
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAbsolute content determination by quantitative NMR (qNMR) spectroscopy: a curious case of aldosterone†
Neeraj Singh *,
Judith Taibon,
Stephan Pongratz and
Christian Geletneky*
*,
Judith Taibon,
Stephan Pongratz and
Christian Geletneky*
Roche Diagnostics, Nonnenwald 2, 82377 Penzberg, Germany. E-mail: neeraj.singh.ns1@roche.com; christian.geletneky@roche.com
First published on 5th July 2021
Abstract
Quantitative NMR spectroscopy has been utilized to calculate the absolute content (g g−1) of aldosterone, which is necessary for electrolyte balance and blood pressure regulation, in commercially available materials. Explanations have been provided for many signals observed in the 1HNMR spectrum, false interpretation of which can have significant effects if such a value is utilized for the primary calibrators in ID-LC-MS/MS (‘gold standard’) reference methods in clinical chemistry.
Aldosterone (Scheme 1), a mineralocorticoid hormone, plays a dominant role (renin–angiotensin system) in maintaining electrolyte balance by regulating Na+ and K+ ion levels in plasma and in homoeostatic regulation of blood pressure.1 It is synthesized by Zona glomuerulosa in the adrenal cortex of suprarenal glands. Adrenal cortex disorders leading to hyper/hypo-aldosteronism, make it an important parameter in clinical chemical diagnostics.1,2
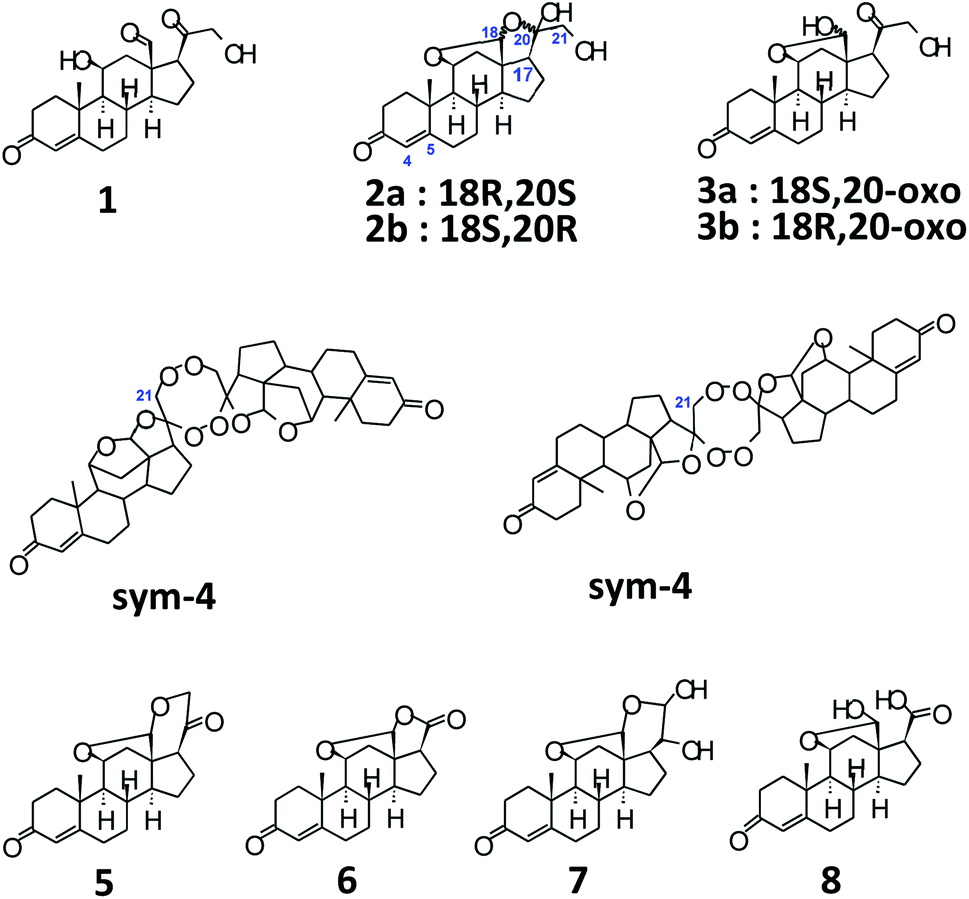 | ||
| Scheme 1 Structures of aldosterone tautomers and some impurities. | ||
Diverse diagnostic assays are utilized for the quantification of clinically relevant analytes such as aldosterone in whole blood, but nowadays, ID-LC-MS/MS based analytics have become the method of choice owing to their higher specificity, sensitivity and accuracy. Normally, an appreciable difference is found between hyphenated-MS based analysis and other assays.3 Moreover, hyphenated-MS methods4 do not suffer from the disadvantage of cross-reactivity with other structurally similar endogenous molecules present in the blood. Additionally, efforts are in progress by JCTLM (Joint Committee for Traceability in Laboratory Medicine) and IFCC (International Federation of Clinical Chemistry and Laboratory Medicine) for the development of ‘ID-LC-MS/MS based Reference Methods' for the standardization of all clinical diagnostic assays.5 One of the most important determinant of the success and trueness of such measurements is the absolute content of the primary calibrator and it's high order traceability to SI units, in order to standardize all such assays. In case of aldosterone, however, no solid-based reference standards are available from metrological institutes.5,6
We, therefore, decided to utilize only quantitative NMR (qHNMR) spectroscopy7 to calculate the ‘absolute content’ of commercially available aldosterone (non-reference standard materials), and to identify any side/by-products, if present. qNMR is the only analytical technique wherein the structure of the organic molecule and it's absolute content (g g−1) can be ascertained in a single non-destructive method. Since the intensity of a signal in 1HNMR is directly proportional to the amount of the resonant nuclei, the content can be calculated by comparison with an internal