
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe function of peptide-mimetic anionic groups and salt bridges in the antimicrobial activity and conformation of cationic amphiphilic copolymers†
Rajani Bhat‡
a,
Leanna L. Foster‡b,
Garima Rani‡ c,
Satyavani Vemparala*de and
Kenichi Kuroda*ab
c,
Satyavani Vemparala*de and
Kenichi Kuroda*ab
aDepartment of Biologic and Materials Sciences & Prosthodontics, School of Dentistry, University of Michigan, Ann Arbor, Michigan 48109, USA. E-mail: kkuroda@umich.edu
bMacromolecular Science and Engineering Center, University of Michigan, Ann Arbor, Michigan 48109, USA
cTIFR Centre for Interdisciplinary Sciences, Tata Institute of Fundamental Research, Hyderabad, 500046, India
dThe Institute of Mathematical Sciences, C. I. T. Campus, Taramani, Chennai 600113, India. E-mail: vani@imsc.res.in
eHomi Bhabha National Institute, Training School Complex, Anushakti Nagar, Mumbai 400094, India
First published on 22nd June 2021
Abstract
Herein we report the synthesis of ternary statistical methacrylate copolymers comprising cationic ammonium (amino-ethyl methacrylate: AEMA), carboxylic acid (propanoic acid methacrylate: PAMA) and hydrophobic (ethyl methacrylate: EMA) side chain monomers, to study the functional role of anionic groups on their antimicrobial and hemolytic activities as well as the conformation of polymer chains. The hydrophobic monomer EMA was maintained at 40 mol% in all the polymers, with different percentages of cationic ammonium (AEMA) and anionic carboxylate (PAMA) side chains, resulting in different total net charge for the polymers. The antimicrobial and hemolytic activities of the copolymer were determined by the net charge of +3 or larger, suggesting that there was no distinct effect of the anionic carboxylate groups on the antimicrobial and hemolytic activities of the copolymers. However, the pH titration and atomic molecular dynamics simulations suggest that anionic groups may play a strong role in controlling the polymer conformation. This was achieved via formation of salt bridges between cationic and anionic groups, transiently crosslinking the polymer chain allowing dynamic switching between compact and extended conformations. These results suggest that inclusion of functional groups in general, other than the canonical hydrophobic and cationic groups in antimicrobial agents, may have broader implications in acquiring functional structures required for adequate antimicrobial activity. In order to explain the implications, we propose a molecular model in which formation of intra-chain, transient salt bridges, due to the presence of both anionic and cationic groups along the polymer, may function as “adhesives” which facilitate compact packing of the polymer chain to enable functional group interaction but without rigidly locking down the overall polymer structure, which may adversely affect their functional roles.
Introduction
Due to the ever-growing bacterial resistance to existing drugs, there is an urgent need of new antimicrobials that can overcome existing microbial resistance mechanisms.1–7 One approach has been to create synthetic polymers which mimic the functions and characteristics of host-defense antimicrobial peptides (AMPs) found in the innate immune system.8–11 Antimicrobial polymers are designed to act by disrupting bacterial cell membranes, which are effective in killing drug-resistant bacteria and are not suspected to contribute to the development of drug resistance in bacteria.12,13 These polymers have both cationic and hydrophobic groups, presenting cationic amphiphilicity. Since bacterial cell membranes are highly negatively charged as compared to human cell membranes, the polymers selectively bind to bacterial cell membranes through electrostatic interactions.14 Subsequently, the hydrophobic groups of the polymer chain are inserted into the nonpolar region of the bacterial cell membranes, causing membrane disruption and ultimately cell death.15,16Traditionally, the binary composition of cationic charge and hydrophobicity has been thought to be a minimal requirement for the antimicrobial mechanism of the antimicrobial polymers. Many efforts have been made to develop antimicrobial polymers based on different polymer backbones, such as methacrylate,17,18 and optimize the monomer compositions and chemical identities of cationic and hydrophobic groups for potent antimicrobial activity and selectivity.19–21 Recently, the approach has been extended to block copolymers with specific sequences,22,23 branched polymers,24–26 comb-like polymers,27,28 polymer assemblies and micelles,29,30 and single-chain polymer nanoparticles.31–33 In addition, polymers with facially cationic amphiphilicity in the side chains have shown to promote effective interactions of the polymer with the bacterial cell membrane, causing subsequent membrane disruption.34
While these cationic amphiphilic copolymers and macromolecules are promising platforms, the sequences of natural AMPs contain functional groups other than cationic and hydrophobic residues, and these functional groups control their stability of active conformations and specificity toward bacterial cell membranes.6,35 For example, the disulfide bond (S–S) covalently crosslinks peptide sequences, which stabilizes the conformation of defensins.36–38 Tryptophan residues of antimicrobial peptides are known to penetrate into the interfacial region of lipid bilayers.39–41 Arginine residues form a cation–π complex with tryptophan residues, which facilitates penetration of arginine into the hydrophobic domain of a lipid bilayer.42 These examples suggest that the current minimalist design of binary copolymers does not take full advantage of the evolutionarily optimized AMP compositions and/or functions.43,44 To that end, previous studies have incorporated different functional groups found in natural AMPs into the design of antimicrobial polymers. Locock and coworkers developed tryptophan-rich polymers,45 which showed antimicrobial activity against S. aureus. Kuroki et al. demonstrated that guanidine-rich polymers can target intracellular S. aureus.46 Tyrosine-rich polymers have been studied for their antimicrobial activity against Shigella.47 Gellman and Wong demonstrated that the incorporation of hydroxyl groups into the side chains of antimicrobial nylon copolymers retained the antimicrobial activity, but significantly reduced the hemolytic activity.48 Our laboratory has systematically investigated the ternary statistical methacrylate copolymers with hydroxyl side chains and showed the hydroxyl monomeric units act as a spacer to tune the numbers of cationic side chains and control the interactions with cell membranes for selectivity.49 These studies demonstrated that the approach beyond the cationic functionality and hydrophobicity would be effective in the development of a new class of potent, selective antimicrobial polymers. However, the scope of these previous studies has been limited to the structure–activity relationship, and the functional roles or mechanisms in the biological activities remains unexplored. If we can implement the inherent functionalities of AMPs to our molecular design, we wonder if it may ultimately lead to more potent antimicrobial activity and selectivity as well as the mimicry of the biological functions of AMPs other than antimicrobial activity.
While naturally occurring AMPs generally display an overall positive charge of around +2 to +11, many AMPs including defensin, magainin, lactoferricin, and LL-37 also contain negatively charged amino acids in their sequences. For example, human cathelicidin antimicrobial peptide LL-37 sequence contains 11 cationic amino acid residues (6 lysine and 5 arginine residues) and 5 anionic amino acid residues (3 glutamic acid and 2 aspartic acid residues), giving net positive charge of +6.50,51 Defensins also contain anionic amino acid residues. α-Defensin has an anionic glutaric acid, which form an electrostatic pair with cationic arginine residue, or a salt bridge, stabilizing a protruding loop in the peptide conformation.52 Interestingly, a salt bridge is conserved in mammalian α-defensins even though this family of AMPs have diverse sequences.53 In the case of de novo design of short helical peptides, it has been shown that the intra-peptide salt bridges tend to stabilize the helical conformation54 and similar implications of stability via salt bridges has also been proposed for other cell-penetrating α-helical and β-sheet antimicrobial peptides.55–58 These examples suggest that the anionic amino acids play functional roles in the AMP conformations and antimicrobial functions.
In this study, we investigate the functional roles of anionic groups of cationic amphiphilic random methacrylate in the antimicrobial activity and selectivity as well as the polymer conformations in solution. The purposes of this study are to determine (1) whether the anionic groups contribute to the antimicrobial activity and selectivity of the copolymers, and/or (2) whether the anionic groups contribute towards adoption or stabilization of specific conformations in solution as found in AMPs by forming salt-bridge(s) with the cationic groups. To that end, we synthesized copolymers with ternary compositions of cationic, anionic, and hydrophobic side chains (Fig. 1) in an attempt to gain better understanding of the roles anionic groups play in the characteristics of these polymers. More specifically, the polymers with different net charges were prepared by altering the compositions of cationic and anionic monomer units. This polymer design contrasts with the conventional antifouling polymers consisting of zwitterionic monomers such as sulfobetaines or carboxy betaines,59,60 which have same number of cationic and anionic groups in the same side chain, and therefore, the net charge of polymers is neutral. First, the antimicrobial activity against Escherichia coli and Staphylococcus aureus as well as the hemolytic activity against human red blood cells (RBCs) was determined. The ionic states of polymer side chains were investigated by conducting potentiometric titrations of selected polymers. Using molecular dynamics simulations, we also studied the conformational landscape of a single polymer chain in solution, in order to probe into the formation of salt bridges between the side chains and its structural role in adopting compact conformations.
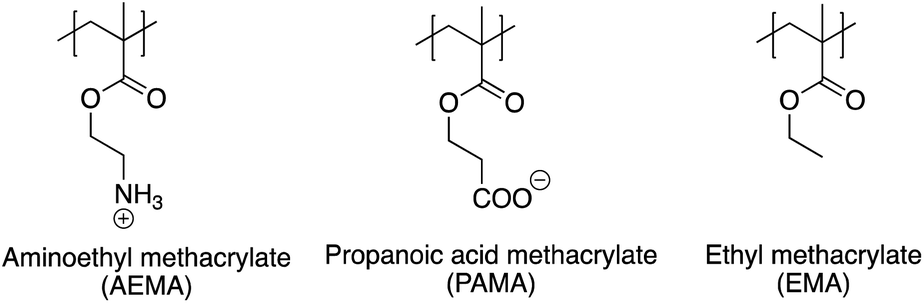 | ||
| Fig. 1 Structures of monomer units in the cationic amphiphilic polymers with anionic groups. | ||
Experimental
Materials
tert-Butyl-3-hydroxypropanoate, methyl 3-mercaptopropionate, and ethyl methacrylate were purchased from Acros Organics. 2,2′-Azobis-(isobutyronitrile) (AIBN) and ethyl methacrylate were purchased from Sigma-Aldrich Co. Trifluoroacetic acid (TFA) and solvents were purchased from Thermo Fisher Scientific, Inc. The chemicals were used without further purification, with the exception of methacryloyl chloride which was distilled before use and AIBN which was recrystallized from hot methanol. Boc-amino ethyl methacrylate was synthesized according to the previous reported procedure.61 1H NMR was performed using a Varian MR400 (400 MHz), 13C NMR was performed using a Bruker NEO600 (600 MHz) and the data was analyzed using VNMRJ 3.2 and MestReNova. Gel permeation chromatography (GPC) analysis was performed using a Waters 1515 HPLC instrument equipped with Waters Styragel (7.8 × 300 mm) HR 0.5, HR 1, and HR 4 columns in sequence and detected by a differential refractometer (RI). Mueller Hinton broth (MHB, BD and Company) and phosphate buffered saline (PBS, pH = 7.4, Gibco) were prepared according to manufacturer instructions and sterilized prior to use. Human red blood cells (RBCs) (leukocytes reduced adenine saline added) were obtained from the American Red Cross Blood Services Southeastern Michigan Region and used prior to the out date indicated on each unit.Synthesis of 3-(tert-butoxy)-3-oxopropyl methacrylate
The protected acidic monomer was prepared from the reaction between t-butyl-3-hydroxypropanoate and methacryloyl chloride (Fig. 2). t-Butyl-3-hydroxypropanoate (3.4 mmol, 0.5 g) was dissolved in 50 mL anhydrous tetrahydrofuran in a round bottom flask, and triethyl amine (5.1 mmol, 0.74 mL) was added to it. The reaction mixture was cooled to 0 °C under nitrogen and stirred for half hour. Freshly distilled methacryloyl chloride (3.7 mmol, 0.36 mL) was added over a period of 20 min and the reaction mixture was stirred at 0 °C for 1 h. The reaction was allowed to gradually warm up to room temperature and was stirred overnight. Next day, the solid was filtered and the filtrate was concentrated. The concentrated liquid was then extracted with dichloromethane and washed with dilute sodium bicarbonate solution twice and once with brine before drying it over magnesium sulfate. The organic layer was filtered, concentrated and purified by column chromatography (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 hexane, ethyl acetate) to get the final pure product. Yield = 0.4 g (53.3%) 1H NMR (CDCl3, 400 MHz) δ 6.07 (s, 1H), 5.53 (s, 1H), 4.38–4.34 (t, 2H), 2.59–2.56 (t, 2H), 1.91 (s, 3H), 1.43 (s, 9H); 13C NMR (CDCl3, 600 MHz) δ 169.91, 167.09, 136.12, 125.65, 80.95, 60.46, 35.23, 28.03, 18.22.
:2 hexane, ethyl acetate) to get the final pure product. Yield = 0.4 g (53.3%) 1H NMR (CDCl3, 400 MHz) δ 6.07 (s, 1H), 5.53 (s, 1H), 4.38–4.34 (t, 2H), 2.59–2.56 (t, 2H), 1.91 (s, 3H), 1.43 (s, 9H); 13C NMR (CDCl3, 600 MHz) δ 169.91, 167.09, 136.12, 125.65, 80.95, 60.46, 35.23, 28.03, 18.22.
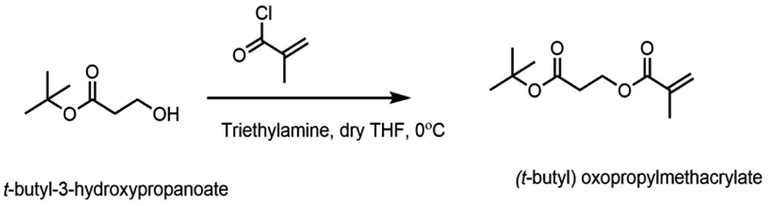 | ||
| Fig. 2 Synthesis of propionate monomer from t-butyl-3-hydroxypropanoate. | ||
Polymerization of ternary copolymers
The synthesis of the copolymer with a monomer composition of amino ethyl methacrylate (AEMA), 39 mol%; propionic acid methacrylate (PAMA), 17 mol%; and ethyl methacrylate (EMA), 44 mol% (AE39PA17E44) is described here as a representative sample.The free radical polymerization is carried out by using methyl mercaptopropionate as chain transfer agent and azoisobisbutyronitrile (AIBN) as the initiator (Fig. 3). 4-((tert-butoxycarbonyl) amino) ethyl methacrylate (Boc-AEMA) (0.48 mmol, 101 mg), 3-(tert-butoxy)-3-oxopropyl methacrylate (t-Bu-PAMA) (0.24 mmol, 51.4 mg), ethyl methacrylate (EMA) (0.48 mmol, 54.8 mg), AIBN (0.012 mmol, 2 mg), and methyl 3-mercaptopropionate (0.12 mmol, 14.4 mg) in acetonitrile (0.4 mL)/DMF (0.1 mL) were mixed in a reaction vessel equipped with a stir rod. Nitrogen gas was purged through for 3 min, and the reaction mixture was dipped in an oil bath at 70 °C for 16 h. The reaction mixture was cooled to room temperature. The solvent was removed by evaporation under reduced pressure, and the residue was dissolved in dichloromethane and precipitated in cold hexanes. It is important to note that as the monomer ratio of tBu-PAMA increased beyond 35%, the polymer was subsequently more difficult to precipitate, and we had to resort to precipitating it in water/methanol mixture (6:3). To simultaneously remove the Boc and tert-butyl groups of the copolymer, the protected polymers were dissolved in trifluoroacetic acid (TFA) and stirred for 20 min. TFA was removed by blowing the solution with nitrogen gas in a closed container and allowing the gas to pass through a basic (NaOH) aqueous solution to trap TFA. The residues were dissolved in methanol and precipitated in ice cold diethyl ether. The degree of polymerization and monomer composition was determined through 1H NMR by comparing the integrated areas of the peaks from the side chains to the end group of the chain transfer agent, methyl-3-mercaptopropionate in the polymer.
 | ||
| Fig. 3 Synthesis of ternary statistical copolymers. The statistical copolymers were prepared by free-radical polymerization in the presence of thiol chain transfer agent methyl 3-mercaptopropionate. The Boc and t-butyl groups were removed using trifluoroacetic acid at room temperature. | ||
Potentiometric titration
Polymers were dissolved in aqueous saline (1 mg mL−1 except 0.5 mg mL−1 for AE39PA17E44; [NaCl] = 150 mM) in a glass scintillation vial and purged with N2 for 10 minutes to remove dissolved carbon dioxide. A nitrogen blanket was maintained over the sample throughout the titration, and the temperature was held at 25 °C using a water bath. Aliquots of standardized 0.100 N sodium hydroxide (2.5–10 μL) were injected using a Hamilton syringe to a rapidly stirring polymer solution. After 2 minutes of rapid stirring to allow thermal and chemical equilibrium, the pH was recorded using a Mettler Toledo FiveEasy Plus pH meter. The titration curve was plotted as pH vs. volume of NaOH added. The pKa is calculated as the half equivalence point or the point where 0.5 mole equivalents of carboxylic or cationic ammonium groups have reacted with the base. We calculated the fraction of cationic groups that are deprotonated at pH of ∼7.4, using the Henderson–Hasselbalch equation (eqn (1)). Subsequently, we could calculate the average corrected charges on the polymer chain.
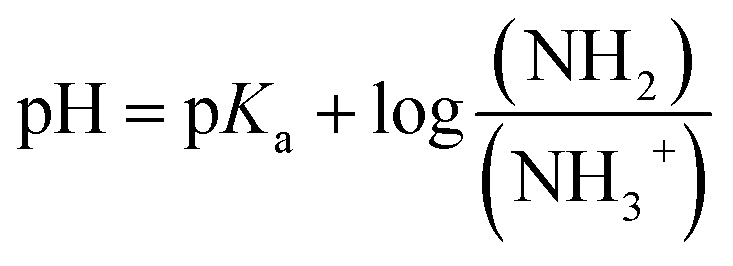 | (1) |
Antimicrobial activity
The minimum inhibitory concentration (MIC) of polymers against E. coli (ATCC 25922) and S. aureus (ATCC 25923) was determined in a standard microbroth dilution assay according to the Clinical and Laboratory Standards Institute guidelines with suggested modifications by R. E. W Hancock Laboratory (University of British Columbia, British Columbia, Canada) and Giacometti et al. Bacteria was grown overnight (∼18 hours) in MHB at 37 °C with orbital shaking (180 rpm), and used as an inoculum by diluting overnight culture in MHB to a concentration of OD600 = 0.1. The inoculated solution was then grown at 37 °C to the exponential phase (OD600 = 0.5–0.7, 2 hours). Final dilution to OD600 = 0.001, ∼4 × 105 CFU mL−1, was made with MHB. Bacterial suspension (OD600 = 0.001, 90 μL per well) was transferred to a 96-well sterile round-bottom polypropylene plate. Polymers were dissolved in dimethyl sulfoxide (DMSO) to achieve stock concentrations of 20 mg mL−1. Serial 2-fold dilutions of polymers were prepared from stock solutions in PBS and transferred to the 96-well sterile round-bottom polypropylene plate for a final concentration of 7.8–1000 μg mL−1 (10 μL per well). PBS and DMSO were used as solvent controls in place of polymer. While we used DMSO as a solvent to dissolve the polymers, DMSO is known for membrane activity and may exert toxic effect to bacteria.62,63 Therefore, we tested DMSO as a control, and the bacterial cultures with DMSO control showed growth after overnight incubation. Plates were sealed with parafilm and incubated for 18 hours at 37 °C without shaking. MIC was defined as the lowest concentration of polymers to completely inhibit bacterial growth, as indicated by lack of turbidity. Assays were repeated a minimum of three times in triplicate on different days.Hemolytic activity
A 10% solution of human red blood cells (RBCs) in PBS was centrifuged at 2000 rpm for 5 minutes and washed with PBS ×2 to remove initial hemoglobin. The number of RBCs in the resulting solution was determined by counting chamber, and the solution diluted in PBS to give a final concentration of 3.33 × 108 cells per mL. Polymers were dissolved in DMSO to achieve stock concentrations of 20 mg mL−1. Serial 2-fold dilutions of polymers were prepared from stock solutions in PBS and polymers (10 μL) were transferred to a 96-well sterile round-bottom polypropylene plate, followed by the RBC suspension (90 μL). Plates were incubated 37 °C with orbital shaking (180 rpm) for 1 hour. Triton X-100 (0.1% v/v in water) was used as the positive lysis control and PBS used as a negative control. Following incubation, the plate was centrifuged at 2000 rpm for 5 minutes. The supernatant (5 μL) from each well diluted in PBS (100 μL) with thorough mixing in a 96-well flat-bottomed polystyrene plate. The absorbance of released hemoglobin (415 nm) was measured using a Varioskan Flash microplate reader (Thermo Fisher). The percentage hemolysis was determined relative to Triton X-100 (100%) and PBS negative control (0%). The polymer concentration causing 50% hemoglobin release (HC50) was determined, and the hemolysis (%) at the highest concentration of polymer (1000 μg mL−1) was reported if below 50%. Assays were repeated a minimum of three times in triplicate.Simulation protocol
Each polymer model (M1 and M2) with DP = 20 (Table 1) was inserted in aqueous phase modelled by TIP3P water.64 Since the net charge of a single polymer chain was +4e, an appropriate number of Na+ and Cl− ions were added to neutralize the system and maintain 150 mM salt concentration for both the model polymers. All simulations were performed with the NAMD 2.9 simulation package.65 Each polymer system was first energy minimized for 1000 steps with the conjugate gradient method and simulations were then performed with a 2 fs timestep in the NPT ensemble for 300 ns. Constant temperature was maintained at 305 K with Langevin dynamics at a collision frequency of 5 ps−1 and a pressure of 1 atm was maintained through Langevin piston.66,67 Electrostatic interactions were calculated by Particle Mesh Ewald method68 and the cut off for non-bonded interactions was set to 12 Å, with smoothing starting from 10 Å. The parameter values for the polymers were adopted from the CHARMM force fields69 and previous simulations.61,70,71 Visual Molecular Dynamics (VMD) with its embedded TCL scripting language71 was used for data visualization and analysis. Structural properties of the polymer can be inferred by measuring the Solvent Accessible Surface Area (SASA).72 The total SASA values of the polymer is calculated with a spherical probe of radius 1.4 A. We compute SASA as a function of time over the 300 ns simulation time.Results and discussion
Polymer design and synthesis
To study the effect of charge balance or net charge on antimicrobial and hemolytic activities, we synthesized a series of ternary statistical methacrylate copolymers with cationic (AEMA), carboxylic (PAMA), and hydrophobic ethyl (EMA) groups in the side chains (Fig. 3). In this study, we used propanoic acid methacrylate (PAMA) instead of the commonly used methacrylic acid, to mimic the side chain of glutamic acid and provide a structural counterpart of the amino ethyl side chain of AEMA. The copolymers were synthesized by free radical polymerization in the presence of 3-methyl mercaptopropionate (MMP) as the chain transfer agent to give low molecular weight polymers (∼2000–3000 g. mol−1) as we demonstrated previously.7 The low molecular weights mimic the small molecular sizes of AMPs. In general, MMP terminates chain propagation, but generate the thiyl radical that initiates new chain propagation. This chain transfer cycle continues to consume all monomers and gives low molecular weight polymers with high yield.7 Our previous studies established that 10 mole percent of MMP relative to the total number of monomers were used, which generally gave the polymer with degree of polymerization (DP) of 15–20.7 This is in good agreement with our current study.14,18,22,43,49,61,70,73 To facilitate the polymerization in organic solvents and avoid side reactions, the protected monomers (N-Boc-AEMA and t-butyl PAMA) were used for the polymerization to give Boc-protected precursor polymers.In our previous study, we demonstrated that ternary copolymers with 40 mol% of hydrophobic ethyl side chains (EMA) showed potent antimicrobial activity against E. coli and low hemolytic activity.49 Therefore, we prepared and studied the copolymers with a fixed 40 mol% EMA, but with different percentages of cationic ammonium (AEMA) and anionic carboxylate (PAMA) side chains, which give different total net charges. The mole percentages of monomers in the resultant protected copolymers were close to the feed compositions, and the percent conversion of the polymerization for selected polymers was >85% (Table S1†). The protected copolymers showed relatively broad molecular weight distributions (dispersity = 1.2–1.7), typical of polymers prepared by a free radical polymerization (Table 2). The N-boc and t-butyl protecting groups of the precursor polymers were simultaneously removed by TFA to yield polymers with ammonium and carboxylic acid groups. The yield for precipitation was decreased from ∼50% to 16% as the mole percentage of PAMA was increased. This is likely because the solubility of the copolymers with high percentages of carboxylic acid in the precipitation solvent (diethyl ether) was increased. However, the monomer compositions and degree of polymerization (DP) of deprotected copolymers are close to those of protected copolymers, suggesting that the precipitation was not selective to monomer compositions or molecular sizes of the copolymers.
| Polymera | The average number of monomers/polymer chains | DP (NMR) | Mnb (NMR) | Mnc (GPC) | Mwc (GPC) | Đc | ||
|---|---|---|---|---|---|---|---|---|
| AEMA | PAMA | EMA | ||||||
|
|
|
||||||
| a The polymers are denoted as AExPAyEz where the numbers x, y, and z represent the average mole percentages of AEMA, PAMA, and EMA in the polymer chain determined by 1H NMR analysis, respectively.b Mn (NMR) was calculated based on the monomer compositions and DP determined by 1H NMR analysis using the molecular weights of monomers and MMP. The molecular weight is based on the chemical formula of copolymer without TFA salt.c The Mn, Mw, and Đ were measured for the protected copolymers in THF solvent. | ||||||||
| AE00PA55E45 | 0.0 | 10.5 | 8.7 | 19.6 | 2770 | 4750 | 6800 | 1.43 |
| AE07PA47E46 | 1.2 | 7.1 | 7.0 | 15.3 | 2190 | 2860 | 3880 | 1.36 |
| AE21PA42E37 | 3.2 | 6.1 | 5.5 | 14.8 | 2130 | 2540 | 3690 | 1.45 |
| AE29PA27E44 | 5.1 | 4.7 | 7.3 | 17.1 | 2360 | 3210 | 3860 | 1.20 |
| AE37PA24E39 | 5.7 | 3.6 | 6.2 | 15.3 | 2140 | 2930 | 3770 | 1.29 |
| AE37PA22E41 | 5.7 | 3.3 | 6.3 | 15.4 | 2100 | 2820 | 5290 | 1.87 |
| AE39PA17E44 | 6.7 | 2.8 | 7.5 | 16.9 | 2290 | 3000 | 5100 | 1.70 |
| AE46PA12E42 | 8.5 | 2.3 | 7.8 | 18.6 | 2480 | 2250 | 3090 | 1.37 |
| AE59PA00E41 | 8.5 | 0.0 | 5.9 | 14.4 | 1900 | 2630 | 3500 | 1.33 |
Antimicrobial activity
The antimicrobial activity of the ternary copolymers was examined by determining the minimum inhibitory concentration (MIC) against Gram-negative Escherichia coli as a model bacterium (Table 3). The copolymers with low mol% (0–30%) of cationic AEMA did not show any antimicrobial activity (MIC >1000 μg mL−1). The MIC values decreased as AEMA mol% was increased or PAMA mol% was decreased but leveled off at AEMA mol% higher than 40%. To better understand the role of charges of the polymers in their antimicrobial activity, the relationship between the MIC values and net charge of the copolymer was examined (Table 3 and Fig. 4). The net charge was determined based on the monomer compositions of the polymers as the net charge = the number of AEMA − the number of PAMA in a polymer chain. The copolymers with net negative charges or the equal number of both charges did not show any activity (MIC > 1000 μg mL−1). It has been reported that the random copolymers with the same number of cationic and anionic groups in the same polymer chains prevented the adhesion of bacteria.74–77 The result of our study is in good agreement with the previous reports. For the copolymers with net positive charges, the MIC values decreased as the number of net positive charges was increased, but leveled off at the MIC of 31 μg mL−1 at the net positive charges larger than +3.9. As we demonstrate later, the pKa values of ammonium groups were ∼8, and thus a fraction of ammonium groups (–NH3+) were deprotonated to give the non-ionic base form (–NH2). The net charge was corrected based on the fraction of cationic ammonium groups at pH 7.4 which is calculated using the pKa values of copolymers (Table 3 and Fig. 5). It is also interesting that the MIC values decreased significantly when the net cationic charge increased from 2 to ∼3. The data suggests that the net positive charge greater than +3 is sufficient and essential to maximize the antimicrobial activity of the copolymers (MIC = 31 μg mL−1). The potentiometric titration and pKa values will be discussed later.| Polymer | MIC (μg mL−1) | HC50 (μg mL−1) | Net chargea | ||
|---|---|---|---|---|---|
| E. coli | S. aureus | Monomer compositionb | Correctedc | ||
| a Expected net charge of polymers in solution pH 7.4.b Net charge is reported as the difference in average number of monomers of AEMA and PAMA in a polymer chain determined by 1H NMR.c Corrected net charge calculated using equation based on the pKa values of AEMA and PAMA of the corresponding copolymer. The pKa values were determined by the titration curve of each copolymer (Fig. 4A and Table 4). “n.d.” indicates that titration curves were not obtained because of low solubility of polymers or low availability. | |||||
| AE0PA55E45 | >1000 | >1000 | n.d. | −10.5 | n.d. |
| AE7PA47E46 | >1000 | >1000 | >1000 (−0.7 ± 0.8%) | −5.9 | n.d. |
| AE21PA42E37 | >1000 | >1000 | >1000 (1.6 ± 1.1%) | −2.9 | −3.0 |
| AE29PA27E44 | >1000 | >1000 | >1000 (4.0 ± 1.7%) | +0.4 | +0.1 |
| AE37PA24E39 | 250 | >1000 | >1000 (3.7 ± 1.0%) | +2.1 | n.d. |
| AE37PA22E41 | 125 | >1000 | >1000 (14.1 ± 4.0%) | +2.4 | +2.0 |
| AE39PA17E44 | 31 | 500 | 740 ± 110 | +3.9 | +3.1 |
| AE46PA12E42 | 31 | 250 | 320 ± 130 | +6.2 | +3.4 |
| AE59PA0E41 | 31 | 125 | n.d. | +8.5 | +6.3 |
| AE34HE29E37 [ref. 49] | 31 | 1000 | 1000 | +6.5 | n.d. |
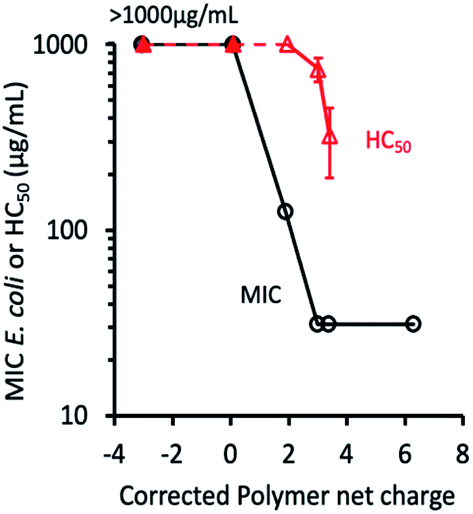 | ||
| Fig. 4 Antimicrobial and hemolytic activities activity of the copolymers. The MIC and HC50 values were plotted as a function of corrected net charges on the polymer. The MIC values for the polymers that measured above the highest polymer concentration used (>1000 μg mL−1) were denoted by (- -●- -). The values HC50 >1000 μg mL−1 were denoted by (- -▲- -). | ||
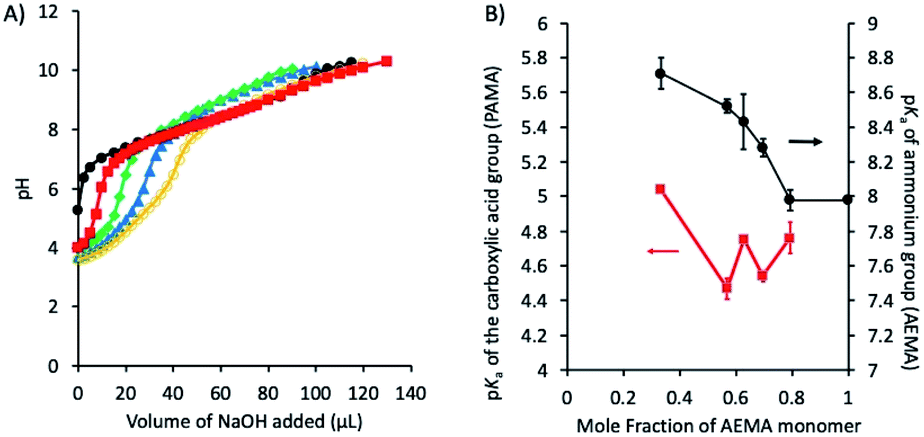 | ||
| Fig. 5 (A) Representative potentiometric titration curves of copolymers AE21PA42E37 (○), AE29PA27E44 (▲), AE37PA22E41 (◆), AE46PA12E42 (■) and AE59PA0E41 (●) with NaOH (0.1 N) at 25 °C in 150 mM NaCl solution. (B) pKa values of PAMA (pKa1) (-■-) and AEMA (pKa2) (-●-) in the polymer side chains as a function of mole fraction of AEMA monomer relative to the total number of monomers defined as AEMA/(AEMA + PAMA). | ||
Yang and coworkers previously designed pH-responsive antimicrobial methacrylate random copolymers having both cationic ammonium and carboxylic acid groups in the side chains.78 These copolymers were inactive at neutral pH, but the COO– groups were protonated to be neutral carboxylic acid (COOH) at low pH, so that the net charge of the copolymers becomes positive due to the cationic ammonium groups, and thus the copolymers showed bactericidal activity. This result supports our finding that the antimicrobial activity of the copolymers depends on their net positive charges. The threshold net charge of +3 of the copolymers for antimicrobial activity are comparable to those of naturally occurring antimicrobial peptides magainin (+4) and LL-37 (+6).79 This may indicate that these net positive charges are optimal for their antimicrobial mechanism including binding to the anionic cell membrane of E. coli and membrane disruption. In addition, the presented result is also in good agreement with our previous study.49 We previously examined the antimicrobial activity of ternary copolymers with ammonium (AEMA), hydroxyl (HEMA) and ethyl (EMA) side chains.49 The MIC value of copolymers with HEMA was also increased as the number of positive charges was increased, and level off at around 4 to 6 positive charges, which is very similar to that of the copolymers with PAMA studied here. The ternary copolymers with PAMA studied here and those with HEMA showed the same MIC value of 32 μg mL−1, indicating that PAMA did not improve the antimicrobial activity of the copolymers as compared to those with HEMA. These results suggest the net positive charge, but not acidic groups, is the determinant factor in the antimicrobial activity against E. coli.
We further examined the antimicrobial activity of the copolymers against Gram-positive S. aureus. The MIC value decreased as the net positive charge was increased (Table 3). However, the MIC values are 125–500 μg mL−1, which are substantially larger than those for E. coli, suggesting these copolymers are not effective in inhibiting S. aureus growth. In our previous study, the copolymers with HEMA in the side chains instead of anionic PAMA showed similar higher MIC values against S. aureus.49
Hemolytic activity
It is important to assess the biocompatibility of antimicrobial agents to mammalian cells for clinical applications. Because the polymers are expected to act by disrupting bacterial cell membranes, we determined lysis of human erythrocytes (haemolysis) caused by the polymers as a measure of toxicity, which reflects the damages to the mammalian cell membrane. We investigated the effect of variation of net charge and monomer composition on the hemolytic activity of the copolymers (Table 4 and Fig. S15†). The copolymers with net negative charges and low net positive charges did not show any significant hemolytic activity. The copolymers with higher net positive charges (AE39PA17E44 and AE46PA12E42) showed significant hemolytic activity, and the HC50 values increased as the net positive charge was increased (Fig. 5B). These results suggest that increasing the net positive charge of copolymers is also a driving force for hemolytic activity. This may be counterintuitive because one may think that the copolymers with more cationic charges are more hydrophilic so that the polymer chains should not bind to the cell membranes of red blood cells (RBCs). However, the RBC membrane contains anionic components such as phosphatidylserine lipids and silylated glycoproteins, which confer a negatively charged surface.80–82 Therefore, it is reasonable that the copolymers with high net positive charges will bind strongly to the RBC surfaces, causing membrane lysis (hemolysis).| Polymers | pKa1 (COOH) | pKa2 (NH3+) | Net charge | Corrected net charge | +/− ratio |
|---|---|---|---|---|---|
| a Titration was measured at polymer concentration of 0.5 mg mL−1 due to low availability. | |||||
| AE21PA42E37 | 5.10 ± 0.16 | 8.70 ± 0.18 | −2.9 | −3.0 | 0.7 |
| AE29PA27E44 | 4.54 ± 0.06 | 8.59 ± 0.03 | +0.4 | +0.1 | 1.1 |
| AE37PA22E41 | 4.75 ± 0.03 | 8.49 ± 0.06 | +2.4 | +2.0 | 1.7 |
| AE39PA17E44a | 4.56 ± 0.06 | 8.31 ± 0.06 | +3.9 | +3.1 | 2.3 |
| AE46PA12E42 | 4.78 ± 0.04 | 7.98 ± 0.10 | +5.2 | +3.4 | 3.8 |
| AE59PA0E41 | — | 7.98 ± 0.02 | +8.5 | +6.3 | — |
We have previously reported that the binary copolymer without the anionic groups PAMA (AE45HE0E55 in ref. 49) that has +6.7 and 8.3 EMA units showed the MIC of 7.8 μg mL−1 against E. coli and HC50 of 35 μg mL−1. Compared to the polymer with the similar numbers of cationic and hydrophobic groups AE39PA17E44, the binary polymer was substantially more activity against E. coli, but very toxic to human cells. However, the ternary copolymers with the anionic groups tend to show higher hemolytic activity as compared to the copolymers with HEMA, instead of PAMA, in our previous report.49 For example, one of the copolymers with HEMA, AE34HE29E37 in our previous study49 has the net charge of +6.5, the number of EMA of 7.0, and DP of 19, which are very similar to the copolymer with PAMA, AE46PA12E42, these copolymers displayed the same MIC value of 31 μg mL−1, but HC50 of AE34HE29E37 was 1000 μg mL−1, while that of AE46PA12E42 in this study was 320 μg mL−1. This result suggests that although the antimicrobial activity against E. coli is primarily determined by their net positive charge, the hemolytic activity of copolymers is dependent on their chemical functionalities of monomer compositions (carboxylic vs. hydroxyl groups).
We previously proposed that the mildly polar hydroxyl groups may not be favorable for the polymer insertion into the RBC membranes, resulting in low hemolytic activity. Although the molecular mechanism is beyond the scope of this study, we speculate that the anionic carboxylic side chains form a salt bridge with the cationic side chains when the copolymers interact with the RBC membrane. In general, the insertion of charged polar side chains into the non-polar hydrophobic region of the RBC membrane is considered unfavorable. However, the salt bridge between a pair of oppositely charged groups would reduce the polarity of the ionic groups, which might enhance the insertion of the polymer chains into the RBC membrane, thus resulting in the higher hemolytic activity than the counterpart polymers with hydroxyl groups. A model peptide study indicated that the salt bridge is more thermodynamically stable when the peptide penetrates deeper into the bilayer,83 which supports our proposed mechanism.
Potentiometric titration
In order to gain a better understanding of the solution properties of the polymers, we first studied the ionization states of primary ammonium and carboxylic groups of the ternary copolymers. We conducted potentiometric titrations of selected polymers using 0.1 N NaOH as a base. The potentiometric titration curve indicated that the pKa of ammonium groups of the binary cationic copolymer (AE59PA0E41) was 7.9, which is close to the reported value (pKa = 7.6) of AEMA homopolymer.84 The potentiometric titration of binary anionic copolymer consisting of EMA and PAMA could not be performed because the copolymer was not sufficiently soluble in water to carry out the titration. The analysis of titration curves showed that the carboxylic acid groups were first deprotonated by the base, then the ammonium groups reacted with the base, resulting in two distinct pKa values (pKa1 and pKa2) (Fig. 5A and Table 4). More specifically, at physiological pH 7.4 used for the antimicrobial and hemolysis assays, the polymer chains have both negative and positive charges although a faction of ammonium groups are deprotonated to be non-ionic basic amine form (–NH2) for those with the pKa values below 8. According to the titration results, the ionization states of copolymers can be described by the following:
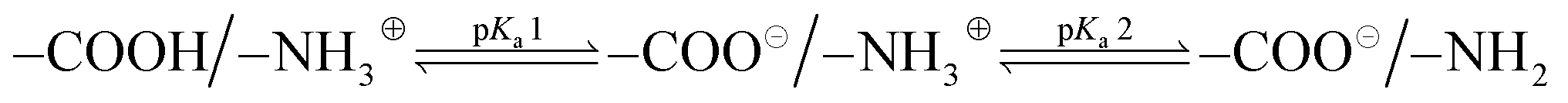 | (2) |
The pKa of the ammonium groups of the ternary polymers (pKa2) was decreased from 8.70 to 7.98 as the mole fraction of the ammonium AEMA monomer was increased from 0.34 to 1. This indicates that the cationic ammonium groups were deprotonated at lower pH as the fraction of AEMA became larger. This can be explained by the electrostatic repulsion between the neighboring cationic side chains, which discourages the protonation of amine groups as they are closer when the mole fraction of AEMA is larger. In addition, because the EMA composition is fixed, the mole fraction of anionic carboxylate groups (PAMA) was decreased as that of the cationic ammonium groups (AEMA) was increased. Therefore, when the mole fraction of AEMA is small, a small fraction of cationic ammonium groups is surrounded by many anionic carboxylate groups. This could enhance the formation of electrostatic association of these groups or salt bridge, which encourages the protonation of amine groups, thus resulting in larger pKa2. Similarly, the pKa of the carboxylic acid (–COOH) groups of the polymers (pKa1) was decreased from 5.10 to 4.54–4.86 as the mole fraction of PAMA was decreased from 0.66 to 0.21. This can be also explained by the repulsive interactions between the anionic side chains and salt bridge as described for the pKa2 of ammonium side chains above. However, we cannot distinguish or determine the contribution of each individual effect to the bulk pKa values.
Computational modeling
We performed atomistic MD simulations with explicit water and ions on the ternary copolymers to investigate the interactions between the cationic and anionic groups of the copolymers and understand their roles in the polymer conformation. Based on the results of the biological assays (Table 3), AE39PA17E44 showed the potent antimicrobial activity and selectivity; we selected this polymer as a model and constructed a model random sequence (M1) consisting of 8 AEMA groups (A), 8 EMA groups (E), and 4 PAMA groups (P): A–E–P–A–E–P–E–A–E–P–E–A–A–E–A–E–A–A–E–P (Fig. 6A). We hypothesized that a pair of oppositely charged carboxylate (PAMA) and ammonium groups (AEMA) forms salt bridge, which stabilizes specific polymer conformations in solution. To that end, we first examined the global shape and stability of the polymer conformation through monitoring the radius of gyration, Rg, defined as the root mean square distance of the constituents of the polymers from its center of mass (Fig. 6B).85 The Rg values and the representative snapshots showed that the polymer dynamically switches between compact and extended conformations, showing a compact ring-like structure in the last 50 ns of simulation time.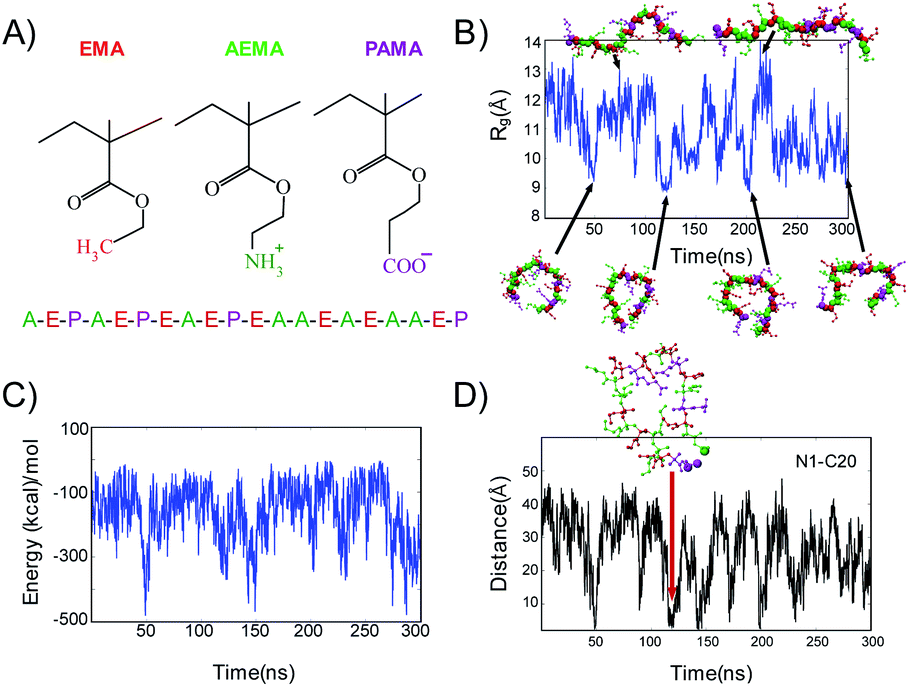 | ||
| Fig. 6 Polymer structure and conformations. (A) Chemical structure of EMA, AEMA and PAMA groups considered in the model polymers. The constituent functional groups of the polymer are illustrated as – hydrophobic (EMA, red); cationic (AEMA, green); anionic (PAMA, magenta). (B) Radius of gyration (Rg) for the model polymers (M1) as a function of simulation time. Representative snapshots to illustrate the conformations of the polymers are also presented, with arrows indicating the corresponding simulation time and Rg values. (C) Total electrostatic energy between the anionic PAMA groups and cationic AEMA groups is plotted as a function of time. (D) Time evolution of distance between the nitrogen atom of NH3+ in AEMA group (N1) and the carboxyl oxygen atoms of COO– (C20). A representative snapshot of the polymer is shown with arrows pointing to the corresponding distance between nitrogen (shown as green ball) and oxygen atoms (shown as magenta balls) and simulation time. | ||
Next, we examined the role of electrostatic interactions between the anionic carboxylate and cationic ammonium groups, which implicates salt bridge formation, and its relationship with the polymer conformation. For this, we first computed the total electrostatic energy between these two groups (Fig. 6C). The total energy indicated negative values (<−100 kcal mol−1) during the entire time of simulation, suggesting that these groups show attractive interactions. The polymer showed relatively consistent energy values of ∼−100 kcal mol−1, but with intermittent steep valleys of larger negative values, which coincide with the times when the polymer conformation is compact. This result strongly suggests that the role of intramolecular electrostatic interactions is most likely to control the formation of the ring-like compact conformation as well as the dynamic switching between the compact and extended conformations.
Salt bridge formation
Accordingly, we hypothesized that a pair of the oppositely charged groups of the polymer chain formed a salt bridge and crosslinked the polymer chain, resulting in the compact conformation. To test this hypothesis, we investigated salt bridge formation along the polymer chain. Given the polymer chain contains 8 positively (AEMA) and 4 negatively (PAMA) charged monomers in the polymer chain, there can be many possible pairs involved in salt bridges. As a representative case, we looked into all the salt bridges formed by the carboxylic group C10, located in the center of the polymer chain, with all cationic groups in the polymer chain. In this study, we defined salt bridge formation when the distance between the nitrogen atom of (NH3+) and the two carboxyl oxygen atoms of (COO−) is less than 4.0 Å.86 C10 formed salt bridges with neighboring cationic groups N8 and N13 frequently, but occasionally also with sequentially distant N4 and N18 for small periods of time (Fig. 7). This result suggests that the salt bridge formation is dependent on the distance between the pair groups and that each salt bridge was intermittently formed, and the pairs changed constantly, resulting in transient crosslinking of the polymer chain.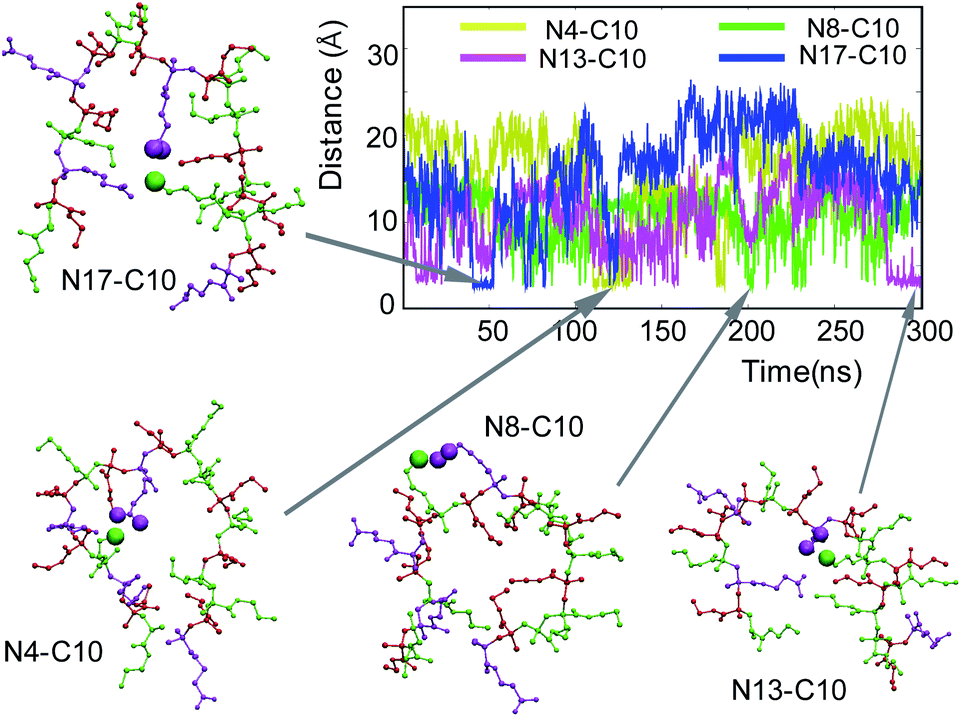 | ||
| Fig. 7 Salt bridge formation between C10 and cationic ammonium groups. | ||
We propose that the formation and stability of salt bridge are determined by the balance between the attractive electrostatic interactions between the oppositely charged groups and the repulsive forces owing to solvation and polymer chain constraint. The favorable formation of salt bridge between the neighboring groups is reasonable because they are spatially close, and thus salt bridge doesn't require large changes in the polymer conformation. However, C10 also formed a salt bridge with sequentially distant N4 and N18, likely due to the compact polymer conformation, which brings these groups close enough to C10, facilitating the salt bridge formation. Alternatively, the salt bridge-mediated crosslinking of polymer chains also contributed to the formation of the compact polymer conformation.
Ring-like compact conformation
The model polymer M1 formed a very characteristic ring-like structure, which is likely to require a specific attraction force between the oppositely charged pair of N1 and C20 at the polymer chain terminal. Indeed, these groups, which are the most sequentially distant pair in the polymer chain, formed a salt bridge (Fig. 6D), but it underwent a periodic cycle of forming and breaking. The compact polymer conformation appears to be formed concomitantly with the salt bridge formation between N1 and C20, suggesting that the salt bridge contributed to the ring-like compact conformation. To further confirm this, we constructed another model sequence M2: A–E–A–A–E–P–E–A–E–P–E–A–A–E–P–E–A–P–E–A, in which, the sequence at the terminal regions were scrambled, and both terminal ends have cationic groups, in order to disturb the N1–C20 salt bridge formation seen in M1. M2 tends to adopt more extended conformation without any salt bridge between the terminal groups (Fig. S17†), as compared to M1, and the salt bridge formation between neighboring groups was more consistent (longer lifetimes), indicating good stability (Fig. S18†). This supports our conclusion on the effect of the salt bridge between the terminal groups on the ring-like conformation. In addition, this result also suggests that the ring-like compact structure is not common to all monomer sequences and is likely to be sequence dependent. However, both M1 and M2 adopted relatively compact conformations and underwent the dynamic switching between the compact and extended conformations. The results from these different sequences suggest that the salt bridge functions as a transient crosslinker to control the polymer conformation. In addition, the plot of the distance between various possible ion-pairs showed overlaps of different pairs (different colors) at <4 Å (Fig. 7). The snapshots of representative polymer conformations indicate that 2–3 salt bridges exist at the same time (Fig. 7). This result suggests that the polymer chain is crosslinked by multiple transient salt bridges. While the crosslinking by salt bridges similar to natural peptides and proteins, the chains of copolymers synthesized in this study are not likely to form any defined conformations such as α-helix or β-sheet because of their flexible backbone and random sequences.87,88Solvent accessible surface area
Solvent accessible surface area. Natural proteins exclude water molecules during folding and form a hydrophobic environment in the inside of their globular conformations. We hypothesized that the polymer chain also might exclude water molecules when the polymer conformation is compact. To test the hypothesis, we determined the time evolution of solvent accessible surface area (SASA) of M1 (Fig. S19†), which represents the surface area of the polymer chain that is accessible by water molecules. Interestingly, the SASA value steeply dropped to much smaller values when the polymer adopted the ring-like compact conformation that was facilitated by the formation of the N1–C20 salt bridge. This result suggests that a larger part of the polymer surface squeezed out water molecules from the compact conformation, which supports our earlier hypothesis of a hydrophobic environment created inside the ring like structure. This behavior is similar to natural globular proteins; however, the formation of compact polymer conformation was only intermittent, and not stable for a long period of time, which may be due to the lack of strong hydrophobicity in the polymer side chains. Because the general solvation of ionic groups may preclude the formation of salt bridges, we speculate that such water exclusion may facilitate the intra-polymer salt bridge formation.The result in Fig. 8 suggests that multiple salt bridges are simultaneously formed in the polymer chain. Taking together these results, the combination of water exclusion and shorter distances between the oppositely charged groups in the compact polymer conformation might enhance the formation and stability of salt bridge, allowing the simultaneous formation of multiple salt bridges. Because the compact polymer conformation is created by salt bridges, and the created polymer environment is favorable to the salt bridge formation, it is also possible that the formation of multiple salt bridges is a result of autocatalytic reactions; the formation of one salt bridge facilitates the formation of another salt bridge.
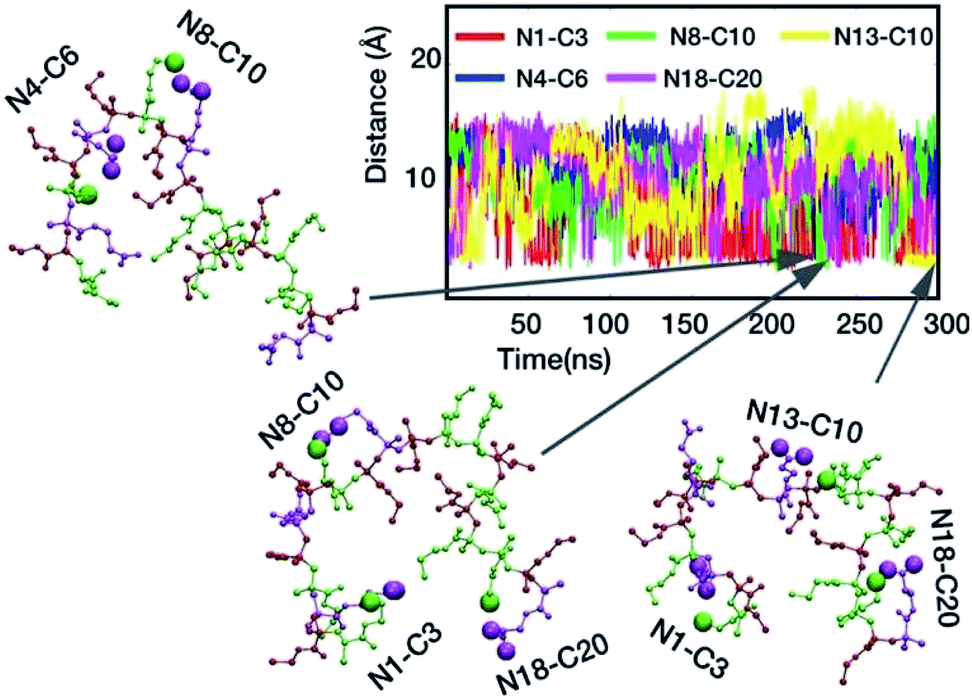 | ||
| Fig. 8 Multiple salt bridge formation. | ||
The functional roles of salt bridge
The biological assay results suggest that there is no distinct effect of the anionic carboxylate groups (PAMA) on the antimicrobial and hemolytic activities of the polymers; the net positive charge was the determinant for these activities. The pH titration and computational simulations suggest that a pair of cationic and anionic groups formed salt bridge and transiently crosslinked the polymer chain to control the polymer conformation and dynamic switching of compact and extended conformations. However, the salt bridges did not render the polymers to adopt a specific polymer conformation that lasts throughout the simulation time scale, though there are clear indications that transient compact structures and formation of transient salt bridges are closely related.The previous study on antimicrobial peptides indicated that, while mammalian α-defensins have diverse sequences, the Arg5–Glu13 salt bridge is highly conserved.89 This study demonstrated that the bactericidal activity of six defensin derivatives was correlated with the number of net positive charges, irrespective of whether or not the salt bridge was removed.89 The same study also showed that the defensins are more susceptible to enzymatic degradation when the salt bridge is destabilized.89,90 Therefore, it has been proposed that the salt bridge in α-defensins is conserved for their in vivo stability of mature α-defensin. Another study on α-defensins also demonstrated that salt-bridge variants retained bactericidal peptide activity, confirming that the salt-bridge does not intrinsically determine bactericidal activity.91 The salt bridge also stabilized the peptide conformation, which confers resistance to degradation by proteases. In addition, the spectroscopy study also suggested that stabilizing salt bridges speeded up α-helix formation.92 This may explain the role of apparently nonfunctional (destabilizing) salt bridges in many proteins.91
Our results of the ternary copolymers on the antimicrobial activity are in good agreement with these previous reports in α-defensins. The random copolymers of this study were not designed to adopt any specific secondary structural conformations, and the salt bridge did not stabilize any specific conformation. In addition, the carbon–carbon backbone of methacrylate polymers is not subject to enzymatic degradation. Therefore, while the salt bridge plays in peptide folding and stability, the salt bridge of the polymer chains may not directly contribute to the antimicrobial activity, which is in good agreement with our results.
Conclusion
In summary, we investigated the functional and structural roles of amino acid-mimetic anionic side chains of cationic amphiphilic ternary copolymers in their antimicrobial and hemolytic activities and polymer conformations. The carboxylate groups did not improve the antimicrobial activity against E. coli as compared to the hydroxyl counterparts, but the hemolytic activity is higher in carboxylate copolymers compared to the hydroxyl ones. MD simulations showed that a pair of oppositely charged side chains formed a salt bridge, and the salt bridge provided transient cross-linking of polymer chains, which controlled dynamics of compact and extended polymer conformations. In addition, the compact polymer conformation excluded water molecules, implying the formation of hydrophobic environment, which is similar to natural proteins. However, the salt bridge did not facilitate adoption or stabilization of a specific conformation. These results suggest that salt bridges do not directly determine their antimicrobial activity and selectivity through their functional and structural roles. The previous study also suggested that salt bridges of α-defensins does not directly control the antimicrobial activity.91 Inclusion of anionic amino acids and salt bridges to simulate the AMPs may not directly contribute to the design of antimicrobial polymers with potent activity and selectivity.One may question whether an anionic monomer and salt bridges are still useful for us to consider as an AMP-mimetic functionality or not. The study on α-defensin demonstrated that the salt bridge facilitated the formation of correct disulfide pairs in the peptide, which are essential for the folding to the active peptide conformation.90 Therefore, we envision that the salt bridge in polymer chains may also work as a provisional “adhesive” which would allow the polymer chains to be compact, but without rigidly locking the structure. This unlocked chain compaction may be able to bring specific groups physically close, facilitating the covalent bond formation. In addition, our result also indicated the compact polymer conformation provide a non-polar environment. Because the molecular size of the copolymers used in this study is relatively small, it may not be feasible to directly observe or quantify the compact form of polymer chains by analytical methods such as electron microscopy93,94 or dynamic light scattering.95 However, it may be possible to use the formation of non-polar environment as a measure of compaction of polymer chains.96 The non-polar environment may also enhance chemical reactions for covalent bond formation because of lack of solvation.97 It has been also reported that the presence of anionic residues can promote hetero-oligomerization between antimicrobial peptides PGLa and magainin-2 on bacterial cell membranes, promoting pore formation,98 which leads to synergistic antimicrobial activity. Similarly, while we investigated in this study the conformations of single polymer chains in solution, we might be able to design the anionic groups of polymers as to enhance the interactions between polymer chains and the formation of polymer assembly for potent antimicrobial activity.
Natural AMP sequences contain many different types of amino acids, and the specific functions of these sequences are orchestrated. However, most of our current biomimetic polymer design is focused only on a few functional groups, the most obvious ones being hydrophobic and cationic. Perhaps we are just scratching the surface of our understating of how to mimic the functions of evolutionarily optimized AMP sequences and the role played by these different groups both in their antimicrobial function and the requisite structural stability for the function. Further studies are needed to implement the knowledge into our AMP-mimetic design of synthetic polymers for potent antimicrobial activity and selectivity.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by National Science Foundation (DMR-2004305 BMAT to K. K.), the Department of Biologic and Materials Sciences & Prosthodontics, School of Dentistry, University of Michigan, Rackham Predoctoral Fellowship from the University of Michigan (L. L. F.), MCube (Cost-effective novel antibiotics to combat multidrug-resistant bacteria) from University of Michigan, and the National Science Graduate Research Fellowship (L. L. F. no. DGE-1315231). Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the National Science Foundation. We also thank School of Dentistry, University of Michigan for bridging support, University of Michigan Office of Research for funding for Small scale and preliminary projects, and Macromolecular Science and Engineering Center, University of Michigan for financial support to L. L. F. The simulations were carried out on the supercomputing machines Annapurna and Nandadevi at the Institute of Mathematical Sciences, Chennai India. We also thank Dr Gregory Caputo at Rowan University for discussion.Notes and references
- S. B. Levy and B. Marshall, Nat. Med., 2004, 10, S122–S129 CrossRef CAS PubMed.
- C. Llor and L. Bjerrum, Ther. Adv. Drug Saf., 2014, 5, 229–241 CrossRef PubMed.
- I. Levin-Reisman, I. Ronin, O. Gefen, I. Braniss, N. Shoresh and N. Q. Balaban, Science, 2017, 355, 826–830 CrossRef CAS PubMed.
- B. Khameneh, R. Diab, K. Ghazvini and B. S. Fazly Bazzaz, Microb. Pathog., 2016, 95, 32–42 CrossRef CAS PubMed.
- O. G. Travkova, H. Moehwald and G. Brezesinski, Adv. Colloid Interface Sci., 2017, 247, 521–532 CrossRef CAS PubMed.
- H. Long, S. F. Miller, C. Strauss, C. Zhao, L. Cheng, Z. Ye, K. Griffin, R. Te, H. Lee, C. C. Chen and M. Lynch, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E2498–E2505 CrossRef CAS PubMed.
- L. L. Foster, M. Mizutani, Y. Oda, E. F. Palermo and K. Kuroda, in Polymers for Biomedicine, 2017, pp. 243–272 Search PubMed.
- M. M. Konai, B. Bhattacharjee, S. Ghosh and J. Haldar, Biomacromolecules, 2018, 19, 1888–1917 CrossRef CAS PubMed.
- C. Ergene, K. Yasuhara and E. F. Palermo, Polym. Chem., 2018, 9, 2407–2427 RSC.
- E. Chee and A. C. Brown, Biomater. Sci., 2020, 8, 1089–1100 RSC.
- X. Zhou, J. He and C. Zhou, Polym. Chem., 2019, 10, 945–953 RSC.
- N. F. Kamaruzzaman, L. P. Tan, R. H. Hamdan, S. S. Choong, W. K. Wong, A. J. Gibson, A. Chivu and M. d. F. Pina, Int. J. Mol. Sci., 2019, 20, 2747 CrossRef CAS PubMed.
- A. C. Engler, N. Wiradharma, Z. Y. Ong, D. J. Coady, J. L. Hedrick and Y.-Y. Yang, Nano Today, 2012, 7, 201–222 CrossRef CAS.
- E. F. Palermo and K. Kuroda, Biomacromolecules, 2009, 10, 1416–1428 CrossRef CAS PubMed.
- B. Bechinger and S. U. Gorr, J. Dent. Res., 2017, 96, 254–260 CrossRef CAS PubMed.
- E. F. Palermo, I. Sovadinova and K. Kuroda, Biomacromolecules, 2009, 10, 3098–3107 CrossRef CAS PubMed.
- R. J. Kopiasz, W. Tomaszewski, A. Kuzminska, K. Chreptowicz, J. Mierzejewska, T. Ciach and D. Janczewski, Macromol. Biosci., 2020, e2000063, DOI:10.1002/mabi.202000063.
- K. Kuroda and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 4128–4129 CrossRef CAS PubMed.
- M. S. Ganewatta and C. B. Tang, Polymer, 2015, 63, A1–A29 CrossRef CAS.
- Y. Yang, Z. Cai, Z. Huang, X. Tang and X. Zhang, Polym. J., 2018, 50, 33–44 CrossRef CAS.
- C. Peng, A. Vishwakarma, S. Mankoci, H. A. Barton and A. Joy, Biomacromolecules, 2019, 20, 1675–1682 CrossRef CAS PubMed.
- Y. Oda, S. Kanaoka, T. Sato, S. Aoshima and K. Kuroda, Biomacromolecules, 2011, 12, 3581–3591 CrossRef CAS PubMed.
- A. Kuroki, P. Sangwan, Y. Qu, R. Peltier, C. Sanchez-Cano, J. Moat, C. G. Dowson, E. G. L. Williams, K. E. S. Locock, M. Hartlieb and S. Perrier, ACS Appl. Mater. Interfaces, 2017, 9, 40117–40126 CrossRef CAS.
- M. Rauschenbach, S. B. Lawrenson, V. Taresco, A. K. Pearce and R. K. O'Reilly, Macromol. Rapid Commun., 2020, 2000190 CrossRef CAS PubMed.
- M. Chen, M. Hu, D. Wang, G. Wang, X. Zhu, D. Yan and J. Sun, Bioconjugate Chem., 2012, 23, 1189–1199 CrossRef CAS PubMed.
- W. J. Yang, K.-G. Neoh, E.-T. Kang, S. Lay-Ming Teo and D. Rittschof, Polym. Chem., 2013, 4, 3105–3115 RSC.
- W. Zheng, M. Anzaldua, A. Arora, Y. Jiang, K. McIntyre, M. Doerfert, T. Winter, A. Mishra, H. Ma and H. Liang, Biomacromolecules, 2020, 21, 2187–2198 CrossRef CAS PubMed.
- A. L. Garle and B. M. Budhlall, Langmuir, 2019, 35, 3372–3382 CrossRef CAS PubMed.
- C. Yang, W. Lou, G. Zhong, A. Lee, J. Leong, W. Chin, B. Ding, C. Bao, J. P. K. Tan, Q. Pu, S. Gao, L. Xu, L. Y. Hsu, M. Wu, J. L. Hedrick, W. Fan and Y. Y. Yang, Acta Biomater., 2019, 94, 268–280 CrossRef CAS.
- S. Laroque, M. Reifarth, M. Sperling, S. Kersting, S. Klöpzig, P. Budach, J. Storsberg and M. Hartlieb, ACS Appl. Mater. Interfaces, 2020, 12(27), 30052–30065 CrossRef CAS.
- T.-K. Nguyen, S. J. Lam, K. K. K. Ho, N. Kumar, G. G. Qiao, S. Egan, C. Boyer and E. H. H. Wong, ACS Infect. Dis., 2017, 3, 237–248 CrossRef CAS PubMed.
- R. Namivandi-Zangeneh, R. J. Kwan, T.-K. Nguyen, J. Yeow, F. L. Byrne, S. H. Oehlers, E. H. H. Wong and C. Boyer, Polym. Chem., 2018, 9, 1735–1744 RSC.
- G. Wei, D. Nguyen, S. Reghu, J. Li, C. Chua, Y. Ishida and M. B. Chan-Park, ACS Appl. Nano Mater., 2020, 3, 2654–2664 CrossRef CAS.
- M. A. Rahman, M. Bam, E. Luat, M. S. Jui, M. S. Ganewatta, T. Shokfai, M. Nagarkatti, A. W. Decho and C. Tang, Nat. Commun., 2018, 9, 5231 CrossRef CAS PubMed.
- J. Koehbach and D. J. Craik, Trends Pharmacol. Sci., 2019, 40, 517–528 CrossRef CAS PubMed.
- R. I. Lehrer and W. Lu, Immunol. Rev., 2012, 245, 84–112 CrossRef CAS PubMed.
- E. Deplazes, Y. K. Y. Chin, G. F. King and R. L. Mancera, Proteins: Struct., Funct., Bioinf., 2020, 88, 485–502 CrossRef CAS PubMed.
- M. Pazgier, D. M. Hoover, D. Yang, W. Lu and J. Lubkowski, Cell. Mol. Life Sci., 2006, 63, 1294–1313 CrossRef CAS PubMed.
- S. Persson, J. A. Killian and G. Lindblom, Biophys. J., 1998, 75, 1365–1371 CrossRef CAS.
- H. Sun, D. V. Greathouse, O. S. Andersen and R. E. Koeppe 2nd, J. Biol. Chem., 2008, 283, 22233–22243 CrossRef CAS PubMed.
- W. M. Yau, W. C. Wimley, K. Gawrisch and S. H. White, Biochemistry, 1998, 37, 14713–14718 CrossRef CAS PubMed.
- D. I. Chan, E. J. Prenner and H. J. Vogel, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1184–1202 CrossRef CAS.
- H. Takahashi, G. A. Caputo, S. Vemparala and K. Kuroda, Bioconjugate Chem., 2017, 28, 1340–1350 CrossRef CAS.
- M. Hartlieb, E. G. L. Williams, A. Kuroki, S. Perrier and K. E. S. Locock, Curr. Med. Chem., 2017, 24, 2115–2140 CrossRef CAS PubMed.
- K. E. S. Locock, T. D. Michl, N. Stevens, J. D. Hayball, K. Vasilev, A. Postma, H. J. Griesser, L. Meagher and M. Haeussler, ACS Macro Lett., 2014, 3, 319–323 CrossRef CAS.
- A. Kuroki, A. Kengmo Tchoupa, M. Hartlieb, R. Peltier, K. E. S. Locock, M. Unnikrishnan and S. Perrier, Biomaterials, 2019, 217, 119249 CrossRef CAS PubMed.
- L. P. Datta, D. Dutta, A. Chakraborty and T. K. Das, Biomater. Sci., 2019, 7, 2611–2622 RSC.
- S. Chakraborty, R. Liu, Z. Hayouka, X. Chen, J. Ehrhardt, Q. Lu, E. Burke, Y. Yang, B. Weisblum, G. C. L. Wong, K. S. Masters and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 14530–14535 CrossRef CAS PubMed.
- H. Mortazavian, L. L. Foster, R. Bhat, S. Patel and K. Kuroda, Biomacromolecules, 2018, 19, 4370–4378 CrossRef CAS PubMed.
- U. H. N. Dürr, U. S. Sudheendra and A. Ramamoorthy, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1408–1425 CrossRef PubMed.
- J. Johansson, G. H. Gudmundsson, M. E. Rottenberg, K. D. Berndt and B. Agerberth, J. Biol. Chem., 1998, 273, 3718–3724 CrossRef CAS.
- A. J. Wommack, S. A. Robson, Y. A. Wanniarachchi, A. Wan, C. J. Turner, G. Wagner and E. M. Nolan, Biochemistry, 2012, 51, 9624–9637 CrossRef CAS PubMed.
- Z. Wu, X. Li, E. de Leeuw, B. Ericksen and W. Lu, J. Biol. Chem., 2005, 280, 43039–43047 CrossRef CAS PubMed.
- S. Marqusee and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8898–8902 CrossRef CAS PubMed.
- P. F. Almeida and A. Pokorny, Biochemistry, 2009, 48, 8083–8093 CrossRef CAS PubMed.
- B. Mojsoska and H. Jenssen, Pharmaceuticals, 2015, 8, 366–415 CrossRef CAS PubMed.
- C. A. Olson, E. J. Spek, Z. S. Shi, A. Vologodskii and N. R. Kallenbach, Proteins, 2001, 44, 123–132 CrossRef CAS PubMed.
- J. S. Smith and J. M. Scholtz, Biochemistry, 1998, 37, 33–40 CrossRef CAS PubMed.
- Y. Pu, Z. Hou, M. M. Khin, R. Zamudio-Vázquez, K. L. Poon, H. Duan and M. B. Chan-Park, Biomacromolecules, 2017, 18, 44–55 CrossRef CAS.
- K. E. B. Doncom, H. Willcock and R. K. O'Reilly, Eur. Polym. J., 2017, 87, 497–507 CrossRef CAS.
- E. F. Palermo, S. Vemparala and K. Kuroda, Biomacromolecules, 2012, 13, 1632–1641 CrossRef CAS PubMed.
- V. I. Gordeliy, M. A. Kiselev, P. Lesieur, A. V. Pole and J. Teixeira, Biophys. J., 1998, 75, 2343–2351 CrossRef CAS PubMed.
- G. Dyrda, E. Boniewska-Bernacka, D. Man, K. Barchiewicz and R. Słota, Mol. Biol. Rep., 2019, 46, 3225–3232 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS.
- G. J. Martyna, D. J. Tobias and M. L. Klein, J. Chem. Phys., 1994, 101, 4177–4189 CrossRef CAS.
- S. E. Feller, Y. H. Zhang, R. W. Pastor and B. R. Brooks, J. Chem. Phys., 1995, 103, 4613–4621 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- I. Ivanov, S. Vemparala, V. Pophristic, K. Kuroda, W. F. DeGrado, J. A. McCammon and M. L. Klein, J. Am. Chem. Soc., 2006, 128, 1778–1779 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph. Model., 1996, 14, 33–38 CrossRef CAS.
- B. Lee and F. M. Richards, J. Mol. Biol., 1971, 55, 379 CrossRef CAS PubMed.
- E. F. Palermo, S. Vemparala and K. Kuroda, ACS Symp. Ser., 2013, 1135, 319–330 CrossRef.
- S. Jiang and Z. Cao, Adv. Mater., 2010, 22, 920–932 CrossRef CAS.
- I. Izquierdo-Barba, S. Sánchez-Salcedo, M. Colilla, M. J. Feito, C. Ramírez-Santillán, M. T. Portolés and M. Vallet-Regí, Acta Biomater., 2011, 7, 2977–2985 CrossRef CAS PubMed.
- M. Colilla, I. Izquierdo-Barba and M. Vallet-Regí, Medicines, 2018, 5, 125 CrossRef CAS PubMed.
- I. Izquierdo-Barba, M. Colilla and M. Vallet-Regí, Acta Biomater., 2016, 40, 201–211 CrossRef CAS PubMed.
- Y. Jiang, X. Yang, R. Zhu, K. Hu, W.-W. Lan, F. Wu and L. Yang, Macromolecules, 2013, 46, 3959–3964 CrossRef CAS.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- H. Jeong, J. Hwang, H. Lee, P. T. Hammond, J. Choi and J. Hong, Sci. Rep., 2017, 7, 9481 CrossRef.
- R. A. Anderson and R. E. Lovrien, Nature, 1984, 307, 655–658 CrossRef CAS PubMed.
- T. Aoki, Membranes, 2017, 7, 56 CrossRef.
- M. P. Aliste, J. L. MacCallum and D. P. Tieleman, Biochemistry, 2003, 42, 8976–8987 CrossRef CAS PubMed.
- L. H. He, E. S. Read, S. P. Armes and D. J. Adams, Macromolecules, 2007, 40, 4429–4438 CrossRef CAS.
- M. Y. Lobanov, N. S. Bogatyreva and O. V. Galzitskaya, Mol. Biol., 2008, 42, 623–628 CrossRef CAS.
- D. J. Barlow and J. M. Thornton, J. Mol. Biol., 1983, 168, 867–885 CrossRef CAS.
- Z.-G. Wang, Macromolecules, 2017, 50, 9073–9114 CrossRef CAS.
- R. D. Ulrich, in Macromolecular Science: Retrospect and Prospect, ed. R. D. Ulrich, Springer US, Boston, MA, 1978, pp. 69–98, DOI:10.1007/978-1-4684-2853-7_5.
- Z. Wu, X. Li, E. de Leeuw, B. Ericksen and W. Lu, J. Biol. Chem., 2005, 280, 43039–43047 CrossRef CAS PubMed.
- M. Rajabi, E. de Leeuw, M. Pazgier, J. Li, J. Lubkowski and W. Lu, J. Biol. Chem., 2008, 283, 21509–21518 CrossRef CAS PubMed.
- H. S. Andersson, S. M. Figueredo, L. M. Haugaard-Kedström, E. Bengtsson, N. L. Daly, X. Qu, D. J. Craik, A. J. Ouellette and K. J. Rosengren, Amino Acids, 2012, 43, 1471–1483 CrossRef CAS PubMed.
- H. Meuzelaar, J. Vreede and S. Woutersen, Biophys. J., 2016, 110, 2328–2341 CrossRef CAS.
- G. H. Michler and W. Lebek, in Polymer Morphology, 2016, pp. 37–53 Search PubMed.
- M. R. Libera and R. F. Egerton, Polym. Rev., 2010, 50, 321–339 CrossRef CAS.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 8, 409–427 CrossRef CAS PubMed.
- A. R. Petcu, E. A. Rogozea, C. A. Lazar, N. L. Olteanu, A. Meghea and M. Mihaly, Arabian J. Chem., 2016, 9, 9–17 CrossRef CAS.
- M. Giedyk, R. Narobe, S. Weiß, D. Touraud, W. Kunz and B. König, Nat. Catal., 2020, 3, 40–47 CrossRef CAS.
- E. Han and H. Lee, RSC Adv., 2015, 5, 2047–2055 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02730a |
| ‡ RB, LF and GR contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |