DOI:
10.1039/D1RA02352G
(Paper)
RSC Adv., 2021,
11, 18458-18467
Influence of cerium doping on Cu–Ni/activated carbon low-temperature CO-SCR denitration catalysts
Received
25th March 2021
, Accepted 13th May 2021
First published on 21st May 2021
Abstract
In this study, to evaluate the effects of two methods for activation of nitric acid, air thermal oxidation and Ce doping were applied to a Cu–Ni/activated carbon (AC) low-temperature CO-SCR denitration catalyst. The Cu–Ni–Ce/AC0,1 catalyst was prepared using the ultrasonic equal volume impregnation method. The physical and chemical structures of Cu–Ni–Ce/AC0,1 were studied using scanning electron microscopy, Brunauer–Emmett–Teller analysis, Fourier-transform infrared spectroscopy, X-ray diffractometry, X-ray photoelectron spectroscopy, CO-temperature programmed desorption (TPD) and NO-TPD characterisation techniques. It was found that the denitration efficiency of 6Cu–4Ni–5Ce/AC1 can reach 99.8% at a denitration temperature of 150 °C, a GHSV of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 and 5% O2. Although the specific surface area of the AC activated by nitric acid was slightly lower than that activated by air thermal oxidation, the pore structure of the AC activated by nitric acid was more developed, and the number of acidic oxygen-containing functional groups was significantly increased. Ce metal ions were inserted into the graphite microcrystalline structure of AC, splitting it into smaller graphene fragments, whereby the dispersibility of Cu and Ni was improved. In addition, many reaction units were formed on the catalyst surface, which could adsorb more CO and NO reaction gases. With the increase in Ce doping, the relative proportions of Cu2+/Cun+, Ni3+/Nin+ and surface adsorbed oxygen (Oα) in the Cu–Ni–Ce/AC0,1 catalyst increased. In addition, after the introduction of Ce into Cu–Ni/AC, the amount of weak and medium acids significantly increased. This may be due to the Ce species or its influence on the Cu/Ni species. Further, the active sites of the acid were more exposed. According to the results of the study, a composite metal oxide CO-SCR denitration mechanism is proposed. Through the oxidation–reduction reaction between the metals, the reaction gas of CO and NO is adsorbed and the incoming O2 is converted into (Oα), which promotes the conversion of NO into NO2. The CO-SCR reaction is accelerated, and the rate of low-temperature denitration was increased. Overall, the results of this study will provide theoretical support for the research and development of low-temperature denitration catalysts for sintering flue gas in iron and steel enterprises.
000 h−1 and 5% O2. Although the specific surface area of the AC activated by nitric acid was slightly lower than that activated by air thermal oxidation, the pore structure of the AC activated by nitric acid was more developed, and the number of acidic oxygen-containing functional groups was significantly increased. Ce metal ions were inserted into the graphite microcrystalline structure of AC, splitting it into smaller graphene fragments, whereby the dispersibility of Cu and Ni was improved. In addition, many reaction units were formed on the catalyst surface, which could adsorb more CO and NO reaction gases. With the increase in Ce doping, the relative proportions of Cu2+/Cun+, Ni3+/Nin+ and surface adsorbed oxygen (Oα) in the Cu–Ni–Ce/AC0,1 catalyst increased. In addition, after the introduction of Ce into Cu–Ni/AC, the amount of weak and medium acids significantly increased. This may be due to the Ce species or its influence on the Cu/Ni species. Further, the active sites of the acid were more exposed. According to the results of the study, a composite metal oxide CO-SCR denitration mechanism is proposed. Through the oxidation–reduction reaction between the metals, the reaction gas of CO and NO is adsorbed and the incoming O2 is converted into (Oα), which promotes the conversion of NO into NO2. The CO-SCR reaction is accelerated, and the rate of low-temperature denitration was increased. Overall, the results of this study will provide theoretical support for the research and development of low-temperature denitration catalysts for sintering flue gas in iron and steel enterprises.
1. Introduction
The nitrogen oxides (NOx) produced by stationary and mobile sources are among the main sources of air pollution, and they have caused many environmental problems, such as acid rain and photochemical smog.1–3 At present, the ammonia selective catalytic reduction (NH3-SCR) denitration technology is the most effective method for removing NOx.4,5 However, this technology has limitations, such as NH3 leakage, catalyst poisoning, pipeline corrosion and air preheating. Therefore, an alternative to NH3-SCR is required.6,7 The selective catalytic reduction method (CO-SCR), in which CO is used as a reducing agent, is considered to be a denitration technology that can be promoted and applied on a large scale, as it can simultaneously remove the CO and NO in industrial exhaust gases. In addition, it is believed that the development of low-temperature CO-SCR denitration catalysts will have various applications. In the process of low-temperature CO-SCR denitration catalyst carrier research, it was found that carriers affect the interactions among NO, CO and metal oxides and that they play a vital role in improving metal dispersion and enhancing catalytic performance. Activated carbon (AC) has received extensive attention in the research of low-temperature SCR denitration catalyst supports, due to its abundant oxygen-containing functional groups and huge specific surface area.8,9 The surface properties of AC have a significant impact on its catalytic performance. Further, the use of HNO3, H2SO4, H2O2, microwaves and other activation methods to treat AC can change its surface chemical properties, thereby affecting its catalytic activity.10,11 For example, Zhou et al.12 studied the influence of HNO3 activation on AC and found that a large number of ester and carbonyl types of oxygen-containing functional groups were formed on the surface of activated carbon. Li et al. used air thermal oxidation activation (973 K) to pretreat AC, and the results showed that the specific surface area and pore volume of activated AC increased.13
In recent years, activated AC-loaded precious metal nanoparticles, such as Pt, Pd, Ru and Au, have been identified as ideal catalysts for NO + CO model reactions and CO reduction.14–17 However, the high price, resource scarcity and low heat stability of precious metals limit their wide application as supported precious metal catalysts.18,19 Therefore, some alternatives to noble metal catalysts have attracted a lot of attention in the design of low-cost and high-efficiency catalysts. Among them, transition metals, such as Cu, Ni and Ce, are very attractive choices. Copper and nickel have high catalytic activity for CO-SCR reactions. The active sites activated by NO molecules may be related to the Cu species,20 and the active sites of CuO show easy redox effects with reactants.21,22 The good redox performance, sufficient surface oxygen vacancies and high oxygen storage/release capabilities of NiO have attracted special attention,23,24 and it is also widely used in CO-SCR denitration catalysts. In previous works,25,26 it was found that in the process of copper–nickel coordinated loading, in addition to affecting the selectivity and denitrification activity of NO gas, Cu can prevent the formation of carbon deposits and the sintering of Ni nanoparticles and reduce the reduction temperature of NiO. In addition, it showed excellent low-temperature denitration activity, which may be due to the synergy between bimetallic oxides. Other studies have found that the doping of cerium can promote the transfer rate of metal ions from low valence states to high valence states and also increase the relative atomic ratio of Cu2+ and Ni3+. Since the conversion between Ce4+/Ce3+ forms oxygen defects, CeO2 has significant oxygen storage capacity and excellent redox performance. By promoting the oxidation of NO to NO2, it can increase the catalytic activity of CO-SCR. Ce doping may have a positive effect on the denitration performance of the AC catalysts supported by copper and nickel. However, after Ce doping, the specific interaction denitration mechanisms between copper, nickel and cerium need to be further revealed.