standard. The qNMR internal standards are either traceable to NIST Benzoic Acid (350b; coulometric) and/or NIST PS1 qNMR standard, thereby, provide unparalleled traceability to SI units.8a Because of the high accuracy, simplicity and precision of the qHNMR analytical method, this approach is gaining fast acceptance by metrological institutes8b such as NIST, NMIJ, NMIA along with chemical companies such as Sigma-Aldrich (Merck), Wako, etc., for the characterization and quantification of reference materials.
In this work, 1 was obtained from two sources (S1 and S2), with ≥95% chromatographic purity (HPLC/TLC; not a mass-fraction (g g−1) value). The NMR experiments were performed in acetonitrile-d3, obtained from Sigma-Aldrich. In order to eliminate solvent based differences, same CD3CN was utilized for S1 and S2. Tecnazene (qNMR Internal Standard) was also bought from Sigma-Aldrich. 1D and 2D measurements were performed, at 300 K, on a JEOL 600 MHz NMR spectrometer equipped with a He-cooled Ultra-Cool cryoprobe and/or JEOL 500 MHz NMR with a N2-cooled Super-Cool cryoprobe. Weighing was accomplished using a METTLER-TOLEDO XPR6U ultra-micro balance. For quantitative NMR experiments (1H{13C} WURST sequence) ca. 1.7–4.84 mg of 1 was weighed together with tecnazene (1.64–2.58 mg) and the mixture was dissolved in ca. 0.75 mL of CD3CN. NMR measurements were performed in 5 mm NMR tubes, and the inter-scan delay was set to 70 s.
Structurally, aldosterone 1 (Scheme 1)9 is represented as 11β,21-dihydroxy-3,20-diketo-4-pregnene-18-al, characterized by the presence of an aldehyde group at C-13. However, it is well known that, steroids with an –OH at C-11, and carbonyl functionalities at C-13 and C-17 tend to form hemiacetals and/or acetals/ketals. Appropriately, 1 has been shown to exist as an equilibrating tautomeric mixture of 18-acetal-20-hemiketal form 2 and 11β,18-oxide form 3 (Scheme 1), in solution of different solvents.10 Furthermore, even XRD findings have confirmed the presence of only tautomer 3, as a monohydrate, in solid crystals.11 As a result, the parent structure 1 has never been confirmed in solution or solid state, plausibly, owing to the high reactivity of the –CHO group at C-13 and the presence of acid/base traces in solvents, thereby, promoting the setup of 2 ↔ 3 equilibrium.
Acetonitrile-d3 was found to be a suitable solvent owing to good dissolution of aldosterone 1 and tecnazene, mainly, because the lipophobic/lipophilic nature of –CH3 group/CN functional group, respectively, promotes solubility of polar as well as apolar solutes. Additionally, excellent dispersion of spin-systems can be observed in Fig. 1, probably, owing to the local diamagnetic anisotropic interaction of the CN sp-hybridized bond with aldosterone functionalities. Since acetonitrile and/or acetonitrile–H2O eluent mixtures are frequently the solvents/eluents of choice for RP-UHPLC separations, the choice of AcN as NMR solvent ensures similar relative concentrations of aldosterone tautomers; although, additional pH factors can alter this equilibrium. Moreover, utilization of a polar aprotic solvent inhibits labile 1H–2H exchange; therefore, the tautomeric equilibrium of 1 would not be affected by kinetic isotope effect.
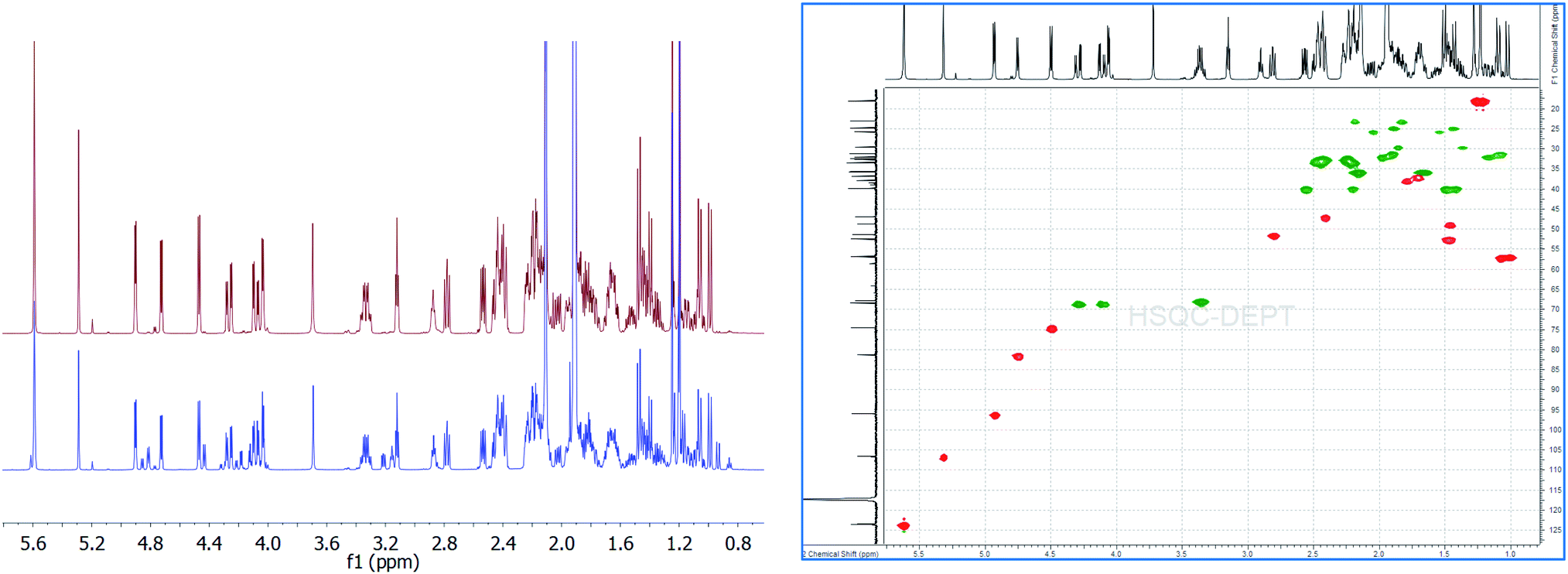 | ||
| Fig. 1 Overlay of 1H{13C}NMR spectra of aldosterone 1 (S1: red; S2: blue) indicating the various diastereomers 2a, 2b, 3a, 3b along with the 2D-gHSQCAD spectrum (S1 material). | ||
We first utilized 1 from commercial source S1 for the quantitation experiments. As can be seen,12 there was no signal at δCHO: 8.5–10 ppm, thereby, aldehyde form of 1 can be completely ruled out. However, we could easily find intense signals from 18R,20S 2a (δH-4 = 5.62 ppm, s; δH-18 = 5.32 ppm, s; δH-21 = 3.36 ppm, ddd, 2H; δH-11 = 4.75 ppm, 3J = 5.54 Hz) and 18S,20-oxo 3a (δH-4 = 5.62 ppm, s; δH-18 = 4.94 ppm, 3JH–OH = 5.14 Hz; δH-21α(R) = 4.29 ppm, dd (ABX), 1H; δH-21β = 4.11 ppm, dd (ABX), 1H; δH-11 = 4.50 ppm, 3J = 6.53 Hz, 1H) in a ratio of 0.784![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (or approx. 3:4) (Fig. 1).13 Both of these major diastereomers exist in a tautomeric equilibrium, and account for the actual structure of aldosterone in solution.10,13 Conveniently, H-4, in both structures 2a and 3a, was found to resonate at the same chemical shift. This signal being a nicely separated singlet with S/N of > 2000:1, was well-suited for utilization as the quantitation signal. Therefore, triplicate experiments were performed using H-4 as the quantification resonance, and an average absolute content of 1 was obtained to be 93.80% (Table 1). However, when 2a (δH-18 = 5.32 ppm) and 3a (δH-18 = 4.94 ppm) were quantified separately, for experiment 1 (Table 1), values of 38.82% and 49.52% were obtained, respectively. This led to a difference of 5.46% in the absolute content of aldosterone 1, when a summation of the count of 2a and 3a was compared to the olefinic signal at δH-4 = 5.62 ppm. Therefore, on a closer look of the qHNMR spectrum, we found more signals which could, potentially, account for this significant discrepancy.
:1 (or approx. 3:4) (Fig. 1).13 Both of these major diastereomers exist in a tautomeric equilibrium, and account for the actual structure of aldosterone in solution.10,13 Conveniently, H-4, in both structures 2a and 3a, was found to resonate at the same chemical shift. This signal being a nicely separated singlet with S/N of > 2000:1, was well-suited for utilization as the quantitation signal. Therefore, triplicate experiments were performed using H-4 as the quantification resonance, and an average absolute content of 1 was obtained to be 93.80% (Table 1). However, when 2a (δH-18 = 5.32 ppm) and 3a (δH-18 = 4.94 ppm) were quantified separately, for experiment 1 (Table 1), values of 38.82% and 49.52% were obtained, respectively. This led to a difference of 5.46% in the absolute content of aldosterone 1, when a summation of the count of 2a and 3a was compared to the olefinic signal at δH-4 = 5.62 ppm. Therefore, on a closer look of the qHNMR spectrum, we found more signals which could, potentially, account for this significant discrepancy.
| Aldosterone source | Analyte weighed (mg) | ISTD weighed (mg) | qNMR absolute content (%) | Standard deviation |
|---|---|---|---|---|
| S1 (experiment 1) | 3.9082 | 1.9430 | 93.91 | |
| S1 (experiment 2) | 4.8432 | 2.5879 | 93.56 | |
| S1 (experiment 3) | 2.5978 | 1.3418 | 93.93 | |
| Average value (n = 3) | 93.80 | 0.2044 |
Before the interpretation of these signals was undertaken, we also analyzed the aldosterone 1 from commercial source S2. Here, also, similar major and minor signals were seen as observed in S1, with the exception of some signals that were not present in the 1HNMR of 1 in S1. However, in S2 aldosterone 1, the signals not belonging to 2a and 3a were more intense when compared to that in S1. A qHNMR experiment for 1 in S2 yielded a value of 81.85%, following the exact same procedure as mentioned for S1.12 The absolute content of S1 aldosterone is clearly much better than S2, and the low content of S1 aldosterone makes it unsuitable to be utilized as a reference compound.
A quick look into already published literature, pointed towards the existence of some possible structures, as illustrated in Scheme 1, such as the dimer 4,14 aldosterone acetal 5,14 aldosterone-γ-lactone 6.14 Additionally, a basic/acidic workup procedure for the synthesis of aldosterone 1, can lead to additional molecules such as vicinal diol 710c,14 and carboxylic acid 814 (Scheme 1). The absence of sym-dimer 4 and asym-dimer 4 was confirmed by the absence of a singlet/dd at δH-21 ≈ 3.7 ppm. The signal at δH-21 = 3.72 ppm in Fig. 1 corresponds to an OH/exchangeable proton, for 1, with no 1JC–H in 2D-HSQC spectrum.12 Since no dd with 2J = 16 Hz can be observed around ca. δH-21 = 4 ppm, the presence of aldosterone-γ-lactone 614 can also be ruled out. Aldosterone acetal 5 is also highly unlikely to be present since the synthesis involves reflux conditions, and has only been postulated to be present in aldosterone synthesis, but not found.14a Also, carboxylic acid 8 is not present since no broad singlet could be observed at δCOOH ≈ 11 ppm.12
Among the signals which are common to both S1 and S2 aldosterone 1, besides that of 2a and 3a, signals at δH-18 = 4.84 ppm, 3J = 6.17 Hz; δH-11 = 4.46 ppm, 3J = 6.41 Hz; δHα-21 = 4.23 ppm, dd, 2J = 19.10 Hz, 3JH–OH = 5.57 Hz; δHβ-21 = 4.15 ppm, dd, 3JH–OH = 4.15 Hz (other signals of the dd spin system overlap with that of 3a), 2H; δH-17 = 3.24 ppm, dd, 3J = 7.83, 7.59 Hz; δOH-21 = 3.18 ppm, can be attributed to the other diastereomer of 3a, i.e., 18R,20-oxo 3b.12–15 Signals of 3b are present in significant amounts in S2, but are present only in traces in S1. Additionally, signals at δH-18 = 5.23 ppm, s; δH-11 = 4.80 ppm, d, 3J = 5.69 Hz; δHα-21 = 3.49 ppm, dd, 2J = 11.48 Hz, 3J = 5.14 Hz; δHβ-21 = 3.41 ppm (overlapping with that of 2a) can be assigned to 2b, the 18R,20R diastereomer.12–15 The intensity of the signals of 2b, in both S1 and S2, is almost similar.
Moreover, in S2, signals are present at δH-4 = 5.64 ppm, s; δH-18 = 4.88 ppm, 3J = 6.41 Hz; δHα-21 = 4.33 ppm, dd, 2J = 18.50 Hz, 3JH–OH = 4.74 Hz; δHβ-21 = complete overlapping with the signals of 3a/3b. These signals are, however, absent in S1 aldosterone. The structure corresponding to these chemical shifts is difficult to ascertain, however; it must be added that similar splitting pattern of H-21 geminal protons and the associated chemical shift region, point towards a third diastereomer of 3. But, since no detailed data is available for this particular diastereomer, we would not like to speculate at this point. Also, the absence of these signals from S1 aldosterone 1, and down-shifted H-4 δ value, could also result from a stereochemical inversion at C-11 or from impurities present from synthesis, such as from 11β-nitrite of corticosterone acetate,9a–9c which is a synthetic precursor for aldosterone. Additionally, a singlet at δ = 7.14 ppm,12 with similar integral area as compared to the above mentioned data, is too down-field shifted for any kind of aldosterone diastereomers. The exclusion of these signals would lead to a reduction in the absolute content of S2 aldosterone 1 by ca. 7%, which can have significant repercussions if this material is utilized as a primary calibrator in hyphenated-MS analytics. Since, primary calibrators should be of ultra-high purity grade (g g−1) and have the highest order of traceability, aldosterone from S2 commercial source was found insuitable. However, it provided important clues when compared to S1 aldosterone, for the interpretation of additional diastereomers in the tautomeric equilibria of 1.
Furthermore, in case of aldosterone 1 from S1 commercial source, the inclusion of the quantitative value for 3b (0.78%) and 2b (3.22%), a total of 4.0% increase is observed when the quantitation signal (H-4) is compared to the sum of absolute values from 2a + 3a + 2b + 3b. This leads to a total value of 92.34%, when calculated for experiment 1. A difference of 1.57% can be explained on the basis of extremely low S/N ratio of the quantitation signals of 3b (20:1) and 2b (200:1). Therefore, in our opinion, qNMR can be utilized to quantify aldosterone, suitable to be a reference compound, and also at the same time, work as a universal detector (for organic components) to analyse the chemical nature of additional entities, if present, something which cannot be said for HPLC-UV based quantitations.
Conclusions
To summarize, we have presented a qHNMR based method for the quantitation of commercially available aldosterone, and also shown the challenges associated with identification of various diastereomers of hemiketal 2 and the hemiacetal 3. A high-order traceability has been clearly established to SI units owing to the traceability of the qNMR internal standard, which makes this method of quantitation appropriately comparable to ‘mass-fraction (g g−1)’ values given for reference materials. Also, in our opinion, LC-UV based quantitation values utilized for the generation of mass-fraction values, should be cross-checked with qNMR, whenever possible. Moreover, aldosterone should be represented as a hemiacetal/hemiketal structure to avoid confusion of it being considered a single molecule characterized by the presence of an aldehyde functional group. We would also like to add that suitable columns should be employed, to ensure the separation/quantification of all the hemiketal/hemiacetal diastereomers of aldosterone tautomers, since diastereomers generally tend to have different physical properties (e.g., λmax, η), and can interact in various ways with functional groups on the surface of a particular column.Conflicts of interest
All the authors are employees of Roche Diagnostics, Nonnenwald 2, 82377 Penzberg, Germany. There are no conflicts to declare.Acknowledgements
Authors would like to thank Ms Katrin Gradl and Dr Tobias Santner for reading the manuscript.Notes and references
- M. Briet and E. L. Schiffrin, Nat. Rev. Nephrol., 2010, 6, 261 CrossRef CAS.
