
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAminations and arylations by direct C–O activation for the design of 7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidines†
Mazarine Laurenta,
Stéphane Bostynb,
Mathieu Marchiviec,
Yves Robind,
Sylvain Routier *a and
Frédéric Buron*a
*a and
Frédéric Buron*a
aInstitut de Chimie Organique et Analytique, ICOA, UMR CNRS 7311, Université d’Orléans, Orléans, France. E-mail: frederic.buron@univ-orleans.fr; sylvain.routier@univ-orleans.fr
bInstitut de Combustion, Aérothermique, Réactivité, et Environnement (ICARE), 1c, Avenue de la Recherche Scientifique, 45071 CEDEX 2 Orléans, France
cICMCB CNRS-UMR 5026, Université de Bordeaux, Bordeaux, France
dISOCHEM, 4 Rue Marc Sangnier, 45300 Pithiviers, BP 16729, France
First published on 28th May 2021
Abstract
The design of some novel disubstituted 7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine derivatives is reported. The series was developed from quinuclidinone, which afforded versatile platforms bearing one lactam function in position C-2 that were then used to create C–N or C–C bonds for SNAr or palladium-catalyzed cross-coupling reactions by in situ C–O activation. The reaction conditions were optimized under microwave irradiation, and a wide range of amines or boronic acids were used to determine the scope and limitations of each method. To complete this study, the X-ray crystallographic data of 7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine derivative 49 were used to formally establish the structures of the products.
Introduction
Exploring chemical space is a major challenge to discover new biologically active small molecules.1–3 This strategy has long been applied in heterocyclic chemistry, in particular through the design and the functionalization of fused polynitrogenated derivatives, which contain hetero-aromatic and aliphatic moieties.4–8 The resulting original structures increase the molecular diversity and find applications in reagents, 3D fine chemical or novel pharmaceutical specialities.9–11 Among those reported in the aliphatic series, quinuclidine holds an important place due to its presence in a number of natural products, such as Cinchona officinalis alkaloids or FDA-approved drugs (Fig. 1).12–17 Moreover, this skeleton has also been used as a catalyst for the development of asymmetric aldolisation, Baylis–Hillman or Diels–Alder reactions.18–23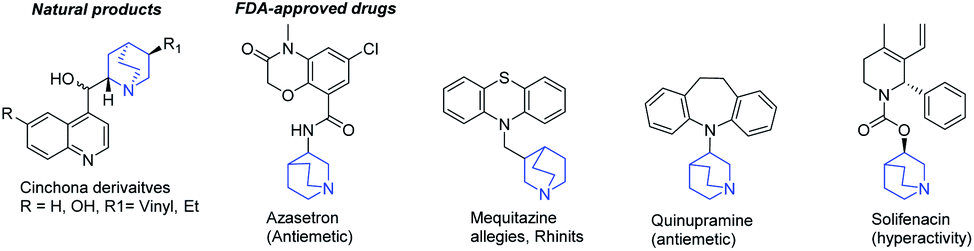 | ||
| Fig. 1 Some examples of the compounds of interest with a quinuclidine moiety. | ||
The fusion of quinuclidine to aromatic and nitrogen-containing heterocycle moieties has seldom been reported. Only a few references describe the synthesis and the reactivity of this skeleton with a pyridine group as an example.24–26 This may be due to the strong Brønsted and Lewis basic character of the nitrogen atom, which is likely to inhibit a large panel of reactions.27 For our own part, our group has developed efficient methodologies to functionalize and obtain biologically active molecules in the pyrimidine series such as pyrido[3-2,d]pyrimidines, pyrido[1′,2′:1,5]pyrazolo[3,4-d]pyrimidines or pyrido[1′,2′:1,5]pyrazolo[4,3-d]pyrimidine.28–31 To escape from flatland and evaluate the replicability of our know-how in this poorly explored area, we propose in this paper the access to a 4-aryl-7,8-dihydro-1H-5,8-ethanopyrido[3,2-d]pyrimidin-2(6H)-one platform and its further substitution at the C-2 position.32 This fused arylated skeleton A resulting from the fusion of a quinuclidine and a pyrimidine (quinuclidino-pyrimidinone) was functionalized by aminations or the Suzuki–Miyaura cross-coupling reaction using in situ C–O activation, an innovative and direct method that is particularly powerful to modulate heteroaromatic structures (Fig. 2).
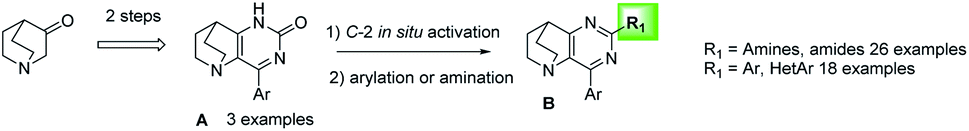 | ||
| Fig. 2 General scheme leading to 2,4 disubstituted quinuclidino pyrimidines under C–O direct activation. | ||
Results and discussion
To perform the required C-2 amination or Suzuki–Miyaura cross-coupling reactions, some ethanopyrido[3,2-d]pyrimidinones of type A were prepared in two steps. First, 4-aryl-7,8-dihydro-1H-5,8-ethanopyrido[3,2-d]pyrimidin-2(6H)one 2–4 were obtained in satisfactory yields after aldol condensation between the commercially available 3-quinuclidinone hydrochloride 1 with three benzaldehydes in presence of sodium hydroxide. Next, a solvent-free condensation with urea was performed to generate bicyclic tetrahydropyrimidinones 5–7.33 Finally, an oxidative hetero aromatization was carried out using manganese dioxide34 in acetone under microwave irradiation to access the desired platforms 8–10 in very good to excellent yields (Scheme 1).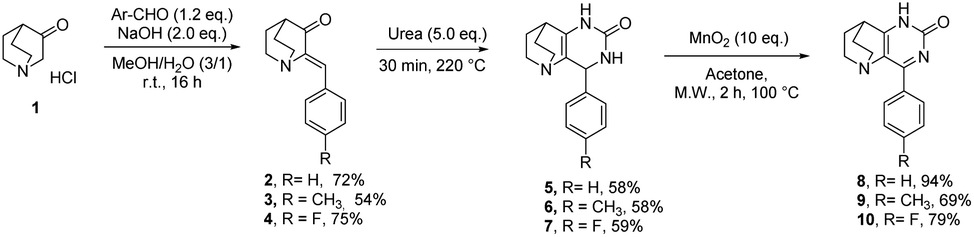 | ||
| Scheme 1 Synthesis of 8–10. | ||
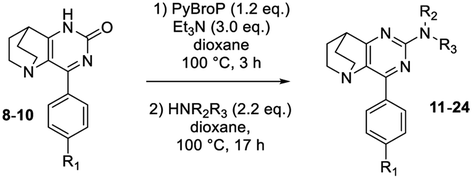
To take advantage of the lactam, we began our methodological study by a one-pot amination at the C-2 position using C–O direct activation involving PyBroP and Et3N.35–37 In this tandem reaction, the first in situ step generated the O-phosphonium leaving group, which was then displaced with the adequate nucleophile. In a first attempt, the reactivity was examined by treating 4-phenyl-pyrido[3,2-d]pyrimidinone 8 with n-propylamine and several reaction parameters (temperature, duration) were screened to reach an acceptable level of reaction efficiency (Table 1).38,39
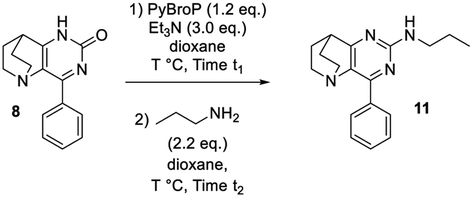
The first step was also performed with 8 in the presence of PyBrOP at a temperature of 80 °C and after 2 h the primary amine (2.2 eq.) was added. After 3 additional hours, only a small proportion of the intermediate had been consumed and the desired product 11 was isolated in low yield whereas a large amount of starting material 8 was recovered. Increasing the temperature to 100 °C slightly enhanced the yield to 32%. A time reaction screening for each step (t1 and t2) led to interesting results. A full conversion of 8 to O-phosphonium was reached after 3 h at 100 °C. The nucleophilic attack of the amine on the activated heterocycle was the rate-limiting step. The second step was achieved during 17 h at 100 °C and led to the desired compound 11 in 72% of yield.
In order to explore the scope and limitations of this tandem sequence, we then condensed previously synthesized ethanopyrido[3,2-d]pyrimidinones A with various amines (Table 2). With 1-pentylamine and 8, the yield decreased slightly (entry 2 versus 1). The same behaviour was observed when benzyl amines were used (entries 8–10). An attempted strengthening of nucleophilicity with secondary cyclic amines such as piperidine, morpholine or thiomorpholine increased the reactivity up to 82% (entries 3–7). Moreover, when piperidine was used as a secondary amine with ethanopyrido[3,2-d]pyrimidinones 9 and 10, the two SNAr reactions were efficiently achieved and compounds 23 and 24 were isolated in 86% and 79% of yield, respectively (entries 15–16).
|
|||
|---|---|---|---|
| Entry | R1 | HNR2R3 | Cpd, yielda |
| a Cpd: compound number; yield is indicated as isolated product.b Not detected. | |||
| 1 | H | Propylamine | 11, 72% |
| 2 | H | Pentylamine | 12, 59% |
| 3 | H | Piperidine | 13, 77% |
| 4 | H | Morpholine | 14, 82% |
| 5 | H | Thiomorpholine | 15, 72% |
| 7 | H | 4,4-Difluoropiperidine | 16, 61% |
| 8 | H | C6H5CH2NH2 | 17, 51% |
| 9 | H | 4-CH3O-C6H4CH2NH2 | 18, 42% |
| 10 | H | 4-CF3-C6H4CH2NH2 | 19, 39% |
| 11 | H | Imidazole | 20, 22% |
| 12 | H | 4-CH3-C6H4NH2 | 21, NDb |
| 13 | H | δ-Valerolactam | 22, NDb |
| 15 | Me | Piperidine | 23, 86% |
| 16 | F | Piperidine | 24, 79% |
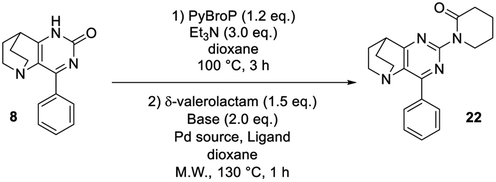
During this investigation, we encountered two limitations when anilines or lactams, which are very weakly nucleophilic species, were used: the final compounds 21 and 22 (entries 12, 13) were never observed. To access these derivatives, it became necessary to develop a one-pot sequence involving in situ CO-activation followed by a Pd-catalyzed C–N bond forming sequence, an original alternative to the previously unsuccessful SNAr method.
First, we used δ-valerolactam as amide to prevent any SNAr competition, Pd2dba3 as the palladium source in the presence of Xantphos as bidentate ligand, K2CO3 as base, and dioxane as solvent. With these conditions, the desired product 22 was isolated in low yield (19%, Table 3, entry 1) despite the total consumption of the starting material, indicating the low reactivity of the O-phosphonium intermediate. When the palladium catalyst was replaced by Pd(OAc)2, the same behaviour was observed and the desired compound 22 was obtained in a similar 21% yield. By increasing the catalytic charge to 10 mol% the O-phosphonium species was consumed, affording 22 in 40% of yield accompanied with degradation. A fine adjustment of the temperature (to 150 °C) drastically decreased the yield (entry 4) and the replacement of Xantphos with well-known Buchwald ligands such as CyJohnPhos or Ruphos totally inhibited the reactivity. Changing the nature of the base indicated that the use of Na2CO3 and Cs2CO3 was not tolerated and showed the highest reaction sensitivity (entries 7, 8).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Ligand (mol%) | Base | Temp. (°C) | Yielda (%) |
| a Yield is indicated as isolated product. | |||||
| 1 | Pd2dba3 (5%) | Xantphos (10%) | K2CO3 | 130 | 19 |
| 2 | Pd(OAc)2 (5%) | Xantphos (10%) | K2CO3 | 130 | 21 |
| 3 | Pd(OAc)2 (10%) | Xantphos (20%) | K2CO3 | 130 | 40 |
| 4 | Pd(OAc)2 (10%) | Xantphos (20%) | K2CO3 | 150 | 33 |
| 5 | Pd(OAc)2 (10%) | CyJohnPhos (20%) | K2CO3 | 130 | 0 |
| 6 | Pd(OAc)2 (10%) | RuPhos (20%) | K2CO3 | 130 | 0 |
| 7 | Pd(OAc)2 (10%) | Xantphos (20%) | Na2CO3 | 130 | Traces |
| 8 | Pd(OAc)2 (10%) | Xantphos (20%) | Cs2CO3 | 130 | 9 |
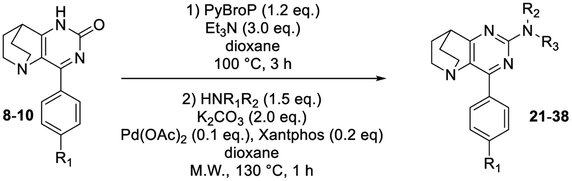
Next, the scope and generality of the Pd-coupling step were examined. The 2-pyrrolidinone reaction gave the desired product 25 with the same reactivity (Table 4, entry 2), whereas using aniline afforded product 26 in a good isolated yield of 56% (Table 4, entry 4). The reactions proceeded with the same efficiency regardless of the nature of the aniline used (i.e., with electron-withdrawing or electron-donating substituents) and the compound 21 was finally synthesized with these conditions (entry 1). Steric hindrance symbolized by the presence of a methyl group on the aniline in ortho vs. meta or para position (entries 1, 6–7) induced a dramatic decrease in yield. With aminopyridines, reactivity was maintained and compounds 33 and 34 were isolated in 34% and 28% of yield, respectively. The sole limitation in this trend involved the use of 4-(4-fluorophenyl)-7,8-dihydro-1H-5,8-ethanopyrido[3,2-d]pyrimidinone 10 as starting material, which inhibited the reactivity whereas the tolyl derivative 9 restored the efficiency.
|
|||
|---|---|---|---|
| Entry | R1 | HNR2R3 | Cpd, yielda |
| a Cpd: compound number; yield is indicated as isolated product.b Not detected. | |||
| 1 | H | 4-CH3-C6H4NH2 | 21, 48% |
| 2 | H | δ-Valerolactam | 22, 40% |
| 3 | H | 2-Pyrrolidinone | 25, 39% |
| 4 | H | C6H5NH2 | 26, 56% |
| 5 | H | 4-CH3O-C6H4NH2 | 27, 48% |
| 6 | H | 3-CH3-C6H4NH2 | 28, 31% |
| 7 | H | 2-CH3-C6H4NH2 | 29, 15% |
| 8 | H | 4-CF3-C6H4NH2 | 30, 56% |
| 9 | H | 4-CN-C6H4NH2 | 31, 35% |
| 10 | H | 4-NO2-C6H4NH2 | 32, 39% |
| 11 | H | 3-NH2-pyridine | 33, 34% |
| 12 | H | 5-NH2-2-MeOpyridine | 34, 28% |
| 13 | Me | C6H5NH2 | 35, 44% |
| 14 | Me | 4-CF3-C6H4NH2 | 36, 49% |
| 15 | F | C6H5NH2 | 37, traces |
| 16 | F | 4-CF3-C6H4NH2 | 38, NDb |
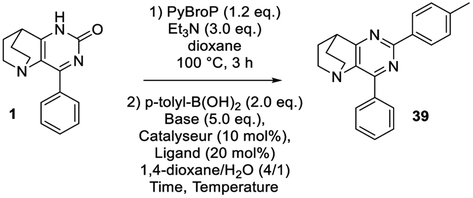
We next focused our attention on using this C–O activation strategy to create a C–C bond instead of a C–N bond under palladium catalysis.28,40,41 In this tandem procedure, the PyBrop activation was achieved as previously described during 3 h and the reagents necessary to perform the cross coupling reaction were then added. Each parameter of the Suzuki–Miyaura reaction was modulated and the results are summarized in Table 5. 24 h of reaction with Pd(OAc)2 at 110 °C in the presence of a bidentate phosphine proved to be a better catalytic system than with PdCl2(dppf)·CH2Cl2 or Pd(PPh3)4 (entries 1–3). Modulation of the base indicated an increase in the yield using K3PO4 instead of classical carbonates (entries 3–6) whereas optimization of the nature of the ligand clearly indicated that Xantphos and Ruphos gave the desired compound 39 in the best 68% yield under thermal conditions (entries 6–8). Finally, optimization under microwave irradiation (not shown) established that in the presence of Ruphos at 150 °C the reaction was achieved in only 1 h to furnish 39 in a 70% yield (entry 9).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst/ligand | Base | Time step 2 | Temp step 2 | Yielda (%) |
| a Yield is indicated as isolated product. | |||||
| 1 | PdCl2(dppf)·CH2Cl2 | Na2CO3 | 24 h | 110 °C | 50 |
| 2 | Pd(PPh3)4 | Na2CO3 | 24 h | 110 °C | 41 |
| 3 | Pd(OAc)2/Xantphos | Na2CO3 | 24 h | 110 °C | 60 |
| 4 | Pd(OAc)2/Xantphos | Cs2CO3 | 24 h | 110 °C | 50 |
| 5 | Pd(OAc)2/Xantphos | K2CO3 | 24 h | 110 °C | 54 |
| 6 | Pd(OAc)2/Xantphos | K3PO4 | 24 h | 110 °C | 68 |
| 7 | Pd(OAc)2/Xphos | K3PO4 | 24 h | 110 °C | 62 |
| 8 | Pd(OAc)2/Ruphos | K3PO4 | 24 h | 110 °C | 68 |
| 9 | Pd(OAc)2/Ruphos | K3PO4 | 1 h | 150 °C | 70 |
| M.W. | |||||
In the last stage of this study, we investigated the modulation of the nature of the boron derivative to identify the potential limitations. In fact, whatever the substituent on the phenyl boronic acid (i.e., electron-donating or withdrawing), or the steric hindrance induced by an ortho substitution, the C–C bond was efficiently generated and products were isolated in fairly good yields ranging from 54% to 82% (Table 6, products 39–50). The only identified limit concerned the presence of an acidic proton which slightly altered the yield of the reaction but this constraint was easily removed by the use of a protective group such as THP (entries 6–7). The use of (Het)arylboronic acids such as thiophene or pyridine was well tolerated and compounds 51 and 52 were isolated in 77 and 60% of yield, respectively. Finally, proportionally, the reactions conducted with substituted ethanopyrido[3,2-d]pyrimidinones 9–10 (entries 15–18) were less efficient than those conducted with 8 (entries 1 vs. 15 or 17, 10 vs. 16 or 18).
|
|||
|---|---|---|---|
| Entry | R1 | R2B(OH)2 | Cpd, yielda |
| a Cpd: compound number; yield is indicated as isolated product. | |||
| 1 | H | 4-CH3-C6H4B(OH)2 | 39, 70% |
| 2 | H | C6H5B(OH)2 | 40, 68% |
| 3 | H | 3-CH3-C6H4B(OH)2 | 41, 73% |
| 4 | H | 2-CH3-C6H4B(OH)2 | 42, 76% |
| 5 | H | 4-CH3O-C6H4B(OH)2 | 43, 69% |
| 6 | H | 4-OH-C6H4B(OH)2 | 44, 54% |
| 7 | H | 4-OTHP-C6H4B(OH)2 | 45, 69% |
| 8 | H | 2-NaphtylB(OH)2 | 46, 67% |
| 9 | H | 4-F-C6H4B(OH)2 | 47, 70% |
| 10 | H | 4-CN-C6H4B(OH)2 | 48, 82% |
| 11 | H | 4-CF3-C6H4B(OH)2 | 49, 72% |
| 12 | H | 4-NO2-C6H4B(OH)2 | 50, 76% |
| 13 | H | 3-ThienylB(OH)2 | 51, 77% |
| 14 | H | 3-PyridylB(OH)2 | 52, 60% |
| 15 | Me | 4-CH3-C6H4B(OH2) | 53, 37% |
| 16 | Me | 4-CN-C6H4B(OH)2 | 54, 52% |
| 17 | F | 4-CH3-C6H4B(OH)2 | 55, 45% |
| 18 | F | 4-CN-C6H4B(OH)2 | 56, 68% |
During our investigation, we were able to obtain single crystals of 49 suitable for X-ray diffraction analysis (Fig. 3). Compound 49 crystalizes in the monoclinic P21 space group with the following cell parameters: a = 15.385(8), b = 5.893(3), c = 22.187(14) Å, β = 110.05(4) ° and V = 1890(3) Å3. The asymmetric unit is composed of two independent molecules leading to four molecules in the unit cell.
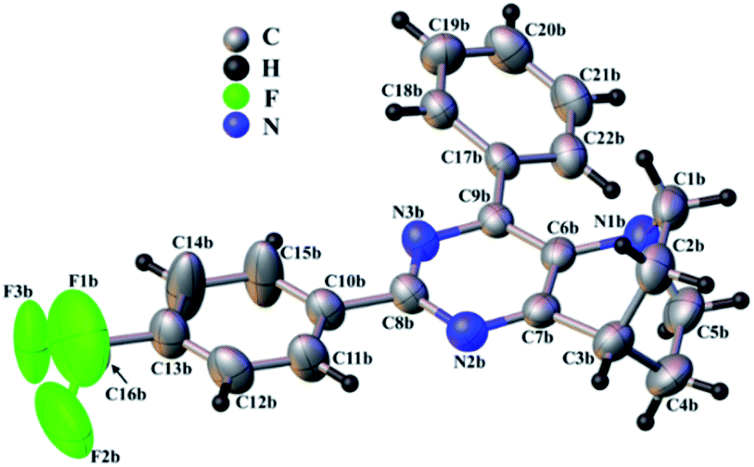 | ||
| Fig. 3 View of the molecular structure of 49. Only one of the two independent molecules of the asymmetric unit is shown (molecule B). The disorder on the CF3 moiety is not represented for clarity. The labelling scheme of molecule A is the same but the suffix “b” has been replaced by suffix “a”. The thermal atomic displacements are represented using ellipsoids at a 50% probability level. | ||
Typical bond lengths are close to the expected values, ranging from 1.352(8) to 1.390(7) Å for the C–C bonds and from 1.332(6) to 1.356(5) for N–C bonds within the aromatic rings, from 1.477(9) to 1.541(8) Å, 1.430(6) to 1.488(7) and 1.309(9) to 1.336(10) for single C–C, C–N and C–F bonds respectively.
The pyrimidine and trifluoro-methyl-phenyl rings are almost coplanar with an angle between planes of 7.3(2) and 7.4(2)° for the two independent molecules. Nevertheless, this angle may be underestimated as the trifluoro-methyl-phenyl ring appears slightly disordered. The second phenyl ring, meanwhile, is significantly tilted from the pyrimidine ring plane (25.4(2)° and 26.9(2)°) for both molecules. Fig. 3 shows one of the two independent molecules of 49 extracted from the crystal structure. The two molecules are very similar (RMSD = 0.452 Å) except as regards the phenyl ring, which is reversely tilted in the two molecules.
Conclusions
In summary, the quick access to variously functionalized ethanopyrido[3,2-d]pyrimidines has been described herein. Aminated or arylated groups were introduced at the C-2 position of the ethanopyrido[3,2-d]pyrimidinones series using a tandem one-pot direct C–O activation sequence involving first PyBroP as activator and next a SNAr or a palladium cross coupling reaction. Aminations were achieved under SNAr reactions which were performed with a large variety of amines or using Buchwald–Hartwig cross-coupling reactions in combination with microwave irradiation. In addition, we have also reported the efficiency of the Suzuki–Miyaura reactions in C-2 position, which is compatible with all the boronylated starting material used. This work afforded a novel class of 1,4 disubstituted 7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidines which will undoubtedly have a major impact on the synthesis of new bioactive compounds that contain the rare ethanopyrido[3,2-d]pyrimidine scaffold as the central skeleton. Efforts to achieve these objectives are currently in progress.Experimental section
Materials and methods
1H NMR and 13C NMR spectra were recorded on a Bruker DPX 250 or 400 Mhz instrument using CDCl3 and DMSO-d6. The chemical shifts are reported in parts per million (δ scale), and all coupling constant (J) values are reported in hertz. The following abbreviations were used for the multiplicities: s (singlet), d (doublet), t (triplet), q (quartet), p (pentuplet), m (multiplet), sext (sextuplet), and dd (doublet of doublets). Melting points are uncorrected. IR absorption spectra were obtained on a PerkinElmer PARAGON 1000 PC, and the values are reported in inverse centimeters. HRMS spectra were acquired in positive mode with an ESI source on a Q-TOF mass by the “Fédération de Recherche” ICOA/CBM (FR2708) platform. Monitoring of the reactions was performed using silica gel TLC plates (silica Merck 60 F 254). Spots were visualized by UV light (254 nm and 356 nm). Column chromatography was performed using silica gel 60 (0.063–0.200 mm, Merck.). Microwave irradiation was carried out in sealed vessels placed in a Biotage Initiator or Biotage Initiator+ system (400 W maximum power). The temperatures were measured externally by IR. Pressure was measured by a non-invasive sensor integrated into the cavity lid. All reagents were purchased from commercial suppliers and were used without further purification. The crystal structure of 49 was solved by single crystal X-ray diffraction at room temperature using a Bruker Apex-II diffractometer with Mo-Kα radiation. CCDC 2074887 contains the supplementary crystallographic data for this paper.Synthetic procedures
(Z)-2-Benzylidenequinuclidin-3-one (2). The reaction was carried out as described in general procedure A using a solution of 3-quinuclidinone hydrochloride (3.99 g, 24.73 mmol, 1.0 eq.) and sodium hydroxide (1.99 g, 49.7 mmol, 2.0 eq.) in a mixture of methanol/water (3/1, 75 mL) and benzaldehyde (3.0 mL, 29.5 mmol, 1.2 eq.). The reaction mixture was stirred 16 h at r.t. to afford 2 (3.82 g, 72%) as a yellow solid. Rf (PE/EA: 90/10): 0.32. Mp: 141–143 °C. IR (ATR diamond, cm−1) ν: 2941, 2873, 1700, 1621, 1096, 688. 1H NMR (400 MHz, CDCl3) δ: 2.03 (td, J = 3.1, 7.9 Hz, 4H, 2xCH2), 2.63 (p, J = 3.0 Hz, 1H, CH), 3.00 (dt, J = 7.6, 12.5 Hz, 2H, N–CH2), 3.16 (dt, J = 7.8, 13.4 Hz, 2H, N–CH2), 7.02 (s, 1H, C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.29–7.42 (m, 3H, 3xCHAr), 7.97–8.09 (m, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 26.3 (2xCH2), 40.7 (CH), 47.9 (2xN–CH2), 125.5 (CCH), 128.8 (2xCHAr), 130.0 (CHAr), 132.6 (2xCHAr), 134.4 (CAr), 145.1 (CCH), 206.8 (CO). HRMS (EI/MS): m/z calculated for C14H16NO: 214.1225 [M + H]+; found 214.1226.
CH), 7.29–7.42 (m, 3H, 3xCHAr), 7.97–8.09 (m, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 26.3 (2xCH2), 40.7 (CH), 47.9 (2xN–CH2), 125.5 (CCH), 128.8 (2xCHAr), 130.0 (CHAr), 132.6 (2xCHAr), 134.4 (CAr), 145.1 (CCH), 206.8 (CO). HRMS (EI/MS): m/z calculated for C14H16NO: 214.1225 [M + H]+; found 214.1226.
(Z)-2-(4-Methylbenzylidene)quinuclidin-3-one (3). The reaction was carried out as described in general procedure A using a solution of 3-quinuclidinone hydrochloride (4.00 g, 24.79 mmol, 1.0 eq.) and sodium hydroxide (2.01 g, 50.27 mmol, 2.0 eq.) in a mixture of methanol/water (3/1, 75 mL) and p-tolualdehyde (3.5 mL, 29.68 mmol, 1.2 eq.). The reaction mixture was stirred 16 h at r.t. to afford 3 (3.05 g, 54%) as a yellow solid. Rf (EP/EA: 90/10): 0.48. Mp: 128–130 °C. IR (ATR diamond, cm−1) ν: 2938, 1700, 1626, 1507, 1251, 1092, 810, 517. 1H NMR (400 MHz, CDCl3) δ: 2.01 (td, J = 7.9, 2.9 Hz, 4H, 2xCH2), 2.36 (s, 3H, CH3), 2.62 (p, J = 2.9 Hz, 1H, CH), 2.91–3.04 (m, 2H, N–CH2), 3.09–3.22 (m, 2H, N–CH2), 7.00 (s, 1H, C
CH), 7.17 (d, J = 7.9 Hz, 2H, 2xCHAr), 7.93 (d, J = 8.0 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 21.5 (CH3), 25.9 (2xCH2), 40.3 (CH), 47.5 (2xN-CH2), 125.2 (=CH), 129.1 (2xCHAr), 131.2 (Cq), 132.1 (2xCHAr), 139.9 (Cq), 144.0 (Cq), 206.4 (CO). HRMS (EI/MS): m/z calculated for C15H18NO: 228.1381; found 228.1383.
(Z)-2-(4-Fluorobenzylidene)quinuclidin-3-one (4). The reaction was carried out as described in general procedure A using a solution of 3-quinuclidinone hydrochloride (4.00 g, 24.76 mmol, 1.0 eq.) and sodium hydroxide (2.00 g, 50.00 mmol, 2.0 eq.) in a mixture of methanol/water (3/1, 75 mL) and 4-fluorobenzaldehyde (3.2 mL, 29.82 mmol, 1.2 eq.). The reaction mixture was stirred 16 h at r.t. to afford 4 (4.33 g, 75%) as a yellow solid. Rf (PE/EA: 90/10): 0.30. Mp: 131–133 °C. IR (ATR diamond, cm−1) ν: 2943, 1698, 1619, 1595, 1502, 1220, 853. 1H NMR (400 MHz, CDCl3) δ: 2.02 (td, J = 7.9, 3.0 Hz, 4H, 2xCH2), 2.63 (p, J = 3.0 Hz, 1H, CH), 2.90–3.04 (m, 2H, N–CH2), 3.07–3.21 (m, 2H, N–CH2), 6.97 (s, 1H,
CH), 6.99–7.10 (m, 2H, 2xCHAr), 7.99–8.10 (m, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 26.0 (2xCH2), 40.3 (CH), 47.5 (2xN-CH2), 115.5 (d, J = 21.5 Hz, 2xCHAr), 123.9 (CH), 130.3 (d, J = 3.4 Hz, C), 134.3 (d, J = 8.1 Hz, 2xCHAr), 144.3 (d, J = 2.6 Hz, CAr), 163.3 (d, J = 251.6 Hz, CAr-F), 206.3 (CO). 19F NMR (376 MHz, CDCl3) δ: −109.84. HRMS (EI/MS): m/z calculated for C14H15FNO: 232.1132 [M + H]+; found 232.1132.
4-Phenyl-3,4,7,8-tetrahydro-1H-5,8-ethanopyrido[3,2-d]pyrimidin-2(6H)-one (5). 2 (4.998 g, 23.43 mmol, 1.0 eq.) and urea (7.050 g, 117.38 mmol, 5.0 eq.) were ground in a mortar and put in a 100 mL-flask. The mixture was then heated at 220 °C in a pre-heated bath. After 30 min, 80 mL of a solution of NaOH 2 N were added, and the resulting mixture was allowed to cool to room temperature. The precipitate formed was filtered, washed with water (80 mL), DCM (80 mL) and acetone (2 × 80 mL) to afford 5 (3.505 g, 58%) as a white solid. Rf (DCM/MeOH: 95/5): 0.44. Mp: >260 °C. IR (ATR diamond, cm−1) ν: 3295, 2951, 1681, 1642, 1454, 1241, 752, 732, 691. 1H NMR (400 MHz, DMSO-d6) δ: 1.30–1.43 (m, 1H, CH), 1.45–1.56 (m, 2H, 2xCH), 1.56–1.65 (m, 1H, CH), 1.65–1.79 (m, 1H, N–CH), 2.50–2.61 (m, 3H, 2xN-CH, CH), 2.76 (ddd, J = 4.6, 8.6, 12.9 Hz, 1H, N–CH), 4.83 (d, J = 2.1 Hz, 1H, CH), 6.98 (s, 1H, NH), 7.20–7.35 (m, 5H, 5xCHAr), 8.35 (s, 1H, NH). 13C NMR (101 MHz, DMSO-d6) δ: 27.9 (CH), 28.6 (CH2), 28.7 (CH2), 49.9 (N–CH2), 50.0 (N–CH2), 57.9 (CH), 118.3 (Cq), 126.7 (2xCHAr), 127.1 (CH), 128.1 (2xCHAr), 137.2 (Cq), 144.4 (Cq), 152.5 (Cq). HRMS (EI/MS): m/z calculated for C15H18N3O: 256.1442 [M + H]+; found: 256.1444.
4-(p-Tolyl)-3,4,7,8-tetrahydro-1H-5,8-ethanopyrido[3,2-d]pyrimidin-2(6H)-one (6). 3 (3.97 g, 17.48 mmol, 1.0 eq.) and urea (5.25 g, 87.51 mmol, 5.0 eq.) were ground in a mortar and put in a 100 mL-flask. The mixture was then heated at 220 °C in a pre-heated bath. After 30 min, 60 mL of a solution of NaOH 2 N were added, and the resulting mixture was allowed to cool to room temperature. The precipitate formed was filtered, washed with water (60 mL), DCM (60 mL) and acetone (2 × 60 mL) to afford 6 (2.76 g, 58%) as a white solid. Rf (DCM/MeOH: 95/5): 0.22. Mp: >260 °C. IR (ATR diamond, cm−1) ν: 3294, 3091, 2941, 1644, 1455, 1340, 1102, 750. 1H NMR (400 MHz, DMSO-d6) δ: 1.31–1.41 (m, 1H, CH), 1.43–1.56 (m, 2H, 2xCH), 1.56–1.64 (m, 1H, CH), 1.70–1.81 (m, 1H, N–CH), 2.27 (s, 3H, CH3), 2.52–2.61 (m, 3H, 2xN-CH, CH), 2.69–2.80 (m, 1H, N–CH), 4.78 (s, 1H, NH–CH), 6.91 (s, 1H, NH), 7.13 (dd, J = 9.0, 10.4 Hz, 4H, 4xCHAr), 8.31 (s, 1H, NH). 13C NMR (101 MHz, DMSO-d6) δ: 20.6 (CH3), 27.8 (CH), 28.6 (CH2), 28.7 (CH2), 49.8 (N–CH2), 50.0 (N–CH2), 57.6 (CH), 118.4 (Cq), 126.6 (2xCHAr), 128.6 (2xCHAr), 136.0 (Cq), 137.0 (Cq), 141.5 (Cq), 152.4 (Cq). HRMS (EI/MS): m/z calculated for C16H20N3O: 270.1601 [M + H]+; found: 270.1596.
4-(4-Fluorophenyl)-3,4,7,8-tetrahydro-1H-5,8-ethanopyrido[3,2-d]pyrimidin-2(6H)-one (7). 4 (2.0 g, 8. mmol, 1.0 eq.) and urea (2.60 g, 43.42 mmol, 5.0 eq.) were ground in a mortar and put in a 50 mL-flask. The mixture was then heated at 220 °C in a pre-heated bath. After 30 min, 30 mL of a solution of NaOH 2 N were added, and the resulting mixture was allowed to cool to room temperature. The precipitate formed was filtered, washed with water (30 mL), DCM (30 mL) and acetone (2 × 30 mL) to afford 7 (1.40 g, 59%) as a white solid. Rf (DCM/MeOH: 95/5): 0.19. Mp: 258–260 °C. IR (ATR diamond, cm−1) ν: 3302, 3093, 2945, 1638, 1505, 1221, 1155, 1098, 748. 1H NMR (400 MHz, DMSO-d6) δ: 1.31–1.43 (m, 1H, CH), 1.44–1.53 (m, 2H, 2xCH), 1.53–1.66 (m, 1H, CH), 1.72 (tt, J = 3.3, 12.8 Hz, 1H, N–CH), 2.52–2.63 (m, 3H, 2xN-CH, CH), 2.70–2.81 (m, 1H, N–CH), 4.85 (s, 1H, NH–CH), 7.00 (s, 1H, NH), 7.14 (t, J = 8.7 Hz, 2H, 2xCHAr), 7.29 (dd, J = 5.6, 8.4 Hz, 2H, 2xCHAr), 8.37 (s, 1H, NH). 13C NMR (101 MHz, DMSO-d6) δ: 27.8 (CH), 28.6 (2xCH2), 49.8 (N–CH2), 50.0 (N–CH2), 57.1 (CH), 114.6 (CHAr), 114.8 (CHAr), 118.1 (Cq), 128.5 (CHAr), 128.6 (CHAr), 137.3 (Cq), 140.6 (d, J = 2.9 Hz, Cq), 152.3 (Cq), 161.3 (d, J = 242.4 Hz, CAr-F). 19F NMR (376 MHz, DMSO-d6) δ: −115.92. HRMS (EI/MS): m/z calculated for C15H17FN3O: 274.1350 [M + H]+; found: 274.1350.
4-Phenyl-7,8-dihydro-1H-5,8-ethanopyrido[3,2-d]pyrimidin-2(6H)-one (8). In a microwave vial of 10–20 mL, MnO2 (873 mg, 10.0 mmol, 10.0 eq.) was added to a solution of 5 (259 mg, 1.0 mmol, 1.0 eq.) in acetone (12 mL). The reaction mixture was heated for 2 h at 100 °C under microwave irradiation. The reaction mixture was filtered through celite and washed with acetone (20 mL), DCM (20 mL) and MeOH (20 mL). The solvents were evaporated to give 8 (237 mg, 94%) as a white solid without further purification. Rf (DCM/MeOH: 95/5): 0.38. Mp: >260 °C. IR (ATR diamond, cm−1) ν: 2967, 2928, 1626, 1551, 1442, 1390, 1324, 1128, 776, 747, 685. 1H NMR (400 MHz, CDCl3) δ: 1.74–1.86 (m, 2H, CH2), 1.94–2.06 (m, 2H, CH2), 2.72 (td, J = 4.7, 11.3, 11.7 Hz, 2H, N–CH2), 3.08–3.19 (m, 3H, N–CH2, CH), 7.44–7.56 (m, 3H, 3xCHAr), 8.02 (d, J = 7.0 Hz, 2H, 2xCHAr), 13.00 (s, 1H, NH). 13C NMR (101 MHz, CDCl3) δ: 27.0 (2xCH2), 33.9 (CH), 49.3 (2xN-CH2), 126.1 (Cq), 128.6 (2xCHAr), 129.8 (2xCHAr), 131.2 (CHAr), 158.8 (Cq). HRMS (EI/MS): m/z calculated for C15H16N3O: 254.1286 [M + H]+; found: 254.1288.
4-(p-Tolyl)-7,8-dihydro-1H-5,8-ethanopyrido[3,2-d]pyrimidin-2(6H)-one (9). In a microwave vial of 10–20 mL, MnO2 (1.11 g, 12.8 mmol, 10.0 eq.) was added to a solution of 6 (331 mg, 1.2 mmol, 1.0 eq.) in acetone (18 mL). The reaction mixture was heated for 2 h at 100 °C under microwave irradiation. The reaction mixture was filtered through celite and washed with acetone (20 mL), DCM (20 mL) and MeOH (20 mL). The solvents were evaporated to give 9 (225 mg, 69%) as a white solid without further purification. Rf (DCM/MeOH: 95/5): 0.27. Mp: >260 °C. IR (ATR diamond, cm−1) ν: 2963, 2942, 2869, 1710, 1633, 1449, 1357, 803, 746. 1H NMR (400 MHz, DMSO-d6) δ: 1.68–1.80 (m, 2H, CH2), 2.02–2.13 (m, 2H, CH2), 2.41 (s, 3H, CH3), 2.68–2.79 (m, 2H, N–CH2), 3.10–3.21 (m, 2H, N–CH2), 3.25 (p, J = 2.6 Hz, 1H, CH), 7.40 (d, J = 8.0 Hz, 2H, 2xCHAr), 7.72 (d, J = 8.2 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, DMSO-d6) δ: 21.2 (CH3), 25.3 (2xCH2), 30.7 (CH), 48.8 (2xN-CH2), 115.2 (q, J = 288.7 Hz, TFA), 124.0 (Cq), 124.6 (Cq), 129.2 (2xCHAr), 130.5 (2xCHAr), 143.5 (Cq), 149.5 (Cq), 156.6 (Cq), 158.5 (q, J = 38.1 Hz, TFA), 177.9 (C
O). HRMS (EI/MS): m/z calculated for C16H18N3O: 268.1444 [M + H]+; found: 268.1448.
4-(4-Fluorophenyl)-7,8-dihydro-1H-5,8-ethanopyrido[3,2-d]pyrimidin-2(6H)-one (10). In a microwave vial of 10–20 mL, MnO2 (1.30 g, 15.0 mmol, 10.0 eq.) was added to a solution of 7 (413 mg, 1.5 mmol, 1.0 eq.) in acetone (18 mL). The reaction mixture was heated for 2 h at 100 °C under microwave irradiation. The reaction mixture was filtered through celite and washed with acetone (20 mL), DCM (20 mL) and MeOH (20 mL). The solvents were evaporated to give 10 (321 mg, 79%) as a white solid without further purification. Rf (DCM/MeOH, 95/5): 0.30. Mp: >260 °C. IR (ATR diamond, cm−1) ν: 2944, 1651, 1590, 1556, 1450, 1359, 1230, 1157, 800, 658. 1H NMR (400 MHz, DMSO-d6) δ: 1.68–1.80 (m, 2H, CH2), 2.02–2.14 (m, 2H, CH2), 2.70–2.81 (m, 2H, N–CH2), 3.12–3.22 (m, 2H, N–CH2), 3.25 (p, J = 3.0 Hz, 1H, CH), 7.43 (t, J = 8.7 Hz, 2H, 2xCHAr), 7.88 (dd, J = 8.7, 5.3 Hz, 2H, 2xCHAr), 14.00 (s, TFA). 13C NMR (101 MHz, DMSO-d6) δ: 25.20 (2xCH2), 30.84 (CH), 48.92 (2xN-CH2), 115.34 (q, J = 290.4 Hz, TFA), 115.66 (CHAr), 115.88 (CHAr), 124.01 (Cq), 124.40 (d, J = 2.7 Hz, Cq), 133.24 (CHAr), 133.33 (CHAr), 150.35 (Cq), 155.47 (Cq), 158.54 (m, TFA), 164.64 (d, J = 251.8 Hz, CAr-F), 177.85 (Cq). 19F NMR (376 MHz, CDCl3) δ: −75.86 (TFA), −100.94. HRMS (EI/MS): m/z calculated for C15H15FN3O: 272.1194 [M + H]+; found: 272.1194.
4-Phenyl-N-propyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (11). Compound 11 was obtained according to the general procedure B using propylamine (0.14 mL, 1.73 mmol, 2.2 eq.). After purification by silica gel flash chromatography (PE/EA, 70/30), 11 (167 mg, 72%) was obtained as a white solid. Rf (PE/EA: 70/30): 0.37. Mp: 149–151 °C. IR (ATR diamond, cm−1) ν: 3264, 3115, 2955, 2869, 1596, 1360, 1131, 770, 687, 626. 1H NMR (400 MHz, CDCl3) δ: 1.01 (t, J = 7.4 Hz, 3H, CH3), 1.67 (h, J = 7.0 Hz, 2H, CH2), 1.71–1.81 (m, 2H, CH2), 1.90–2.02 (m, 2H, CH2), 2.72 (td, J = 4.7, 11.2 Hz, 2H, N–CH2), 3.04 (p, J = 3.0 Hz, 1H, CH), 3.104–3.21 (m, 2H, N–CH2), 3.47 (q, J = 6.6 Hz, 2H, CH2), 5.03 (t, J = 5.8 Hz, 1H, NH), 7.36–7.49 (m, 3H, 3xCHAr), 8.25 (d, J = 7.2 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 11.7 (CH3), 23.1 (CH2), 27.8 (2xCH2), 33.7 (CH), 43.7 (CH2), 49.4 (2xN-CH2), 128.1 (2xCHAr), 129.5 (CHAr), 129.8 (2xCHAr), 131.4 (Cq), 136.7 (Cq), 155.6 (Cq), 160.1 (Cq), 175.6 (Cq). HRMS (EI/MS): m/z calculated for C18H23N4: 295.1916 [M + H]+; found: 295.1917.
N-Pentyl-4-phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (12). Compound 12 was obtained according to the general procedure B using amylamine (0.20 mL, 1.71 mmol, 2.2 eq.). After purification by silica gel flash chromatography (PE/EA, 70/30), 12 (150 mg, 59%) was obtained as a white solid. Rf (PE/AE: 70/30): 0.38. Mp: 138–140 °C. IR (ATR diamond, cm−1) ν: 3256, 2947, 2867, 1569, 1359, 1130, 773, 688, 625. 1H NMR (400 MHz, CDCl3) δ: 0.92 (t, J = 6.8 Hz, 3H, CH3), 1.31–1.47 (m, 4H, CH2), 1.66 (q, J = 7.1 Hz, 2H, CH2CH2CH2CH2CH3), 1.69–1.81 (m, 2H, CH2), 1.90–2.02 (m, 2H, CH2), 2.72 (td, J = 5.0, 11.4 Hz, 2H, N–CH2), 3.04 (p, J = 3.1 Hz, 1H, CH), 3.09–3.21 (m, 2H, N–CH2), 3.49 (q, J = 6.6 Hz, 2H, CH2), 5.02 (t, J = 5.7 Hz, 1H, NH), 7.36–7.49 (m, 3H, 3xCHAr), 8.25 (d, J = 7.1 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 14.2 (CH3), 22.6 (CH2), 27.8 (2xCH2), 29.3 (CH2), 29.6 (CH2), 33.7 (CH), 41.9 (CH2), 49.4 (2xN-CH2), 128.1 (2xCHAr), 129.5 (CHAr), 129.8 (2xCHAr), 131.3 (Cq), 136.7 (Cq), 155.5 (Cq), 160.0 (Cq), 175.6 (Cq). HRMS (EI/MS): m/z calculated for C20H27N4: 323.2230 [M + H]+; found: 323.2230.
4-Phenyl-2-(piperidin-1-yl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (13). Compound 13 was obtained according to the general procedure B using piperidine (0.17 mL, 1.71 mmol, 2.2 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 13 (195 mg, 77%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.41. Mp: 207–209 °C. IR (ATR diamond, cm−1) ν: 2939, 2863, 1571, 1503, 1386, 1290, 776, 694, 624. 1H NMR (400 MHz, CDCl3) δ: 1.60–1.69 (m, 6H, 3xCH2), 1.70–1.82 (m, 2H, CH2), 1.89–2.01 (m, 2H, CH2), 2.71 (td, J = 4.8, 11.2 Hz, 2H, N–CH2), 3.07 (p, J = 3.1 Hz, 1H, CH), 3.10–3.21 (m, 2H, N–CH2), 3.87 (t, J = 4.9 Hz, 4H, 2xN-CH2), 7.36–7.48 (m, 3H, 3xCHAr), 8.25–8.32 (m, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 25.1 (CH2), 26.0 (2xCH2), 27.7 (2xCH2), 33.9 (CH), 45.19 (2xCH2), 49.5 (2xN-CH2), 128.05 (CHAr), 129.4 (CHAr), 129.91 (2xCHAr), 130.1 (Cq), 137.1 (Cq), 154.9 (Cq), 159.6 (Cq), 175.2 (Cq). HRMS (EI/MS): m/z calculated for C20H25N4: 321.2074 [M + H]+; found: 321.2070.
4-(4-Phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-yl)morpholine (14). Compound 14 was obtained according to the general procedure B using morpholine (0.15 mL, 1.73 mmol, 2.2 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 14 (210 mg, 82%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.20. Mp: 212–214 °C. IR (ATR diamond, cm−1) ν: 2949, 2858, 1552, 1483, 1385, 1263, 1115, 774, 688, 624. 1H NMR (400 MHz, CDCl3) δ: 1.69–1.81 (m, 2H, CH2), 1.91–2.03 (m, 2H, CH2), 2.72 (td, J = 4.7, 11.1 Hz, 2H, N–CH2), 3.09 (p, J = 3.1 Hz, 1H, CH), 3.17 (ddd, J = 4.8, 9.6, 13.8 Hz, 2H, N–CH2), 3.81 (q, J = 3.9, 4.4 Hz, 4H, 2xCH2-O), 3.88 (q, J = 3.4, 4.0 Hz, 4H, 2xN-CH2), 7.37–7.49 (m, 3H, 3xCHAr), 8.26–8.35 (m, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.9 (2xCH2), 33.8 (CH), 44.8 (2xN-CH2), 49.4 (2xN-CH2), 67.17 (2xCH2-O), 128.1 (2xCHAr), 129.7 (CHAr), 129.92 (2xCHAr), 131.5 (Cq), 136.8 (Cq), 155.0 (Cq), 159.5 (Cq), 175.5 (Cq). HRMS (EI/MS): m/z calculated for C19H23N4O: 323.1866 [M + H]+; found: 323.1868.
4-(4-Phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-yl)thiomorpholine (15). Compound 15 was obtained according to the general procedure B using thiomorpholine (0.17 mL, 1.68 mmol, 2.2 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 15 (193 mg, 72%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.36. Mp: 211–213 °C. IR (ATR diamond, cm−1) ν: 2951, 2926, 1555, 1481, 1387, 1258, 771, 696, 624. 1H NMR (400 MHz, CDCl3) δ: 1.72–1.83 (m, 2H, CH2), 1.93–2.05 (m, 2H, CH2), 2.68–2.80 (m, 6H, 2xS-CH2, N–CH2), 3.09 (p, J = 3.0 Hz, 1H, CH), 3.13–3.24 (m, 2H, N–CH2), 4.24–4.30 (m, 4H, CH2–N), 7.39–7.51 (m, 3H, 3xCHAr), 8.27–8.36 (m, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.2 (2x S–CH2), 27.9 (2xCH2), 33.8 (CH), 46.6 (2xCH2-N), 49.4 (2xN-CH2), 128.0 (2xCHAr), 129.6 (CHAr), 129.8 (2xCHAr), 130.9 (Cq), 136.8 (Cq), 155.0 (Cq), 158.9 (Cq), 175.56 (Cq). HRMS (EI/MS): m/z calculated C19H23N4S: 339.1638 [M + H]+; found: 339.1637.
2-(4,4-Difluoropiperidin-1-yl)-4-phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (16). Compound 16 was obtained according to the general procedure B using 4,4-difluoropiperidine hydrochloride (271 mg, 1.72 mmol, 2.2 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 16 (172 mg, 61%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.32. Mp: 212–214 °C. IR (ATR diamond, cm−1) ν: 2954, 2871, 1568, 1489, 1356, 1108, 1048, 777, 690, 624, 511. 1H NMR (400 MHz, CDCl3) δ: 1.61–1.85 (m, 2H, CH2), 1.83–2.23 (m, 6H, 3xCH2), 2.72 (td, J = 5.1, 11.5 Hz, 2H, N–CH2), 3.09 (p, J = 3.1 Hz, 1H, CH), 3.12–3.23 (m, 2H, N–CH2), 4.07 (t, J = 5.8 Hz, 4H, 2xN-CH2), 7.27–7.57 (m, 3H, 3xCHAr), 8.29 (d, J = 7.8 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.8 (CH), 34.0 (t, J = 22.6 Hz, 2xCH2-CF2), 41.3 (t, J = 5.2 Hz, 2xN-CH2), 49.3 (2xN-CH2), 122.8 (t, J = 241.7 Hz, CF2), 128.1 (2xCHAr), 129.7 (CHAr), 129.9 (2xCHAr), 131.4 (Cq), 136.7 (Cq), 155.1 (Cq), 158.8 (Cq), 175.7 (Cq). 19F NMR (376 MHz, CDCl3) δ: −96.89. HRMS (EI/MS): m/z calculated C20H23F2N4: 357.1885 [M + H]+; found: 357.1886.
N-Benzyl-4-phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (17)
Compound 17 was obtained according to the general procedure B using benzylamine (0.19 mL, 1.73 mmol, 2.2 eq.). After purification by silica gel flash chromatography (PE/EA, 60/40), 17 (141 mg, 51%) was obtained as a white solid. Rf (PE/EA: 70/30): 0.45. Mp: 182–184 °C. IR (ATR diamond, cm−1) ν: 3252, 3084, 2940, 2869, 1570, 1543, 1347, 1133, 774, 689, 626. 1H NMR (400 MHz, CDCl3) δ: 1.69–1.82 (m, 2H), 1.92–2.03 (m, 2H), 2.73 (td, J = 5.1, 11.4 Hz, 2H), 3.06 (p, J = 3.1 Hz, 1H, CH), 3.11–3.22 (m, 2H), 4.73 (d, J = 5.8 Hz, 2H, NH–CH2), 5.35 (t, J = 5.7 Hz, 1H, NH), 7.27 (t, J = 7.3 Hz, 1H, CHAr), 7.34 (t, J = 7.4 Hz, 2H, 2xCHAr), 7.39–7.48 (m, 5H, 5xCHAr), 8.26 (d, J = 8.1 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 46.0 (NH–CH2), 49.4 (2xN-CH2), 127.2 (CHAr), 127.8 (2xCHAr), 128.1 (2xCHAr), 128.6 (2xCHAr), 129.6 (CHAr), 129.9 (2xCHAr), 131.9 (Cq), 136.6 (Cq), 139.9 (Cq), 159.7 (Cq), 175.9 (Cq). HRMS (EI/MS): m/z calculated C22H23N4: 343.1917 [M + H]+; found: 343.1919.
N-(4-Methoxybenzyl)-4-phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (18). Compound 18 was obtained according to the general procedure B using 4-methoxybenzylamine (0.23 mL, 1.75 mmol, 2.2 eq.). After purification by silica gel flash chromatography (PE/EA, 80/20 to 70/30), 18 (126 mg, 42%) was obtained as a white solid. Rf (PE/EA: 70/30): 0.26. Mp: 204–206 °C. IR (ATR diamond, cm−1) ν: 3252, 2945, 1595, 1569, 1544, 1509, 1242, 1172, 1031, 808, 690. 1H NMR (400 MHz, CDCl3) δ: 1.68–1.81 (m, 2H, CH2), 1.91–2.03 (m, 2H, CH2), 2.66–2.79 (m, 2H, N–CH2), 3.06 (p, J = 2.9 Hz, 1H, CH), 3.11–3.22 (m, 2H, N–CH2), 3.80 (s, 3H, OCH3), 4.65 (d, J = 5.7 Hz, 2H, NH–CH2), 5.32 (t, J = 5.8 Hz, 1H, NH–CH2), 6.88 (dt, J = 3.0, 8.8 Hz, 2H, 2xCHAr), 7.35 (dt, J = 2.6, 8.8 Hz, 2H, 2xCHAr), 7.39–7.48 (m, 3H, 3xCHAr), 8.27 (dd, J = 1.7, 7.8 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 45.5 (NH–CH2), 49.4 (2xN-CH2), 55.4 (CH3), 114.0 (2xCHAr), 128.1 (2xCHAr), 129.1 (2xCHAr), 129.6 (CHAr), 129.9 (2xCHAr), 131.8 (Cq), 131.9 (Cq), 136.6 (Cq), 158.9 (Cq), 159.7 (Cq), 175.8 (Cq). HRMS (EI/MS): m/z calculated for C23H25N4O: 373.2023 [M + H]+; found: 373.2020.
4-Phenyl-N-(4-(trifluoromethyl)benzyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (19). Compound 19 was obtained according to the general procedure B using 4-(trifluoromethyl)benzylamine (0.25 mL, 1.75 mmol, 2.2 eq.). After purification by silica gel flash chromatography (PE/EA, 80/20 to 70/30), 19 (129 mg, 39%) was obtained as a white solid. Rf (PE/EA: 70/30): 0.25. Mp: 180–182 °C. IR (ATR diamond, cm−1) ν: 3255, 2948, 1566, 1326, 1112, 1061, 691. 1H NMR (400 MHz, CDCl3) δ: 1.67–1.81 (m, 2H, CH2), 1.92–2.04 (m, 2H, CH2), 2.66–2.79 (m, 2H, N–CH2), 3.05 (p, J = 3.0 Hz, 1H, CH), 3.11–3.22 (m, 2H, N–CH2), 4.79 (d, J = 6.1 Hz, 2H, NH–CH2), 5.52 (t, J = 6.1 Hz, 1H, NH), 7.37–7.47 (m, 3H, 3xCHAr), 7.53 (d, J = 8.1 Hz, 2H, 2xCHAr), 7.59 (d, J = 8.2 Hz, 2H, 2xCHAr), 8.19–8.26 (m, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 45.5 (NH–CH2), 49.3 (2xN-CH2), 124.3 (d, J = 271.9 Hz, Cq-CF3), 125.5 (q, J = 3.7 Hz, 2xCHAr), 127.8 (2xCHAr), 128.1 (2xCHAr), 129.4 (d, J = 32.2 Hz, CF3), 129.7 (CHAr), 129.8 (2xCHAr), 132.2 (Cq), 136.4 (Cq), 144.3 (Cq), 155.5 (Cq), 159.5 (Cq), 176.1 (Cq). 19F NMR (376 MHz, CDCl3) δ: −62.37. HRMS (EI/MS): m/z calculated for C23H22F3N4: 411.1791 [M + H]+; found: 411.1792.
2-(1H-Imidazol-1-yl)-4-phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (20). Compound 20 was obtained according to the general procedure B using imidazole (123 mg, 1.80 mmol, 2.2 eq.). After purification by silica gel flash chromatography (PE/EA, 70/30), 20 (55 mg, 22%) was obtained as a white solid. Rf (PE/EA: 70/30): 0.10. Mp: 198–200 °C. IR (ATR diamond, cm−1) ν: 2963, 2926, 2868, 1561, 1468, 1435, 1387, 1050, 691. 1H NMR (400 MHz, CDCl3) δ: 1.73–1.85 (m, 2H, CH2), 2.02–2.15 (m, 2H, CH2), 2.75 (td, J = 5.1, 11.6 Hz, 2H, N–CH2), 3.21–3.32 (m, 2H, N–CH2), 3.34 (p, J = 3.1 Hz, 1H, CH), 7.18 (s, 1H, CHimidazole), 7.51 (dd, J = 2.5, 4.5 Hz, 3H, 3xCHAr), 8.00 (s, 1H, CHimidazole), 8.41–8.49 (m, 2H, 2xCHAr), 8.70 (s, 1H, CHimidazole). 13C NMR (101 MHz, CDCl3) δ: 27.7 (2xCH2), 33.6 (CH), 48.8 (2xN-CH2), 116.9 (CHimidazole), 128.4 (2xCHAr), 130.2 (2xCHAr), 130.4 (CHimidazole), 130.8 (CHAr), 135.0 (Cq), 136.4 (CHimidazole), 138.9 (Cq), 151.3 (Cq), 156.2 (Cq), 177.7 (Cq). HRMS (EI/MS): m/z calculated for C18H18N5: 304.1557 [M + H]+; found: 304.1557.
2-(Piperidin-1-yl)-4-(p-tolyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (23). Compound 23 was obtained according to the general procedure B using 9 (214 mg, 0.80 mmol, 1.0 eq.) and piperidine (0.17 mL, 1.71 mmol, 2.2 eq.). After purification by silica gel flash chromatography (PE/EA, 100/0 to 90/10), 23 (230 mg, 86%) was obtained as a light yellow solid. Rf (PE/EA: 90/10): 0.51. Mp: 186–188 °C. IR (ATR diamond, cm−1) ν: 2929, 2848, 1589, 1564, 1549, 1496, 1385, 1291, 810. 1H NMR (400 MHz, CDCl3) δ: 1.60–1.69 (m, 6H, 3xCH2), 1.69–1.79 (m, 2H, CH2), 1.90–1.99 (m, 2H, CH2), 2.39 (s, 3H, CH3), 2.65–2.77 (m, 2H, N–CH2), 3.06 (p, J = 3.0 Hz, 1H, CH), 3.09–3.20 (m, 2H, N–CH2), 3.83–3.90 (m, 4H, 2xN-CH2), 7.24 (d, J = 8.0 Hz, 2H, 2xCHAr), 8.20 (d, J = 8.2 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 21.6 (CH3), 25.1 (CH2), 26.0 (2xCH2), 28.0 (2xCH2), 33.9 (CH), 45.1 (2xN-CH2), 49.5 (2xN-CH2), 128.8 (2xCHAr), 129.8 (2xCHAr), 130.2 (Cq), 134.4 (Cq), 139.5 (Cq), 154.9 (Cq), 159.6 (Cq), 175.0 (Cq). HRMS (EI/MS): m/z calculated for C21H27N4: 335.2230 [M + H]+; found: 355.2229.
4-(4-Fluorophenyl)-2-(piperidin-1-yl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (24). Compound 24 was obtained according to the general procedure B using 10 (214 mg, 0.79 mmol, 1.0 eq.) and piperidine (0.17 mL, 1.71 mmol, 2.2 eq.). After purification by silica gel flash chromatography (PE/EA, 100/0 to 90/10), 24 (212 mg, 79%) was obtained as a beige solid. Rf (PE/EA: 90/10): 0.43. Mp: 196–198 °C. IR (ATR diamond, cm−1) ν: 2936, 2863, 1578, 1503, 1384, 1225, 817, 796. 1H NMR (400 MHz, CDCl3) δ: 1.61–1.69 (m, 6H, 3xCH2), 1.70–1.81 (m, 2H, CH2), 1.89–2.01 (m, 2H, CH2), 2.63–2.76 (m, 2H, N–CH2), 3.06 (p, J = 2.9 Hz, 1H, CH), 3.10–3.20 (m, 2H, N–CH2), 3.83–3.90 (m, 4H, 2xN-CH2), 7.10 (t, J = 8.8 Hz, 2H, 2xCHAr), 8.39 (dd, J = 8.9, 5.8 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 25.0 (CH2), 26.0 (2xCH2), 27.9 (2xCH2), 33.8 (CH), 45.1 (2xN-CH2), 49.4 (2xN-CH2), 114.8 (CHAr), 115.0 (CHAr), 130.1 (Cq), 131.9 (CHAr), 132.0 (CHAr), 133.3 (Cq), 153.5 (Cq), 159.5 (Cq), 163.7 (d, J = 249.1 Hz, Cq-F), 175.3 (Cq). 19F NMR (376 MHz, CDCl3) δ: −111.9. HRMS (EI/MS): m/z calculated for C20H24FN4: 339.1980 [M + H]+; found: 339.1976.
1-(4-Phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-yl)piperidin-2-one (22). Compound 22 was obtained according to the general procedure C using 2-piperidone (0.14 mL, 1.51 mmol, 1.5 eq.). After purification by silica gel flash chromatography (DCM/MeOH, 95–5), 22 (133 mg, 40%) was obtained as a light yellow solid. Rf (DCM/MeOH: 95/5): 0.43. Mp: 216–218 °C. IR (ATR diamond, cm−1) ν: 2943, 1670, 1562, 1377, 1153, 838, 776, 696, 624. 1H NMR (400 MHz, CDCl3) δ: 1.79 (m, 2H, CH2), 1.91–2.10 (m, 6H, CH2, 2xCH2,lactame), 2.63 (t, J = 6.4 Hz, 2H, C(O)–CH2), 2.73 (m, 2H, N–CH2), 3.17–3.28 (m, 2H, N–CH2), 3.34 (p, J = 2.9 Hz, 1H, CH), 3.97 (t, J = 5.6 Hz, 2H, N–CH2), 7.39–7.51 (m, 3H, 3xCHAr), 8.25–8.39 (m, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 21.4 (CH2), 23.3 (CH2), 27.7 (2xCH2), 33.5 (CH), 33.6 (C(O)–CH), 48.8 (2xN-CH2), 49.2 (N–CH2), 128.2 (2xCHAr), 130.2 (2xCHAr), 130.2 (CHAr), 135.5 (Cq), 138.3 (Cq), 156.3 (Cq), 157.7 (Cq), 170.9 (C
O), 176.5 (Cq). HRMS (EI/MS): m/z calculated for C20H23N4O: 335.1866 [M + H]+; found: 335.1865.
1-(4-Phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-yl)pyrrolidin-2-one (25). Compound 23 was obtained according to general procedure C using 2-pyrrolidone (134 mg, 1.57 mmol, 1.5 eq.). After purification by silica gel flash chromatography (EA/PE, 80/20 to 90/10), 25 (125 mg, 39%) was obtained as a light yellow solid. Rf (EA/PE: 90/10): 0.18. Mp: 211–213 °C. IR (ATR diamond, cm−1) ν: 2942, 1727, 1557, 1384, 1350, 1131, 775, 693, 622. 1H NMR (400 MHz, CDCl3) δ: 1.72–1.85 (m, 2H, CH2), 1.96–2.08 (m, 2H, CH2), 2.14 (p, J = 7.7 Hz, 2H, CH2), 2.65–2.77 (m, 4H, N–CH2, C(O)–CH2), 3.16–3.27 (m, 2H, N–CH2), 3.36 (p, J = 2.9 Hz, 1H, CH), 4.18 (t, J = 7.1 Hz, 2H, N–CH2), 7.40–7.51 (m, 3H, 3xCHAr), 8.37–8.46 (m, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 17.9 (CH2), 27.7 (2xCH2), 33.6 (CH), 33.8 (C(O)–CH2), 48.6 (N–CH2), 48.9 (2xN-CH2), 128.3 (2xCHAr), 130.2 (2xCHAr), 130.3 (CHAr), 135.8 (Cq), 136.9 (Cq), 154.5 (Cq), 155.2 (Cq), 174.7 (C
O), 176.5 (Cq). HRMS (EI/MS): m/z calculated for C19H21N4: 321.1711 [M + H]+; found: 321.1710.
N,4-Diphenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (26). Compound 26 was obtained according to general procedure C using aniline (0.14 mL, 143 mg, 1.53 mmol, 1.5 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10 to 80/20), 26 (186 mg, 56%) was obtained as a white solid. Rf (PE/EA: 80/20): 0.27. Mp: 191–193 °C. IR (ATR diamond, cm−1) ν: 2956, 1563, 1529, 1446, 1424, 1364, 768, 743, 685. 1H NMR (400 MHz, CDCl3) δ: 1.72–1.85 (m, 2H, CH2), 1.96–2.08 (m, 2H, CH2), 2.69–2.82 (m, 2H, N–CH2), 3.12–3.26 (m, 3H, N–CH2, CH), 7.02 (t, J = 7.4 Hz, 1H, CH), 7.20 (s, 1H, NH), 7.35 (t, J = 7.8 Hz, 2H, 2xCH), 7.40–7.53 (m, 3H, 3xCHAr), 7.74 (d, J = 7.7 Hz, 2H, 2xCHAr), 8.33 (dd, J = 1.8, 8.0 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 49.3 (2xN-CH2), 118.8 (2xCHAr), 122.0 (CHAr), 128.2 (2xCHAr), 129.0 (2xCHAr), 129.9 (CHAr), 130.0 (2xCHAr), 133.3 (Cq), 136.3 (Cq), 140.3 (Cq), 155.6 (Cq), 157.2 (Cq), 176.0 (Cq). HRMS (EI/MS): m/z calculated for C21H21N4: 329.1761 [M + H]+; found: 329.1758.
N-(4-Methoxyphenyl)-4-phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (27). Compound 27 was obtained according to general procedure C using p-anisidine (187 mg, 1.51 mmol, 1.5 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10 to 80/20), 27 (173 mg, 48%) was obtained as a dark red solid. Rf (PE/EA: 80/20): 0.19. Mp: 223–225 °C. IR (ATR diamond, cm−1) ν: 3242, 2920, 1566, 1508, 1357, 12
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 369, 1038, 822, 696, 625. 1H NMR (400 MHz, CDCl3) δ: 1.71–1.83 (m, 2H, CH2), 1.94–2.07 (m, 2H, CH2), 2.68–2.81 (m, 2H, N–CH2), 3.13 (p, J = 2.9 Hz, 1H, CH), 3.15–3.25 (m, 2H, N–CH2), 3.81 (s, 3H, CH3), 6.91 (d, J = 9.2 Hz, 2H, 2xCHAr), 7.04 (s, 1H, NH), 7.39–7.51 (m, 3H, 3xCHAr), 7.61 (d, J = 9.0 Hz, 2H, 2xCHAr), 8.31 (dd, J = 1.6, 7.9 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 49.3 (2xN-CH2), 55.7 (CH3), 114.3 (2xCHAr), 120.9 (2xCHAr), 128.2 (2xCHAr), 129.8 (CHAr), 129.9 (2xCHAr), 132.9 (Cq), 133.6 (Cq), 136.6 (Cq), 155.1 (Cq), 155.6 (Cq), 157.5 (Cq), 176.0 (Cq). HRMS (EI/MS): m/z calculated for C22H23N4O: 359.1866 [M + H]+; found: 359.1863.
369, 1038, 822, 696, 625. 1H NMR (400 MHz, CDCl3) δ: 1.71–1.83 (m, 2H, CH2), 1.94–2.07 (m, 2H, CH2), 2.68–2.81 (m, 2H, N–CH2), 3.13 (p, J = 2.9 Hz, 1H, CH), 3.15–3.25 (m, 2H, N–CH2), 3.81 (s, 3H, CH3), 6.91 (d, J = 9.2 Hz, 2H, 2xCHAr), 7.04 (s, 1H, NH), 7.39–7.51 (m, 3H, 3xCHAr), 7.61 (d, J = 9.0 Hz, 2H, 2xCHAr), 8.31 (dd, J = 1.6, 7.9 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 49.3 (2xN-CH2), 55.7 (CH3), 114.3 (2xCHAr), 120.9 (2xCHAr), 128.2 (2xCHAr), 129.8 (CHAr), 129.9 (2xCHAr), 132.9 (Cq), 133.6 (Cq), 136.6 (Cq), 155.1 (Cq), 155.6 (Cq), 157.5 (Cq), 176.0 (Cq). HRMS (EI/MS): m/z calculated for C22H23N4O: 359.1866 [M + H]+; found: 359.1863.
4-Phenyl-N-(p-tolyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (21). Compound 21 was obtained according to general procedure C using p-toluidine (165 mg, 1.54 mmol, 1.5 eq.). After purification by silica gel flash chromatography (PE/EA, 100/0 to 80/20), 21 (165 mg, 48%) was obtained as a light orange solid. Rf (PE/EA: 80/20): 0.20. Mp: 222–224 °C. IR (ATR diamond, cm−1) ν: 3294, 2943, 2867, 1589, 1519, 1425, 1355, 780, 698, 624. 1H NMR (400 MHz, CDCl3) δ: 1.72–1.83 (m, 2H, CH2), 1.95–2.07 (m, 2H, CH2), 2.34 (s, 3H, CH3), 2.69–2.81 (m, 2H, N–CH2), 3.11–3.25 (m, 3H, N–CH2, CH), 7.13 (s, 1H, NH), 7.15 (d, J = 8.2 Hz, 2H, 2xCHAr), 7.40–7.52 (m, 3H, 3xCHAr), 7.61 (d, J = 8.4 Hz, 2H, 2xCHAr), 8.32 (dd, J = 1.7, 8.0 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 20.9 (CH3), 27.8 (2xCH2), 33.7 (CH), 49.3 (2xN-CH2), 119.0 (2xCHAr), 128.2 (2xCHAr), 129.4 (2xCHAr), 129.8 (CHAr), 129.9 (2xCHAr), 131.5 (Cq), 133.1 (Cq), 136.4 (Cq), 137.7 (Cq), 155.6 (Cq), 157.3 (Cq), 175.9 (Cq). HRMS (EI/MS): m/z calculated for C22H23N4: 343.1917 [M + H]+; found: 343.1917.
4-Phenyl-N-(m-tolyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (28). Compound 28 was obtained according to the general procedure C using m-toluidine (0.16 mL, 1.47 mmol, 1.5 eq.). After purification by silica gel flash chromatography (DCM/PE, 80/20), 28 (106 mg, 31%) was obtained as a light yellow solid. Rf (DCM/PE: 80/20): 0.14. Mp: 230–232 °C. IR (ATR diamond, cm−1) ν: 3226, 2940, 1562, 1531, 1488, 1422, 1357, 1165, 770, 690, 625. 1H NMR (400 MHz, CDCl3) δ: 1.73–1.85 (m, 2H, CH2), 1.96–2.08 (m, 2H, CH2), 2.38 (s, 3H, CH3), 2.69–2.82 (m, 2H, N–CH2), 3.13–3.26 (m, 3H, N–CH2, CH), 6.84 (d, J = 7.4 Hz, 1H, CHAr), 7.12 (s, 1H, NH), 7.23 (t, J = 7.8 Hz, 1H, CHAr), 7.40–7.51 (m, 3H, 3xCHAr), 7.54 (s, 1H, CHAr), 7.58 (dd, J = 8.1, 2.2 Hz, 1H, CHAr), 8.31–8.38 (m, 2H, CHAr). 13C NMR (101 MHz, CDCl3) δ: 21.8 (CH3), 27.8 (2xCH2), 33.7 (CH), 49.3 (2xN-CH2), 115.9 (CHAr), 119.5 (CHAr), 122.9 (CHAr), 128.2 (2xCHAr), 128.8 (CHAr), 129.9 (CHAr), 130.0 (2xCHAr), 133.2 (Cq), 136.4 (Cq), 138.7 (Cq), 140.2 (Cq), 155.5 (Cq), 157.2 (Cq), 176.0 (Cq). HRMS (EI/MS): m/z calculated for C22H23N4: 343.1917 [M + H]+; found: 343.1914.
4-Phenyl-N-(o-tolyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (29). Compound 29 was obtained according to the general procedure C using o-toluidine (0.10 mL, 1.46 mmol, 1.5 eq.). After purification by silica gel flash chromatography (DCM/PE, 70/30 to 80/20), 29 (54 mg, 15%) was obtained as a beige solid. Rf (DCM/PE: 70/30): 0.16. Mp: 208–210 °C. IR (ATR diamond, cm−1) ν: 3212, 2946, 1564, 1529, 1361, 744, 684, 625. 1H NMR (400 MHz, CDCl3) δ: 1.72–1.85 (m, 2H, CH2), 1.96–2.07 (m, 2H, CH2), 2.37 (s, 3H, CH3), 2.70–2.82 (m, 2H, N–CH2), 3.11–3.26 (m, 3H, N–CH2, CH), 6.88 (s, 1H, NH), 6.99 (td, J = 7.4, 1.3 Hz, 1H, CHAr), 7.21 (d, J = 7.3 Hz, 1H, CHAr), 7.27 (td, J = 7.8, 1.6 Hz, 1H, CHAr), 7.39–7.51 (m, 3H, 3xCHAr), 8.29–8.36 (m, 3H, 2xCHAr, CHAr). 13C NMR (101 MHz, CDCl3) δ: 18.3 (CH3), 27.8 (2xCH2), 33.7 (CH), 49.3 (2xN-CH2N-CH2), 120.4 (CHAr), 122.6 (CHAr), 126.7 (CHAr), 127.2 (Cq), 128.2 (2xCHAr), 129.8 (CHAr), 130.0 (2xCHAr), 130.4 (CHAr), 133.2 (Cq), 136.3 (Cq), 138.3 (Cq), 155.6 (Cq), 157.5 (Cq), 176.0 (Cq). HRMS (EI/MS): m/z calculated for C22H23N4: 343.1917 [M + H]+; found: 343.1916.
4-Phenyl-N-(4-(trifluoromethyl)phenyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (30). Compound 30 was obtained according to general procedure C using 4-(trifluoromethyl)aniline (0.19 mL, 243 mg, 1.50 mmol, 1.5 eq.). After purification by silica gel flash chromatography (DCM/PE, 80/20), 30 (212 mg, 56%) was obtained as a beige solid. Rf (DCM/PE: 80/20): 0.20. Mp: 182–184 °C. IR (ATR diamond, cm−1) ν: 3280, 2957, 1534, 1415, 1321, 1109, 1066, 836, 625. 1H NMR (400 MHz, CDCl3) δ: 1.73–1.84 (m, 2H, CH2), 1.98–2.09 (m, 2H, CH2), 2.69–2.82 (m, 2H, N–CH2), 3.14–3.28 (m, 3H, N–CH2, CH), 7.36 (s, 1H, NH), 7.44–7.55 (m, 3H, 3xCHAr), 7.59 (d, J = 8.6 Hz, 2H, 2xCHAr), 7.85 (d, J = 8.5 Hz, 2H, 2xCHAr), 8.29–8.36 (m, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 49.2 (2xN-CH2), 117.9 (2xCHAr), 123.3 (Cq), 124.8 (d, J = 241.0 Hz, Cq-CF3), 126.3 (q, J = 3.9 Hz, 2xCHAr), 128.3 (2xCHAr), 130.0 (2xCHAr), 130.1 (CHAr), 134.1 (Cq), 136.0 (Cq), 143.4 (d, J = 0.9 Hz, CF3), 155.8 (Cq), 156.6 (Cq), 176.3 (Cq). 19F NMR (376 MHz, CDCl3) δ: −61.65. HRMS (EI/MS): m/z calculated for C22H20F3N4: 397.1635 [M + H]+; found: 397.1633.
4-((4-Phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-yl)amino)benzonitrile (31). Compound 31 was obtained according to the general procedure C using 4-aminobenzonitrile (177 mg, 1.50 mmol, 1.5 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10 to 70/30), 31 (124 mg, 35%) was obtained as a light yellow solid. Rf (DCM/PE: 80/20): 0.16. Mp: 234–236 °C. IR (ATR diamond, cm−1) ν: 3399, 2923, 2214, 1566, 1517, 1413, 1357, 1171, 834, 687, 535. 1H NMR (400 MHz, CDCl3) δ: 1.71–1.84 (m, 2H, CH2), 1.98–2.11 (m, 2H, CH2), 2.68–2.81 (m, 2H, N–CH2), 3.15–3.28 (m, 3H, N–CH2, CH), 7.43–7.55 (m, 4H, 3xCHAr, NH), 7.60 (d, J = 8.8 Hz, 2H, 2xCHAr), 7.85 (d, J = 8.8 Hz, 2H, 2xCHAr), 8.25–8.35 (m, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 49.1 (2xN-CH22xN-CH2), 104.0 (Cq), 118.0 (2xCHAr), 119.7 (Cq), 128.3 (2xCHAr), 129.9 (2xCHAr), 130.2 (CHAr), 133.3 (2xCHAr), 134.5 (Cq), 135.8 (Cq), 144.3 (Cq), 155.9 (Cq), 156.2 (Cq), 176.5 (Cq). HRMS (EI/MS): m/z calculated for C22H20N5: 354.1713 [M + H]+; found: 354.1710.
N-(4-Nitrophenyl)-4-phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (32). Compound 32 was obtained according to general procedure C using 4-nitroaniline (208 mg, 1.50 mmol, 1.5 eq.). The obtained solid was washed with DCM and dried under vacuo to afford 32 (148 mg, 39%) as a yellow solid. Rf (PE/EA: 80/20): 0.17. Mp: >260 °C. IR (ATR diamond, cm−1) ν: 3386, 2940, 1530, 1488, 1416, 1324, 1111, 840, 686, 584. 1H NMR (400 MHz, CDCl3) δ: 1.74–1.85 (m, 2H, CH2), 2.01–2.12 (m, 2H, CH2), 2.70–2.81 (m, 2H, N–CH2), 3.13–3.29 (m, 3H, N–CH2, CH), 7.45–7.54 (m, 3H, 3xCHAr), 7.56 (s, 1H, NH), 7.89 (d, J = 9.2 Hz, 2H, 2xCHAr), 8.24 (d, J = 9.2 Hz, 2H, 2xCHAr), 8.32 (dd, J = 2.3, 7.3 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 49.1 (2xN-CH2), 117.3 (2xCHAr), 125.5 (2xCHAr), 128.4 (2xCHAr), 130.0 (2xCHAr), 130.3 (CHAr), 134.9 (Cq), 135.7 (Cq), 141.6 (Cq), 146.3 (Cq), 156.0 (Cq), 156.0 (Cq), 176.6 (Cq). HRMS (EI/MS): m/z calculated for C21H20N5O2: 374.1612 [M + H]+; found: 374.1607.
4-Phenyl-N-(pyridin-3-yl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (33). Compound 33 was obtained according to the general procedure C using 3-aminopyridine (142 mg, 1.5 mmol, 1.5 eq.). After purification by silica gel flash chromatography (EA/PE, 70/30), 33 (115 mg, 34%) was obtained as a light yellow solid. Rf (EA/PE: 70/30): 0.30. Mp: 211–213 °C. IR (ATR diamond, cm−1) ν: 2954, 1531, 1428, 1386, 1353, 692, 623. 1H NMR (400 MHz, CDCl3) δ: 1.71–1.83 (m, 2H, CH2), 1.97–2.10 (m, 2H, CH2), 2.68–2.81 (m, 2H, N–CH2), 3.12–3.26 (m, 3H; N–CH2, CH), 7.27 (dd, J = 8.3, 4.8 Hz, 1H, CHAr), 7.36 (s, 1H, NH), 7.40–7.54 (m, 3H, 3xCHAr), 8.20–8.37 (m, 4H, 2xCHAr, 2xCHAr), 8.83 (d, J = 2.2 Hz, 1H, CHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 49.2 (2xN-CH2), 123.5 (CHAr), 125.4 (CHAr), 128.3 (2xCHAr), 130.0 (2xCHAr), 130.1 (CHAr), 133.9 (Cq), 136.0 (Cq), 137.0 (Cq), 140.7 (CHAr), 143.0 (CHAr), 155.8 (Cq), 156.8 (Cq), 176.3 (Cq). HRMS (EI/MS): m/z calculated for C20H20N5: 330.1713 [M + H]+; found: 330.1714.
N-(6-Methoxypyridin-3-yl)-4-phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (34). Compound 34 was obtained according to the general procedure C using 5-amino-2-methoxypyridine (0.12 mL, 189 mg, 1.52 mmol, 1.5 eq.). After purification by silica gel flash chromatography (PE/EA, 50/50), 34 (102 mg, 28%) was obtained as a light orange solid. Rf (PE/EA: 50/50): 0.52. Mp: 183–185 °C. IR (ATR diamond, cm−1) ν: 1942, 1537, 1490, 1348, 1278, 1035, 831, 691, 624. 1H NMR (400 MHz, CDCl3) δ: 1.73–1.81 (m, 2H, CH2), 1.95–2.07 (m, 2H, CH2), 2.68–2.80 (m, 2H, N–CH2), 3.13 (p, J = 2.9 Hz, 1H, CH), 3.15–3.25 (m, 2H, N–CH2), 3.94 (s, 3H, O–CH3), 6.77 (d, J = 8.9 Hz, 1H, CHAr), 6.95 (s, 1H, NH), 7.39–7.51 (m, 3H, 3xCHAr), 8.04 (dd, J = 8.9, 2.8 Hz, 1H, CHAr), 8.22–8.31 (m, 2H, 2xCHAr), 8.42 (d, J = 2.7 Hz, 1H, CHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 49.3 (2xN-CH2), 53.6 (O–CH3), 110.5 (CHAr), 128.2 (2xCHAr), 129.9 (2xCHAr), 129.9 (CHAr), 131.0 (Cq), 131.7 (CHAr), 133.4 (Cq), 136.1 (Cq), 137.8 (CHAr), 155.8 (Cq), 157.3 (Cq), 160.0 (Cq), 176.2 (Cq). HRMS (EI/MS): m/z calculated for C21H22N5O: 360.1819 [M + H]+; found: 360.1819.
N-Phenyl-4-(p-tolyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (35)
Compound 35 was obtained according to general procedure C using 9 (274 mg, 1.02 mmol, 1.0 eq.) and aniline (0.14 mL, 1.53 mmol, 1.5 eq.). After purification by silica gel flash chromatography (PE/EA: 90/10 to 80/20), 35 (156 mg, 44%) was obtained as a beige solid. Rf (PE/EA: 80/20): 0.50. Mp: 195–197 °C. IR (ATR diamond, cm−1) ν: 3229, 2960, 1574, 1531, 1429, 1362, 1171, 812, 742, 688. 1H NMR (400 MHz, CDCl3) δ: 1.71–1.84 (m, 2H, CH2), 1.95–2.07 (m, 2H, CH2), 2.42 (s, 3H, CH3), 2.68–2.81 (m, 2H, N–CH2), 3.12–3.25 (m, 3H, N–CH2, CH), 7.01 (tt, J = 7.3, 1.2 Hz, 1H, CHAr), 7.15 (s, 1H, NH), 7.29 (d, J = 7.9 Hz, 2H, 2xCHAr), 7.30–7.38 (m, 2H, 2xCHAr), 7.70–7.77 (m, 2H, 2xCHAr), 8.24 (d, J = 8.3 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 21.6 (CH3), 27.8 (2xCH2), 33.7 (CH), 49.3 (2xN-CH2), 118.7 (2xCHAr), 121.9 (CHAr), 129.0 (2xCHAr), 129.0 (2xCHAr), 129.9 (2xCHAr), 133.1 (Cq), 133.5 (Cq), 140.0 (Cq), 140.4 (Cq), 155.7 (Cq), 157.1 (Cq), 175.8 (Cq). HRMS (EI/MS): m/z calculated for C22H23N4: 343.1917 [M + H]+; found: 343.1916.
4-(p-Tolyl)-N-(4-(trifluoromethyl)phenyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-amine (36). Compound 36 was obtained according to general procedure C using 9 (264 mg, 0.98 mmol, 1.0 eq.) and 4-(trifluoromethyl)aniline (0.19 mL, 1.50 mmol, 1.5 eq.). After purification by silica gel flash chromatography (PE/EA: 90/10 to 80/20), 36 (200 mg, 49%) was obtained as a beige solid. Rf (PE/EA: 80/20): 0.20. Mp: 195–197 °C. IR (ATR diamond, cm−1) ν: 3270, 2962, 1576, 1536, 1318, 1100, 1064, 838. 1H NMR (400 MHz, CDCl3) δ: 1.71–1.84 (m, 2H, CH2), 1.97–2.09 (m, 2H, CH2), 2.43 (s, 3H, CH3), 2.68–2.81 (m, 2H, N–CH2), 3.13–3.27 (m, 3H, N–CH2, CH), 7.30 (d, J = 8.0 Hz, 2H, 2xCHAr), 7.37 (s, 1H, NH), 7.58 (d, J = 8.4 Hz, 2H, 2xCHAr), 7.85 (d, J = 8.5 Hz, 2H, 2xCHAr), 8.24 (d, J = 8.3 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 21.5 (CH3), 27.7 (2xCH2), 33.6 (CH), 49.0 (2xN-CH2), 117.7 (2xCHAr), 124.6 (d, J = 252.1 Hz, Cq-CF3), 126.1 (q, J = 3.8 Hz, 2xCHAr), 128.9 (2xCHAr), 129.8 (2xCHAr), 133.1 (Cq), 133.8 (Cq), 140.2 (Cq), 143.3 (d, J = 0.6 Hz, CF3), 155.7 (Cq), 156.4 (Cq), 176.0 (Cq). 19F NMR (376 MHz, CDCl3) δ: −61.63. HRMS (EI/MS): m/z calculated for C23H22F3N4: 411.1791 [M + H]+; found: 411.1791.
4-Phenyl-2-(p-tolyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (39). Compound 39 was obtained according to the general procedure D using p-tolylboronic acid (239 mg, 1.60 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 39 (182 mg, 70%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.40. Mp: 192–194 °C. IR (ATR diamond, cm−1) ν: 2963, 2924, 2871, 1550, 1387, 1170, 772, 689, 623. 1H NMR (400 MHz, CDCl3) δ: 1.76–1.87 (m, 2H, CH2), 2.01–2.13 (m, 2H, CH2), 2.43 (s, 3H, CH3), 2.77 (td, J = 4.9, 11.5 Hz, 2H, N–CH2), 3.20–3.32 (m, 2H, N–CH2), 3.38 (p, J = 2.8, 3.2 Hz, 1H, CH), 7.30 (d, J = 7.8 Hz, 2H, 2xCHAr), 7.43–7.56 (m, 3H, 3xCHAr), 8.47 (dd, J = 8.1, 12.2 Hz, 4H, 4xCHAr). 13C NMR (101 MHz, CDCl3) δ: 21.6 (CH3), 27.9 (2xCH2), 33.7 (CH), 48.9 (2xN-CH2), 128.2 (2xCHAr), 128.3 (2xCHAr), 129.3 (2xCHAr), 130.0 (CHAr), 130.2 (2xCHAr), 135.7 (Cq), 136.4 (Cq), 139.0 (Cq), 140.3 (Cq), 154.7 (Cq), 161.2 (Cq), 175.1 (Cq). HRMS (EI/MS): m/z calculated for C22H22N3: 328.1808 [M + H]+; found: 328.1808.
2,4-Diphenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (40). Compound 40 was obtained according to general procedure D using phenylboronic acid (195 mg, 1.60 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 40 (171 mg, 68%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.30. Mp: 195–197 °C. IR (ATR diamond, cm−1) ν: 2945, 2921, 1553, 1389, 768, 692, 624. 1H NMR (400 MHz, CDCl3) δ: 1.75–1.89 (m, 2H, CH2), 2.02–2.14 (m, 2H, CH2), 2.72–2.84 (m, 2H, N–CH2), 3.21–3.32 (m, 2H, N–CH2), 3.40 (p, J = 2.9 Hz, 1H, CH), 7.42–7.56 (m, 6H, 6xCHAr), 8.50 (dd, J = 1.7, 8.1 Hz, 2H, 2xCHAr), 8.57 (dd, J = 1.8, 8.0 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 48.9 (2xN-CH2), 128.2 (2xCHAr), 128.3 (2xCHAr), 128.5 (2xCHAr), 130.0 (CHAr), 130.2 (2xCHAr), 136. (Cq), 138.4 (Cq), 139.3 (Cq), 154.8 (Cq), 161.1 (Cq), 175.2 (Cq). HRMS (EI/MS): m/z calculated for C21H20N3: 314.1652 [M + H]+; found: 314.1655.
4-Phenyl-2-(m-tolyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (41). Compound 41 was obtained according to general procedure D using m-tolylboronic acid (215 mg, 1.58 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 41 (189 mg, 73%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.27. Mp: 191–193 °C. IR (ATR diamond, cm−1) ν: 2961, 2946, 2869, 1553, 1385, 1376, 771, 733, 690, 624. 1H NMR (400 MHz, CDCl3) δ: 1.75–1.88 (m, 2H, CH2), 2.02–2.14 (m, 2H, CH2), 2.47 (s, 3H, CH3), 2.71–2.84 (m, 2H, N–CH2), 3.21–3.32 (m, 2H, N–CH2), 3.39 (p, J = 3.0 Hz, 1H, CH), 7.28 (d, J = 7.5 Hz, 1H, CHAr), 7.39 (t, J = 7.9 Hz, 1H, CHAr), 7.43–7.56 (m, 3H, 3xCHAr), 8.36 (d, J = 6.9 Hz, 2H, 2xCHAr), 8.48 (dt, J = 1.5, 2.3, 6.4 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 21.7 (CH3), 27.9 (2xCH2), 33.7 (CH), 48.9 (2xN-CH2), 125.6 (CHAr), 128.3 (2xCHAr), 128.5 (CHAr), 128.8 (CHAr), 130.0 (CHAr), 130.2 (2xCHAr), 131.1 (CHAr), 136.3 (Cq), 138.1 (Cq), 138.3 (Cq), 139.2 (Cq), 154.9 (Cq), 161.3 (Cq), 175.1 (Cq). HRMS (EI/MS): m/z calculated for C22H22N3: 328.1808 [M + H]+; found: 328.1813.
4-Phenyl-2-(o-tolyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (42). Compound 42 was obtained according to general procedure D using o-tolylboronic acid (215 mg, 1.58 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 42 (196 mg, 76%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.22. Mp: 228–230 °C. IR (ATR diamond, cm−1) ν: 2944, 2869, 1552, 1387, 1131, 844, 767, 736, 695, 623. 1H NMR (400 MHz, CDCl3) δ: 1.79–1.88 (m, 2H, CH2), 2.03–2.14 (m, 2H, CH2), 2.66 (s, 3H, CH3), 2.75–2.87 (m, 2H, N–CH2), 3.22–3.34 (m, 2H, N–CH2), 3.38 (p, J = 2.9 Hz, 1H, CH), 7.27–7.38 (m, 3H, 3xCHAr), 7.38–7.55 (m, 3H, 2xCHAr, CHAr), 7.87–7.94 (m, 1H, CHAr), 8.44 (dd, J = 1.8, 8.0 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 21.5 (CH3), 27.9 (2xCH2), 33.6 (CH), 48.9 (2xN-CH2), 126.0 (CHAr), 128.3 (2xCHAr), 129.1 (CHAr), 130.0 (CHAr), 130.2 (2xCHAr), 130.6 (CHAr), 131.3 (CHAr), 136.2 (Cq), 137.3 (Cq), 138.5 (Cq), 138.9 (Cq), 154.6 (Cq), 164.1 (Cq), 174.9 (Cq). HRMS (EI/MS): m/z calculated for C22H22N3: 328.1808 [M + H]+; found: 328.1813.
2-(4-Methoxyphenyl)-4-phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (43). Compound 43 was obtained according to general procedure D using 4-methoxyphenylboronic acid (244 mg, 1.60 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 43 (188 mg, 69%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.24. Mp: 204–206 °C. IR (ATR diamond, cm−1) ν: 2960, 2935, 1549, 1387, 1247, 1031, 772, 694. 1H NMR (400 MHz, CDCl3) δ: 1.75–1.87 (m, 2H, CH2), 2.01–2.12 (m, 2H, CH2), 2.77 (td, J = 5.0, 11.4 Hz, 2H, N–CH2), 3.20–3.31 (m, 2H, N–CH2), 3.36 (p, J = 3.0 Hz, 1H, CH), 3.89 (s, 3H, O–CH3), 7.01 (d, J = 8.5 Hz, 2H, 2xCHAr), 7.42–7.55 (m, 3H, 3xCHAr), 8.47 (d, J = 7.7 Hz, 2H, 2xCHAr), 8.52 (d, J = 8.4 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.9 (2xCH2), 33.7 (CH), 49.0 (2xN-CH2), 55.4 (O–CH3), 113.9 (2xCHAr), 128.2 (2xCHAr), 129.9 (2xCHAr), 129.9 (CHAr), 130.2 (2xCHAr), 131.1 (Cq), 136.4 (Cq), 138.6 (Cq), 154.7 (Cq), 160.9 (Cq), 161.5 (Cq), 175.0 (Cq). HRMS (EI/MS): m/z calculated for C22H22N3O: 344.1757 [M + H]+; found: 344.1759.
4-(4-Phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-yl)phenol (44). Compound 44 was obtained according to general procedure D using 4-hydroxyphenylboronic acid (219 mg, 1.59 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 80/20), 44 (141 mg, 54%) was obtained as a white solid. Rf (PE/EA: 80/20): 0.17. Mp: 254–256 °C. IR (ATR diamond, cm−1) ν: 3059, 2942, 2675, 1610, 1552, 1400, 1238, 1166, 774, 688, 625. 1H NMR (400 MHz, DMSO-d6) δ: 1.61–1.75 (m, 2H, CH2), 1.97–2.11 (m, 2H, CH2), 2.58–2.71 (m, 2H, N–CH2), 3.14–3.21 (m, 2H, N–CH2), 3.23 (p, J = 2.3 Hz, 1H, CH), 6.89 (td, J = 1.7, 8.7 Hz, 2H, 2xCHAr), 7.45–7.57 (m, 3H, 3xCHAr), 8.34 (td, J = 2.0, 8.7 Hz, 2H, 2xCHAr), 8.39 (dd, J = 1.9, 7.8 Hz, 2H, 2xCHAr), 9.88 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6) δ: 27.0 (2xCH2), 33.0 (CH), 48.2 (2xN-CH2), 115.3 (2xCHAr), 128.0 (2xCHAr), 128.6 (Cq), 129.5 (2xCHAr), 129.8 (CHAr), 129.8 (CHAr), 135.7 (Cq), 138.2 (Cq), 153.5 (Cq), 159.6 (Cq), 159.8 (Cq), 175.1 (Cq). HRMS (EI/MS): m/z calculated for C21H20N3O: 330.1601 [M + H]+; found: 330.1602.
4-Phenyl-2-(4-((tetrahydro-2H-pyran-2-yl)oxy)phenyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (45). Compound 45 was obtained according to general procedure D using 4-(tetrahydro-2H-pyran-2-yloxy)phenylboronic acid (358 mg, 1.61 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 80/20), 45 (230 mg, 69%) was obtained as a white solid. Rf (PE/EA: 80/20): 0.34. Mp: 210–212 °C. IR (ATR diamond, cm−1) ν: 2940, 2872, 1551, 1386, 1236, 1163, 1113, 958, 920, 768, 693. 1H NMR (400 MHz, CDCl3) δ: 1.57–1.75 (m, 3H, 3xCHTHP), 1.76–1.85 (m, 2H, CH2), 1.86–1.95 (m, 2H, 2xCHTHP), 1.97–2.12 (m, 3H, CH2, CHTHP), 2.70–2.83 (m, 2H, N–CH2), 3.19–3.31 (m, 2H, N–CH2), 3.36 (p, J = 2.9 Hz, 1H, CH), 3.59–3.68 (m, 1H, CHTHP), 3.89–3.99 (m, 1H, CHTHP), 5.52 (t, J = 3.3 Hz, 1H, O–CH–O), 7.15 (d, J = 8.8 Hz, 2H, 2xCHAr), 7.44–7.55 (m, 3H, 3xCHAr), 8.43–8.53 (m, 4H, 2xCHAr, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 18.9 (CH2,THP), 25.3 (CH2,THP), 27.9 (2xCH2), 30.4 (CH2,THP), 33.7 (CH), 48.9 (2xN-CH2), 62.2(CH2,THP), 96.3 (O–CH–O), 116.3 (2xCHAr), 128.2 (2xCHAr), 129.8 (2xCHAr), 129.9 (CHAr), 130.2 (2xCHAr), 131.9 (Cq), 136.4 (Cq), 138.7 (Cq), 154.7 (Cq), 159.0 (Cq), 160.9 (Cq), 175.0 (Cq). HRMS (EI/MS): m/z calculated for C26H28N3O2: 414.2176 [M + H]+; found: 414.2174.
2-(2-Naphthalen-2-yl)-4-phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (46). Compound 46 was obtained according to general procedure D using 2-naphthylboronic acid (275 mg, 1.60 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 46 (196 mg, 67%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.35. Mp: 210–212 °C. IR (ATR diamond, cm−1) ν: 2949, 2868, 1552, 1391, 1380, 773, 755, 700, 625. 1H NMR (400 MHz, CDCl3) δ: 1.80–1.92 (m, 2H, CH2), 2.05–2.16 (m, 2H, CH2), 2.74–2.87 (m, 2H, N–CH2), 3.23–3.34 (m, 2H, N–CH2), 3.45 (p, J = 2.9 Hz, 1H, CH), 7.46–7.60 (m, 5H, 3xCHAr, 2xCHAr), 7.86–7.93 (m, 1H, CHAr), 7.97 (d, J = 8.7 Hz, 1H, CHAr), 8.01–8.09 (m, 1H, CHAr), 8.50–8.57 (m, 2H, 2xCHAr), 8.71 (dd, J = 1.7, 8.6 Hz, 1H, CHAr), 9.10 (s, 1H, CHAr). 13C NMR (101 MHz, CDCl3) δ: 27.9 (2xCH2), 33.7 (CH), 48.9 (2xN-CH2), 125.6 (CHAr), 126.2 (CHAr), 126.9 (CHAr), 127.8 (CHAr), 128.1 (CHAr), 128.3 (2xCHAr), 128.3 (CHAr), 129.3 (CHAr), 130.1 (CHAr), 130.3 (CHAr), 133.5 (Cq), 134.6 (Cq), 135.7 (Cq), 136.3 (Cq), 139.3 (Cq), 155.0 (Cq), 161.0 (Cq), 175.3 (Cq). HRMS (EI/MS): m/z calculated for C25H22N3: 364.1808 [M + H]+; found: 364.1812.
2-(4-Fluorophenyl)-4-phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (47). Compound 47 was obtained according to general procedure D using 4-fluorophenylboronic acid (224 mg, 1.60 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 47 (186 mg, 70%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.42. Mp: 178–180 °C. IR (ATR diamond, cm−1) ν: 2922, 2864, 1601, 1555, 1389, 1151, 832, 768, 688, 622. 1H NMR (400 MHz, CDCl3) δ: 1.75–1.87 (m, 2H, CH2), 2.02–2.14 (m, 2H, CH2), 2.71–2.83 (m, 2H, N–CH2), 3.21–3.32 (m, 2H, N–CH2), 3.37 (p, J = 2.9 Hz, 1H, CH), 7.12–7.22 (m, 2H, 2xCHAr), 7.45–7.56 (m, 3H, 3xCHAr), 8.47 (dd, J = 1.7, 8.0 Hz, 2H, 2xCHAr), 8.57 (dd, J = 5.9, 9.3 Hz, 2H,2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 48.9 (2xN-CH2), 115.3 (CHAr), 115.5 (CHAr), 128.3 (2xCHAr), 130.1 (CHAr), 130.2 (2xCHAr), 130.3 (CHAr), 130.4 (CHAr), 134.6 (d, J = 2.9 Hz, Cq), 136.1 (Cq), 139.2 (Cq), 154.9 (Cq), 160.19 (Cq), 164.5 (d, J = 249.5 Hz, CAr-F), 175.3 (Cq). 19F NMR (376 MHz, CDCl3) δ: −111.46. HRMS (EI/MS): m/z calculated for C21H19FN3: 332.1558 [M + H]+; found: 332.1563.
4-(4-Phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-yl)benzonitrile (48). Compound 48 was obtained according to general procedure D using 4-cyanophenylboronic acid (236 mg, 1.60 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 48 (224 mg, 82%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.22. Mp: 202–204 °C. IR (ATR diamond, cm−1) ν: 2956, 2922, 2227, 1549, 1389, 771, 693, 622, 541. 1H NMR (400 MHz, CDCl3) δ: 1.74–1.87 (m, 2H, CH2), 2.05–2.16 (m, 2H, CH2), 2.71–2.83 (m, 2H, N–CH2), 3.23–3.34 (m, 2H, N–CH2), 3.40 (p, J = 2.9 Hz, 1H, CH), 7.47–7.57 (m, 3H, 3xCHAr), 7.79 (d, J = 8.5 Hz, 2H, 2xCHAr), 8.43–8.54 (m, 2H, 2xCHAr), 8.69 (d, J = 8.5 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 48.8 (2xN-CH2), 113.5 (Cq), 119.1 (Cq), 128.4 (2xCHAr), 128.8 (2xCHAr), 130.2 (2xCHAr), 130.4 (2xCHAr), 132.4 (2xCHAr), 135.8 (Cq), 140.3 (Cq), 142.5 (Cq), 155.1 (Cq), 159.1 (Cq), 175.7 (Cq). HRMS (EI/MS): m/z calculated for C22H19N4: 339.1604 [M + H]+; found: 339.1608.
4-Phenyl-2-(4-(trifluoromethyl)phenyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (49). Compound 49 was obtained according to general procedure D using 4-(trifluoromethyl)phenylboronic acid (303 mg, 1.60 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 49 (222 mg, 72%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.40 Mp: 166–168 °C. IR (ATR diamond, cm−1) ν: 2953, 1552, 1389, 1320, 1116, 1061, 774, 690. 1H NMR (400 MHz, CDCl3) δ: 1.76–1.89 (m, 2H, CH2), 2.04–2.16 (m, 2H, CH2), 2.78 (td, J = 5.1, 11.7 Hz, 2H, N–CH2), 3.23–3.34 (m, 2H, N–CH2), 3.41 (p, J = 3.2 Hz, 1H, CH), 7.45–7.58 (m, 3H, 3xCHAr), 7.75 (d, J = 8.1 Hz, 2H, 2xCHAr), 8.50 (dt, J = 1.6, 7.5 Hz, 2H, 2xCHAr), 8.69 (d, J = 8.0 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 48.8 (2xN-CH2), 124.4 (d, J = 272.2 Hz, Cq-CF3), 125.5 (q, J = 3.8 Hz, 2xCHAr), 128.3 (2xCHAr), 128.6 (2xCHAr), 130.2 (2xCHAr), 130.3 (CHAr), 131.8 (d, J = 32.2 Hz, CF3), 135.9 (Cq), 140.0 (Cq), 141.7 (d, J = 1.7 Hz, Cq), 155.0 (Cq), 159.6 (Cq), 175.6 (Cq). 19F NMR (376 MHz, CDCl3) δ: −62.59. HRMS (EI/MS): m/z calculated for C22H19F3N3: 382.1526 [M + H]+; found: 382.1525.
2-(4-Nitrophenyl)-4-phenyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (50). Compound 50 was obtained according to general procedure D using 4-nitrophenylboronic acid (268 mg, 1.60 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 50 (206 mg, 76%) was obtained as an orange solid. Rf (PE/EA: 90/10): 0.40. Mp: 191–193 °C. IR (ATR diamond, cm−1) ν: 2925, 2872, 1555, 1519, 1388, 1350, 839, 688. 1H NMR (400 MHz, CDCl3) δ: 1.75–1.88 (m, 2H, CH2), 2.06–2.18 (m, 2H, CH2), 2.72–2.84 (m, 2H, N–CH2), 3.24–3.35 (m, 2H, N–CH2), 3.41 (p, J = 2.8 Hz, 1H, CH), 7.47–7.58 (m, 3H, 3xCHAr), 8.34 (dt, J = 2.0, 9.0 Hz, 2H, 2xCHAr), 8.44–8.54 (m, 2H, 2xCHAr), 8.75 (dt, J = 2.3, 9.0 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.7 (CH), 48.8 (2xN-CH2), 123.8 (2xCHAr), 128.4 (2xCHAr), 129.2 (2xCHAr), 130.2 (2xCHAr), 130.4 (CHAr), 135.7 (Cq), 140.4 (Cq), 144.2 (Cq), 149.1 (Cq), 155.2 (Cq), 158.8 (Cq), 175.8 (Cq). HRMS (EI/MS): m/z calculated for C21H19N4O2: 359.1503 [M + H]+; found: 359.1506.
4-Phenyl-2-(thiophen-3-yl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (51). Compound 51 was obtained according to general procedure D using 3-thienylboronic acid (208 mg, 1.62 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 51 (195 mg, 77%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.19. Mp: 189–191 °C. IR (ATR diamond, cm−1) ν: 2956, 2867, 1553, 1386, 1342, 819, 689, 623. 1H NMR (400 MHz, CDCl3) δ: 1.75–1.86 (m, 2H, CH2), 2.00–2.13 (m, 2H, CH2), 2.76 (td, J = 5.5, 12.2 Hz, 2H, N–CH2), 3.19–3.31 (m, 2H, N–CH2), 3.35 (p, J = 3.3 Hz, 1H, CH), 7.38 (t, J = 3.7 Hz, 1H, CHAr), 7.42–7.55 (m, 3H, 3xCHAr), 8.00 (d, J = 5.0 Hz, 1H, CHAr), 8.31–8.36 (m, 1H, CHAr), 8.45 (d, J = 7.9 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.6 (CH), 48.9 (2xN-CH2), 125.8 (CHAr), 127.3 (CHAr), 127.8 (CHAr), 128.2 (2xCHAr), 130.0 (CHAr), 130.2 (2xCHAr), 136.1 (Cq), 138.8 (Cq), 142.3 (Cq), 154.9 (Cq), 158.5 (Cq), 175.2 (Cq). HRMS (EI/MS): m/z calculated forC19H18N3S: 320.1216 [M + H]+; found: 320.1223.
4-Phenyl-2-(pyridin-3-yl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (52). Compound 52 was obtained according to general procedure D using 3-pyridinylboronic acid (200 mg, 1.63 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 50/50), 52 (152 mg, 60%) was obtained as a white solid. Rf (PE/EA: 50/50): 0.25. Mp: 231–233 °C. IR (ATR diamond, cm−1) ν: 2926, 2864, 1552, 1387, 1371, 1023, 843, 692, 620. 1H NMR (400 MHz, CDCl3) δ: 1.75–1.87 (m, 2H, CH2), 2.02–2.15 (m, 2H, CH2), 2.77 (td, J = 5.1, 11.6 Hz, 2H, N–CH2), 3.22–3.33 (m, 2H, N–CH2), 3.40 (p, J = 3.1 Hz, 1H, CH), 7.42 (dd, J = 4.8, 8.0 Hz, 1H, CHAr), 7.45–7.56 (m, 3H, 3xCHAr), 8.50 (d, J = 7.6 Hz, 2H, 2xCHAr), 8.70 (d, J = 4.8 Hz, 1H, CHAr), 8.80 (d, J = 7.8 Hz, 1H, CHAr), 9.76 (s, 1H, CHAr). 13C NMR (101 MHz, CDCl3) δ: 27.8 (2xCH2), 33.6 (CH), 48.8 (2xN-CH2), 123.3 (CHAr), 128.3 (2xCHAr), 130.2 (2xCHAr), 130.3 (CHAr), 133.8 (Cq), 135.5 (CHAr), 135.9 (Cq), 139.9 (Cq), 150.1 (CHAr), 151.0 (CHAr), 155.0 (Cq), 159.2 (Cq), 175.6 (Cq). HRMS (EI/MS): m/z calculated for C20H19N4: 315.1604 [M + H]+; found: 315.1602.
2,4-Di-p-tolyl-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (53). Compound 53 was obtained according to general procedure D using 9 (215 mg, 0.80 mmol, 1.0 eq.) and p-tolylboronic acid (215 mg, 1.58 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 53 (101 mg, 37%) was obtained as a beige solid. Rf (PE/EA: 90/10): 0.34. Mp: 193–195 °C. IR (ATR diamond, cm−1) ν: 2965, 2934, 2869, 1547, 1387, 1179, 812, 803, 744. 1H NMR (400 MHz, CDCl3) δ: 1.74–1.87 (m, 2H, CH2), 2.00–2.12 (m, 2H, CH2), 2.43 (s, 6H, 2xCH3), 2.70–2.83 (m, 2H, N–CH2), 3.19–3.31 (m, 2H, N–CH2), 3.37 (p, J = 2.9 Hz, 1H, CH), 7.31 (dd, J = 5.9, 8.0 Hz, 4H, 4xCHAr), 8.36–8.42 (m, 2H, 2xCHAr), 8.42–8.48 (m, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 21.6 (CH3), 21.6 (CH3), 27.9 (2xCH2), 33.7 (CH), 48.9 (2xN-CH2), 128.3 (2xCHAr), 129.0 (2xCHAr), 129.3 (2xCHAr), 130.1 (2xCHAr), 133.6 (Cq), 135.7 (Cq), 138.8 (Cq), 140.1 (Cq), 140.2 (Cq), 154.8 (Cq), 161.1 (Cq), 174.8 (Cq). HRMS (EI/MS): m/z calculated for C23H24N3: 342.1965 [M + H]+; found: 342.1962.
4-(4-(p-Tolyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-yl)benzonitrile (54). Compound 54 was obtained according to general procedure D using 9 (215 mg, 0.80 mmol, 1.0 eq.) and 4-cyanophenylboronic acid (238 mg, 1.62 mmol, 2.0 eq.). After purification by silica gel flash chromatography (DCM/PE, 60/40 to 80/20), 54 (148 mg, 52%) was obtained as a beige solid. Rf (DCM/PE: 80/20): 0.55. Mp: 236–238 °C. IR (ATR diamond, cm−1) ν: 2940, 2227, 1548, 1387, 1167, 816, 612. 1H NMR (400 MHz, CDCl3) δ: 1.73–1.86 (m, 2H, CH2), 2.03–2.15 (m, 2H, CH2), 2.44 (s, 3H, CH3), 2.70–2.82 (m, 2H, N–CH2), 3.22–3.33 (m, 2H, N–CH2), 3.38 (p, J = 2.8 Hz, 1H, CH), 7.33 (d, J = 8.0 Hz, 2H, 2xCHAr), 7.78 (d, J = 8.6 Hz, 2H, 2xCHAr), 8.39 (d, J = 8.3 Hz, 2H, 2xCHAr), 8.68 (d, J = 8.5 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 21.6 (CH3), 27.8 (2xCH2), 33.7 (CH), 48.8 (2xN-CH2), 113.4 (Cq), 119.1 (Cq), 128.8 (2xCHAr), 129.1 (2xCHAr), 130.2 (2xCHAr), 132.4 (2xCHAr), 133.0 (Cq), 140.0 (Cq), 140.7 (Cq), 142.6 (Cq), 155.2 (Cq), 159.0 (Cq), 175.5 (Cq). HRMS (EI/MS): m/z calculated for C23H21N4: 353.1761 [M + H]+; found: 353.1759.
4-(4-Fluorophenyl)-2-(p-tolyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidine (55). Compound 55 was obtained according to general procedure D using 10 (110 mg, 0.40 mmol, 1.0 eq.) and p-tolylboronic acid (109 mg, 0.80 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 55 (63 mg, 45%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.49. Mp: 164–166 °C. IR (ATR diamond, cm−1) ν: 2926, 2869, 1550, 1505, 1386, 1220, 1155, 819. 1H NMR (400 MHz, CDCl3) δ: 1.74–1.87 (m, 2H, CH2), 2.01–2.13 (m, 2H, CH2), 2.43 (s, 3H, CH3), 2.70–2.82 (m, 2H, N–CH2), 3.19–3.31 (m, 2H, N–CH2), 3.37 (p, J = 2.9 Hz, 1H, CH), 7.18 (t, J = 8.8 Hz, 2H, 2xCHAr), 7.30 (d, J = 8.0 Hz, 2H, 2xCHAr), 8.44 (d, J = 8.2 Hz, 2H, 2xCHAr), 8.59 (dd, J = 5.8, 8.8 Hz, 2H, 2xCHAr). 13C NMR (101 MHz, CDCl3) δ: 21.6 (CH3), 27.8 (2xCH2), 33.6 (CH), 48.8 (2xN-CH2), 115.1 (CHAr), 115.3 (CHAr), 128.2 (2xCHAr), 129.3 (2xCHAr), 132.3 (CHAr), 132.4 (CHAr), 132.5 (d, J = 3.2 Hz, Cq), 135.5 (Cq), 138.7 (Cq), 140.4 (Cq), 153.4 (Cq), 161.1 (Cq), 164.1 (d, J = 250.5 Hz, Cq-F), 175.2 (Cq). 19F NMR (376 MHz, CDCl3) δ: −110.90. HRMS (EI/MS): m/z calculated for C22H21FN3: 346.1714 [M + H]+; found: 346.1710.
4-(4-(4-Fluorophenyl)-7,8-dihydro-6H-5,8-ethanopyrido[3,2-d]pyrimidin-2-yl)benzonitrile (56). Compound 56 was obtained according to general procedure D using 10 (109 mg, 0.40 mmol, 1.0 eq.) and 4-cyanophenylboronic acid (118 mg, 0.80 mmol, 2.0 eq.). After purification by silica gel flash chromatography (PE/EA, 90/10), 56 (97 mg, 68%) was obtained as a white solid. Rf (PE/EA: 90/10): 0.20. Mp: 237–239 °C. IR (ATR diamond, cm−1) ν: 2950, 2917, 2221, 1602, 1544, 1504, 1382, 1157, 824, 568. 1H NMR (400 MHz, CDCl3) δ: 1.74–1.86 (m, 2H, CH2), 2.05–2.17 (m, 2H, CH2), 2.70–2.81 (m, 2H, N–CH2), 3.22–3.34 (m, 2H, N–CH2), 3.39 (p, J = 2.8 Hz, 1H, CH), 7.19 (t, J = 8.7 Hz, 2H, 2xCHAr-F), 7.79 (d, J = 8.5 Hz, 2H, 2xCHAr-CN), 8.60 (dd, J = 5.6, 8.9 Hz, 2H, 2xCHAr-F), 8.67 (d, J = 8.5 Hz, 2H, 2xCHAr-CN). 13C NMR (101 MHz, CDCl3) δ: 27.7 (2xCH2), 33.7 (CH), 48.7 (2xN-CH2), 113.6 (Cq), 115.3 (CHAr-F), 115.5 (CHAr-F), 119.1 (Cq), 128.8 (2xCHAr-CN), 131.9 (d, J = 3.1 Hz, Cq), 132.4 (2xCHAr-CN), 132.4 (CHAr-F), 132.5 (CHAr-F), 139.9 (Cq), 142.4 (Cq), 153.8 (Cq), 159.1 (Cq), 164.3 (d, J = 251.4 Hz, Cq-F), 175.8 (Cq). 19F NMR (376 MHz, CDCl3) δ: −110.06. HRMS (EI/MS): m/z calculated for C22H18FN4: 357.1510 [M + H]+; found: 357.1505.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank the Région Centre Val de Loire (RTR Motivhealth, APR-IR FLOWSYN and INFLUX), FEDER under projects Valbiocosm FEDER-FSE 2017-EX003202, CHembio FEDER-FSE 2014-2020-EX003677, Techsab FEDER-FSE 2014-2020-EX011313 and Mistic FEDER-FSE 2014-2020-EX003242; the SFR neuroimagerie (SFR FED 4224), the GDR Synth_Flux, the Labex programs SYNORG (ANR-11-LABX-0029) and IRON (ANR-11-LABX-0018-01), the Ligue contre le Cancer du Grand Ouest (comités des Deux Sèvres, du Finistère, de l’Ile et Villaine, du Loir et Cher, de Loire Atlantique, du Loiret, de la Vienne) for their financial support.Notes and references
- E. J. Llanos, W. Leal, D. H. Luu, J. Jost, P. F. Stadler and G. Restrepo, Proc. Natl. Acad. Sci., 2019, 116, 12660–12665 CrossRef CAS PubMed.
- J.-L. Reymond, R. van Deursen, L. C. Blum and L. Ruddigkeit, MedChemComm, 2010, 1, 30–38 RSC.
- K. K. Duncan, D. D. Rudnicki, C. P. Austin and D. A. Tagle, Front. Robot. AI, 2020, 6, 1–6 Search PubMed.
- M. Aldeghi, S. Malhotra, D. L. Selwood and A. W. E. Chan, Chem. Biol. Drug Des., 2014, 83, 450–461 CrossRef CAS PubMed.
- C. Cabrele and O. Reiser, J. Org. Chem., 2016, 81, 10109–10125 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- F. Lovering, MedChemComm, 2013, 4, 515–519 RSC.
- T. J. Ritchie and S. J. F. Macdonald, Drug Discovery Today, 2009, 14, 1011–1020 CrossRef CAS PubMed.
- K. C. Bharadwaj, T. Gupta and R. M. Singh, in Synthesis of Medicinal Agents from Plants, ed. A. Tewari and S. Tiwari, Elsevier, 2018, pp. 205–227, DOI:10.1016/B978-0-08-102071-5.00009-X.
- J. Achan, A. O. Talisuna, A. Erhart, A. Yeka, J. K. Tibenderana, F. N. Baliraine, P. J. Rosenthal and U. D'Alessandro, Malar. J., 2011, 10, 144 CrossRef CAS PubMed.
- S. Tsukagoshi, Cancer Chemother., 1999, 26, 1001–1008 CAS.
- N. Ramírez Chanona, B. E. del Rio Navarro and J. Pérez Martín, Rev. Alerg. Mex., 2005, 52, 221–225 Search PubMed.
- H. Sakamoto, N. Yokoyama, S. Kohno and K. Ohata, Jpn. J. Pharmacol., 1984, 36, 455–460 CrossRef CAS PubMed.
- S. Kobayashi, K. Ikeda, M. Suzuki, T. Yamada and K. Miyata, Jpn. J. Pharmacol., 2001, 86, 281–288 CrossRef CAS PubMed.
- S. France, D. J. Guerin, S. J. Miller and T. Lectka, Chem. Rev., 2003, 103, 2985–3012 CrossRef CAS PubMed.
- S.-K. Tian, Y. Chen, J. Hang, L. Tang, P. McDaid and L. Deng, Acc. Chem. Res., 2004, 37, 621–631 CrossRef CAS PubMed.
- M. J. Gaunt and C. C. C. Johansson, Chem. Rev., 2007, 107, 5596–5605 CrossRef CAS PubMed.
- T. Ooi and K. Maruoka, Angew. Chem., Int. Ed., 2007, 46, 4222–4266 CrossRef CAS PubMed.
- C. E. Song, in Cinchona Alkaloids in Synthesis and Catalysis, 2009, pp. 1–10, DOI:10.1002/9783527628179.ch1.
- P. J. Boratyński, M. Zielińska-Błajet and J. Skarżewski, in Alkaloids Chem. Biol., ed. H.-J. Knölker, Academic Press, 2019, vol. 82, pp. 29–145 Search PubMed.
- R. Madhav, Synthesis, 1982, 1982, 27 CrossRef.
- Y. Besidsky, K. Luthman, A. Claesson, C. J. Fowler, I. Csöregh and U. Hacksell, J. Chem. Soc., Perkin Trans. 1, 1995, 465–474, 10.1039/p19950000465.
- G. W. V. Cave and C. L. Raston, Tetrahedron Lett., 2005, 46, 2361–2363 CrossRef CAS.
- W. S. Hamama, O. M. A. El-Magid and H. H. Zoorob, J. Heterocycl. Chem., 2006, 43, 1397–1420 CrossRef CAS.
- A. Ejjoummany, R. Belaroussi, A. El Hakmaoui, M. Akssira, G. Guillaumet, F. Buron and S. Routier, Molecules, 2018, 23, 2740 CrossRef PubMed.
- T. Saurat, F. Buron, N. Rodrigues, M.-L. de Tauzia, L. Colliandre, S. Bourg, P. Bonnet, G. Guillaumet, M. Akssira, A. Corlu, C. Guillouzo, P. Berthier, P. Rio, M.-L. Jourdan, H. Benedetti and S. Routier, J. Med. Chem., 2014, 57, 613–631 CrossRef CAS PubMed.
- F. Buron, J. Y. Merour, M. Akssira, G. Guillaumet and S. Routier, Eur. J. Med. Chem., 2015, 95, 76–95 CrossRef CAS PubMed.
- R. Belaroussi, A. Ejjoummany, A. El Hakmaoui, M. Akssira, G. Guillaumet and S. Routier, RSC Adv., 2018, 8, 732–741 RSC.
- WO2019231942A1, 2019.
- G. Lavecchia, S. Berteina-Raboin and G. Guillaumet, Tetrahedron Lett., 2005, 46, 5851–5855 CrossRef CAS.
- P. A. Solovyev and A. D. Shutalev, Chem. Heterocycl. Compd., 2009, 45, 809 CrossRef CAS.
- F.-A. Kang, Z. Sui and W. V. Murray, Eur. J. Org. Chem., 2009, 2009, 461–479 CrossRef.
- R. Belaroussi, A. El Hakmaoui, M. Akssira, G. Guillaumet and S. Routier, Eur. J. Org. Chem., 2016, 2016, 3550–3558 CrossRef CAS.
- B. Jismy, G. Guillaumet, M. Akssira, A. Tikad and M. Abarbri, RSC Adv., 2021, 11, 1287–1302 RSC.
- B. Jismy, A. Tikad, M. Akssira, G. Guillaumet and M. Abarbri, Molecules, 2020, 25, 2062 CrossRef CAS PubMed.
- B. Jismy, H. Allouchi, G. Guillaumet, M. Akssira and M. Abarbri, Synthesis, 2018, 50, 1675–1686 CrossRef CAS.
- C. Guo-Jun, H. Jie, G. Lian-Xun and H. Fu-She, Chem.–Eur. J., 2011, 17, 4038–4042 CrossRef PubMed.
- F.-A. Kang, Z. Sui and W. V. Murray, J. Am. Chem. Soc., 2008, 130, 11300–11302 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2074887. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03092b |
| This journal is © The Royal Society of Chemistry 2021 |