In this research work, two activation methods of air thermal oxidation and nitric acid were used to activate the activated carbon support to improve the catalytic performance of CO-SCR. The Cu–Ni/AC0,1 catalyst was doped with cerium by the ultrasonic initial wet impregnation method to obtain the Cu–Ni–Ce/AC0,1 composite oxide catalyst. The doping of Ce improves the NOx conversion rate, the tolerance and thermal stability in the presence of oxygen. SEM, BET, FTIR, XRD, XPS, CO-TPD, NO-TPD characterization techniques were used to further study the effects of two activation methods and Ce doping on Cu–Ni–Ce/AC0,1. After the activation of nitric acid, although the specific surface area of the activated carbon carrier is reduced, due to the corrosion of nitric acid, more small pores are created for metal to enter, and through the oxidation of nitric acid, the surface acidic oxygen-containing functional groups increase significantly. The doping of Ce plays a variety of roles in the CO-SCR reaction, including promoting the dispersion of Cu and Ni, increasing the ratio of Cu2+ and Ni3+, increasing the adsorption of CO and NOx in the presence of oxygen and producing more surface adsorbed oxygen (Oα) content, thereby providing more active sites for the CO-SCR reaction.
2. Experiment
2.1 Catalyst preparation
The AC particles (coconut shell AC, particle size of 20–40 mesh, Henan Gongyi Lantian Water Purification Technology Co., Ltd.) were washed with deionised water to remove ash and suspended matter. Then, they were dried in a drying oven at 110 °C for 12 h to obtain the AC divided into two parts. One part was placed in a tube furnace and fired in an air atmosphere at 350 °C for 2 h to obtain AC0. The other part was soaked in an equal volume of 30% HNO3 solution, heated under reflux with magnetic stirring in a three-necked flask at 80 °C for 2 h and dried in a drying oven at 110 °C for 12 h to prepare AC1. With Cu(NO3)2·3H2O, C4H6O4Ni·4H2O and Ce(NO3)3·6H2O (AR, Tianjin Kemiou Chemical Reagent Co., Ltd.) as precursors with different molar ratios, they were mixed and dissolved in a certain amount of deionised water, and the mixed solution was immersed in AC0 under ultrasonic vibration for 160 min. Then, the sample was dried at 110 °C for 12 h and then finally calcined at 450 °C under nitrogen protection for 4 h. The resulting catalysts were marked as Cu–Ni–Ce/AC0 and Cu–Ni–Ce/AC1, where the molar ratios of the Cu–Ni oxide to Ce oxide were 10:3, 10:4 and 10:5, and the molar ratio of the Cu oxide to Ni oxide was 6:4, the mass of copper element accounts for 6% of the catalyst.
2.2 Catalytic performance
A catalyst performance test was carried out in a fixed bed reactor, and the denitrification activity was measured using 10 g Cu–Ni–Ce/AC0,1 at different temperatures. Before denitration, N2 was flushed in situ at 200 °C for 1 h. The furnace temperature was adjusted to 150 °C, the reaction temperature was determined and the simulated flue gas was passed into the reactor. The simulated flue gas (NO) flow rate was 4 mL min−1, and the CO flow rate was 40 mL min−1. O2 was 5%, N2 was the balance gas, the total gas flow rate was 1000 mL min−1 and the GHSV was 30000 h−1. The final exhaust gas was detected using a Testo-340 flue gas analyser (Germany Testo Instruments) for 60 minutes. The conversion rate of NOx was calculated using the following formula:
where, Cin(NO) is the NO concentration at the reactor inlet, ppm; Cout(NO+NO2) is the NO + NO2 concentration at the reactor outlet, ppm; and η is the denitration rate, %.
2.3 Catalyst characterisation
The surface microstructure change of Cu–Ni–Ce/AC0,1 was observed using Scanning electron microscope test (SEM, Tescan VEGAS SBH), nitrogen adsorption and a desorption tester, and the N2 adsorption isotherm was used to observe (BET, QDS-evo). The specific surface area, pore volume and average pore diameter of Cu–Ni–Ce/AC0,1 were measured at 77 K, and the micropore volume was calculated using the Dubinin–Radushkevich method. Before the N2 adsorption, each sample was degassed under vacuum at 200 °C for 4 h. The Cu–Ni–Ce/AC0,1 surface functional group changes were measured using Fourier transform infrared spectroscopy (FTIR, Nicolet iS 10) in the range of 4000–400 cm−1 internal determination. The load metal oxide was estimated using a rotating target X-Ray Diffraction (XRD, TTR18 kW Cu target) to analyse the crystal phase pattern of Cu–Ni–Ce/AC0,1, and the surface atomic state of the catalyst was analysed using X-ray Photoelectron Spectroscopy (XPS, PHI5000 Versaprobe-II) was measured using AlKα X-ray (hν = 1486.6 eV) radiation at 50 W. The binding energy (BE) was calibrated using a C1s BE value of 284.8 eV. The temperature programmed adsorption and desorption of the CO analysis (CO-TPD, Autochem II 2920) was carried out in a flow reactor equipped with a thermal conductivity detector (TCD). The TPD of CO and NO was carried out in a flow reactor equipped with a TCD. 0.1 g catalyst was heated from room temperature to 300 °C in He flow (30 mL min−1) for 60 min and then cooled to room temperature in the He atmosphere. Then, the gas was subjected to 5 vol% NO/He and 10 vol% CO/He for the NO-TPD and CO-TPD detection and it was kept for 30 min. Afterwards, the catalyst was purged by flow for 30 min to remove the physically adsorbed CO and NO. Finally, the sample heating to 800 °C (NO-TPD) and 600 °C (CO-TPD) at a rate of 10 °C min−1 was performed in He flow. TCD was used to continuously monitor the consumption of NO and CO.
3. Results and discussion
3.1 Catalyst CO-SCR denitration activity
To explore the effects of the different activation methods and different Ce doping levels on the CO-SCR denitration activity of the Cu–Ni–Ce/AC0,1 catalyst, take 10 g of catalyst to test the denitration performance of simulated sintering flue gas within 1 h. It can be seen from Fig. 1 that the activation method has a significant effect on the CO-SCR denitrification activity of Cu–Ni–Ce/AC0,1 with the same loading. When the Ce doping amount is a fixed value, the activity of the air thermal oxidation activation catalyst is significantly lower, and the denitration rate is lower than that after nitric acid activation, which is activated by air thermal oxidation, was lowest. This may be due to the activation of nitric acid to form new oxygen-containing functional groups, which promotes the adsorption and oxidation of NO.27 Also, due to the strong oxidation of nitric acid, more pores were formed for the metal ions on the surface of the AC, which is beneficial to the dispersion of the metal oxides in AC. It can be seen from Fig. 1 that under the premise of the same activation method, the low-temperature CO-SCR activity of the Cu–Ni/AC catalyst is improved by the doping of cerium. When there is no cerium doping, the lowest denitration rate of the catalyst is only 67.9%, when the cerium doping amount is 5%, the denitration rate can reach 99.8%. Because Ce doping improves the selectivity of N2 and has certain resistance in the presence of O2, compared with the previous Cu–Ni/AC, the denitration rate in the presence of oxygen is significantly improved. According to relevant literature reports,28 Ce metal ions can be inserted into the graphite microcrystalline structures of AC and then split them into smaller graphene fragments, thereby forming a large number of reaction units on the catalyst surface, so the ratio of the adsorbed oxygen (Oα) on the surface increases. Overall, the reducibility of the catalyst was enhanced, thus promoting the CO-SCR reaction.29 Therefore, under the premise of nitric acid activation, the 6Cu–4Ni–5Ce/AC1 catalyst can reach the highest denitration rate of 99.8%.
 |
| | Fig. 1 NOx conversion with different Cu–Ni–Ce/AC catalysts. | |
3.2 SEM analysis
The SEM of Fig. 2 shows the morphology of the Cu–Ni–Ce/AC0,1 catalyst. Fig. 2a, c and e shows the Cu–Ni–Ce/AC0 activated using air thermal oxidation, where Fig. 2b, d and f shows the catalyst activated using nitric acid activation. After comparing Cu–Ni–Ce/AC1, it can be seen that the nitric acid activation destroyed the physical structure of the original activated carbon, caused corrosion to it and generated new pore structures, which is important for the adsorption of reaction gases and for the effect of metal oxides. The loading process was advantageous, and it could also provide more necessary places for the denitration reaction.30 From Fig. 2c and d, it can be seen that the amount of metal oxides supported by AC1 was significantly more than that supported by AC0, and the spherical particles of the metal oxides were significantly smaller. This is because more metal ion channels are created after the activation of nitric acid. It can be seen from Fig. 2d and f that when the Ce loading was small, large spherical clusters could be observed on the surfaces of AC0 and AC1. As the Ce loading increased, the spherical metal oxide particles became even smaller. This special spherical structure change can be attributed to the mixed crystal effect.31 Due to the doping of Ce, the exerted tensile force on the grain boundaries of the copper–nickel metal oxides reduced the particle size of the spherical structure and improved its dispersibility on the AC,32 which increased the number of active sites for denitration and made the denitration effect more significant.
 |
| | Fig. 2 SEM micrographs of different catalysts: (a) 6Cu–4Ni–3Ce/AC0; (b) 6Cu–4Ni–3Ce/AC1; (c) 6Cu–4Ni–4Ce/AC0; (d) 6Cu–4Ni–4Ce/AC1; (e) 6Cu–4Ni–5Ce/AC0; (f) 6Cu–4Ni–5Ce/AC1. | |
3.3 BET analysis
The specific surface area and average pore volume of the catalysts prepared using different activation methods and the Ce doping amounts are listed in Table 1, which shows that the specific surface area of the AC after the nitric acid activation treatment was less than that after the air thermal oxidation treatment. However, it can also be seen that the average pore size of the AC after the nitric acid activation was reduced and that its average pore volume increased. This is because nitric acid activation takes away the ash in original pores while creating new pores. It is also worth noting that with the increase in Ce loading, the specific surface area of the catalyst activated using the two activation methods increased. This is because the co-doping of metal could reduce the loss of the catalyst's specific surface area during the calcination process.33 As shown in Fig. 3a, the average pore size of the catalyst materials activated using the two activation methods was mainly concentrated around 0.92 nm, indicating that the pore size distribution of all the samples was relatively uniform. Fig. 3b shows that the relative pressures of the adsorption–desorption curves of Cu–Ni–Ce/AC were between 0.35 and 0.95 and that they were very similar and can be classified as type IV isotherms with H1 type hysteresis loops,9 which shows. These catalysts have highly ordered microporous channel structures that are similar to those of the SEM results. To a certain extent, the specific surface area and pore structure of the catalyst affected its SCR denitration activity, and the larger specific surface area and abundant pore structure were beneficial for mass transfer, adsorption and activation of the reactants during the reactions.34 Therefore, increasing the contact area between reaction gases and catalyst surfaces and improving the internal pore structures of catalysts can effectively promote denitration reactions.
Table 1 Specific surface area and pore structure of Cu–Ni–Ce/AC0,1
| Cu–Ni–Ce/AC0,1 (%) |
Specific surface (m2 g−1) |
Pore volume (cm3 g−1) |
Pore size (nm) |
| 6Cu–4Ni–3Ce/AC0 |
756.406 |
0.362 |
2.231 |
| 6Cu–4Ni–4Ce/AC0 |
704.329 |
0.394 |
2.256 |
| 6Cu–4Ni–5Ce/AC |
704.329 |
0.388 |
2.218 |
| 6Cu–4Ni–3Ce/AC1 |
654.713 |
0.421 |
2.223 |
| 6Cu–4Ni–4Ce/AC1 |
681.449 |
0.389 |
2.194 |
| 6Cu–4Ni–5Ce/AC1 |
701.959 |
0.378 |
2.210 |
 |
| | Fig. 3 (a) Pore size distribution chart (b) N2 isotherm adsorption–desorption curve. | |
3.4 FTIR analysis
The chemical properties of the surface of the AC were mainly determined by the type and number of surface functional groups, and the AC surface mostly had oxygen-containing functional groups, such as carboxyl, hydroxyl, carbonyl, ketone and ether groups.35 After the AC was impregnated with a mixed solution of copper nitrate, cerium nitrate and nickel acetate, the functional groups changed during the roasting process, releasing H2O, CO, CO2 and other gases. Fig. 4 shows the FTIR spectrum after the nitric acid activation, air thermal oxidation and loading of metal oxides. According to previous studies,28,36,37 the infrared band of 3600–3200 cm−1 is assigned to the –OH stretching vibration, the infrared band of 1740–1480 cm−1 is attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or CC and the infrared band of 1300–1000 cm−1 is attributed to the stretching vibration of C–O–C or C–O. It can be seen from Fig. 4 that compared with the air thermal oxidation activation method, the AC after activation with nitric acid is 3454 cm−1 (–OH tensile vibration), 1635 cm−1 (–CO tensile vibration) and 1093 cm−1 (–CO tensile vibration) is obviously in an enhanced state, and two new absorption peaks appeared at 1700 cm−1 (–CO stretching vibration) and 1400 cm−1 (–CH) after nitric acid activation. It can be inferred that after the nitric acid activation, more acidic oxygen-containing functional groups that can easily react with NO were formed on the AC surface, such as carboxyl groups, lactone groups and hydroxyl groups.38 It can also be seen from Fig. 4 that under the premises of different activation methods, with the increase in Ce loading, the peak absorption intensities at 3454 cm−1 (–OH stretching vibration) and 1700 cm−1 (–CO stretching vibration) continued to increase. The increase in the oxygen-containing functional groups provided more active sites for the denitration reaction, could provide more high adsorbed oxygen (Oα) for the first stage of the denitration reaction and promoted the progress of the CO-SCR denitration reaction.
O or CC and the infrared band of 1300–1000 cm−1 is attributed to the stretching vibration of C–O–C or C–O. It can be seen from Fig. 4 that compared with the air thermal oxidation activation method, the AC after activation with nitric acid is 3454 cm−1 (–OH tensile vibration), 1635 cm−1 (–CO tensile vibration) and 1093 cm−1 (–CO tensile vibration) is obviously in an enhanced state, and two new absorption peaks appeared at 1700 cm−1 (–CO stretching vibration) and 1400 cm−1 (–CH) after nitric acid activation. It can be inferred that after the nitric acid activation, more acidic oxygen-containing functional groups that can easily react with NO were formed on the AC surface, such as carboxyl groups, lactone groups and hydroxyl groups.38 It can also be seen from Fig. 4 that under the premises of different activation methods, with the increase in Ce loading, the peak absorption intensities at 3454 cm−1 (–OH stretching vibration) and 1700 cm−1 (–CO stretching vibration) continued to increase. The increase in the oxygen-containing functional groups provided more active sites for the denitration reaction, could provide more high adsorbed oxygen (Oα) for the first stage of the denitration reaction and promoted the progress of the CO-SCR denitration reaction.
 |
| | Fig. 4 FTIR spectrum of the Cu–Ni–Ce/AC0,1 catalysts. | |
3.5 XRD analysis
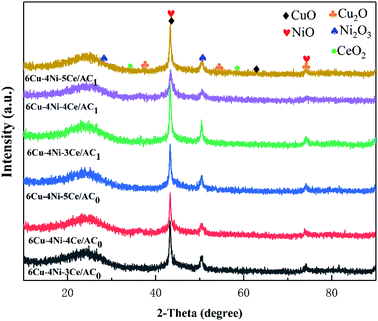
Fig. 5 shows the XRD patterns of the Cu–Ni–Ce/AC0,1 catalysts under different Ce doping levels. CuO had a monoclinic phase diffraction peak of 42.4°, 61.3° at 2θ, and NiO had a monoclinic phase diffraction peak of 43.8°, 76.0° at 2θ. A small amount of Ce (a = 0.3–0.5) was added to the Cu–Ni/AC sample, leading to the reduction of the diffraction peaks of CuO, NiO and the related copper–nickel metal oxides. These results indicate that the cerium doping inhibited the crystallisation of these two phases. When the cerium loading was relatively small, there were no diffraction peaks related to Ce, indicating that Ce is amorphous or poor in crystallinity.39,40 From the XRD diagram, it can be seen that a small amount of Cu2O was on the (111) and (211) crystal planes corresponding to the 2θ of 36.5° and 52.7°. This is due to the reaction of C and CuO during the calcination process, which led to the conversion of Cu2+ into Cu+. It can be seen from Fig. 5 that after the nitric acid was activated, the crystallisation performance of the metal oxides became significantly better than that of the coconut shell AC after the air thermal oxidation. The highly dispersed state and the peak after the air thermal oxidation activation were narrow and sharp, indicating that the supported metal oxide agglomeration was more serious. The reason for this was explained using SEM. Due to the corrosion and pore expansion effects of nitric acid, the precursor solution was easier to pass through the rich pore structure into the inside of the AC, so the metal oxides decomposed during the roasting process and had higher dispersibility. The diffraction peak at 20°–30° indicates the presence of layered graphite crystallites, while the diffraction peak at 40°–45° reflects the graphite-like layer structure. The graphite microcrystalline structure on the surface of the catalyst helped oxidise NO to NO2 in an oxidising atmosphere at low temperatures,41,42 and the simultaneous presence of NO and NO2 induced a ‘fast SCR’ reaction of NO, NO2 and CO, which significantly improved the denitration efficiency.5
 |
| | Fig. 5 XRD pattern of the Cu–Ni–Ce/AC0,1 catalysts. | |
3.6 XPS analysis
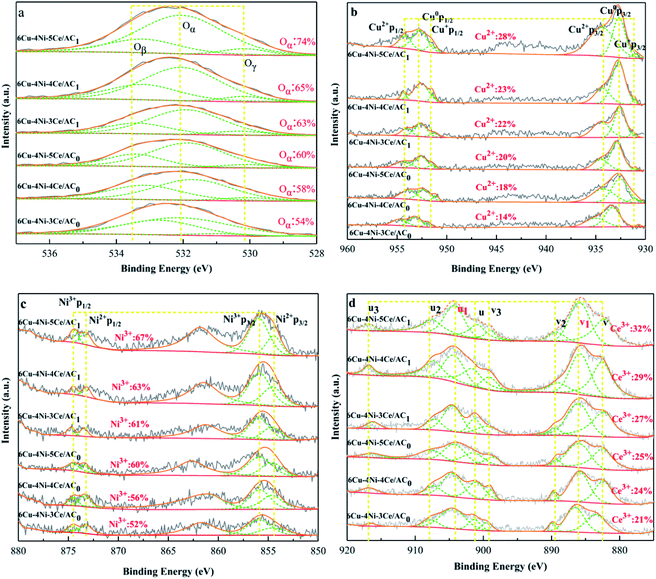
The Cu–Ni–Ce/AC0,1 catalysts activated using the two methods were characterised using XPS before and after denitration. The results of Cu2p, Ni2p, Ce3d and O1s of the samples are shown in Fig. 6. Fig. 6a depicts the O1s spectra of the different samples. Due to the special surface structure of AC, the surface oxygen species could be divided into three components: lattice oxygen (Oβ), surface adsorbed oxygen (Oα) and oxygen species in the hydroxyl group (Oγ).43 According to previous studies,41 Oα was derived with a BE of 531.1–530.4 eV from hydroxyl species or from the O22− and O− on a catalyst's surface, which are defective oxides. Also, the Oγ-derived catalyst surface with a BE of 533.5–535.5 eV the adsorbed H2O, and the BE of 529.06–530.4 eV usually comes from the lattice oxygen of metal oxides. As shown in Fig. 6d, after nitric acid was activated and doped with Ce, the ratio of Oα (Oα/Oα + Oβ + Oγ) increased more obviously. On one hand, as shown in the FTIR diagram, this is due to the production of more hydroxyl groups after the nitric acid activation. On the other hand, oxygen-containing functional groups, such as carbonyl and carbonyl groups, because Ce substances cause charge imbalance, vacancies and unsaturated chemical bonds on catalyst surfaces, which is conducive to the generation of Oα.44 According to the literature, Oα is the most active oxygen species. Due to its high mobility, it plays a key role in CO-SCR denitration reactions. In addition, Oα can promote the oxidation of NO and the production of amides.45 Therefore, the use of nitric acid activation and the addition of Ce can increase the ratio of Oα on catalyst surfaces, thus facilitating CO-SCR reactions.
 |
| | Fig. 6 XPS survey spectrum of the Cu–Ni–Ce/AC catalysts, (a) O1s (b) Cu2p, (c) Ni2p, (d) Ce3d. | |
As shown in Fig. 6b, the components at (∼932.1 eV) and (∼951.5 eV) can be attributed to the peaks of Cu2p3/2 and Cu2p1/2 of Cu2O at (∼933.8 eV) and (∼953.8 eV), which correspond to the Cu2p3/2 and Cu2p1/2 peaks of CuO.36 For the catalyst activated using nitric acid, the ratio of Cu2+/Cu+ was higher than that of in the case of air thermal oxidation activation, and with the increase in Ce doping, the ratio of Cu2+ also increased. This is due to the AC surface formation during the activation of nitric acid. More oxygen-containing functional groups were added, which led to a decrease in the degree of reduction of the metal oxides during the catalyst calcination.46–48 As shown in Fig. 6c, the two peaks at (∼854.6 eV) and (∼855.8 eV) correspond to Ni2+2p3/2 and Ni3+2p3/2,49 respectively, at (∼873.3 eV) and (∼874.5 eV). Also, the two peaks correspond to Ni2+2p1/2 and Ni3+2p1/2, respectively. Due to the increase in Ce doping, the intensity of the Ni3+ peak also increased.
Fig. 6d shows the Ce3d XPS spectra of all the samples containing Ce. Each spectrum was parsed into eight peaks. The peaks at u1 and v1 represent the initial electronic state of 3d104f1 corresponding to Ce3+. According to previous studies, the peaks labelled u, u2, u3, v, v2 and v3 indicate the 3d104f0 state of Ce4+.49–51 It is well known that the conversion between Ce3+ and Ce4+ produces reactive oxygen vacancies (4Ce4+ + O2− → 2Ce4+ + 2Ce3+ + □ + 0.5O2, where □ represents an adsorbed oxygen vacancy).52 Thus, it can be inferred that the higher the proportion of Ce3+, the higher the density of the adsorbed oxygen vacancies. To highlight this problem, the ratio of Ce3+/(Ce3+ + Ce4+) was calculated according to the peak area in the XPS spectrum, and it was then demonstrated, as shown in Fig. 6d. In all the samples, the 6Cu–4Ni–5Ce/AC1 catalyst had the highest Ce3+ ratio (32%), indicating that it contains the most adsorbed oxygen vacancies, which is conducive to the adsorption of NO and contributes to the CO-SCR denitrification activity.50,53
3.7 CO-TPD and NO-TPD analyses
Based on the above characterization results, the Cu–Ni–Ce/AC0,1 catalyst under the conditions of air thermal oxidation and nitric acid activation was analyzed for CO-TPD and NO-TPD. CO-TPD analysis is used to study CO adsorption and desorption on the surface of the catalyst. The results are shown in Fig. 7a. The CO-TPD curve of Cu–Ni–Ce/AC0,1 contains two peaks, which shows different types of active ingredients, which have different adsorption strengths than CO. From the figure, it can be found that the analytical peak temperature of CO after nitric acid activation is significantly lower than that of the catalyst prepared after air thermal oxidation. This is because the pore structure of the AC surface becomes more developed after nitric acid activation, the gas easily enters the catalyst and reacts with the metal oxide at low temperature. Different Ce loadings affected the CO-TPD analytical behaviour of the catalyst. An analytical peak of CO was observed at 145 °C. This low-temperature desorption peak was attributed to the desorption of CO on the CuO/Cu2O position on the surface,54,55 and the XPS results proved this situation. There was a large CO desorption peak at 258.1 °C, and with the increase in the Ce content, this peak shifted to 189.5 °C. Compared with 6Cu–4Ni–3Ce/AC1, 6Cu–4Ni–5Ce/AC1 showed a lower desorption temperature, which might be attributed to the high Ni2O3 on the catalyst surface. Compared with the other Cu–Ni–Ce/AC0,1 catalysts in the CO oxidation and NO + CO reactions, the catalyst activity of 6Cu–4Ni–5Ce/AC1 was higher.56,57
 |
| | Fig. 7 (a) CO-TPD profiles of catalysts with Cu–Ni–Ce/AC0,1; (b) NO-TPD profiles of catalysts with Cu–Ni–Ce/AC0,1. | |
NO-TPD analysis is used to study the adsorption of NO on the catalyst surface. NO is adsorbed on different adsorption sites. As shown in Fig. 7b, the catalyst activated by nitric acid has three desorption peaks at 262.1, 337.3 and 621.7 °C. It can be seen that only two absorption peaks appear after air thermal oxidation activation. The reason is that after the activation of nitric acid, the oxygen-containing functional groups increase, and the oxygen adsorption sites of NO increase, which leads to the adsorption of NO at low temperatures. With the increase of Ce doping, the position of the desorption peak moved to low temperature. It can also be inferred that the Ce addition improved the adsorption capacity of NO at low temperature, which is consistent with the research performed by previous scholars.58,59 The desorption peak above 400 °C was attributed to the decomposition of the nitrite and nitrate with the higher thermal stability.60,61 It was reported that with the increase in the Ce content, more nitrates and nitrites were formed on the catalyst surfaces, which can participate in low-temperature NO + CO reactions. The NO-TPD results showed that the increase in Ce doping can increase the ability to adsorb and decompose NO at low temperatures, thereby improving the catalytic activity of CO-SCR. The reason is that catalysts with high Ce doping have more unpaired electrons. The electrons feedback and the dilution to the vacant antibonding orbitals of the adsorbed species weakened the NO bond.55 As described by XPS, more adsorbed oxygen vacancies are formed with higher Ce3+ concentrations, where the adsorbed oxygen vacancies are combined by the weakened NO bond, achieving the denitration effect.
3.8 Analysis of the denitration mechanism
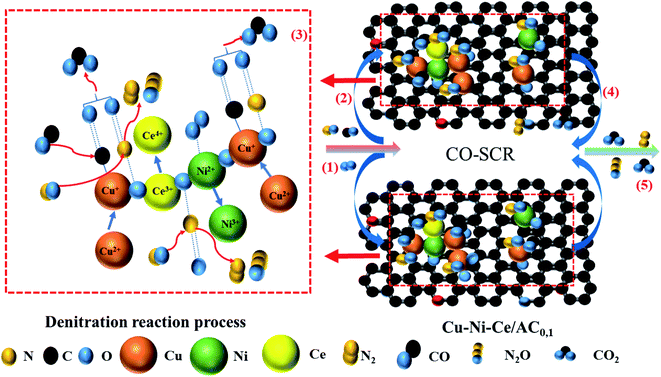
Based on the above characterisation observations, the adsorption and catalytic mechanisms of the catalyst were proposed. As shown in Fig. 8, the CO-SCR low-temperature denitration reaction on the surface of the Cu–Ni–Ce/AC0,1 catalyst is a representative heterogeneous. The reaction can be divided into the following five steps. (1) The CO, NO and O2 reactant gases are continuously diffused from the outside of the catalyst to the outer surface of Cu–Ni–Ce/AC. Since AC has a large specific surface area and a rich pore structure, a large amount of reactant gas could be cached inside the catalyst. A large number of reaction units were produced on the graphite microcrystalline structure.33 (2) The reaction gas is adsorbed on the catalyst surface and in an adsorbed state on the active sites of the catalyst surface and then activated by the active sites to form activated molecules. A large number of acidic oxygen-containing functional groups were detected by FTIR, and defects could be detected in the XPS map. The adsorbed oxygen (Oα) preferentially adsorbed NO. It is generally believed that the presence of the Cu2+ and oxygen-containing groups can enhance the adsorption of CO.54,59 The oxidation–reduction reaction between Ni2+ and Ni3+ (4NiO + 5O2 → 2Ni2O3 + 4O2−) could convert O2 into adsorbed oxygen (Oα), and Ni had a good oxygen storage/release capacity, which could significantly promote the adsorption of NO on the catalyst surface. (3) The reaction between NO and CO (NO + CO → N2 + CO2). During the denitration process, an oxidation–reduction reaction between copper, nickel and cerium (Cu2+ + Ce3+ → Cu+ + Ce4+, Ni3+ + Ce3+ → Ni2+ + Ce4+) occurred. As shown in the XPS diagram, after the Ce doping amount increased, although the oxygen species on the surface of the catalyst did not increase, the Cu–Ni–Ce/AC0,1 surface's adsorbed oxygen content significantly increased due to the high mobility of the surface adsorbed oxygen, which promoted the oxidation of NO to NO2, thus converting the standard SCR reaction to a fast SCR reaction (NO2 + CO → N2 + CO2).5 (4) The gases (N2, N2O and CO2) generated from the surface of the catalyst were desorbed and diffused to the inner surface through the AC channels. (5) The generated gas was diffused from the inner surface of the AC to the outer surface of the Cu–Ni–Ce/AC0,1 catalyst. Based on the above conclusions, with the increase in Ce doping, the valence states of the copper and nickel ions on the Cu–Ni/AC catalyst surface increased and the adsorbed oxygen O(a) on the surface increased, particularly with the increase in the surface acidic functional groups and in the number of reactions. Gas adsorption and the accumulation of all the factors made 6Cu–4Ni–5Ce/AC obtain a significant CO-SCR denitration efficiency.
 |
| | Fig. 8 Denitration mechanism diagram of the Cu–Ni–Ce/AC catalyst. | |
4. Conclusion
In this study, a Cu–Ni–Ce/AC low-temperature CO-SCR denitrification catalyst was prepared using the ultrasonic equal volume co-impregnation method. Also, the nitric acid and air thermal oxidation activation methods, different Ce doping amount pairs and the activated carbon-supported metal catalyst structure were studied. Moreover, the effect of the CO reduction on the NO catalytic performance was investigated. On this basis, the following conclusions could be drawn. (1) In the presence of a denitration temperature of 150 °C, a GHSV of 30000 h−1 and 5% O2, the denitration efficiency of 6Cu–4Ni–5Ce/AC1 could reach 99.8%. (2) Although the specific surface area of the AC activated by nitric acid was slightly lower than that activated by air thermal oxidation, the pore structure of the AC activated by nitric acid was more developed, the AC pore structure after nitric acid activation is more developed and the number of oxygen-containing functional groups increases significantly. (3) By increasing the amount of Ce doping, uniformly dispersed Cu and Ni metal oxides can be obtained, the average valence state of Cu and Ni metal ions and the adsorbed oxygen content on the surface all increase, especially with the increase of surface acid sites, a large number of reaction units are formed on the surface of the catalyst, and the adsorption is more CO and NO reactive gas. (4) Based on the results of this research, a mechanism was proposed to convert the incoming O2 into (Oα) through the oxidation–reduction reaction between metals, promote the conversion of NO into NO2, accelerate the CO-SCR reaction and improve the denitration efficiency. Based on the above research, this paper hopes to optimise the preparation conditions of copper–nickel–cerium catalysts and to explore the best CO-SCR denitration efficiency so as to provide a theoretical reference for the research and development of CO-SCR low-temperature denitrification catalysts for sintering flue gases in iron and steel enterprises.
Funding information
This work was supported by the General Project of Science and Technology Plan of Yunnan Science and Technology Department (Grant No. 2019FB077, 202001AT070029), the Ten Thousand Talents Plan Young Top Talents of Yunnan Province (Grant No. YNWR-QNBJ-2019-263), the Open Fund of Key Laboratory of Ministry of Education for Metallurgical Emission Reduction and Comprehensive Utilization of Resources (Grant No. JKF19-08), and the Open Fund of Key Laboratory for Ferrous Metallurgy and Resources Utilization of Ministry of Education (Grant No. FMRUlab-20-4).
Conflicts of interest
In this article, all authors have no conflicts of interest.
References
- H. Chen, Y. Xia, H. Huang, Y. Gan, X. Tao, C. Liang, J. Luo, R. Fang, J. Zhang, W. Zhang and X. Liu, Chem. Eng. J., 2017, 330, 1195–1202 CrossRef CAS.
- P. Forzatti, Appl. Catal., A, 2001, 222, 221–236 CrossRef CAS.
- F. D. Liu, W. P. Shan and Z. H. Lian, Appl. Catal., B, 2018, 230, 165–176 CrossRef CAS.
- K. Skalska, J. S. Miller and S. Ledakowicz, Sci. Total Environ., 2010, 408, 3976–3989 CrossRef CAS PubMed.
- B. Zhao, R. Rui, X. Guo, C. Li and W. Duan, Appl. Catal., A, 2017, 545, 64–71 CrossRef CAS.
- X. Cheng and X. T. Bi, Chem. Eng. J., 2012, 211–212(47), 453–462 CrossRef CAS.
- T. T. Yang, H. T. Bi and X. Cheng, Appl. Catal., B, 2011, 102(1–2), 163–171 CrossRef CAS.
- Y. F. Qu, J. X. Guo, Y. H. Chu, M. C. Sun and H. Q. Yin, Appl. Surf. Sci., 2013, 282, 425–431 CrossRef CAS.
- L. J. Jiang, Q. C. Liu, Q. Zhao, S. Ren, M. Kong, L. Yao and F. Meng, Res. Chem. Intermed., 2018, 44, 1729–1744 CrossRef CAS.
- A. Boyano, M. E. Gálvez, R. Moliner and M. J. Lázaro, Fuel, 2008, 87, 2058–2068 CrossRef CAS.
- W. Xu, J. Zhou, H. Li, P. Yang, Z. You and Y. Luo, Fuel Process. Technol., 2014, 127, 1–6 CrossRef CAS.
- J. H. Zhou, Z. J. Sui, J. Zhu, P. Li, D. Chen, Y. C. Dai and W. K. Yuan, Carbon, 2007, 45, 785–796 CrossRef CAS.
- Z. Y. Li, M. S. Akhtar, D. H. Kwak and O. B. Yang, Appl. Surf. Sci., 2017, 404, 88–93 CrossRef CAS.
- H. U. Shin, L. Dinesh, Z. Nikolov and G. G. Chase, J. Ind. Eng. Chem., 2016, 33, 91–98 CrossRef CAS.
- K. Huang, L. L. Lin, K. Yang, W. X. Dai, X. Chen and X. Z. Fu, Appl. Catal., B, 2015, 179, 395–406 CrossRef CAS.
- F. Benjamin and P. Alphonse, Appl. Catal., B, 2016, 180, 395–406 Search PubMed.
- W. Y. Hernández, M. A. Centeno, S. Ivanova, P. Eloy, E. M. Gaigneaux and J. A. Odriozola, Appl. Catal., B, 2012, 123–124, 27–35 CrossRef.
- Y. Mahara, H. Ishikawa, J. Ohyama, K. Sawabe and A. Satsuma, Catal. Today, 2016, 265, 2–6 CrossRef CAS.
- X. Yao, Y. Xiong, J. Sun, F. Gao, D. Yu, C. Tang and L. Dong, J. Rare Earths, 2014, 2, 131–138 CrossRef.
- N. Liu, X. Chen, J. Zhang and J. W. Schwank, Catal. Today, 2015, 258, 139–147 CrossRef CAS.
- D. Gamarra, C. Belver, M. Fernandez-García and A. Martínez-Arias, J. Am. Chem. Soc., 2007, 129, 12064–12065 CrossRef CAS PubMed.
- R. Zhang, W. Y. Teoh, R. Amal, B. Chen and S. Kaliaguine, J. Catal., 2010, 272, 210–219 CrossRef CAS.
- P. Li, L. Feng, F. Yuan, D. Wang, Y. Dong, X. Niu and Y. Zhu, Catalysts, 2016, 6, 124 CrossRef.
- Y. H. Chu, J. X. Guo, J. Liang, Q. B. Zhang and H. Q. Yin, Sci. China: Chem., 2010, 53, 846–850 CrossRef CAS.
- X. Lin, P. Ning, K. Li, L. H. Tang, J. M. Hao, X. Song, G. J. Zhang and C. Wang, Russ. J. Phys. Chem. A, 2017, 91, 490–499 CrossRef.
- H. L. Jiang and Q. Xu, J. Mater. Chem., 2011, 21(36), 13705–13725 RSC.
- J. P. S. Sousa, M. F. R. Pereira and J. L. Figueiredo, Fuel Process. Technol., 2013, 106, 727–733 CrossRef CAS.
- Z. Gholami and G. H. Luo, Ind. Eng. Chem. Res., 2018, 57, 8871–8883 CrossRef CAS.
- S. Vivek, S. Preethi, K. T. H. Vignesh, A. K. Sundramoorthy and K. S. Babu, J. Alloys Compd., 2020, 816, 152608 CrossRef CAS.
- S. Zhan, H. Zhang, Y. Zhang, Q. Shi, Y. Li and X. J. Li, Appl. Catal., B, 2017, 203, 199–209 CrossRef CAS PubMed.
- S. Nishigaki, S. Yano, H. Kato, T. Hirai and T. Nonomura, J. Am. Ceram. Soc., 1988, 71, 11–17 CrossRef.
- X. J. Yao, F. Gao, Q. Yu, L. Qi, C. Tang, L. Dong and Y. Chen, Catal. Sci. Technol., 2013, 3, 1355–1366 RSC.
- J. Yang, S. Ren, T. S. Zhang, Z. H. Su, H. M. Long and M. Kong, Chem. Eng. J., 2017, 379, 1–11 Search PubMed.
- R. T. Guo, M. Y. Li, S. Peng, W. G. Pan and S. W. Liu, J. Phys. Chem. C, 2017, 200, 297–308 Search PubMed.
- Z. Liu, Z. J. Wang, S. J. Qing, N. N. Xue and S. P. Jia, Appl. Catal., B, 2018, 232, 86–92 CrossRef CAS.
- A. Sharma, R. K. Dutta, A. Roychowdhury, D. Das, A. Goyal and A. Kapoor, Appl. Catal., A, 2017, 543, 257–265 CrossRef CAS.
- L. Yu, H. Zhou, J. Sun, F. Qin, F. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2017, 10, 1820–1827 RSC.
- Y. Lin, Y. Li, Z. Xu, J. Xiong and T. Zhu, Fuel, 2018, 223, 312–323 CrossRef CAS.
- L. J. France, Q. Yang, W. Li, Z. Chen and X. Li, Appl. Catal., B, 2017, 206, 203–215 CrossRef CAS.
- T. Boningari, P. R. Ettireddy and A. Somogyvari, J. Catal., 2015, 325, 145–155 CrossRef CAS.
- J. Wang, Z. Yan, L. Liu, Y. Chen and X. Wang, Appl. Surf. Sci., 2014, 313, 660–669 CrossRef CAS.
- I. Mochida, S. Kisamori, M. Hironaka, S. Kawano, Y. Matsumura and M. Yoshikawa, Energy Fuels, 1994, 8, 611–624 Search PubMed.
- L. Wang, X. Cheng, Z. Wang, C. Ma and Y. Qin, RSC Adv., 2017, 201, 636–651 CAS.
- A. Shaabani, A. Rahmati and Z. Badri, Catal. Commun., 2008, 9, 13–16 CrossRef CAS.
- F. Cao, S. Su, J. Xiang, P. Wang, S. Hu, L. Sun and A. C. Zhang, Fuel, 2015, 139, 232–239 CrossRef CAS.
- H. H. Tseng, H. Y. Lin, Y. F. Kuo and Y. T. Su, Chem. Eng. J., 2010, 160, 13–19 CrossRef CAS.
- C. Ge, L. Liu, X. Yao, C. Tang and D. Lin, Catal. Sci. Technol., 2013, 3, 1547–1557 RSC.
- Y. Xiong, X. Yao, C. Tang, L. Zhang, Y. Cao, Y. Deng, F. Gao and D. Lin, Catal. Sci. Technol., 2014, 4, 4416–4425 RSC.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, Appl. Catal., B, 2012, 115–116, 100–106 CrossRef CAS.
- P. Maitarad, R. Gao and D. Zhang, J. Phys. Chem. C, 2018, 57, 12736–12741 Search PubMed.
- P. Wang, S. Chen, S. Gao, J. Zhang and Z. Wu, Appl. Catal., B, 2018, 231, 299–309 CrossRef CAS.
- X. Yao, L. Chen, J. Cao, Y. Chen and L. Dong, Chem. Eng. J., 2019, 369, 46–56 CrossRef CAS.
- H. Yu, J. Li, Y. Zhang, S. Yang, K. Han, F. Dong, T. Ma and H. W. Huang, Angew. Chem., 2019, 58, 3880–3884 CrossRef CAS PubMed.
- D. Liu, L. Yang, M. Meng, J. Zhang, L. Zheng, S. Chu and T. Hu, Chem. Eng. J., 2013, 234, 88–98 CrossRef.
- C. Deng, B. Li, L. Dong, F. Zhang and X. Zhou, Phys. Chem. Chem. Phys., 2015, 17, 16092–16109 RSC.
- Q. W. Qian, X. X. Cheng, M. H. Tahir, D. X. Su, Z. Q. Wang and S. Y. Chen, Int. J. Hydrogen Energy, 2020, 45, 16469–16481 CrossRef.
- B. Thirupathi and P. G. Smirniotis, J. Catal., 2012, 288, 74–83 CrossRef CAS.
- W. Shen, D. Mao, Z. Luo and J. Yu, RSC Adv., 2017, 7, 27689–27698 RSC.
- H. He, M. Liu, H. Dai, W. Qiu and X. H. Zi, Catal. Today, 2007, 126, 290–295 CrossRef CAS.
- S. Lai, D. Meng, W. Zhan, Y. Guo and G. Lu, RSC Adv., 2015, 5, 90235–90244 RSC.
- D. Ciuparu, A. Bensalem and L. Pfefferle, Appl. Catal., B, 2000, 26, 241–255 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Bangfu Huang*ab,
Zhe Shiab,
Hongming Longc,
Lu Liab,
Zhengyu Yangab and
Meng Daiab
ab,
Bangfu Huang*ab,
Zhe Shiab,
Hongming Longc,
Lu Liab,
Zhengyu Yangab and
Meng Daiab








