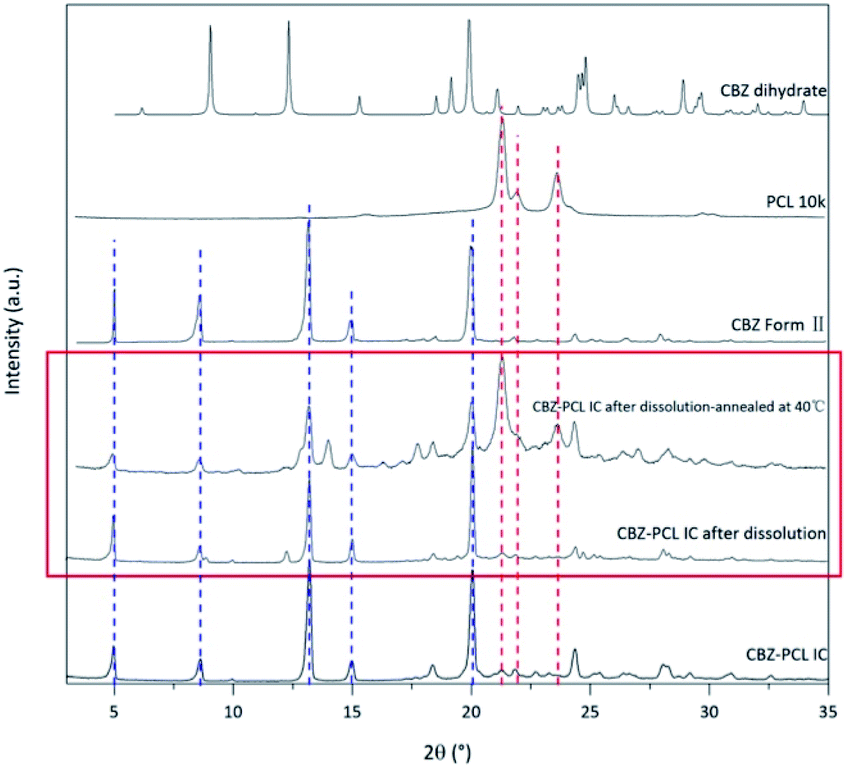 | ||
| Fig. 8 The powder X-ray diffraction patterns of residual CBZ–PCL IC from the 4 h non-sink dissolution study. | ||
Conclusion
Drug–polymer inclusion complex crystal is an interesting but less-studied category of pharmaceutical solids. Though not all drugs can form such complex with polymers, there have been enough proved examples so far and it is reasonable to expect more drug–polymer ICs will be identified in the future. In addition, the guest polymers such as PEG and PCL are widely used in pharmaceutical products, which ensures that drug–polymer ICs can be practically useful in the real world (i.e., not only for academic interests). Therefore, it is important to study the structure–property relationship of the drug–polymer ICs, especially the effect of the guest polymers on their dissolution profile.In this study, we used carbamazepine as the model drug, and confirmed its form II, CBZ–PEG IC, and CBZ–PCL IC crystals all had the same crystal structure. CBZ form II could be regarded as an IC with no guest in the channel voids, while in the other two crystals, the guest polymers were the water-soluble PEG and water-insoluble PCL, respectively. Therefore, for the first time, we were able to directly compare these three systems and study the effect of the guest polymers on the dissolution profile of drug–polymer ICs.
CBZ–PEG ICs were found to dissolve faster than CBZ form II both in the sink and non-sink conditions, suggesting drug–PEG ICs could be used to improve the dissolution rate of insoluble drugs. In contrast, ICs with the hydrophobic and insoluble PCL as the guest polymer initially dissolved at a similar rate to CBZ form II, but CBZ–PCL ICs seemed more stable against crystal transformation to less-soluble crystal forms (i.e., CBZ dihydrate) in the aqueous solution.
As the drug molecules dissolve away from the drug–PCL IC crystals, the insoluble PCL polymer chains coalesced and formed a porous shell structure at the particle surface. As PCL is a biodegradable and safe pharmaceutical excipient, it would be interesting to study how to control the porosity and thickness (even the crystallinity as in the current study) of the PCL shell structure, and potentially used the drug–PCL IC particles as a new controlled released device.
Author contributions
Yanbin Huang contributed to the conceptualization and Lu Chen contributed to the investigation of the current study. Both authors wrote the original manuscript and revised it together.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Natural Science Foundation of China (Project No. 21434008). We are also grateful to Professor Feng Qian, Professor Jun Xu, Dr Chengyu Liu, Dr Xiaotong Yang and Dr Zhi Zhong for the experimental help.References
- L. S. Taylor and F. W. Langkilde, J. Pharm. Sci., 2000, 89, 1342–1353 CrossRef CAS PubMed.
- N. K. Duggirala, M. L. Perry, O. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640–655 RSC.
- R. K. Khankari, D. Law and D. J. W. Grant, Int. J. Pharm., 1992, 82, 117–127 CrossRef CAS.
- A. Nangia and G. R. Desiraju, Chem. Commun., 1999, 605–606 RSC.
- E. Grothe, H. Meekes, E. Vlieg, J. H. Ter Horst and R. de Gelder, Cryst. Growth Des., 2016, 16, 3237–3243 CrossRef CAS.
- A. T. Serajuddin, Adv. Drug Delivery Rev., 2007, 59, 603–616 CrossRef CAS PubMed.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharm., 2007, 4, 323–338 CrossRef CAS PubMed.
- A. Avdeef, Adv. Drug Delivery Rev., 2007, 59, 568–590 CrossRef CAS PubMed.
- C. C. Sun, Expert Opin. Drug Delivery, 2013, 10, 201–213 CrossRef CAS PubMed.
- J. W. Steed, Trends Pharmacol. Sci., 2013, 34, 185–193 CrossRef CAS PubMed.
- X. Yang, Z. Zhong, J. Xu and Y. Huang, Chin. Chem. Lett., 2017, 28, 2099–2104 CrossRef CAS.
- T. Higuchi and J. L. Lach, J. Am. Pharm. Assoc., Sci. Ed., 1954, 43, 465–470 CrossRef CAS PubMed.
- Y. Nakai, K. Yamamoto, K. Terada and K. Ozawa, Yakugaku Zasshi, 1981, 101, 1016–1022 CrossRef CAS PubMed.
- J. J. Point and C. Coutelier, J. Polym. Sci., Polym. Phys. Ed., 1985, 23, 231–239 CrossRef CAS.
- C. C. Sun, PCT Pat. Appl., 2006/024930 A1, 2006 Search PubMed.
- X. Yang, Z. Zhong and Y. Huang, Int. J. Pharm., 2016, 508, 51–60 CrossRef CAS PubMed.
- Z. Zhong, C. Guo, L. Chen, J. Xu and Y. Huang, Chem. Commun., 2014, 50, 6375–6378 RSC.
- Z. Zhong, C. Guo, X. Yang, B. Guo, J. Xu and Y. Huang, Cryst. Growth Des., 2016, 16, 1181–1186 CrossRef CAS.
- Z. Zhong, X. Yang, X.-B. Fu, Y.-F. Yao, B.-H. Guo, Y. Huang and J. Xu, Chin. Chem. Lett., 2017, 28, 1268–1275 CrossRef CAS.
- Z. Zhong, X. Yang, B.-h. Guo, J. Xu and Y. Huang, Cryst. Growth Des., 2016, 17, 355–362 CrossRef.
- X. Yang, B. Yu, Z. Zhong, B. H. Guo and Y. Huang, Int. J. Pharm., 2018, 543, 121–129 CrossRef CAS PubMed.
- Z. Zhong, X. Yang, B.-H. Wang, Y.-F. Yao, B. Guo, L. Yu, Y. Huang and J. Xu, CrystEngComm, 2019, 21, 2164–2173 RSC.
- P. Chappa, A. Maruthapillai, R. Voguri, A. Dey, S. Ghosal and M. A. Basha, Cryst. Growth Des., 2018, 18, 7590–7598 CrossRef CAS.
- J. Lu, P. A. Mirau and A. E. Tonelli, Prog. Polym. Sci., 2002, 27, 357–401 CrossRef CAS.
- A. E. Tonelli, Polymer, 1994, 35, 573–579 CrossRef CAS.
- A. E. Tonelli, Polym. Int., 1997, 43, 295–309 CrossRef CAS.
- A. L. Grzesiak, M. D. Lang, K. Kim and A. J. Matzger, J. Pharm. Sci., 2003, 92, 2260–2271 CrossRef CAS PubMed.
- C. Kim and Y. L. Hsieh, Colloids Surf., A, 2001, 187, 385–397 CrossRef.
- A. J. C. Cabeza, G. M. Day, W. D. S. Motherwell and W. Jones, Chem. Commun., 2007, 9, 1600–1602 RSC.
- F. P. A. Fabbiani, L. T. Byrne, J. J. McKinnon and M. A. Spackman, CrystEngComm, 2007, 9, 728–731 RSC.
- R. K. Harris, P. Y. Ghi, H. Puschmann, D. C. Apperley, U. J. Griesser, R. B. Hammond, C. Y. Ma, K. J. Roberts, G. J. Pearce, J. R. Yates and C. J. Pickard, Org. Process Res. Dev., 2005, 9, 902–910 CrossRef CAS.
- D. H. Doshi, W. R. Ravis and G. V. Betageri, Drug Dev. Ind. Pharm., 2008, 23, 1167–1176 CrossRef.
- D. P. Medarevic, K. Kachrimanis, M. Mitric, J. Djuris, Z. Djuric and S. Ibric, Pharm. Dev. Technol., 2016, 21, 268–276 CrossRef CAS PubMed.
- M. Moneghini, I. Kikic, D. Voinovich, B. Perissutti and J. Filipovic-Grcic, Int. J. Pharm., 2001, 222, 129–138 CrossRef CAS PubMed.
- J. Deng, S. Staufenbiel and R. Bodmeier, Eur. J. Pharm. Sci., 2017, 105, 64–70 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |