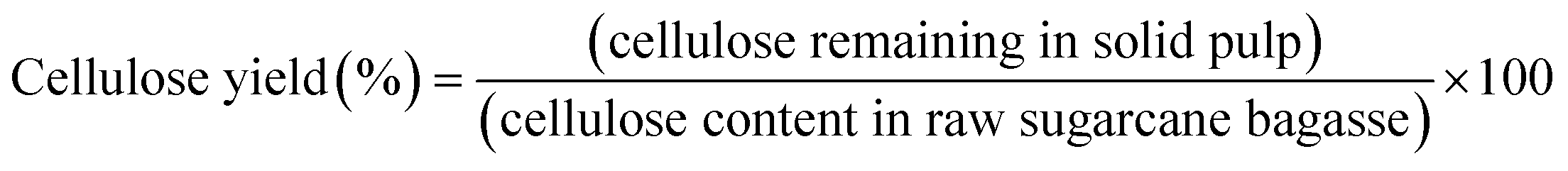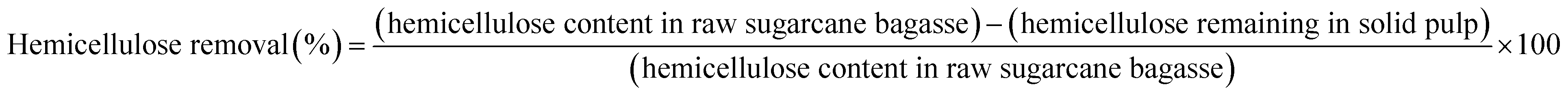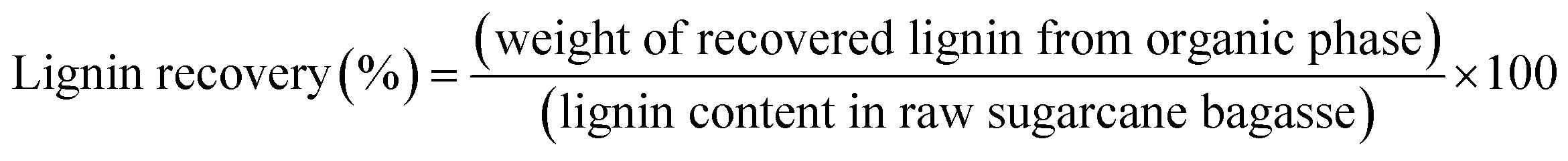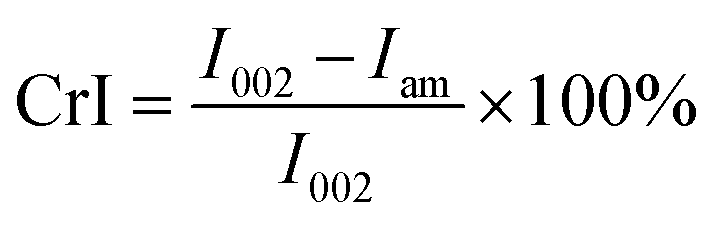DOI:
10.1039/D1RA03237B
(Paper)
RSC Adv., 2021,
11, 26773-26784
Fractionation and characterization of lignin from sugarcane bagasse using a sulfuric acid catalyzed solvothermal process†
Received
26th April 2021
, Accepted 26th July 2021
First published on 5th August 2021
Introduction
Currently, the energy crisis due to the decrease in fossil fuels and the atmospheric pollution caused by the combustion of fossil fuels is an important issue worldwide.1 ln addition, the increase in population and industrial development resulted in increased energy demand and a decrease in fossil fuel reserves. For this reason, many countries have tried to find alternative energy resources to replace fossil fuels. Lignocellulosic biomass is the one alternative renewable energy source that can be considered.2 Agricultural biomass mainly consists of cellulose (30–50%), hemicellulose (20–40%), lignin (10–30%), and other extractive products.3 Cellulose is an organic compound that strengthens plants. It consists of a polysaccharide unit linked together with beta 1–4 glycosidic bonds and has great importance in the chemical industry.4 The natural structure of hemicellulose is one of a number of heteropolymers and generally consist of five-carbon sugars, such as pentose sugar, arabinose sugar, and xylans sugar. Generally, the lignin composition of the softwood type is 24–33%, 19–28% in hardwood type, and 15–25% in the grass type.5 The typical structure of lignin consists of three basic phenylpropanoid monomers, i.e., p-hydroxyphenyl alcohol (H-unit), coniferyl alcohol (G-unit), and sinapyl alcohol (S-unit),6 and the most common linkages are namely, aryglycerol-β-ether dimer (β-O-4), aryglycerol-α-ether dimer (α-O-4), diaryl ether (4-O-5), resinol (β-5), diphenylethane (β-1), phenylcoumaran (β–β′), phenylcoumaran (β–β), and biphenyl (5–5).7 Moreover, the lignin content can be converted into different value-added products, such as phenol, aromatic sources, and vanillin using various conversion technologies.8
The clean fractionation (CF) process is one of the most promising technologies for fractionation-based biorefineries of lignocellulosic material. Nowadays, there are available studies about the one-step clean fractionation process utilizing a ternary solvent mixture (i.e., organic solvent, water and alcohol) in order to carry out one-step isolation of agricultural components. In recent years, the organosolv fractionation process for the degradation and isolation of lignin was considered to be one of the most promising processes for bioproduction, bioenergy, and biofuel.9,10 We have previously examined lignin fractionation by the organosolv fractionation process from sugarcane bagasse, para-rubber wood sawdust, palm fiber, and cassava fiber. The previously studied combination of hydrothermal and organosolv process was selected to fractionate sugarcane bagasse using the solvent of MIBK![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water in the ratio of 80:20 with 5% v/v formic acid promoter at 180 °C for 80 min. The results showed that a cellulose yield of 64.69% was obtained with less contamination from other components. Moreover, the recovered lignin obtained from the organic phase was 65.36%.11 Chutikan et al., 2018 reported the solvent mixture consists of methyl isobutyl ketone:ethanol:water (25%:42%:33% v/v) with a H2SO4 concentration of 0.025 M. In addition, the highest lignin removal efficiencies of 88.2%, 70.6%, 67.3%, and 71.7% were found for bagasse, para-rubber wood sawdust, and palm fiber and cassava fiber, respectively. Lignin recovered through the organic phase was identified as containing amounts of phenolic compounds. The highest lignin recovery of 89.8% was obtained from the organic phase, and 16.9% of phenolic compounds were identified through pyrolysis-GCMS. However, a less studied of the structural feature of isolated lignin was obtained from bagasse after the fractionation process using a ternary mixture of MIBK:methanol:water using H2SO4 as promoter.13 Herein, a one-step organosolv fractionation process is of interest in order to provides a high quality of organosolv lignin for developing biorefinery aspect. An insight of lignin macromolecule characteristics is essential to consider the opportunities and challenges for further lignin conversion into the advance applications.
:water in the ratio of 80:20 with 5% v/v formic acid promoter at 180 °C for 80 min. The results showed that a cellulose yield of 64.69% was obtained with less contamination from other components. Moreover, the recovered lignin obtained from the organic phase was 65.36%.11 Chutikan et al., 2018 reported the solvent mixture consists of methyl isobutyl ketone:ethanol:water (25%:42%:33% v/v) with a H2SO4 concentration of 0.025 M. In addition, the highest lignin removal efficiencies of 88.2%, 70.6%, 67.3%, and 71.7% were found for bagasse, para-rubber wood sawdust, and palm fiber and cassava fiber, respectively. Lignin recovered through the organic phase was identified as containing amounts of phenolic compounds. The highest lignin recovery of 89.8% was obtained from the organic phase, and 16.9% of phenolic compounds were identified through pyrolysis-GCMS. However, a less studied of the structural feature of isolated lignin was obtained from bagasse after the fractionation process using a ternary mixture of MIBK:methanol:water using H2SO4 as promoter.13 Herein, a one-step organosolv fractionation process is of interest in order to provides a high quality of organosolv lignin for developing biorefinery aspect. An insight of lignin macromolecule characteristics is essential to consider the opportunities and challenges for further lignin conversion into the advance applications.
This research focuses on the optimization of isolated lignin yield from sugarcane bagasse using the solvothermal fractionation process. Lignin isolation was carried out with the ternary mixture solvents (i.e. MIBK:methanol:water) in the presence of sulfuric acid in order to enhance the recovery of lignin from the sugarcane bagasse. Moreover, various characterized approaches including X-ray diffraction analysis (XRD), scanning electron microscope (SEM), elemental analysis, fourier transform infrared spectroscopy (FTIR), thermogravimetric analysis (TGA), gel permeation chromatography (GPC), nuclear magnetic resonance spectroscopy (2D HSQC NMR), and pyrolysis-gas chromatography-mass spectrometry (Py-GC/MS) were adopted to document the structural features and the changes in solid fraction and lignin recovery. The solvothermal fractionation process for improving isolation lignin efficiency was investigated for further conversion of lignin into high yield and high purity products according to the application in integrated biorefineries.
Material and methods
Material
Sugarcane bagasse was obtained from Mae Chan, Chiang rai, Thailand. The sugarcane bagasse was dried at 70 °C for 24 h in a hot air oven. The dried sugarcane bagasse was ground and sieved to obtain the range of particles from 0.5 to 2.0 mm with 0.25–0.5 cm thickness. The final moisture content of the milled sugarcane bagasse was 5%, determined using the weight loss after oven drying at 105 °C for a constant time (5 h). The processed sugarcane bagasse was stored in sealed plastic bags at room temperature for the experiments. The chemical composition was determined according to the analytical laboratory procedures of the National Renewable Energy Laboratory.14 The chemical composition of the sugarcane bagasse included cellulose (37.2%), hemicellulose (22.1%), lignin (26.5%), ash (8.2%), and extractives (6%). Commercial organosolv lignin was obtained from Chemical Point UG, Germany. The sample was used as a benchmark lignin.
Procedure for isolation of lignin from sugarcane bagasse using the solvothermal fractionation process
Organosolv fractionation of sugarcane bagasse was performed in a 600 mL stainless steel reactor, and heated by an electric jacket with an inside thermocouple to measure temperature (Parr Reactor 4560, Parr Instrument, Moline, IL, USA). Initially, the reaction contained 15 g of screened sugarcane bagasse mixed with 150 mL of the single phase solvent mixture comprising MIBK/methanol/water (35%:25%:40% v/v) with various concentrations of H2SO4 catalyst (0.01–0.02 M). The reaction temperature and time were 180–200 °C and 30–90 min, respectively. Nitrogen gas (N2) was purged into the reactor, and the initial pressure was adjusted at 20 bar. The reaction was stirred at 100 rpm to maintain a homogeneous system. After the desired temperature and time were reached, the reaction was immediately stopped in a water bath. The solid cellulose-enriched fraction was separated by filtration and washed with MIBK and water (1:2 v/v) mixture. The liquid fraction was combined and placed into a separatory funnel. Distilled water was added to the organic phase until the phase separation. The mixture was stirred and then incubated at room temperature for 20 min to complete phase separation. The liquid phase containing hemicellulose and soluble products was recovered. The separated organic phase was dried at 105 °C to obtain the isolated lignin. The yield of the isolated solid was determined on a weight basis. The chemical composition of the isolated solid was determined using the National Renewable Energy Laboratory (NREL method). The cellulose yield, hemicellulose removal, lignin removal, and lignin recovery were calculated using the following equations:| |
 | (1) |
| |
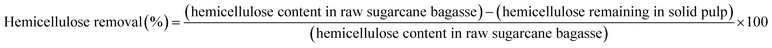 | (2) |
| |
 | (3) |
| |
 | (4) |
Statistical design and optimization for the target responses using response surface method
The experimental design and statistical analysis were performed according to Box–Behnken design (BBD) method using the Design-Expert software (version 10.0.1). The design of the experiment was explored with three independent factors based on the three level. The three factors comprised reaction time (X1, 30–90 min), reaction temperature (X2, 180–200 °C), and concentration (X3, 0.01–0.02 M) with three coded levels for each factor (−1, 0, 1). A total of 15 experiments with three replications at the center point were run in random order. The assessed variance of each factor was partitioned into offset, linear, interaction, and quadratic components. All parameters were evaluated using a second order polynomial equation as follows.| | |
Y = β0 + β1X1 + β2X2 + β3X3 + β12X1X2 + β13X1X3 + β23X2X3 + β11X12 + β22X22 +β33X32
| (5) |
where Y is the predicted response; X1, X2, and X3 are the independent variables; β0 is a constant; β1, β2, and β3 are the linear coefficients; β12, β13, and β23 are the interaction coefficients; and β11, β22, and β33 are the quadratic coefficients.
Characterization of the isolated solid fraction
The microstructure of the native sugarcane bagasse and fractionated bagasse under the optimal condition was analyzed by SEM instrument (JSM-6301F, JEOL, Japan) with electron beam energy of 20 kV. The sugarcane bagasse samples were dried and coated with gold for SEM analysis. The absorption bands of solid fraction were determined on a Shimadzu FTIR (Model: IRTracer-100, Shimadzu, Kyoto, Japan) with an attenuated total reflectance (ATR) mode. The region between 4000 and 400 cm−1 at a resolution of 4 cm−1 for 64 scans were recorded. Crystallinities of solid fractions were determined by XRD using an X'Pert PRO diffractometer (Panalytical, Almelo, Netherland). The samples were scanned in a range of 2θ = 10°–30° with a step size of 0.02° at 40 kV and 30 mA and Cu Kα radiation (λ = 1.54 A°). Crystallinity was calculated according to the following equation.| |
 | (6) |
where I002 is the scattered intensity at the main peak of cellulose which typically lies around 002 plane and Iam is the scattered intensity of amorphous portion evaluated as the minimum intensity diffraction between the main and secondary (101 and 002 planes).
Analysis of aqueous phase
Soluble product profiles of the aqueous fraction were analyzed using a high-performance liquid chromatograph (LDC Model 4100, Shimadzu, Kyoto, Japan) equipped with refractive index and ultraviolet-visible spectroscopy detectors and an Aminex HPX-87H column (Bio-Rad, Hercules, CA, USA). The operating condition was performed at 65 °C with 5 mM H2SO4 as the mobile phase at a flow rate of 0.5 mL min−1. The number of oligosaccharides was determined according to the NREL method.15
Characterization of lignin samples
The elemental composition of lignin recovery and commercial organosolv lignin was estimated using an elemental analyzer CHNS-628 (LECO, Saint Joseph, MI, USA). Lignin samples were dried at 60 °C under vacuum to remove moisture. Then, 0.1 g of lignin sample was encapsulated in a container to determine the amount of carbon, hydrogen, and nitrogen in the sample. Then, for sulfur analysis, 0.2 g lignin was placed in a ceramic boat furnace. The sample was incinerated at 1350 °C using a sulfur IR cell to calculate the amount of sulfur. The chemical structures of lignin samples were determined using FTIR technique. FTIR was performed with an instrument from PerkinElmer (Waltham, MA, USA). The samples were prepared by using the KBr pellet method. The region between 4000 and 400 cm−1 at a resolution of 4 cm−1 for 32 scans were recorded. The peaks made by lignin were compared with the peaks of the standard functional groups. The thermal stability of lignin samples was measured by TGA. The specimens were performed at a rate of 5 °C per minute under the flow of nitrogen gas at 20–35 mL min−1. The weight of a specimen was monitored as a function of temperature using an instrument from TA Instruments Inc. (Model TA Q50). Approximately 30–50 mg of recovered lignin was weighed in an aluminum pan and heated at a rate of 5 °C min−1 from ambient temperature to approximately 1000 °C for determination of the total loss of mass loss. A curve depicting the weight loss against temperature was constructed from the obtained data. A derivative of this curve (DTG) was calculated to determine the temperature corresponding to the maximum rate of weight loss. The glass transition temperature (Tg) of lignin samples was analysed by differential scanning calorimeter (DSC) machine, Mettler Toledo DSC1 instruments (Mettler Toledo, Ohio, US). The DSC was performed under nitrogen at flow rate of 60 mL min−1. The temperature program was in range of 25 °C to 120 °C, then cooled down from 120 °C to 25 °C, and heated from 25 °C to 250 °C. The lignin samples were subjected by GPC analysis to determine the average molecular weight (Mw) and the polydispersity index (Mw/Mn) using a Jasco instrument unit equipped with an interface (LC-NetII/ADC) and a UV detector (254 nm). Lignin (0.1 g) was dissolved in tetrahydrofuran (THF) and the test was carried out using two PolarGel-M 803 columns (300 mm × 7.5 mm) and a PolarGel-M guard (50 mm × 7.5 mm); the tetrahydrofuran was used as a mobile phase. The flow rate was 0.5 mL min−1. The column temperature was 40 °C. Calibration was performed using polystyrene standards (Sigma-Aldrich) ranging from 266 to 55000 g mol−1. The molecular structures of lignin samples were determined using 2D HSQC NMR analysis (Bruker Advance 600 spectrometer, Bruker, Germany). The experiment began with the determination of 1H- and 13C- dimensions at 10–0 ppm and 220–5 ppm, respectively; 0.5 mg of lignin samples was dissolved in 0.5 mL of DMSO-d6. Pyrolysis-GCMS was used to characterize the chemical composition of lignin samples. The pyrolysis of recovered lignin was performed at 500 °C in an EGA/PY-3030D microfurnace pyrolyzer (Frontier Laboratories Ltd., Fukushima, Japan) connected to a GC 7820A system (Agilent Technologies, Inc., Santa Clara, CA) and an Agilent 5975 mass-selective detector (EI of 70 eV). The A DB-1701 column was 30 m × 0.25 mm, 0.25 μm film thickness (J&W Scientific, Folsom, CA). The oven temperature was programmed to increase from 50 °C (1 min) to 100 °C at 20 °C min−1 and then to 280 °C (5 min) at 6 °C min−1. Helium was used as the carrier gas (1 mL min−1). The released compounds were identified by comparison of their mass spectra with those provided by the Wiley and NIST libraries, and with the spectra reported in the literature.16 and, whenever possible, with the standard retention times and mass spectra. Molar peak areas were calculated for each released lignin degradation product. The summarized areas were normalized, and the data for two replicates were averaged and expressed as percentages.17
Results and discussion
The interactive effects of residence time, reaction temperature, and acid concentration on the cellulose yield, hemicellulose removal, lignin removal, and lignin recovery
All responses were evaluated based on the quality and quantity of the remaining solid and derived liquid phase, which depends on the degree of independent factors in the solvothermal fractionation process. The outputs corresponded to the relative responses including cellulose yield, hemicellulose removal, lignin removal, and lignin recovery. These responses were generated by feeding the experimental results into the RSM method to study the interactive effects of the process parameters on the responses.
The effect of the three parameters, which includes residence time, reaction temperature, and acid concentration on the major components of sugarcane bagasse, were investigated using the Design-Expert software. The designs of 15 experiments are shown in Table 1. For optimizing the fractionation condition, the residence time 30–90 min, reaction temperature 180–200 °C, and acid concentration from 0.01 to 0.02 M were assessed to determine the relative responses. The results showed that the cellulose yield, hemicellulose removal, lignin removal, and lignin recovery after the solvothermal fractionation process varied in the range of 70.1–86.1%, 88.4–98.6%, 72.7–87.2%, and 71.5–86.2%, respectively. As the results, the increasing of reaction parameters (temperature, time, and acid concentration) enhanced hemicellulose and lignin removal from native sugarcane bagasse into the liquid phase, while the cellulose yield in the remaining solid decreased under the harsh conditions. According to the procedure of fractionation process, the aqueous phase contains mainly of hemicellulose fraction in form of soluble products (Table S1†). This could be described that the harsh condition resulted in the degradation of carbohydrate fractions (hemicellulose and cellulose) into by-products (e.g. organic acid, HMF, and furfural). In this study, the soluble phase was considered as co-product. Then, the target cellulose and lignin were further evaluated for maximized fractionation of the individual response.
Table 1 The effect of reaction condition based on BBD
| Factors |
Responses |
| Run no. |
Time (min) |
T (°C) |
Conc. (M) |
Cellulose yielda (%) |
Hemicellulose removala (%) |
Lignin removala (%) |
Lignin recoveryb (%) |
| Based on the respective content of each fraction in the native biomass. Based on the relative weight of lignin in organic phase compared with lignin content in raw material. |
| 1 |
60 |
190 |
0.015 |
85.9 |
94.3 |
81.5 |
81.0 |
| 2 |
90 |
190 |
0.010 |
78.7 |
89.6 |
76.7 |
76.8 |
| 3 |
60 |
200 |
0.020 |
86.1 |
96.7 |
83.4 |
83.1 |
| 4 |
30 |
190 |
0.020 |
78.4 |
89.1 |
79.8 |
79.2 |
| 5 |
90 |
180 |
0.015 |
84.0 |
97.2 |
87.2 |
86.2 |
| 6 |
60 |
190 |
0.015 |
85.9 |
95.9 |
82.5 |
81.1 |
| 7 |
90 |
200 |
0.015 |
83.1 |
93.1 |
78.6 |
77.9 |
| 8 |
90 |
190 |
0.020 |
79.3 |
98.6 |
86.3 |
86.1 |
| 9 |
30 |
200 |
0.015 |
82.0 |
89.8 |
76.8 |
76.4 |
| 10 |
30 |
180 |
0.015 |
74.8 |
93.3 |
77.7 |
77.1 |
| 11 |
60 |
200 |
0.010 |
76.1 |
92.7 |
72.7 |
72.0 |
| 12 |
60 |
180 |
0.020 |
79.5 |
94.1 |
85.1 |
84.0 |
| 13 |
60 |
180 |
0.010 |
77.2 |
95.6 |
81.5 |
80.1 |
| 14 |
60 |
190 |
0.015 |
85.0 |
96.4 |
82.5 |
81.6 |
| 15 |
30 |
190 |
0.010 |
70.1 |
88.4 |
72.9 |
71.5 |
The results obtained after the solvothermal fractionation process were optimized based on three main responses. It was found that the determined regression equations for the optimization of cellulose yield (Y1, %), lignin removal (Y2, %), and lignin recovery (Y3, %) were the functions of the residence time (X1), reaction temperature (X2), and acid concentration (X3). The predicted equation was fitted to the multiple second-order polynomial regression, which is defined by the eqn (7)–(9).
| | |
Cellulose yield (%) = Y1 = −271 + 2.072X1 + 2.87X2 + 120X3 − 0.004292X12 − 0.00763X22 − 204500X32 − 0.00675X12− 12.83X13 + 38.50X23
| (7) |
| | |
Lignin removal (%) = Y2 = 54 + 1.498X1 + 0.36X2 − 4722X3 − 0.002139X12 − 0.00200X22 − 54000X32 − 0.006417X12 + 4.50X13 + 36.00X23
| (8) |
| | |
Lignin recovery (%) = Y3 = 44.4 + 1.478X1 + 0.427X2 − 4736X3 − 0.001793X12 − 0.00214X22 − 48792X32 − 0.006354X12 + 2.6X13 + 36.00X23
| (9) |
A variance analysis (ANOVA) of the quadratic regression model provided the equations of the cellulose yield (Y1), lignin removal (Y2), and lignin recovery (Y3) responses. It was proved to be a statistically significant model for determining the optimal conditions of solvothermal process. All quadratic models were statistically significant at the confidence interval of 95%. In addition, this consistency indicated the high accuracy of the model based on a comparison of the predicted and experimental values resulting in the R-square of cellulose yield (99.31%), lignin removal (99.49%), and lignin recovery (99.63%). According to p-value (Table 2), all parameters (time, temperature, and acid concentration) in term of linear, quadratic and interaction were statistically significant for all responses.
Table 2 Resultant regression models of responses based on ANOVA analysis
| Source |
p-Valuea |
| Cellulose yield |
Lignin removal |
Lignin recovery |
| The significant p-value are highlighted. |
| Model |
| Linear |
| Time |
0.0000 |
0.0000 |
0.0000 |
| Temperature |
0.0001 |
0.0000 |
0.0000 |
| Concentration |
0.0000 |
0.0000 |
0.0000 |
|
| Quadratic |
| Time × time |
0.0000 |
0.0001 |
0.0001 |
| Temperature × temperature |
0.0076 |
0.0503 |
0.0405 |
| Concentration × concentration |
0.0000 |
0.0005 |
0.0003 |
|
| 2-Way interaction |
| Time × temperature |
0.0002 |
0.0001 |
0.0000 |
| Time × concentration |
0.0002 |
0.0052 |
0.0136 |
| Temperature × concentration |
0.0002 |
0.0001 |
0.0000 |
Optimization of reaction parameters for solvothermal fractionation process
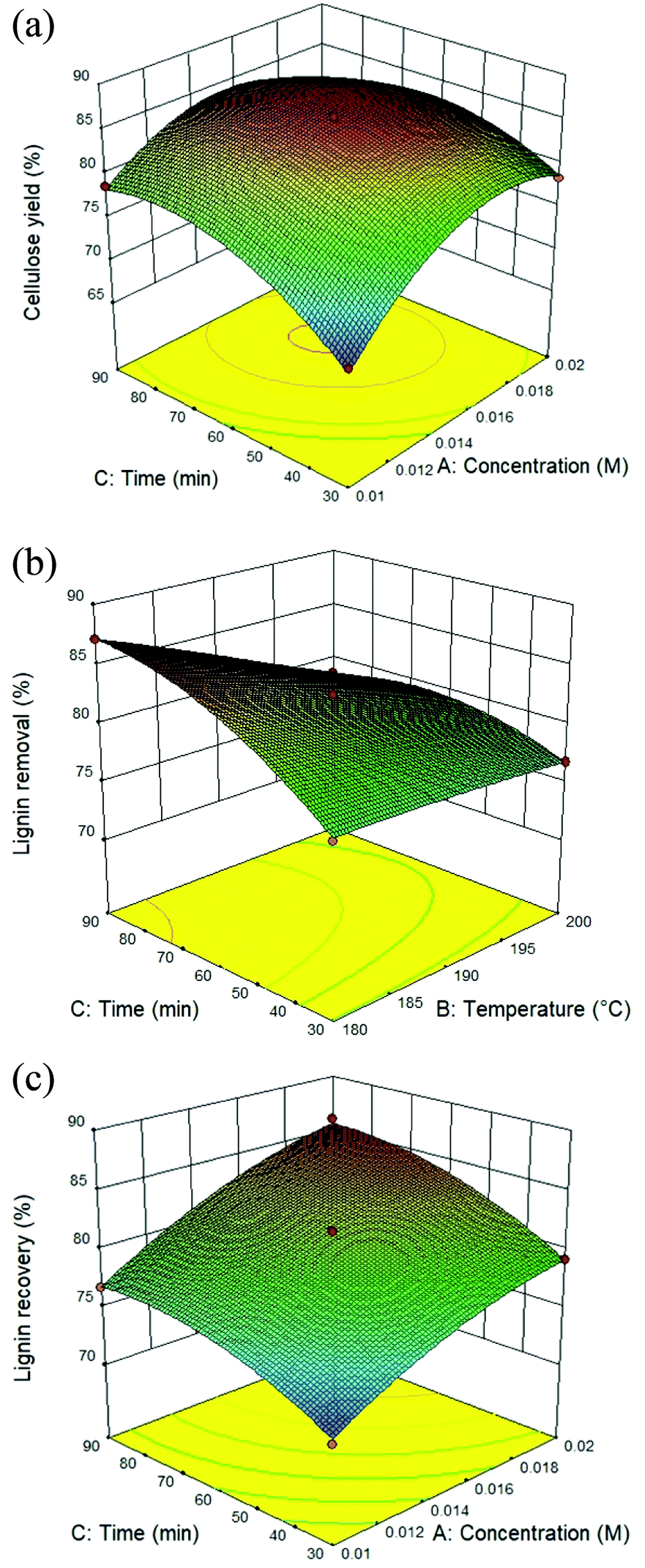
The utilization of isolated lignin from sugarcane bagasse using the solvothermal fractionation process and the subsequent lignin recovery from the organic fraction is an attentive method for lignocellulose treatment. Therefore, the fractionation conditions need to be optimized for maximum lignin recovery. The response surface plot for all the target responses (Fig. 1A–C) showed the influence of residence time, reaction temperature, and acid concentration on cellulose yield, lignin removal, and lignin recovery in solvothermal fractionation process of sugarcane bagasse. It was found that the optimal conditions for maximum cellulose yield of 87.4%, the process was performed at 0.017 M of sulfuric acid at 180 °C for 38.5 min. In addition, the maximum lignin removal from the solid fraction (88.6%) and lignin recovery from the organic fraction (87.8%) was achieved at 0.02 M, 200 °C for 90 min. In this work, the highest lignin recovery is considered as the main criteria for optimized solvothermal fractionation process. Under the selected optimal condition (0.02 M, 200 °C for 90 min), the experimental results gave the reaction efficiency of cellulose yield (86.1%) with maximum of lignin removal (88.0%) and lignin recovery (87.1%).
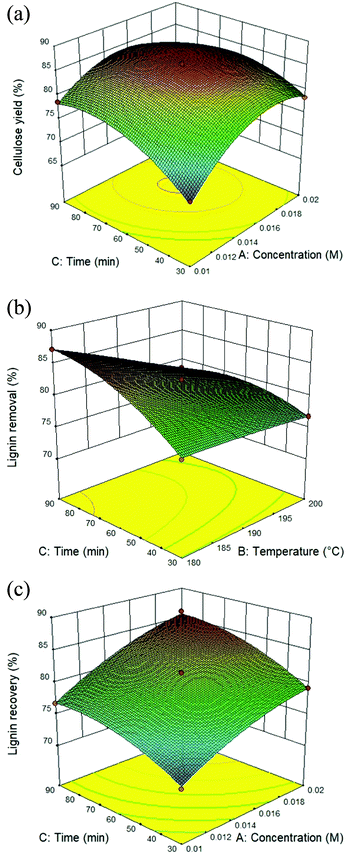 |
| | Fig. 1 Response surface plot of the solvothermal fractionation process: effect of residence time (30–90 min) and reaction temperature (180–200 °C) in the presence of 0.01–0.02 M of sulfuric acid on (A) cellulose yield, (B) lignin removal, and (C) lignin recovery. | |
Characterization of the native sugarcane bagasse and remaining pulp under the optimal conditions of solvothermal fractionation
The analysis of the physio-chemical properties of the remaining pulp after the solvothermal fractionation process under the optimal conditions was compared with the native sugarcane bagasse using SEM, XRD, and FTIR techniques. The morphological appearances of the native sugarcane bagasse and remaining pulp are shown in Fig. 2. The morphological features of native sugarcane bagasse showed a smooth surface because of the coverage of embedded recalcitrant components and wax layer. According to Fig. 2b, the disruption of sugarcane bagasse was observed during the solvothermal fractionation. This related to the obvious alteration on their morphology resulting in the roughness of cover surface and exposure the internal structures. The SEM results are well agreement with the entire extraction of hemicellulose and lignin in fractionation process, resulting in highly pure cellulose fibers.
 |
| | Fig. 2 Scanning electron microscope images of (a) native sugarcane bagasse and (b) remaining pulp under the optimal condition of solvothermal fractionation process. | |
In addition, the crystallinity index of native sugarcane bagasse and remaining pulp were examined using XRD patterns (Table 3). It was observed that the removal of hemicellulose and lignin during solvothermal fractionation leads to increased crystallinity of remaining pulp (78.5%) compared with native sugarcane bagasse (68.8%). In addition, the structural feature of cellulose is present in both of amorphous and crystalline forms. Crystalline cellulose is more resist under acid hydrolysis; thus, crystallinity increased due to an increasing the relative amount of crystalline cellulose in the remaining pulp compared to native biomass as previously observed in the literature using mild conditions.18 As a consequence, the solvothermal process should also remove (almost complete) amorphous cellulose.
Table 3 The crystallinity index of native sugarcane bagasse and remaining pulp under the optimal condition of solvothermal fractionation process
| Sample |
Crystallinity index (%) |
| Native sugarcane bagasse |
68.8 |
| Remaining pulp |
78.5 |
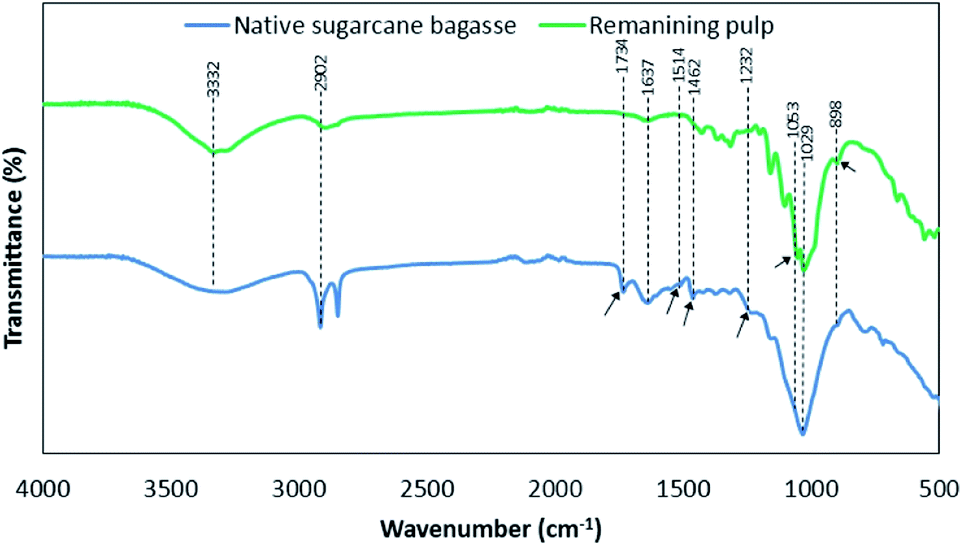
Fig. 3 shows the FTIR spectra of native sugarcane bagasse and remaining pulp. The absorption bands were summarized according to previous research as shown in Table S2.†19–21 It was observed that the assigned peak in the range of 3100–3700 cm−1 was attributed to O–H stretching vibrations of the –OH groups in cellulose molecules. The characteristic signal of 2902 cm−1 in both samples was assigned to C–H stretching vibrations of cellulose, hemicellulose and lignin. The vibrations at 1637 cm−1 was related to the O–H bending vibrations of absorbed waters for all samples. The bands at 1734 cm−1 and 1514 cm−1 were attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester group vibrations of the acetyl and uronic ester groups and aromatic CC in plane stretching of the aromatic rings, respectively, in lignin and/or hemicellulose fraction. Besides, the vibration band at 1462 cm−1 was related to the CC stretching vibrations of the aromatic rings in lignin. The band at 1232 cm−1 was associated with the acetyl groups as branched groups in polymeric hemicellulose fraction. These peaks disappeared after solvothermal fractionation process. This result is in agreement with the removal of hemicellulose and lignin in fractionation process.19 On the other hand, the peaks at 1053 cm−1 and 898 cm−1 were found after fractionation process.20 These peaks were assigned to the C–O–H stretching of alcohols in crystalline structure of cellulose and glycosidic C–H stretching vibration out of the plane of the aromatic ring (β-glycosidic linkages), respectively, in cellulose. This result is well agreement with the increasing of crystallinity index according to XRD analysis.
O ester group vibrations of the acetyl and uronic ester groups and aromatic CC in plane stretching of the aromatic rings, respectively, in lignin and/or hemicellulose fraction. Besides, the vibration band at 1462 cm−1 was related to the CC stretching vibrations of the aromatic rings in lignin. The band at 1232 cm−1 was associated with the acetyl groups as branched groups in polymeric hemicellulose fraction. These peaks disappeared after solvothermal fractionation process. This result is in agreement with the removal of hemicellulose and lignin in fractionation process.19 On the other hand, the peaks at 1053 cm−1 and 898 cm−1 were found after fractionation process.20 These peaks were assigned to the C–O–H stretching of alcohols in crystalline structure of cellulose and glycosidic C–H stretching vibration out of the plane of the aromatic ring (β-glycosidic linkages), respectively, in cellulose. This result is well agreement with the increasing of crystallinity index according to XRD analysis.
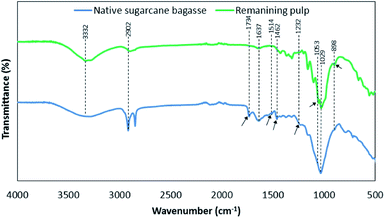 |
| | Fig. 3 The functional groups based on FTIR analysis of native sugarcane bagasse and remaining pulp under the optimal condition of solvothermal fractionation process. | |
Characterization of lignin samples
In this present work, the physio-chemical properties of bagasse lignin recovery (BGL) in the organic phase under the optimal conditions were analyzed in comparison to that of commercial organosolv lignin (COL).
FTIR analysis
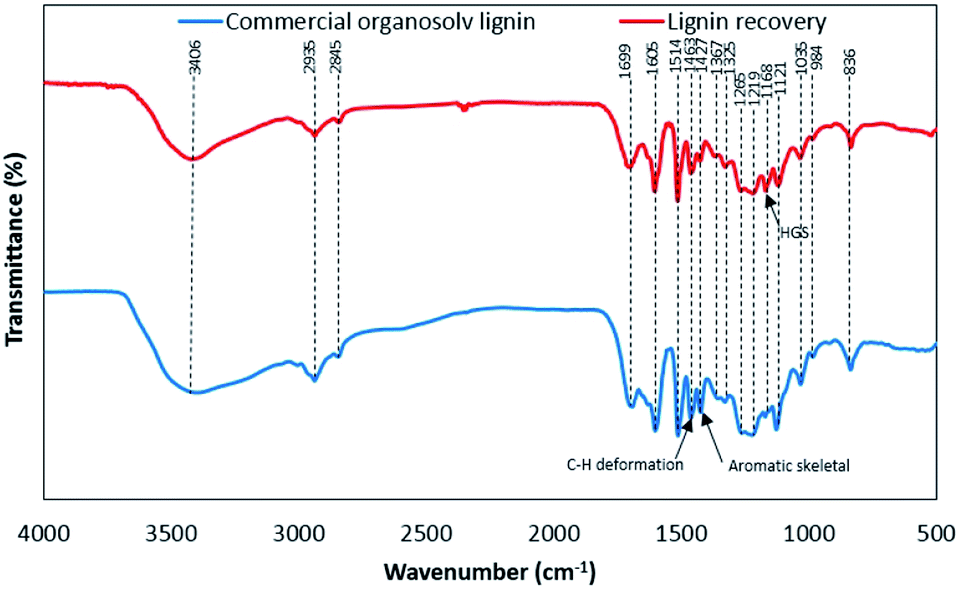
The FTIR spectra of lignin samples are shown in Fig. 4. The functional groups of lignin samples were assigned according to FTIR spectra of lignin as reported earlier (Table S3†).12,22,23 The lignin samples showed the similar of the main functional groups. The relative functional groups showed the vibration of O–H stretching at 3406 and 3410 cm−1.24 The characteristic signal of C–H stretching in CH2 and CH3 were assigned to 2935 and 2845 cm−1, respectively.25 The vibrations at 1699–1704 cm−1 were related to CO stretching, unconjugated.22 The characteristic signals of aromatic skeletal vibrations S > G (1602–1605 cm−1), aromatic skeletal vibrations G > S (1514 cm−1), C–H deformations in –CH2– and –CH3 (1459–1463 cm−1), aromatic skeletal vibrations (1422–1427 cm−1), aliphatic C–H stretch in CH3, not in OCH3 (1362–1367 cm−1), S ring breathing (1325–1330 cm−1), G ring breathing (1261–1265 cm−1), C–C and C–O stretch, G condensed > G (1219–1224 cm−1), C–O stretch in ester groups HGS (1168–1173 cm−1), aromatic C–H in plane deformation S (1121–1127 cm−1), aromatic C–H in plane deformation G > S (1030–1035 cm−1), CHCH-out-of-plane deformation trans (984 cm−1) and aromatic C–H out-of-plane deformation S + H (835–836 cm−1) could be observed in both lignin samples. It was found that both derived lignin samples resulted in no substantial alteration of the core of lignin structure under different extraction conditions.26 In comparison, COL showed the obvious peak of C–H deformations in –CH2– and –CH3 and aromatic skeletal vibrations, while BGL contains an intense peak of C–O stretch in ester groups HGS. According to the relative functional groups SGH units in BGL, the FTIR spectra is well agreement with the source of BGL as grass type. Since lignin from grass is composed mainly of three monolignols of S, G, and H units.23,27 The proportion of SGH units could be determined according to the results in section of Py-GC/MS.
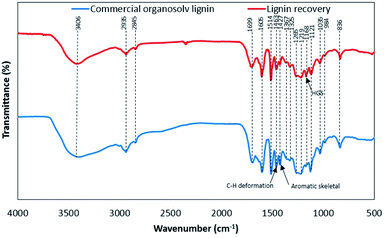 |
| | Fig. 4 The functional groups based on FTIR analysis of lignin recovery and commercial organosolv lignin. | |
Molecular weight distribution
The effect of lignin extraction after solvothermal fractionation process on the molecular weight distributions and polydispersity index (Mw/Mn) of lignin samples are presented in Table 4. The outputs were analyzed in term of molecular weight (Mw), number average (Mn), and PDI. It was found that the Mw of COL was slightly higher than that of BGL. The different condition would cause the condensation reaction of isolated lignin which resulted in a higher Mw.20 The PDI value of BGL and COL were 1.75 and 2.12 (g mol−1), respectively. This result corresponded to a lower PDI of BGL compared to COL. As previously reported, lignin isolation from bagasse was carried out under optimal conditions using ethanol–water fractionation. It was found that the PDI of lignin extraction after ethanol–water fractionation process was 1.58, which is slightly lower than that of alcell lignin (2.18).28
Table 4 Mw, Mn, and polydispersity (Mw/Mn) of lignin recovery and commercial organosolv lignin
| Sample |
Mwa (g mol−1) |
Mnb (g mol−1) |
Mw/Mnc |
| Weight-average molecular weight. Number-average molecular weight. Polydispersity index (PDI). |
| BGL |
1374 |
785 |
1.75 |
| COL |
1558 |
733 |
2.12 |
Elemental analysis
The chemical composition of lignin samples is demonstrated in Table 5. The composition content of the lignin samples included the element of carbon (C), hydrogen (H), oxygen (O), nitrogen (N), and sulfur (S), which in range of 62.45–63.72%, 5.21–5.30%, 29.79–31.4%, 0.78–0.8%, and 0.13–0.4%, respectively. As the results, the BGL showed a relative high content of carbon resulting in a good candidate for valorization of high-quality lignin. Typically, a low number of contaminated elements (e.g. nitrogen and sulfur) is an aspect of lignin properties. However, the BGL showed a slightly higher of sulfur than COL. The sulfur element could be gained from the used of sulfuric acid as promoter during the solvothermal fractionation process.29
Table 5 Proportion of elemental in the lignin recovery and commercial organosolv lignin (wt%)
| Lignin sample |
C |
H |
O |
N |
S |
| BGL |
63.72 |
5.30 |
29.79 |
0.78 |
0.40 |
| COL |
62.45 |
5.21 |
31.4 |
0.80 |
0.13 |
Thermal decomposition
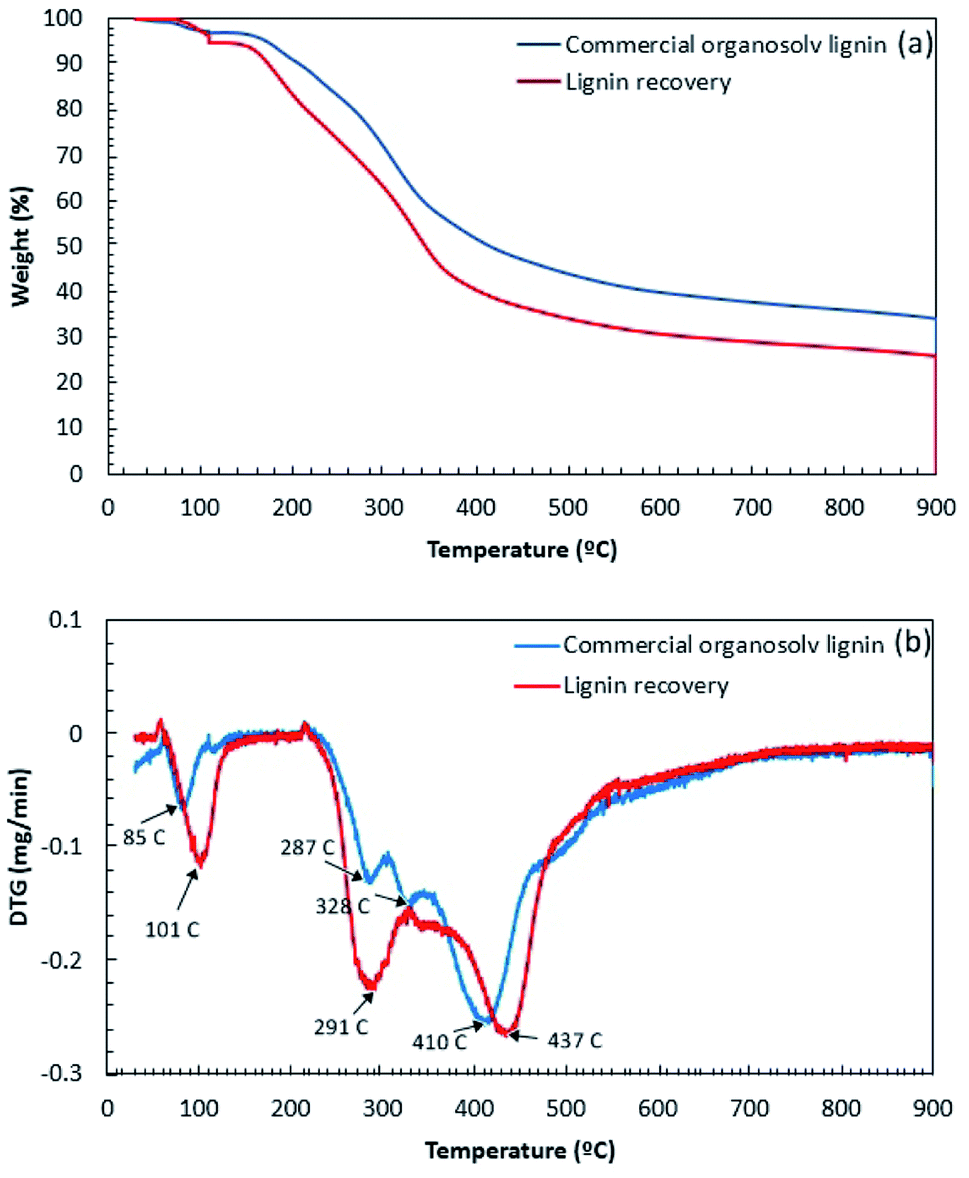
The thermal decomposition of lignin samples is illustrated in Fig. 5. The results showed that the thermal degradation of COL and BGL could be divided into four stages. Initially, the first stage was observed at 50–150 °C due to moisture loss from the lignin sample.30 The second stage, showing the decomposition of volatile substances (i.e., CO, CO2, CH4, and β′-O-4′ linkages due to the cleaving of the side chains) was observed at 150–380 °C.31 The third stage was observed in the temperature range from 400 to 450 °C; the decomposition of the aromatic rings and the cleavage of the C–C linkages and 5–5′ linkages of the lignin sample took place in this stage.32 In the final stage, the degradation of the aromatic ring and the condensation reaction occurred above 450 °C.33 In comparison (Fig. 5a), it was observed that the thermal stability of COL was slightly higher than BGL. This could be due to the different chemical structure according to FTIR analysis. Fig. 5b displays the degradation of weight loss of lignin samples. In details, the first stage of COL and BGL was observed at 85 °C and 101 °C, respectively. The COL showed two degradation at 287 °C and 328 °C during the second stage, whereas BGL was found at 291 °C. This could be described that the different quantity of side chains lignin samples, which related to the degradation of β-O-4 cross-links.34,35 Since the solvothermal process presume to obtain a greater number of β-O-4 linkage as previously observed.36 The major degradation temperatures of BGL and COL were observed in the third region. The DTGmax of COL and BGL was estimated at 410 °C and 437 °C, respectively. Above 450° (up to 900 °C), COL and BGL showed a gradual weight loss. The final solid residue yield at 900 °C of COL and BGL was 34% and 26%, respectively. This could be due to the presence of different inorganic compounds. According to Table S4,† it was observed that COL showed a relative higher of glass temperature (154.1 °C) compared with BGL (111.6 °C). This indicated that the working temperature for valorization of organosolv lignin-based polymer blends should be performed under these temperatures. Further specific lignin features were characterized by 2D-HSQC NMR and Py-GC/MS.
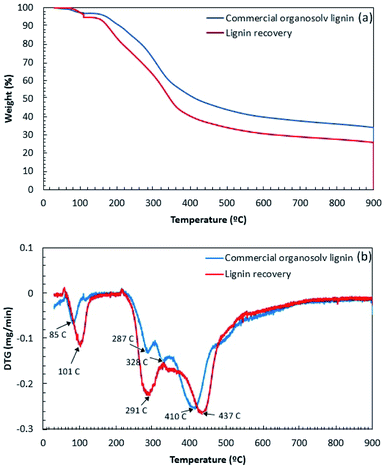 |
| | Fig. 5 The thermal decomposition of lignin recovery and commercial organosolv lignin: (a) TG curve and (b) DTG curve. | |
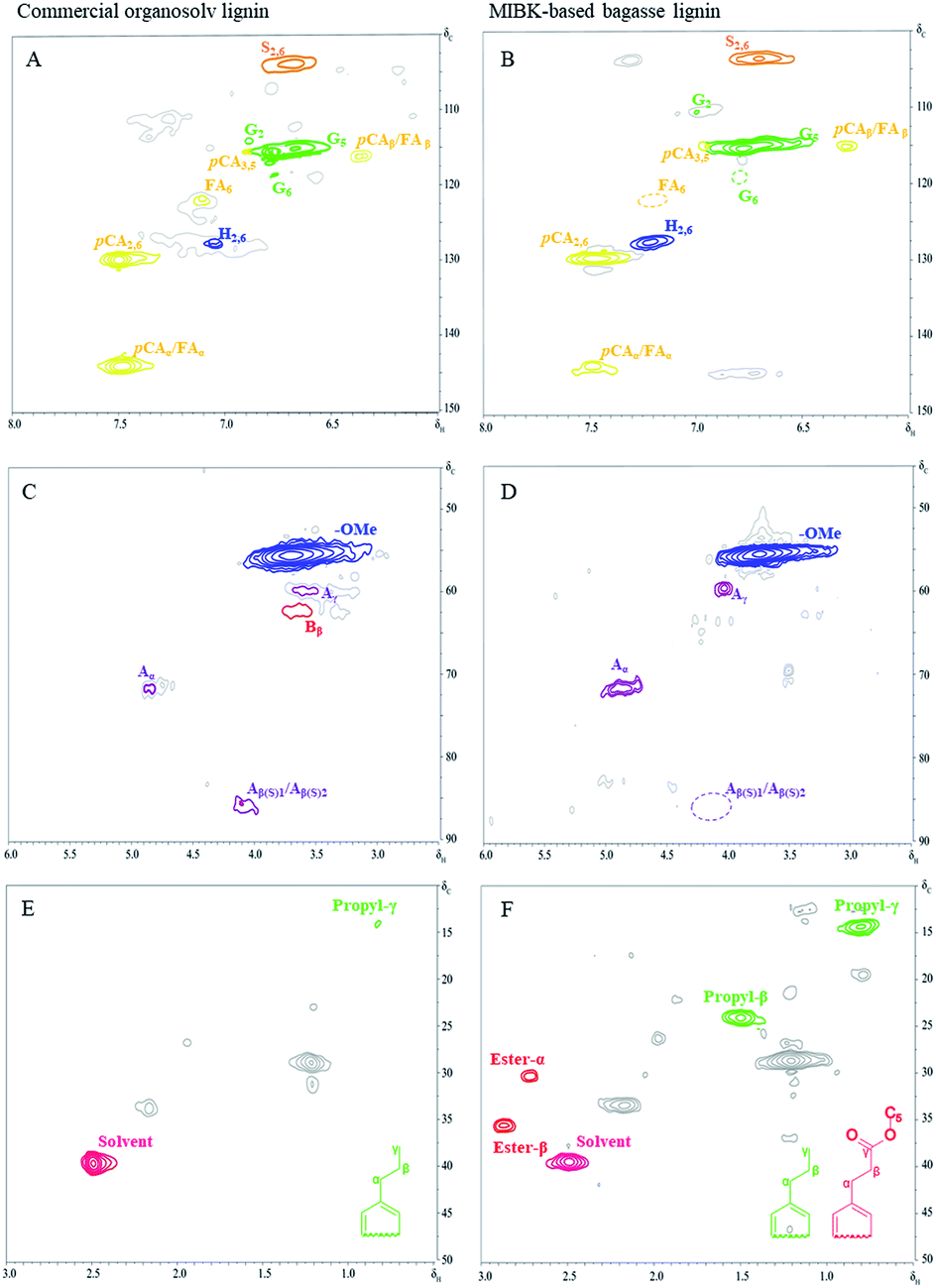
Structural features through 2D-HSQC NMR
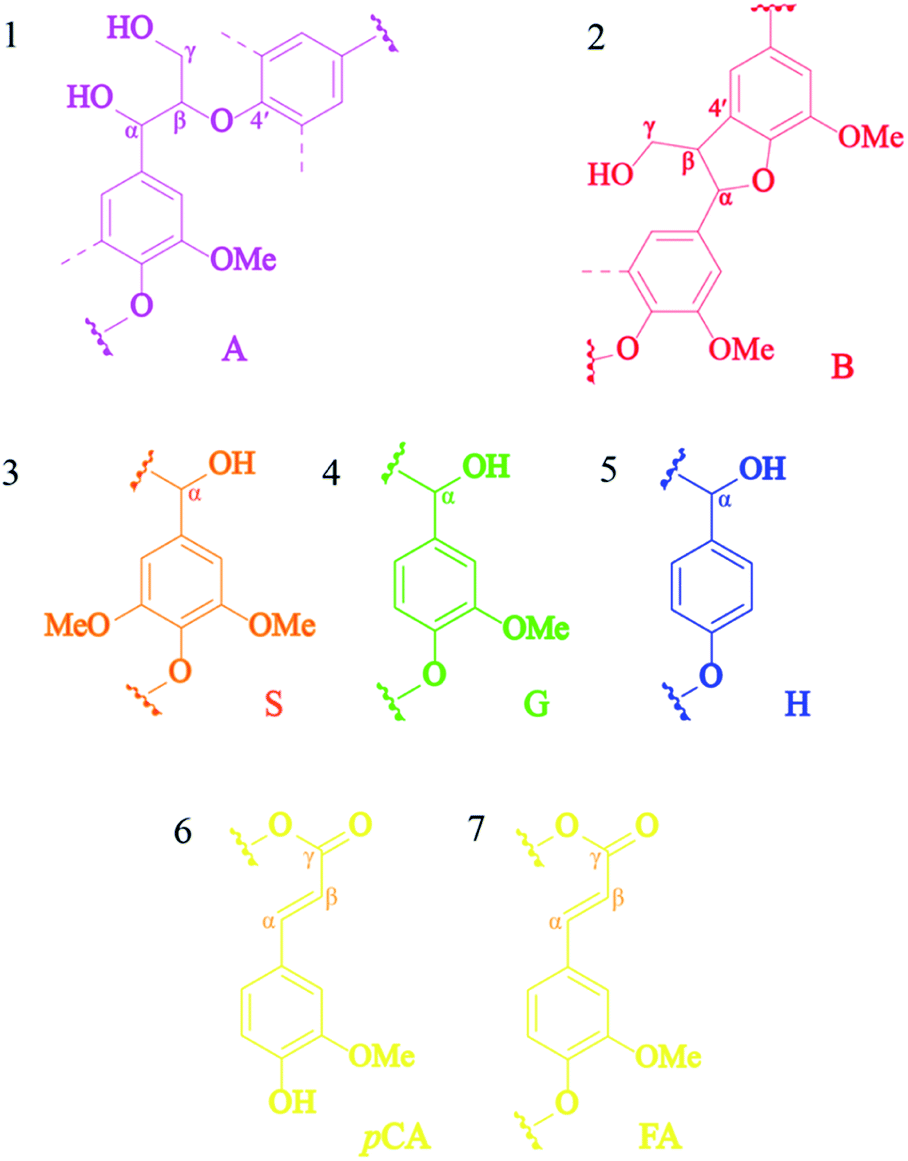
The obtained lignin samples were further investigated the structural features by 2D NMR. It should be mentioned that the detailed assignment of the correlation signals in the spectra (Table 6) is based on previous publications.37 Fig. 6 shows the side-chain (δC/δH 50.0–90.0/2.5–6.0) and aromatic/unsaturated side-chain regions (δC/δH 100.0–150.0/6.0–8.0) of the HSQC spectra of the lignins. The main substructures found are also depicted in Fig. 7. In the side-chain region of the HSQC spectra, the most prominent correlation signals represent the methoxyl groups (δC/δH 55.7/3.71) in both BGL and COL. In addition to the methoxyl groups, the signals for Cα/Hα correlations of the β-O-4 ayrl ether linkages (A) structures at δC/δH 71.3/4.81 were also observed in all lignins, though they were weaker in COL. This result is in agreement with Moghaddam et al.37 who reported that this signal could be detected when mild extraction conditions were employed. Besides, the Cγ/Hγ correlations of β-O-4 structures (A) at 59.9/3.61 were also observed in the lignins, these are commonly present in significant amounts in the HSQC spectra of native lignin from bagasse.38 The signals from the Cγ/Hγ correlations of γ-acylated lignin structures (A) in the lignins arise from p-coumarates acetylating at γ-OH of the lignin side-chain, as already published by del Río et al.39 There is only one interunit linkage visible corresponding to the Cβ(S)1/Hβ(S)1 and Cβ(S)2/Hβ(S)2 correlations at δC/δH 85.9/4.10 and 86.2/4.08, respectively, in COL; however, these were absent in the spectrum of BGL. Signals for β–β′ resinol substructures (B) were also observed in the HSQC spectra.
Table 6 Assignment of main lignin recovery signals in the 13C–1H 2D HSQC NMR spectra shown in Fig. 6
| |
Label |
δC/δH |
Assignments |
| Alkyl groups region |
C3H7-γ |
13.7/0.83 |
Propyl groups of phenolic monomers at γ position |
| C3H7-β |
24.2/1.47 |
Propyl groups of phenolic monomers at β position |
| Ester-α |
30.3/2.71 |
Ester groups of phenolic monomers at α position |
| Ester- β |
35.7/2.87 |
Propyl groups of phenolic monomers at β position |
| Side-chain region |
–OCH3 |
55.7/3.71 |
C–H in methoxyls |
| Aγ |
59.9/3.61 |
Cγ–Hγ in β-O-4′ substructure (A) |
| Bγ |
62.4/3.67 |
Cβ–Hβ in β–β (resinol) substructures (B) |
| Aα |
71.3/4.81 |
Cα–Hα in β-O-4′ substructure (A) |
| Aβ(S)1 |
85.9/4.10 |
Cβ–Hβ in β-O-4′ substructures (A, erythro form) |
| Aβ(S)2 |
86.2/4.08 |
Cβ–Hβ in β-O-4′ substructures (A, threo form) |
| Aromatic region |
S2,6 |
103.7/6.67 |
C2,6–H2,6 in syringyl units (S) |
| G2 |
111.3/6.96 |
C2–H2 in guaiacyl units (G) |
| G5 |
115.4/6.78 |
C5–H5 in guaiacyl units (G) |
| pCA3,5 |
115.6/6.93 |
C3,5–H3,5 in p-coumarate |
| pCAβ/FAβ |
116.3/6.38 |
Cα–Hα in p-coumarate and ferulate |
| G6 |
118.6/6.84 |
C6–H6 in guaiacyl units (G) |
| FA6 |
123.3/7.11 |
C6–H6 in ferulate |
| H2,6 |
127.7/7.21 |
C2,6–H2,6 in p-hydroxyphenyl units (H) |
| pCA2,6 |
130.0/7.48 |
C2,6–H2,6 in p-coumarate |
| pCA2,6 |
130.0/7.48 |
C2,6–H2,6 in p-coumarate |
| pCAα/FAα |
144.0/7.48 |
Cα–Hα in p-coumarate and ferulate |
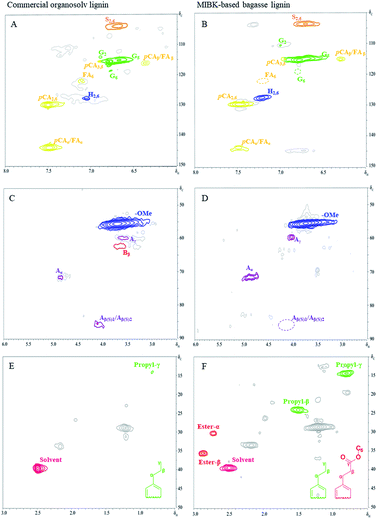 |
| | Fig. 6 The 2D HSQC NMR spectra of commercial organosolv lignin (A, C, and E) and lignin recovery (B, D, and F) extracted with MIBK. | |
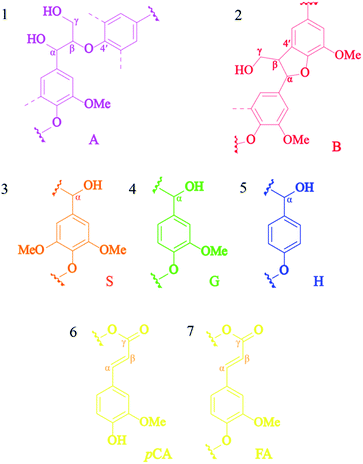 |
| | Fig. 7 The lignin substructures observed in the HSQC spectra of the lignins: (A) β-O-4′ linkages, (B) resinol structures formed by β–β, (S) syringyl unit, (G) guaiacyl unit, (H) p-hydroxyphenyl unit, (pCA) p-coumarate, and (FA) ferrulate. | |
The 13C–1H correlations in a range of δC/δH 100.0–150.0/6.0–8.0 (Fig. 6A–C) correspond to the aromatic/unsaturated side-chain regions of p-hydroxyphenyl (H), syringyl (S), and guaiacyl (G) lignin units, as well as the pCA and FA pendant units that are associated with the lignin. Prominent signals at δC/δH 103.7/6.67 were present in all lignin samples due to C2,6–H2,6 correlations in S units. Strong signals at δC/δH 111.3/6.96 and 115.4/6.78 corresponding to C2–H2 and C5–H5 correlations in G units were also observed in all the lignins, while the signal at δC/δH 118.6/6.84 related to C6–H6 in G-units was only observed for COL (Fig. 7, substructure 3). Notably, the C3,5–H3,5 correlations of p-coumarates (pCA) overlapped with the C5–H5 correlations of G units and were observed for both COL and BGL. The signals for H-lignin units at δC/δH 127.7/7.21 associated with C2,6–H2,6 correlations (Fig. 7, substructure 5) were also observed in all samples, including COL and BGL. Strong Cα–Hα and Cβ–Hβ correlation signals between pCA and ferulates (FA) at δC/δH 144.0/7.48 and 116.3/6.38 were also presented by all samples (Fig. 7, substructure 6 and 7). In addition to the signals of the pCA units, minor C6–H6 correlation signals in ferulates (FA) at δC/δH 123.3/7.11 were only presented by COL. Besides, BGL shows relatively strong signals between the regions of δC/δH 10.0–50.0/0.5–3.0 (Fig. 6E and F); these can be attributed to methyl groups attached to methyl-esters as well as propyl and ester groups of phenolic monomers, indicating the formation of new functional groups of lignin during the acid-catalyzed MIBK pretreatment. As reported by Wanmolee et al.,17 MIBK did not show the addition of a replacement of the solvent incorporated in the lignin-derived products due to the steric hindrance of isobutyl group; it thus also provides more stability during fractionation whereas methanol plays a major role by facilitating alkylation reaction on lignin. In conclusion, given the location in the HSQC spectra substructures during the pretreatment.39 The results of the of these signals, the absence of oxidized S (CαO) corresponding to C2,6–H2,6 (S′) correlations and pCAβ and FAβ clearly indicate changes in lignin substructures during the pretreatment.39 The results of the phenolic monomers attribution using the HSQC spectra were further confirmed by the Py-GC/MS.
The identification of relative mass compounds
The relative molar abundances of compounds of lignin samples were analyzed using Py-GC/MS (Table 7). The results show that COL was composed of 30.53% of H-units, 44.42% of G-units, and 25.05 of S-units, while the BGL from the organic fraction shows a predominance of G-unit, which accounted for 42.19% and is composed of 32.69% of H-units and 25.12% of the S-type. However, the S/G ratio of lignin recovery was approximately 0.60 and 0.56 for COL and BGL, respectively. As a result, it was found that the major products obtained from BGL were guaiacol (18.9%), syringol (17.2%), and 4-ethyl phenol (14.2%). Moreover, the pyrograms compound showed the products derived from p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) lignin. The results showed that BGL contained mainly aromatic hydrocarbon groups, alkoxy groups, and phenolic compounds. BGL comprised mainly of the guaiacol types (G), such guaiacol unit (1), 4-methylguaiacol (8), and 4-vinylguaiacol (11) or products in the hydroxylphenyl group (H), consisting of phenol (2), 4-ethyl phenol (7), and 4-vinylphenol (12). However, low amounts of syringol (13) and acetosyringone (18) compounds were derived from syringol type (S). Thus, the effect of organosolv fractionation lead to the change of sub-structure of derived lignin, and obviously depolymerization and repolymerization occurred during the process.40
Table 7 Py-GC/MS analysis results of the identification and relative molar abundance (%) of commercial organosolv lignin and lignin recovery
| Label |
Compound |
Origin |
COL |
BGL |
| Calculated by using all the H: p-hydroxyphenyl units; G: guaiacyl units and S: syringyl units; -lignin derived products. |
| 1 |
Guaiacol |
G |
15.2 |
18.9 |
| 2 |
Phenol |
H |
4.5 |
7.3 |
| 3 |
4-Propylguaiacol |
G |
0.3 |
0.4 |
| 4 |
3-Methylphenol |
H |
0.71 |
0.59 |
| 5 |
4-Methylphenol |
H |
1.8 |
2.1 |
| 6 |
2-Methylphenol |
H |
0.5 |
0.4 |
| 7 |
4-Ethyl phenol |
H |
3.6 |
14.2 |
| 8 |
4-Methylguaiacol |
G |
5.2 |
8.6 |
| 9 |
4-Ethylguaiacol |
G |
2.12 |
3.53 |
| 10 |
3-Methoxyphenol |
H |
2.32 |
4.5 |
| 11 |
4-Vinylguaiacol |
G/FA |
17.9 |
5.8 |
| 12 |
4-Vinylphenol |
H/PCA |
17.1 |
8.1 |
| 13 |
Syringol |
S |
13.5 |
17.2 |
| 14 |
Syringyl vinyl ketone |
S |
0.9 |
0.5 |
| 15 |
trans-Isoeugenol |
G |
1.2 |
1.2 |
| 16 |
4-Allylguaiacol |
G |
0.9 |
0.8 |
| 17 |
4-Ethylsyringol |
S |
1.7 |
1.3 |
| 18 |
Acetosyringone |
S |
2.5 |
1.4 |
| 19 |
Syringaldehyde |
S |
1.2 |
1 |
| 20 |
Syringylacetone |
S |
0.8 |
1.4 |
| 21 |
Propiosyringone |
S |
1.37 |
0.24 |
| 22 |
Vanillin |
G |
0.8 |
1.6 |
| 23 |
Eugenol |
G |
0.3 |
0.5 |
| 24 |
Homosyringaldehyde |
S |
0.12 |
0.15 |
| 25 |
Syringic acid |
S |
1.98 |
0.47 |
| 26 |
Dihydroconiferyl alcohol |
G |
0.5 |
0.86 |
| 27 |
trans-Sinapaldehyde |
S |
0.98 |
0.26 |
| 28 |
Hexadecanoic acid, ethyl ester |
FA |
0.95 |
0.72 |
| 29 |
p-Coumaric acid ethyl ester |
PCA |
1.06 |
0.6 |
| 30 |
Ferulic acid ethyl ester |
FA |
0.37 |
0.98 |
| |
|
%Ha |
30.5 |
32.6 |
| |
|
%Sa |
25.0 |
25.1 |
| |
|
%Ga |
44.4 |
42.1 |
| |
|
S/Ga |
0.56 |
0.60 |
In summary, the isolated lignin was further compared with the recent research of different solvothermal process and various biomass resources based on their structural features (Table S5†).22,35,37,41–46 It was observed that the isolated lignin showed a relatively low of Mw, Mn, PDI of isolated lignin compared with others lignin. In contrast, the DTGmax and Tg showed a relatively high. This result demonstrated a good candidate for working temperature of lignin-based polymer blends. The ratio of lignin monomers (S:G:H) of lignin samples was found in different proportion. This could be described that the different ratio is a function of genotypes and fractionation process. For lignin substructures, the lignin samples showed a similar linkages and subunits via solvothermal process. Therefore, the fractionation strategies and their conditions are considered as the main criteria on the different of lignin properties.
Conclusions
This study aimed to find the optimal conditions for the modified solvothermal fractionation process of sugarcane bagasse for maximized extraction of lignin. As the results, the optimal conditions were performed at 200 °C for 90 min in the presence of acid concentration with 0.02 M. It was observed that 88.0% of lignin fraction was removed from the solid resulting in 87.1% of lignin recovery in organic phase. The maximum lignin decomposition obtained from the TGA analysis due to the cleavage of β-aryl structures (β-O-4), resinol (β–β), and phenylcoumaran (β-5). In addition, HSQC NMR spectra revealed the formation of alkyl-ester groups of the lignin side-chain structures during acid-catalyzed MIBK pretreatment. The chemical composition of lignin derivatives composed mainly of G-structures according to Py-GC/MS. The GPC analysis showed a low molecular weight and PDI of lignin recovery compared to commercial organosolv lignin. This study provides a deep insight the structural differences of the produced lignin during solvothermal fractionation process for further conversion into high value-added products in industrial biorefineries.
Author contributions
Conceptualization, S. I., N. L., and N. S.; methodology, S. I., P. K., and N. S.; validation, W. W. and T. K.; formal analysis, S. I., P. K., and N. S.; investigation, T. K., C. S., and C. C; writing – original draft, S. I., P. K., and N. S.; writing – review and editing, W. W., N. L., T. K., C. S., and C. C.; funding acquisition, S. I., and N. L., supervision, S. I., W. W., and N. S.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Navadol Laosiripojana was supported by a research grant (RTA 6280003) from the Thailand Research Fund. Nopparat Suriyachai and Saksit Imman were supported by a Unit of Excellence (UOE64001) from the University of Phayao.
References
- F. Perera, Int. J. Environ. Res. Public Health, 2018, 15(1), 16 CrossRef
 .
. - A. Tursi, Biofuel Res. J., 2019, 22, 962–979 CrossRef .
- X. Zhao, L. Zhang and D. Liu, Biofuels, Bioprod. Biorefin., 2012, 6, 465–482 CrossRef CAS .
- A. Zoghlami and G. Paës, Front. Chem., 2019, 7, 874 CrossRef CAS PubMed .
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS .
- Y. Lu, Y. C. Lu, H. Q. Hu, F. J. Xie, X. Y. Wei and X. Fan, J. Spectrosc., 2017, 8951658 Search PubMed .
- T. B. Rawal, M. Zahran, B. Dhital, O. Akbilgic and L. Petridis, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864(5), 129547 CrossRef CAS PubMed .
- E. Paone, T. Tabanelli and F. Mauriello, Curr. Opin. Green Sustainable Chem., 2020, 24, 1–6 CrossRef .
- M. Chen, F. Malaret, A. E. J. Firth, P. Verdía, A. R. Abouelela, Y. Chen and J. P. Hallett, Green Chem., 2020, 22, 5161 RSC .
- F. Xue, W. Li, S. An, C. Li, X. Li, M. Wu and X. Wei, RSC Adv., 2021, 11, 14140 RSC .
- P. Bancha, N. Laosiripojana, V. Champreda and M. Raita, J. Sustainable Energy Environ., 2018, 9, 51–57 Search PubMed .
- C. Inkrod, M. Raita, V. Champreda and N. Laosiripojana, BioEnergy Res., 2018, 11(3), 277–290 CrossRef CAS .
- I. Cybulska, G. Brudecki, K. Rosentrater, J. L. Julson and H. Lei, Bioresour. Technol., 2012, 118, 30–36 CrossRef CAS .
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, NREL/TP-510-42618, 2012.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, and D. Templeton, NREL/TP-510-42623, 2008.
- P. Hang, S. J. Dong, H. H. Ma, B. X. Zhang, Y. F. Wang and X. M. Hu, Ind. Crops Prod., 2015, 76, 688–696 CrossRef .
- W. Wanmolee, N. Laosiripojana, P. Daorattanachai, L. Moghaddam, J. Rencoret, J. C. del Río and W. O. S. Doherty, ACS Sustainable Chem. Eng., 2018, 6(3), 3010–3018 CrossRef CAS .
- D. Nobrega, I. V. Pedrosa, C. R. R. Fernandes, A. Lachgar, M. Neli, R. Garrett, Y. L. Lam and M. M. Pereira, Sustainable Energy Fuels, 2020, 4, 4158–4169 RSC .
- G. Vanitjinda, T. Nimchua and P. Sukyai, Int. J. Biol. Macromol., 2019, 122, 503–516 CrossRef CAS PubMed .
- R. G. Candido and A. R. Gonçalves, Ind. Crops Prod., 2019, 142, 111616 CrossRef CAS .
- Y. Yue, J. Han, G. Han, Q. Zhang, A. D. French and Q. Wu, Carbohydr. Polym., 2015, 133, 438–447 CrossRef CAS .
- M. Fodil Cherif, D. Trache, N. Brosse, F. Benaliouche and A. F. Tarchoun, Waste Biomass Valorization, 2020, 11, 6541–6553 CrossRef .
- J. J. Liao, N. H. Abd Latif, D. Trache, N. Brosse and M. Hazwan Hussin, Int. J. Biol. Macromol., 2020, 162, 985–1024 CrossRef CAS .
- M. Fodil Cherif, D. Trache, F. Benaliouche, S. Chelouche, A. F. Tarchoun and A. Mezroua, Thermochim. Acta, 2020, 692, 178732 CrossRef CAS .
- M. H. Hussin, A. A. Rahim, M. N. Mohamad Ibrahim and N. Brosse, Ind. Crops Prod., 2013, 49, 23–32 CrossRef CAS .
- M. Yanez-S, B. Matsuhiro, C. Nunez, S. Pan, C. A. Hubbell, P. Sannigrahi and A. J. Ragauskas, Polym. Degrad. Stab., 2014, 110, 184–194 CrossRef CAS .
- T. Dutta, G. Papa, E. Wang, J. Sun, N. G. Isern, J. R. Cort, B. A. Simmons and S. Singh, ACS Sustainable Chem. Eng., 2018, 6(3), 3079–3090 CrossRef CAS .
- F. E. F. María, A. S. Aprigio, M. D. Curvelo, L. R. Zambon and A. María, BioResources, 2011, 6(2), 1158–1171 Search PubMed .
- Y. Huang, H. Liu, H. Yuan, X. Zhuang, S. Yuan, X. Yin and C. Wu, J. Anal. Appl. Pyrolysis, 2018, 134, 25–34 CrossRef CAS .
- L. Azzari, E. Domingos, L. Silva, A. Kuznetsov, W. Romão and J. Araujo, Polimeros, 2019, 29(4), 1678–5169 Search PubMed .
- Y. Guo, J. Zhou, J. Wen, G. Sun and Y. Sun, Ind. Crops Prod., 2015, 76, 522–529 CrossRef CAS .
- C. L. Waters, R. R. Janupala, R. G. Mallinson and L. L. Lobban, J. Anal. Appl. Pyrolysis, 2017, 126, 380–389 CrossRef CAS .
- S. Chu, A. V. Subrahmanyam and G. W. Huber, Green Chem., 2013, 15(1), 125–136 RSC .
- J. Kim, S. Oh, H. Hwang, U. Kim and J. W. Choi, Polym. Degrad. Stab., 2013, 98, 1671–1678 CrossRef CAS .
- C. Xu, F. Liu, Md. Asraful Alam, H. Chen, Y. Zhang, C. Liang, H. Xu, S. Huang, J. Xu and Z. Wang, Int. J. Biol. Macromol., 2020, 146, 132–140 CrossRef CAS .
- Z. Qin, X. Wang, H. Liu, D. Wang and G. Qin, Bioresour. Technol., 2018, 262, 212–220 CrossRef CAS .
- L. Moghaddam, Z. Zhang, R. M. Wellard, J. P. Bartley, I. M. O’ Hara and W. O. S. Doherty, Biomass Bioenergy, 2014, 70, 498–512 CrossRef CAS .
- T. Tana, Z. Zhang, J. Beltramini, H. Zhu, K. Ostrikov, J. Bartley and W. Doherty, Green Chem., 2019, 21, 861–873 RSC .
- J. Río, A. Linoa, J. Colodettec, C. Lima, A. Gutiérreza, A. T. Martinez and J. Rencoret, Biomass Bioenergy, 2015, 81, 322 CrossRef .
- M. Wu, J. Liu, Z. Yan, B. Wang, X. Zhang, F. Xu and R. Sun, RSC Adv., 2016, 6, 6196 RSC .
- M. Hassanpour, M. Abbasabadi, L. Gebbie, V. S. J. Teo, I. M. O'Hara and Z. Zhang, ACS Sustainable Chem. Eng., 2020, 8(28), 10380–10388 CrossRef CAS .
- M. Hassanpour, M. Abbasabadi, L. Moghaddam, F. F. Sun, L. Gebbie, V. S. J. Teo, I. M. O'Hara and Z. Zhang, Bioresour. Technol., 2020, 318, 124059 CrossRef CAS PubMed .
- F. G. C. Pinheiro, A. K. L. Soares, S. T. Santaella, L. M. A. Silva, K. M. Canuto, C. A. Cáceres, M. de Freitas Rosa and J. P. de Andrade Feitosa, Ind. Crops Prod., 2017, 96, 80–90 CrossRef CAS .
- X. Cheng, X. Guo, Z. Qin, X. Wang, H. Liu and Y. Liu, Int. J. Biol. Macromol., 2020, 164, 4348–4358 CrossRef CAS PubMed .
- G. Kim and B. Um, Int. J. Biol. Macromol., 2020, 158, 443–451 CrossRef CAS .
- T. Klamrassamee, T. Tana, N. Laosiripojana, L. Moghaddam, Z. Zhang, J. Rencoret, A. Gutierrez, J. C. del Rio and W. O. S. Doherty, RSC Adv., 2016, 6, 92638–92647 RSC .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03237b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a