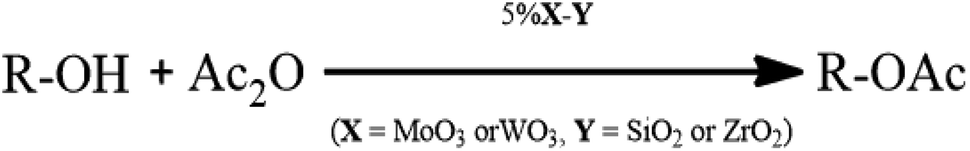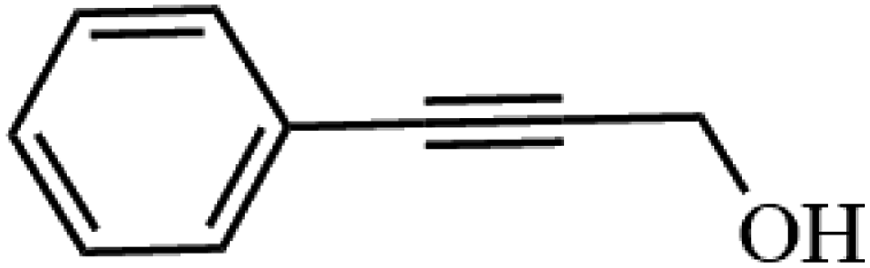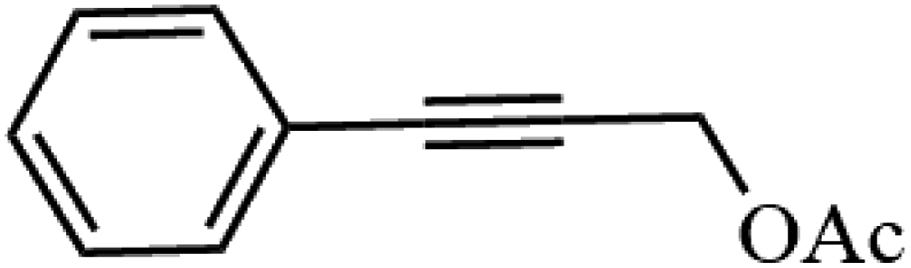DOI:
10.1039/D1RA02134F
(Paper)
RSC Adv., 2021,
11, 16468-16477
Molybdenum-modified mesoporous SiO2 as an efficient Lewis acid catalyst for the acetylation of alcohols†
Received
17th March 2021
, Accepted 26th April 2021
First published on 5th May 2021
Abstract
A suitable, expeditious and well-organized approach for the acetylation of alcohols with acetic anhydride in the presence of 5%MoO3–SiO2 as an optimum environmentally benign heterogeneous catalyst was developed. The high surface area obtained for 5%MoO3–SiO2, 101 m2 g−1 compared to other catalysts, 22, 23, and 44 m2 g−1 for 5%WO3–ZrO2, 5%WO3–SiO2, and 5%MoO3–ZrO2, respectively, appears to be the driving force for better catalytic activity. Amongst the two dopants used, molybdenum oxide is the better dopant compared to its tungsten oxide counterpart. High yields of up to 86% were obtained with MoO3 doping while WO3 containing catalysts did not show any activity. Other reaction parameters such as reactor stirring speed, and solvent variation were studied and revealed that the optimum stirring speed is 400 rpm and cyclohexane is the best solvent. Thus, the utilization of affordable and nontoxic materials, short reaction times, reusability, and producibility of excellent yields of the desired products are the advantages of this procedure.
1. Introduction
The acetylation of hydroxyl groups is a fundamental process that is frequently utilized in the transformation of organic compounds as it provides an alternative route that is affordable and efficient in organic synthesis.1,2 In organic synthesis, the protection of functional groups is essential.3 Thus, through the various transformations of functional groups, the protection of such functional groups that results in the acetylated species is highly important.4 The obtained acetylated species can often be utilized in a range of ways such as through protection of functional groups in multistep synthetic processes.5,6 Generally, the acetylated species are obtained from the most frequently utilized reagent combination which uses an acid anhydride in the presence of an acid7–15 or base.16–18 Furthermore, the acetylated groups are also more frequently found in pharmaceuticals, solvents, cosmetics, fragrances and foodstuffs as well as in plasticizers.19,20
Numerous methods for the acetylation reactions have been previously reported.21–23 However, countless of these reported methods suffer from several limitations such as drastic reaction conditions, long reaction time, formation of toxic or undesirable byproducts, use of expensive reagents, utilization of catalysts which are moisture sensitive or toxic, tedious workup procedures and poor yields of the desired products.5,21–25 Therefore, the developments of new catalysts and mild methods for the acetylation of alcohols are still in great demands. The transformation of various chemicals under heterogeneous catalysis has gained a great deal of interest over the past years due to the following findings; the efficiency of chemical transformations is greatly improved, improvement of the desired pure products, easier work-up, and usage of less toxic and affordable catalysts.26,27
In the recent years, there were some developments made to instigate solid reagents or solid reagents supports for uncomplicated work-ups which comprise silica gel supported TaCl5,28 NaHSO4,29 KSF,30 Sc(III) or La(III) triflates.31 Nonetheless, several of these catalysts contained some disadvantages like the production of unwanted byproducts, expeditious catalyst deactivation, and severe reaction conditions.32 There are still some serious concerns with regards to green chemistry, hence numerous efforts of replacing ordinary catalysts with new solid acids that contain few advantages such as easily separable after the reaction, reusability, and presenting minimal toxicity are applied worldwide.6,32 Thus, extensive exploitation of mesoporous metal oxides could be a breakthrough for the development of environmentally benign solid Lewis acid catalysts.33
More recently, investigations revealed that molybdenum or tungsten doped zirconium is an alternative efficient catalyst essential for reactions requiring vigorous acid site.34,35 Bearing this in mind, the development of well-organized effective green catalysts is still part of ongoing work.36,37 Herein, we wish to report the use of 5%MoO3–SiO2 as an efficient solid acid catalyst for the acetylation of alcohols (Scheme 1) under mild reaction conditions.
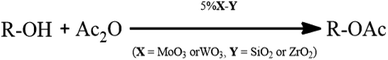 |
| | Scheme 1 Catalytic acetylation of alcohols with acetic anhydride. | |
2. Experimental
2.1. Materials
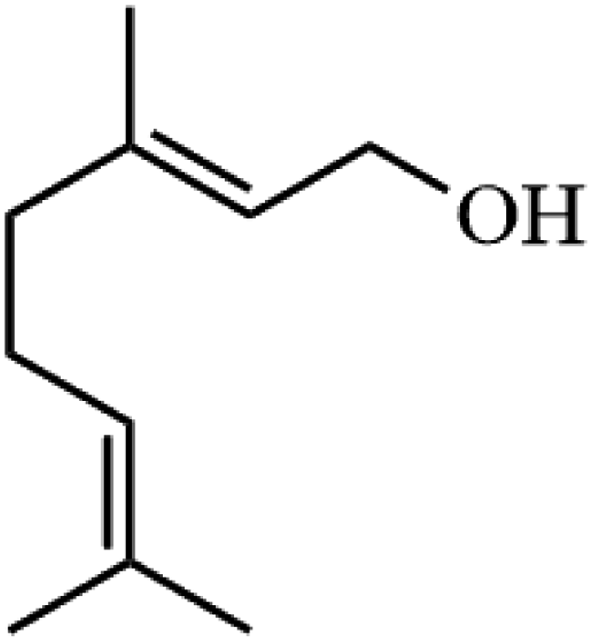
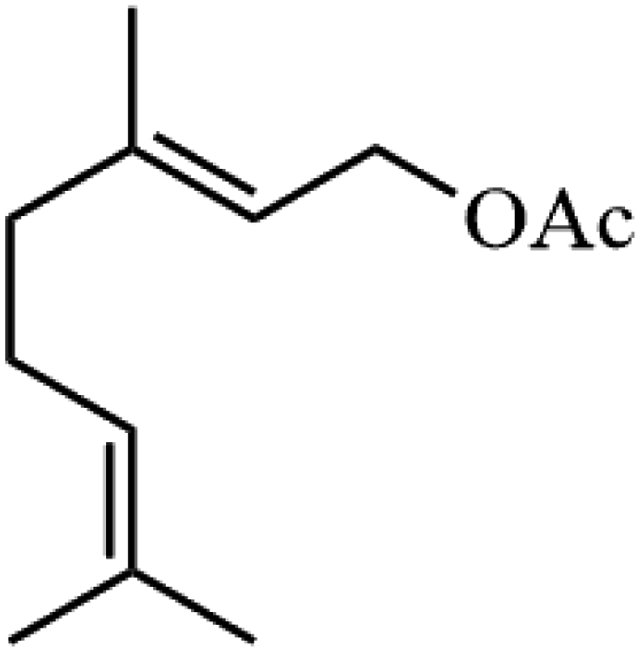
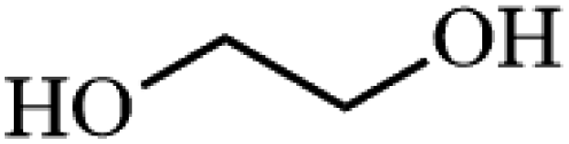
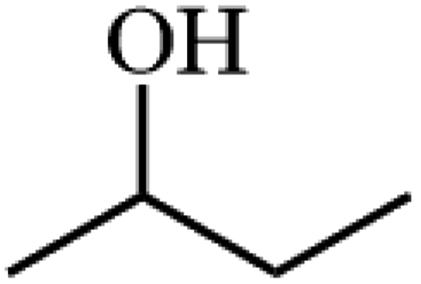
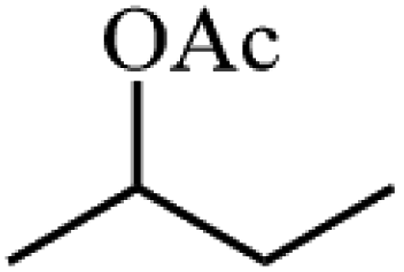
Ammonium metatungstate hydrate (H26N6O40W12·xH2O) (≥99.0%), ammonium molybdate tetrahydrate (H24Mo7N6O24·4H2O) (81.0–83.0%), tetraethyl orthosilicate (C8H20O4Si) (≥99.0%), zirconium(IV) oxynitrate hydrate (N2O7Zr·xH2O) (99%), decane (C10H22) (≥99%), dichloromethane (CH2Cl2) (≥99.8%), nitric acid (HNO3) (70%), geraniol (C10H18O) (98%), ethylene glycol (C2H6O2) (99.8%), 2-phenylethanol (C8H10O) (≥99.0%), 3-phenyl-2-propyn-1-ol (C9H8O) (96%), and poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol), nonyl acetate (C11H22O2) (≥97.0%), pentyl acetate (CH3COO(CH2)4CH3) (99.0%), 2-phenethyl acetate (CH3COOCH2CH2C6H5) (≥99.0%), benzyl acetate (CH3COOCH2C6H5) (≥99.0%), hexyl acetate (CH3COO(CH2)5CH3) (99.0%), sec-butyl acetate (CH3CO2CH(CH3)C2H5) (99.0%), tert-butyl acetate (CH3COOC(CH3)3) (≥99.0%), and geranyl acetate (CH3CO2CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)CH2CH2CHC(CH3)2) (≥97.0%) were all purchased from Sigma Aldrich. Toluene (C6H5CH3) (99.8%) and cyclohexane (C10H12) (98%) were purchased from Rochelle Chemicals. Acetic anhydride [(CH3CO)2O] (97%) was purchased from UniLAB. All chemicals were used as received.
C(CH3)CH2CH2CHC(CH3)2) (≥97.0%) were all purchased from Sigma Aldrich. Toluene (C6H5CH3) (99.8%) and cyclohexane (C10H12) (98%) were purchased from Rochelle Chemicals. Acetic anhydride [(CH3CO)2O] (97%) was purchased from UniLAB. All chemicals were used as received.
2.2. Catalyst synthesis
The solid acid catalysts were synthesized by the sol–gel method as described by Poyraz et al.,38 following the approach of Corma.39 430 μmol of P123 and 0.02 mol of each metal precursor were added into two separate 300 mL beaker, and then 0.19 mol of n-butanol and 0.04 mol of HNO3 were added to the mixtures. The mixtures were stirred at 25 °C until transparent gels were acquired. Two other mixtures of 0.001 mol of ammonium molybdate and ammonium metatungstate were separately prepared in 300 mL beakers. Thus, each salt was added inside a stirring beaker which contained 2.5 g of Milli-Q water plus 2.5 g of ethanol. As soon as the salt solution became clear, it was transferred into the acquired clear gel under vigorous stirring, and the mixture was further stirred vigorously for 20 minutes. The amounts added of both molybdenum and tungsten were 5 mol% (molX/molY × 100, where X = MoO3 or WO3 and Y = ZrO2 or SiO2). The obtained mixture was placed inside an oven set at 120 °C for six hours and immediately calcined for 1 hour at 600 °C under air. The generated brittle materials were crushed and marked 5%X–Y.
2.3. Instrumentation
Prior to nitrogen sorption experiments (BET analysis) on Micromeritics ASAP 2460 surface area and porosity analyzer at −196 °C, the acquired powder samples were degassed under nitrogen gas for 8 hours at 90 °C and further degassed under vacuum at 250 °C for another 8 hours. A Rigaku MiniFlex-600 diffractometer with Cu Kα radiation (λ = 1.5406 Å) was utilized at room temperature for carrying out analysis of wide and low-angle powder X-ray diffraction (p-XRD). SDT Q600 thermogravimetric analyzer (TGA) was utilized to scrutinize the thermal behavior of our prepared catalysts under nitrogen flow, with a range of 10 °C min−1 from 30 to 1000 °C. Samples were firstly carbon-coated using an Agar Turbo Carbon Coater and then analyzed Tescan Vega 3LMH Scanning Electron Microscopy (SEM) for imaging, morphology and elemental mapping. JEOL JEM-2100F electron microscope consisting of 200 kV accelerating voltage was used to capture Transmission Electron Microscopy (TEM) images of our synthesized catalysts for the estimation of the particle sizes. The acid sites of the as synthesized catalysts were verified by temperature programed desorption method (TPD) using a Chemisorption Analyzer Micromeritics AutoChem II 2920. The Carousel 12 plus reaction station from Radleys TECH was utilized for monitoring the stirring speed and the temperature of the reactions. For qualitative analysis, Shimadzu GC-MS equipped with the mass selective detector and the capillary column was utilized for the qualitative analysis. Shimadzu GC-2010 equipped with Restek-800-356-1688 capillary column (30 m × 0.25 mm × 0.25 μm) and flame ionization detector (FID) was utilized for quantitative analysis. Temperatures of 250 °C and 300 °C were utilized for FID and injection port respectively. For the separation process during catalyst recycling, a NEYA 8 benchtop centrifuge was successfully utilized.
2.4. Catalytic evaluation
To a solution of alcohol (2 mmol) and acetic anhydride (3 mmol), 5%X–Y (X = MoO3 or WO3, Y = ZrO2 or SiO2) was added. The reaction solution was stirred (400 rpm) at 35 °C for 30 minutes and consecutively sampled after every 5 minutes. Thus, the reaction progress was monitored by GC. The controlled experiments revealed that no reaction took place in the absence of the catalyst and the tungsten promoter was inactive in this reaction. The results showed that the reaction is favorable towards MoO3 as the catalyst promoter. 5%MoO3–SiO2 was revealed to be an ideally suited solid acid catalyst for the acetylation of alcohols. For catalyst recyclability, the optimum catalyst (30 mg) was transferred into a Falcon tube (500 mL) and thoroughly washed three times with cyclohexane (15 mL). After each wash, the solution was centrifuged at 4500 rpm for 15 minutes.
3. Results and discussions
3.1. Synthesis and characterization
Thermally stable solid Lewis acid catalysts were synthesized by the sol–gel method described by Poyraz et al.38 Due to the presence of nitrate ions, color changes such as molybdenum solution turning dark yellow and tungsten solution becoming opaque were observed. This is in agreement with the report in the literature.40 The acquired parameters such as surface areas, pore diameters, pore volumes, acidic sites, and crystallite sizes are summarized in Table 1. Nitrogen sorption analysis confirmed that the prepared materials were within mesoporous materials range of 2–50 nm. Mesoporous metal oxides have remarkable features like high surface areas, easily tunable, chemical and thermal stability. Generally, materials with higher surface areas turn to be more active and become favorable for most reactions.41,42 Looking at 5%MoO3–Y catalysts on Table 1, it was observed that SiO2 gave higher surface area (100.8 m2 g−1) compared to ZrO2 (44.1 m2 g−1). There is no clear correlation between the surface area these materials possess and the acidic sites on their surface.
Table 1 BET, XRD and TPD characterization of the solid Lewis acid catalysts
| Catalyst |
ABET (m2 g−1) |
Pv (cm3 g−1) |
PD (nm) |
Crystallite size (nm) |
Acidic sites (a.u.) |
| 5%MoO3–SiO2 |
101 |
0.29 |
11.6 |
104.9 |
0.05 |
| 5%MoO3–ZrO2 |
44 |
0.13 |
11.7 |
14.5 |
0.03 |
| 5%WO3–SiO2 |
23 |
0.02 |
3.7 |
8.1 |
0.04 |
| 5%WO3–ZrO2 |
22 |
0.07 |
12.8 |
37.7 |
0.06 |
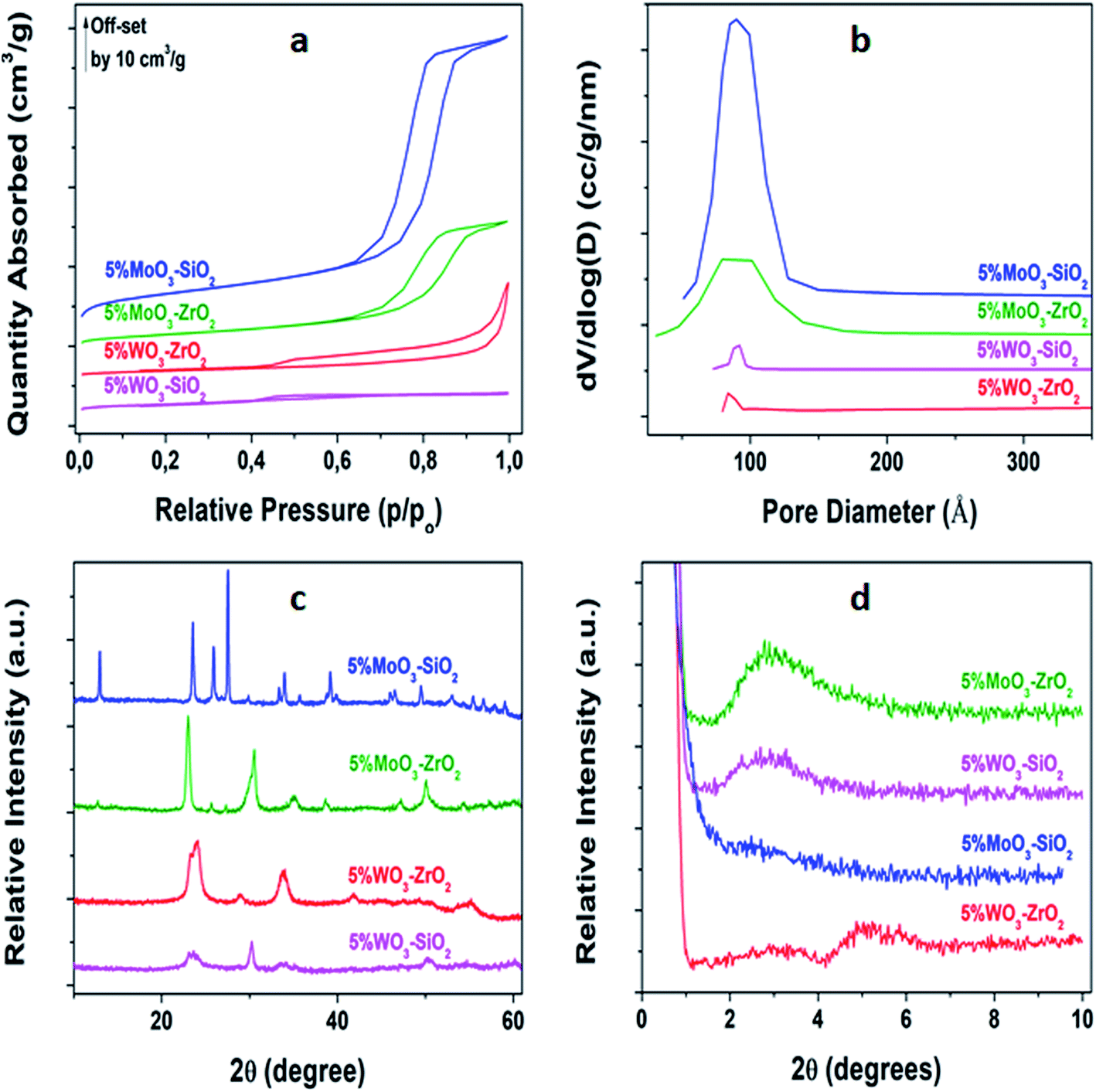
As shown in Fig. 1(a), nitrogen sorption of the synthesized solid Lewis acid catalysts aged at 600 °C conveyed type IV isotherms with hysteresis loop associated with capillary condensation taking place in the mesopores, confirming the mesoporosity of the as-synthesized materials.43,44 Fig. 1(b) depicts the corresponding pore size distributions, acquired from utilizing BJH calculation procedure. It can be observed that the MoO3-based catalysts have wide pore size distribution, while the WO3-based catalysts possess narrow pore size distribution of uniform size. X-ray diffraction patterns of the synthesized solid Lewis acid catalysts calcined at 600 °C are displayed in Fig. 1(c). Moreover, for MoO3 doping shaper peaks were observed indicating non-crystalline characteristic of 5%MoO–SiO2 and 5%MoO3–ZrO2 catalysts. However, the WO3 promoter revealed broad peaks because of the amorphous nature of 5%WO3–SiO2 and 5%WO3–ZrO2 catalysts. As shown in Fig. 1(d), all the synthesized mesoporous metal oxides catalysts except 5%MoO3–SiO2 exhibited diffraction peaks at low angles. The 5%MoO3–SiO2 depicted broad peak-like characteristic which was unclear due to low intensity indicating that the obtained pores did not indicate the regular mesostructured.45
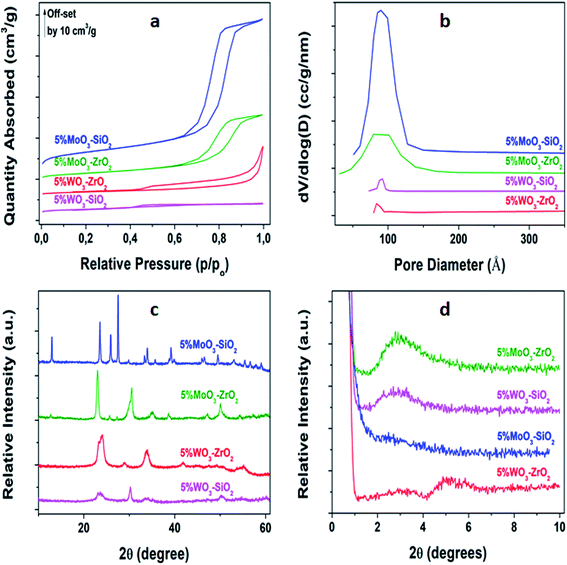 |
| | Fig. 1 Characterization of the synthesized MMO's (a) BET N2-sorption isotherms, (b) BJH desorption pore volume distribution, (c) wide-angle p-XRD, and (d) low-angle p-XRD. | |
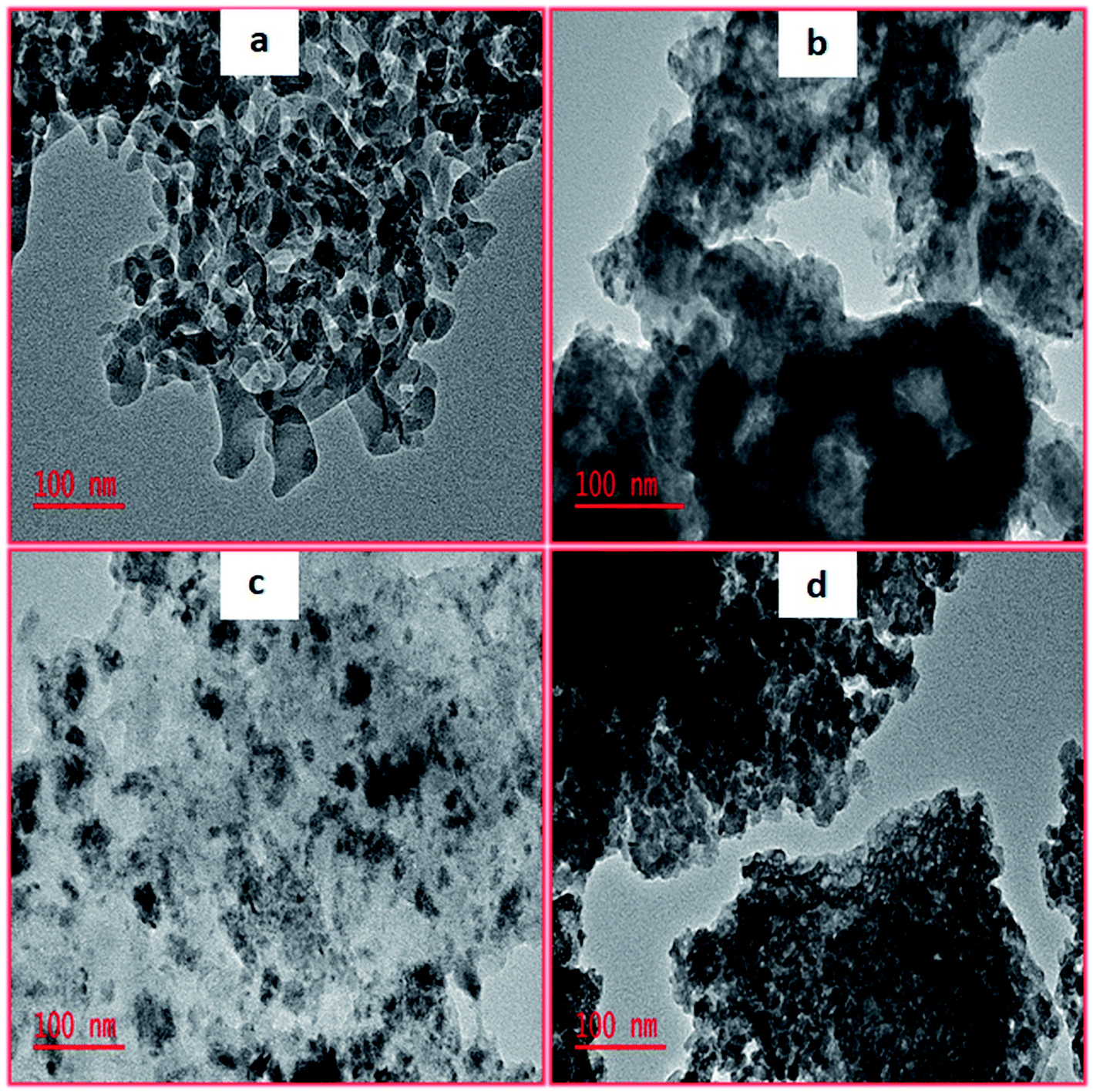
As illustrated in Fig. 2, TEM images provided some insights into mesopore structural features. These acquired images illustrated the arrangement of crystallite which clearly indicated that pores were present. Thus, it was observed that upon using molybdenum as a promoter intensified the thermal stability and also enhanced the surface areas as shown in Table 1. However, utilizing tungsten as a promoter resulted in the amorphous materials which had dark spots that were previously ascribed to tungsten clusters from the literature.46,47 Fig. S1† shows the representative SEM images of the calcined mesoporous metal oxides. Moreover, a closer inspection of the synthesized materials revealed that they contained uneven surfaces with countless pores irregularly shaped. Thus, the EDX analysis conducted (Fig. S2†) confirmed that no impurities were present in our synthesized mesoporous metal oxides catalysts.
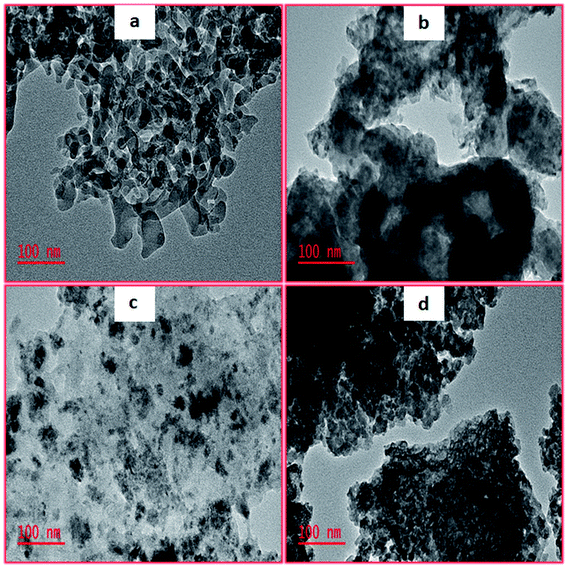 |
| | Fig. 2 TEM images of (a) 5%MoO3–SiO2, (b) 5%WO3–ZrO2, (c) 5%WO3–SiO2 and (d) 5%MoO3–ZrO2. | |
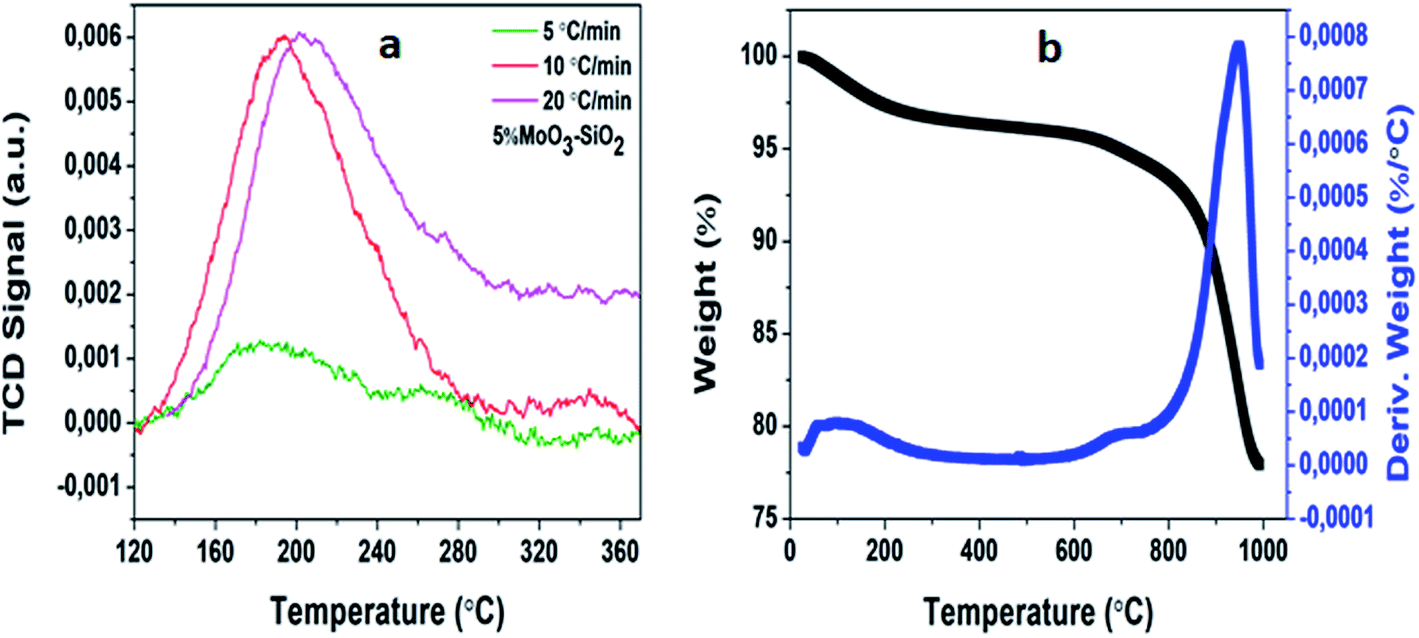
In most cases, the acidity of mesoporous metal oxides plays an essential role in numerous reactions. Consequently, the acidity of mesoporous metal oxides needs to be investigated. The properties of solid acids also consist of strength, type, and a number of acid sites. Thus, NH3-TPD is an ideal method which is utilized extensively for determining the active site on a catalyst.48,49 Fig. 3(a) depicts peak areas of 5%MoO3–SiO2. It was clearly observed that elevating the heating rate from 5 to 20 °C min−1 resulted in the increment of the peak areas, where the peak area is equivalent to the active site. For all the synthesized catalysts it was observed that the heating rates were directly proportional to the peak areas (see Fig. S3†). Thus, TPR experiments were conducted to acquire a deep understanding of the synthesized mesoporous metal oxides.
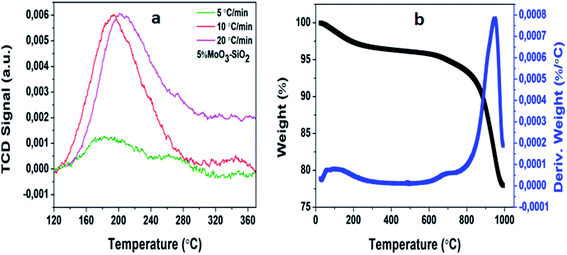 |
| | Fig. 3 The depiction of 5%MoO3–SiO2 via (a) NH3-TPD and (b) thermogravimetric analysis and its derivative. | |
Fig. S4† shows the acquired profiles which exhibit multiple reduction peaks. Looking at molybdenum promoter, it was previously reported that it had two peaks at 767 and 997 °C, respectively.49 Furthermore, it was observed that upon promoting silica and zirconium, the reduction peaks migrated to lower temperatures, demonstrating excessive reducibility.50 Generally, tungsten undergoes three reduction steps as previously reported.51 The obtained results of 5%WO3–Y only revealed two reduction peaks shifted to lower temperatures instead of three possible peaks which might be due to the limitation of temperature range on the instrument used. Fig. 3(b) depicts the thermogravimetric analysis curve of 5%MoO3–SiO2 and its derivation curve. For this optimum catalyst, it was observed that three decomposition steps took place. The dehydroxylation52 from 50 up to 73 °C, followed by the weight loss at 734 °C, and the third decomposition at 948 °C corresponding to the detachment of oxygen molecules.53 The total weight loss observed for this specific catalyst was 23 wt%. The rest of the synthesized solid Lewis acid catalysts are shown in Fig. S5† with 5%MoO3–ZrO2 exhibiting the average weight loss of 2.75 wt%, 5%WO3–SiO2 revealing the average weight loss of 24 wt%, and lastly, 5%WO3–ZrO2 depicting the average weight loss of about 18.5 wt%.
3.2. Optimization of the catalytic variables
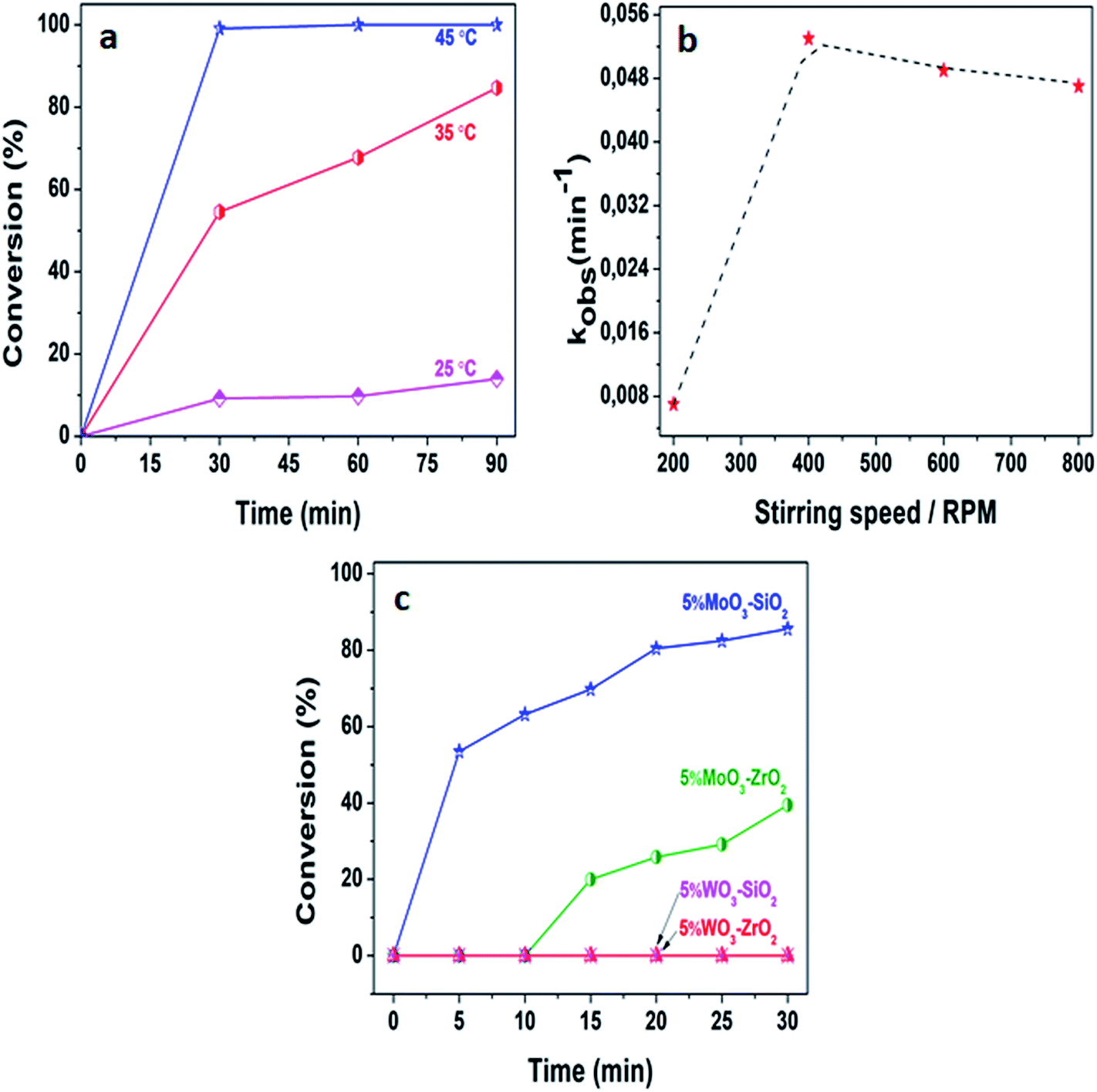
Generally, the exploitation of mesoporous metal oxides is considered as one of the promising developments of the desired solid Lewis acid catalysts which are environmentally friendly and suitable for a variety of sustainable chemistry.33 Herein, we delineate a mild, convenient and efficient approach for the acetylation of various alcohols that is typically performed utilizing acetic anhydride in the presence of solid Lewis acid catalysts. To maximize the desired yields, the reaction conditions were firstly optimized. We investigated the capability of 5%MoO3–SiO2 in the acetylation of 2-phenylethanol, in which an insignificant activity was acquired at room temperature, whereby toluene was utilized as a solvent. Thus, certain parameters such as temperatures, stirring speeds, solvents, substrates, and catalyst amount were intensely investigated in order to enhance the activity and the producibility of the desired products. Fig. 4(a) demonstrate the impact of distinct temperatures on the acetylation of 2-phenylethanol. It was clearly illustrated that the reaction temperature is directly proportional to the activity. Thus, after establishing the optimum temperature of 35 °C, the stirring speed was also investigated as illustrated in Fig. 4(b). It was clearly observed that the diffusion zone whereby the reaction was independent of the stirring speed appeared after 400 rpm, hence we deduced that the optimum stirring speed for further investigation was 400 rpm. Fig. 4(c) depicts the activity of different catalysts with 5%MoO3–SiO2 being the most favourable catalyst for the acetylation of 2-phenylethanol. This is a result of the high surface area this catalyst possess coupled with the fairly acceptable acidity, see Table 1. No activity was observed upon using 5%WO3–Y catalysts, indicating that the tungsten promoter was inactive towards this specific reaction. The reaction was also performed with similar reaction parameters to investigate the formation of the product in the absence of the catalyst and it was observed that no traces of the product were acquired from the GC analysis. Furthermore, we compared these obtained results with literature as noted in Table 2. It follows that most reported catalysts such as fluorous distannoxane56 required longer reaction time for the acetylation of 2-phenylethanol and gave lower yield. Thus, 5%MoO3–SiO2 afforded great catalytic improvement on the acetylation of alcohols under mild reaction conditions, hence it was considered as an efficient Lewis acid catalyst. Interestingly, 86% of 2-phenylethanol was converted within 30 minutes under mild reaction condition.
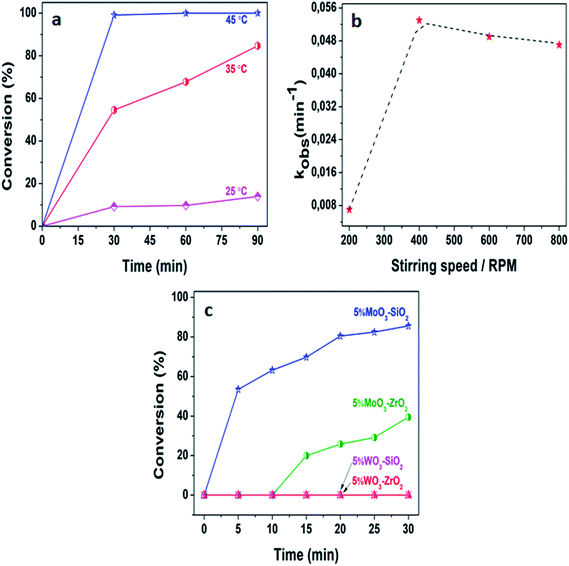 |
| | Fig. 4 (a) The effect of temperature upon the acetylation of 2-phenylethanol, (b) the influence of stirring speed at 35 °C, and (c) performance of different catalysts at 35 °C and 400 rpm. The reaction conditions were: acetic anhydride = 3 mmol, toluene = 6 mL, decane = 0.132 mmol, 2-phenylethanol = 2 mmol, and catalyst amount = 25 mg. | |
Table 2 Comparison of the effectiveness of 5%MoO3–SiO2 with other catalysts for the acetylation of 2-phenylethanola
| Catalyst |
Temp. (°C) |
Time (min) |
Yield (%) |
Solvent |
Ref. |
| TW: this work. |
| 5%MoO3–SiO2 |
35 |
30 |
86 |
Toluene |
TW |
| 5%MoO3–ZrO2 |
35 |
30 |
40 |
Toluene |
TW |
| 5%WO3–SiO2 |
35 |
30 |
— |
Toluene |
TW |
| 5%WO3–ZrO2 |
35 |
30 |
— |
Toluene |
TW |
| Catalyst-free |
35 |
30 |
— |
Toluene |
TW |
| H14[NaP5W30O110] |
25 |
90 |
95 |
Solvent-free |
54 |
| Cp2ZrCl2 |
25 |
840 |
90 |
Solvent-free |
55 |
| Fluorous distannoxane |
25 |
360 |
72 |
Toluene |
56 |
| 1,3-Dibromo-5,5-dimethylhydantoin |
25 |
1800 |
90 |
Dichloromethane |
57 |
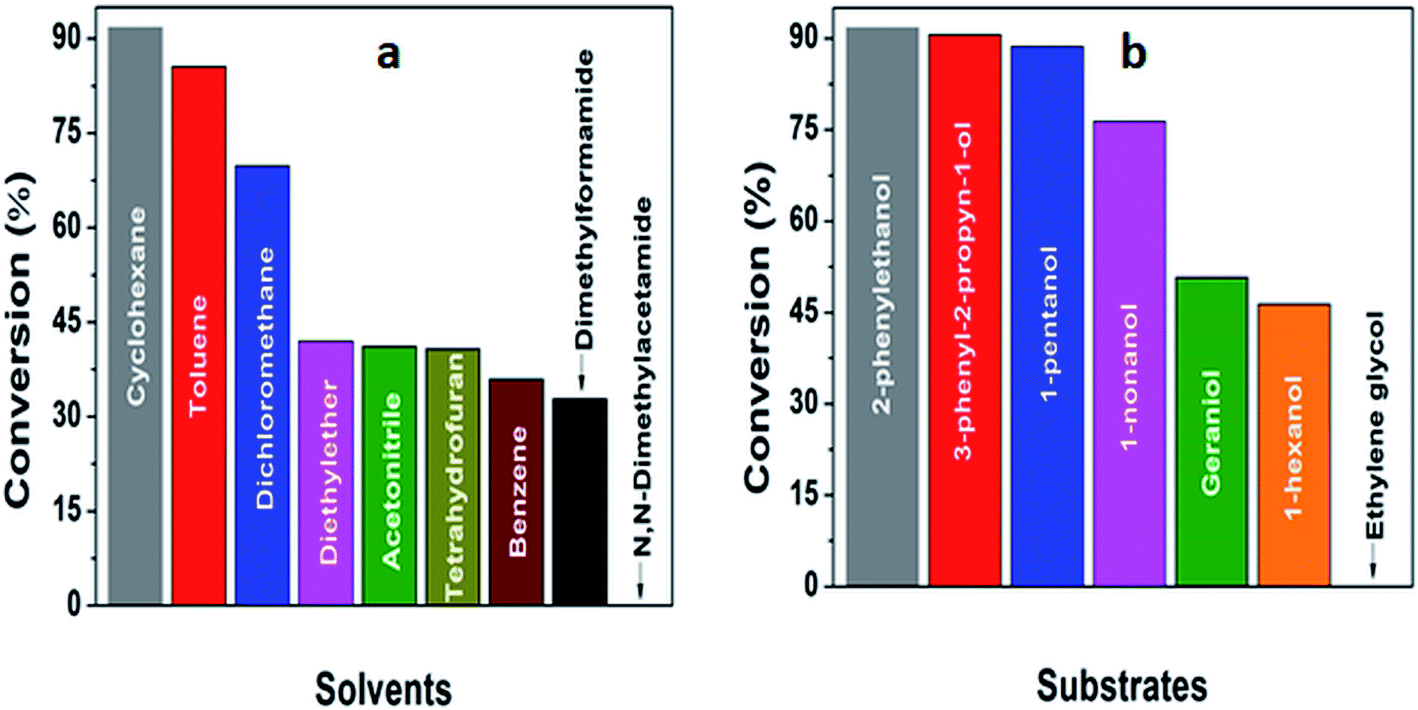
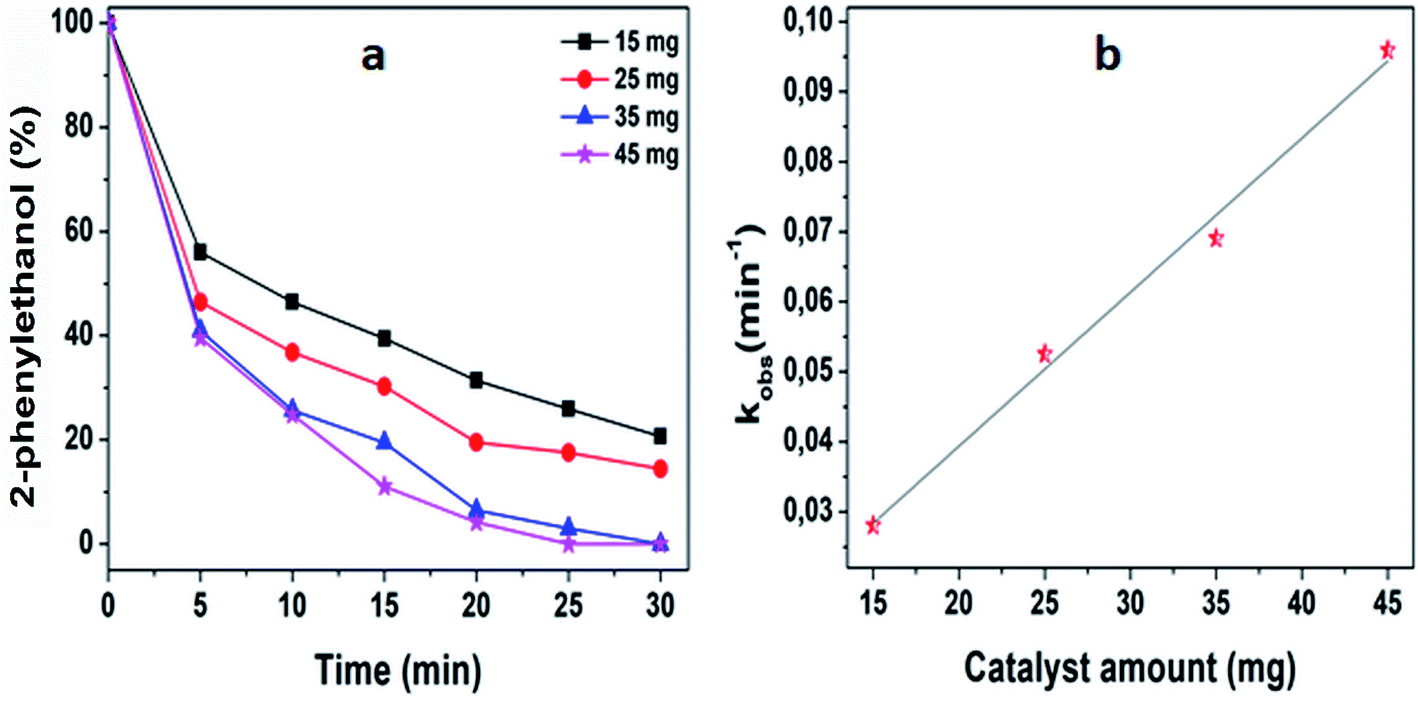
The ability of 5%MoO3–SiO2 catalyst was investigated on several solvents as illustrated in Fig. 5(a). Cyclohexane was revealed to be the best solvent, followed by toluene and dichloromethane, while the use of N,N-dimethylacetamide as solvent did not display any conversion. Thus, the choice of an appropriate solvent is crucial for the efficiency of the study reaction. Consequently, cyclohexane which afforded the yield of 91.8% was deduced as the optimum solvent for further investigation. Later, a variety of substrates was investigated under the same reaction conditions using cyclohexane as solvent. It was observed that the best yield (91.8%) was obtained when 2-phenylethanol was used as substrate, followed by 3-phenyl-2-propyn-1-ol with a yield of 90.6% (see Fig. 5(b) and Table 3). Furthermore, it is noteworthy that secondary and tertiary alcohols such as 2-butanol and tert-butanol (entry 9 and 10, Table 3) can be acetylated indicating that the catalyst was not only limited to primary alcohols. Additionally, we investigated the impact of the mass transfer limitation whereby a variety of catalyst amounts were utilized as illustrated in Fig. 6(a). The data presented in Fig. 6(a), the change in concentration of the substrate with time was used to study the kinetics of the reaction. It is clear from Fig. 6(b) that the rate of conversion of the substrate increases with increase in the catalysts surface area available in the reactor. From these findings, as depicted in Fig. 6(b) it can be deduced that the mass transport limitation did not affect any conversion. The initial rate was directly proportional to the catalyst amount, and the linear increasing confirmed the absence of the mass transfer limitation.
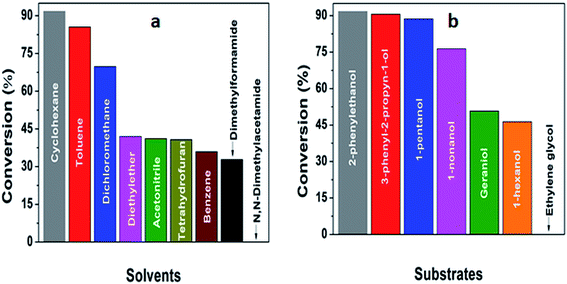 |
| | Fig. 5 (a) The investigation of an effective solvent, (b) deducing an optimum substrate for further investigation. The reaction conditions were: solvents = 6 mL, decane = 0.132 mmol, acetic anhydride = 3 mmol, substrates = 2 mmol, stirring speed = 400 rpm, and catalyst amount = 25 mg. | |
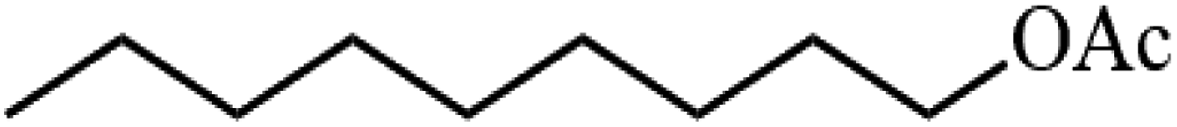
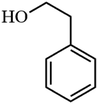
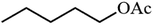
Table 3 Acetylation of selected alcohols with acetic anhydride over 5%MoO3–SiO2 under mild reaction conditions
| Entrya |
Substrate |
Productb |
Yield (%) |
| Reaction conditions: substrate = 2 mmol, Ac2O = 3 mmol, cyclohexane = 0.056 mol, 5%MoO3–SiO2 = 25 mg, temperature = 35 °C, time = 30 min. Products: they were all confirmed with GC-MS and the standards were used as well. |
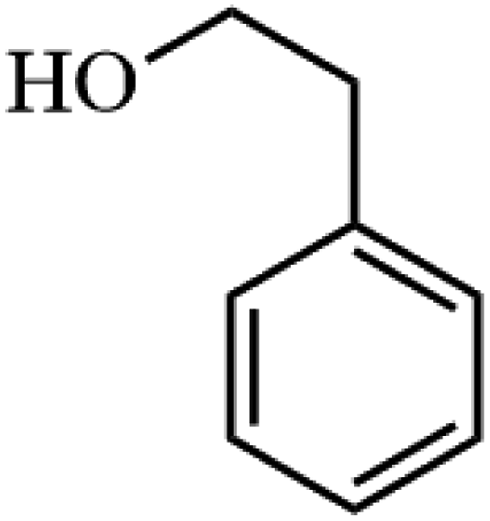
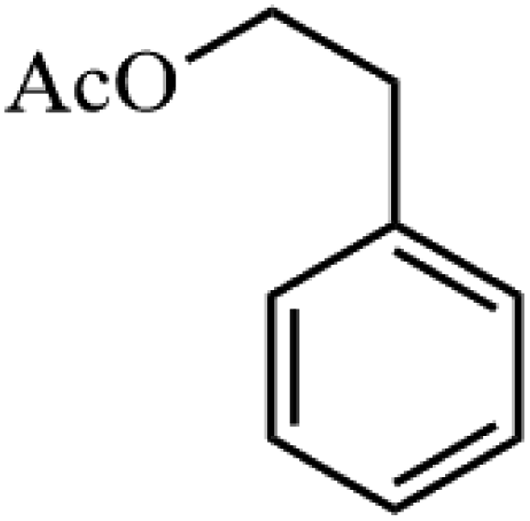
| 1 |
 |
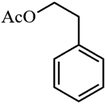 |
91.8 |
| 2 |
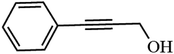 |
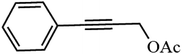 |
90.6 |
| 3 |
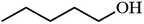 |
 |
88.6 |
| 4 |
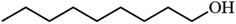 |
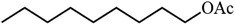 |
76.4 |
| 5 |
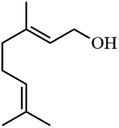 |
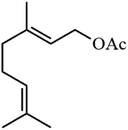 |
50.8 |
| 6 |
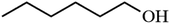 |
 |
46.4 |
| 7 |
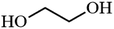 |
— |
— |
| 8 |
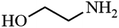 |
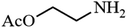 |
86.9 |
| 9 |
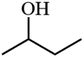 |
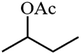 |
65.4 |
| 10 |
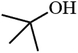 |
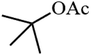 |
31.7 |
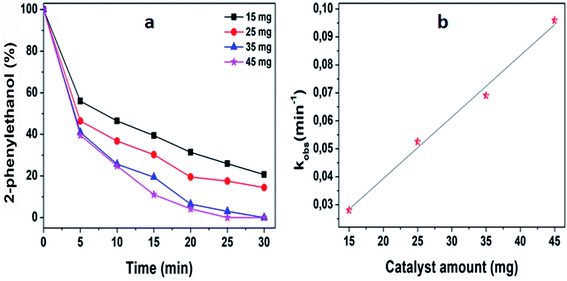 |
| | Fig. 6 (a) Various amount of optimum catalyst, (b) the study of mass transfer limits. The reaction conditions were: cyclohexane = 6 mL, decane = 0.132 mmol, acetic anhydride = 3 mmol, 2-phenylethanol = 2 mmol, stirring speed = 400 rpm, and catalyst amount = 25 mg. | |
3.3. Catalyst recyclability
Moreover, after deducing the kinetic zone, it follows that we took 30 mg of the catalyst which was within the kinetic zone for the catalyst recycling tests. Fig. 7 illustrates the reusability of 5%MoO3–SiO2 on the acetylation of 2-phenylethanol up to the fifth run from Fig. 7(a), it can be observed that the catalyst was recyclable but the activity decreases as the number of reaction cycles increases. This is due to some loss of the catalyst during the separation process between each recycling test. However, to confirm the stability of the catalyst, the conversion was normalized by the amount of catalyst used in each reaction cycle, and it was observed that the catalyst was very stable with only minor loss in activity (Fig. 7(c)). TEM analysis was performed after the 5th catalytic cycle and it was observed that the mesostructure of the catalyst was not affected as illustrated in Fig. 7(b). The stability of the catalysts could be due to the stable structure of the as-synthesized materials, and this feature could make them good catalysts for industrial application.
 |
| | Fig. 7 (a) The catalytic recycle test of the optimum catalyst, (b) TEM image and (c) the normalized by mass conversion. Reaction conditions: 6 mL of cyclohexane, temperature = 35 °C, decane = 0.132 mmol, acetic anhydride = 3 mmol, 2-phenylethanol = 2 mmol, stirring speed = 400 rpm. | |
4. Conclusion
In conclusion, highly stable meso-structured solid acid nanocatalysts such as 5%MoO3–SiO2 can be synthesized using a sol–gel method. We have demonstrated that 5%MoO3–SiO2 is a flexible and very effective inexpensive heterogeneous catalyst for the acetylation of alcohols with acetic anhydride. The results show that the as-synthesized catalyst was highly selective towards the desired products. The notable advantages of this catalyst are the producibility of excellent yields, shorter reaction times, and lower reaction temperatures. Also, the catalyst was revealed to be environmentally friendly and could be reused a number of times without loss of activity and selectivity.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We acknowledge the financial support from South African NRF (Grant specific number 117997 and 111710), Analytical division (spectrum), Sasol R&D and research fund from the University of Johannesburg. We would also like to acknowledge Mr D. Hariss and Dr R. Meyer from Shimadzu South Africa for the usage of their equipment throughout this study.
References
- T. W. Greene and P. G. M. Wuts, Protective groups in organic synthesis, Wiley, 1999 Search PubMed.
- P. J. Kocienski, Protecting groups, Thieme, Stuttgart, 1994, vol. 1 Search PubMed.
- T. W. Green and P. G. M. Wuts, Protective Groups in Organic Synthesis, New York, 1999, vol. 27, pp. 708–711 Search PubMed.
- M. Moghadam, S. Tangestaninejad, V. Mirkhani, I. Mohammadpoor-Baltork and S. A. Taghavi, J. Mol. Catal. A: Chem., 2007, 274, 217–223 CrossRef CAS.
- B. Das and P. Thirupathi, J. Mol. Catal. A: Chem., 2007, 269, 12–16 CrossRef CAS.
- L. Ma, I. Jia, X. Guo and L. Xiang, Chin. J. Catal., 2014, 35, 108–119 CrossRef CAS.
- T. Mukaiyama, I. Shiina and M. Miyashita, Chem. Lett., 1992, 21, 625–628 CrossRef.
- K. Ishihara, M. Kubota, H. Kurihara and H. Yamamoto, J. Org. Chem., 1996, 61, 4560–4567 CrossRef CAS PubMed.
- A. Orita, C. Tanahashi, A. Kakuda and J. Otera, Angew. Chem., Int. Ed., 2000, 39, 2877–2879 CrossRef CAS PubMed.
- B. Karimi and J. Maleki, J. Org. Chem., 2003, 68, 4951–4954 CrossRef CAS PubMed.
- R. Dalpozzo, A. De Nino, L. Maiuolo, A. Procopio, M. Nardi, G. Bartoli and R. Romeo, Tetrahedron Lett., 2003, 44, 5621–5624 CrossRef CAS.
- S. Velusamy, S. Borpuzari and T. Punniyamurthy, Tetrahedron, 2005, 61, 2011–2015 CrossRef CAS.
- T. S. Reddy, M. Narasimhulu, N. Suryakiran, K. C. Mahesh, K. Ashalatha and Y. Venkateswarlu, Tetrahedron Lett., 2006, 47, 6825–6829 CrossRef CAS.
- N. Ahmed and J. E. van Lier, Tetrahedron Lett., 2006, 47, 5345–5349 CrossRef CAS.
- A. Kamal, M. N. A. Khan, K. S. Reddy, Y. V. V. Srikanth and T. Krishnaji, Tetrahedron Lett., 2007, 48, 3813–3818 CrossRef CAS.
- W. Steglich and G. Höfle, Angew. Chem., Int. Ed. Engl., 1969, 8, 981 CrossRef CAS.
- E. F. V. Scriven, Chem. Soc. Rev., 1983, 12, 129–161 RSC.
- E. Vedejs and S. T. Diver, J. Am. Chem. Soc., 1993, 115, 3358–3359 CrossRef CAS.
- H.-J. Yoon, S.-M. Lee, J.-H. Kim, H.-J. Cho, J.-W. Choi, S.-H. Lee and Y.-S. Lee, Tetrahedron Lett., 2008, 49, 3165–3171 CrossRef CAS.
- S. A. Taghavi, M. Moghadam, I. Mohammadpoor-Baltork, S. Tangestaninejad, V. Mirkhani and A. R. Khosropour, Inorg. Chim. Acta, 2011, 377, 159–164 CrossRef CAS.
- A. Kamal, M. N. A. Khan, K. S. Reddy, Y. V. V. Srikanth and T. Krishnaji, Tetrahedron Lett., 2007, 48, 3813–3818 CrossRef CAS.
- V. Mirkhani, S. Tangestaninejad, M. Moghadam, B. Yadollahi and L. Alipanah, Monatsh. Chem., 2004, 135, 1257–1263 CrossRef CAS.
- F. Rajabi, Tetrahedron Lett., 2009, 50, 395–397 CrossRef CAS.
- F. Shirini, M. A. Zolfigol and M. Abedini, Monatsh. Chem., 2004, 135, 279–282 CrossRef CAS.
- J. R. Satam and R. V. Jayaram, Catal. Commun., 2008, 9, 2365–2370 CrossRef CAS.
- P. Laszlo, Acc. Chem. Res., 1986, 19, 121–127 CrossRef CAS.
- F. Bigi, S. Carloni, R. Maggi, C. Muchetti and G. Sartori, J. Org. Chem., 1997, 62, 7024–7027 CrossRef CAS.
- S. Chandrasekhar, T. Ramachander and M. Takhi, Tetrahedron Lett., 1998, 39, 3263–3266 CrossRef CAS.
- G. W. Breton, J. Org. Chem., 1997, 62, 8952–8954 CrossRef CAS.
- A. Arienti, F. Bigi, R. Maggi, E. Marzi, P. Moggi, M. Rastelli, G. Sartori and F. Tarantola, Tetrahedron, 1997, 53, 3795–3804 CrossRef CAS.
- A. G. M. Barrett and D. C. Braddock, Chem. Commun., 1997, 351–352 RSC.
- B. M. Reddy and P. M. Sreekanth, Synth. Commun., 2002, 32, 2815–2819 CrossRef CAS.
- B. Tang, W. Dai, G. Wu, N. Guan, L. Li and M. Hunger, ACS Catal., 2014, 4, 2801–2810 CrossRef CAS.
- M. Hino and K. Arata, Appl. Catal., A, 1998, 169, 151–155 CrossRef CAS.
- B. M. Reddy and V. R. Reddy, Synth. Commun., 1999, 29, 2789–2794 CrossRef CAS.
- A. R. Hajipour and G. Azizi, Green Chem., 2013, 15, 1030–1034 RSC.
- A. R. Hajipour and H. Karimi, Appl. Catal., A, 2014, 482, 99–107 CrossRef CAS.
- A. S. Poyraz, C.-H. Kuo, E. Kim, Y. Meng, M. S. Seraji and S. L. Suib, Chem. Mater., 2014, 26, 2803–2813 CrossRef CAS.
- A. Corma, A. Martinez and C. Martinez, Appl. Catal., A, 1996, 144, 249–268 CrossRef CAS.
- N. Masunga, G. S. Tito and R. Meijboom, Appl. Catal., A, 2018, 552, 154–167 CrossRef CAS.
- J. Xu, A. Zheng, J. Yang, Y. Su, J. Wang and D. Zeng, J. Phys. Chem. B, 2006, 110, 10662–10671 CrossRef CAS PubMed.
- R. G. Pearson and J. Songstad, J. Am. Chem. Soc., 1967, 39, 1827–1836 CrossRef.
- K. S. W. Sing, J. Porous Mater., 1995, 2, 5–8 CrossRef CAS.
- S. Storck, H. Bretinger and W. F. Maier, Appl. Catal., A, 1998, 174, 137–146 CrossRef CAS.
- J. R. Matos, M. Kruk, L. P. Mercuri, M. Jaroniec, L. Zhao, T. Kamiyama, O. Terasaki, T. J. Pinnavaia and Y. Liu, J. Am. Chem. Soc., 2003, 125, 821–829 CrossRef CAS PubMed.
- W. Zhou, N. Soultanidis, H. Xu, M. S. Wong, M. Neurock, C. J. Kiely and I. E. Wachs, ACS Catal., 2017, 7, 2181–2198 CrossRef CAS.
- T. Yamamoto, A. Teramachi, A. Orita, A. Kurimoto, T. Motoi and T. Tanaka, J. Phys. Chem. C, 2016, 120, 19705–19713 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS PubMed.
- L. Damjanović and A. Auroux, in Zeolite characterization and catalysis, Springer, 2009, pp. 107–167 Search PubMed.
- W. Shan, W. Shen and C. Li, Chem. Mater., 2003, 15, 4761–4767 CrossRef CAS.
- D. C. Vermaire and P. C. Van Berge, J. Catal., 1989, 116, 309–317 CrossRef CAS.
- M. J. Ndolomingo and R. Meijboom, Appl. Surf. Sci., 2016, 390, 224–235 CrossRef CAS.
- X. Shen, A. M. Morey, J. Liu, Y. Ding, J. Cai, J. Durand, Q. Wang, W. Wen, W. A. Hines, J. C. Hanson, J. Bai, A. I. Frenkel, W. Rei, M. Aindow and S. L. Suib, J. Phys. Chem. C, 2011, 115, 21610–21619 CrossRef CAS.
- M. M. Heravi, F. K. Behbahani and F. F. Bamoharram, J. Mol. Catal. A: Chem., 2006, 253, 16–19 CrossRef CAS.
- M. Lakshmi Kantam, K. Aziz and P. R. Likhar, Catal. Commun., 2006, 7, 484–487 CrossRef.
- Z. Peng, A. Orita, D. An and J. Otera, Tetrahedron Lett., 2005, 46, 3187–3189 CrossRef CAS.
- M. A. Zolfigol, A. Khazaei, A. G. Choghamarani, A. Rostami and M. Hajjami, Catal. Commun., 2006, 7, 399–402 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Scanning electron microscopy images (a) 5%WO3–ZrO2, (b) 5%MoO3–SiO2, (c) 5%MoO3–ZrO2, and (d) 5%WO3–SiO2 (Fig. S1). EDX spectrums of (a) 5%MoO3–SiO2, (b) 5%MoO3–ZrO2, (c) 5%WO3–SiO2, and (d) 5%WO3–ZrO2 (Fig. S2). NH3-TPD of (a) 5%MoO3–ZrO2, (b) 5%WO3–SiO2, and (c) 5%WO3–ZrO2 (Fig. S3). TPR of the synthesized solid Lewis acid catalysts (Fig. S4). Thermogravimetric analysis and their derivatives: (a) 5%WO3–SiO2, (b) 5%WO3–ZrO2, and (c) 5%MoO3–ZrO2 (Fig. S5). See DOI: 10.1039/d1ra02134f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Reinout Meijboom
* and
Reinout Meijboom