- R. Horton, Metabolism, 1973, 22, 1525 CrossRef CAS PubMed.
- T. Nishikawa, Ma. Omura, M. Kawaguchi, A. Takatsu, F. Satoh, S. Ito, I. Kurihara, H. Itoh, T. Yanase, H. Shibata, Y. Oki, M. Naruse, K. Sakurai, H. Sasamoto and K. Kuwa, Endocr. J., 2016, 63, 1065 CrossRef CAS PubMed.
- (a) J. J. Pitt, Clin. Biochem. Rev., 2009, 30, 19 Search PubMed; (b) S. Grebe and R. J. Singh, Clin. Biochem. Rev., 2011, 32, 5 Search PubMed.
- http://jctlm.org.
- R. F. Greaves, C. S. Ho, K. E. Hoad, J. Joseph, B. McWhinney, J. P. Gill, T. Koal, C. Fouracre, H. P. Iu, B. R. Cooke, C. Boyder, H. T. Pham and L. M. Jolly, Clin. Biochem. Rev., 2016, 37, 63 Search PubMed.
- (a) L. Griffiths and A. M. Irving, Analyst, 1998, 123, 1061 RSC; (b) S. K. Bharti and R. Roy, Trends Anal. Chem., 2012, 35, 5 CrossRef CAS.
- (a) M. A. Nelson, J. F. Waters, B. Toman, B. E. Lang, A. Rück, K. Beitruck, M. Obkircher and K. A. Lippa, Anal. Chem., 2018, 90, 10510 CrossRef CAS; (b) NIST (National Institute of Standards and Technology); NMIJ (National Metrology Institute of Japan); NMIA (National Measurement Institute of Australia).
- (a) D. H. R. Barton, N. K. Basu, M. J. Day, R. H. Hesse, M. M. Pechet and A. N. Starratt, J. Chem. Soc., Perkin Trans. 1, 1975, 2243 RSC; (b) D. H. R. Barton and J. M. Beaton, J. Am. Chem. Soc., 1960, 82, 2641 CrossRef CAS; (c) D. H. R. Barton and J. M. Beaton, J. Am. Chem. Soc., 1961, 83, 4083 CrossRef CAS; (d) W. S. Johnson, J. C. Collins, R. Pappo and M. B. Rubin, J. Am. Chem. Soc., 1958, 80, 2585 CrossRef CAS.
- (a) P. Genard, Org. Magn. Reson., 1971, 3, 759 CrossRef CAS; (b) B. G. Carter, D. N. Kirk and P. J. Burke, J. Chem. Soc., Perkin Trans. 2, 1987, 1247 RSC; (c) S. A. latif, D. J. Morris, L. Wei, D. N. Kirk, P. J. Burke, H. C. Toms and C. H. L. Shackelton, J. Steroid Biochem., 1989, 33, 1119 CrossRef CAS; (d) P. Genard, Org. Magn. Reson., 1978, 11, 478 CrossRef CAS.
- (a) W. L. Duax and H. Hauptman, J. Am. Chem. Soc., 1972, 94, 5467 CrossRef CAS PubMed; (b) W. L. Duax, H. Hauptman, C. M. Weeks and D. A. Norton, Chem. Commun., 1971, 1055 RSC.
- For details, see ESI.†.
- D. N. Kirk and M. S. Rajagopalan, J. Chem. Soc., Perkin Trans. 1, 1987, 1339 RSC.
- (a) K. Lichtwald and M. Przybylski, Angew. Chem., Int. Ed., 1985, 24, 130 CrossRef; (b) D. N. Kirk and B. W. Miller, J. Steroid Biochem., 1982, 16, 269 CrossRef CAS; (c) M. Harnik, Y. Kashman, M. Cojocaru, S. Lewicka and P. Vecsei, Steroids, 1989, 54, 11 CrossRef CAS.
- K. Yamashita, Y. Tadokoro, M. Takahashi and M. Numazawa, Chem. Pharm. Bull., 2008, 56, 873 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03472c |
| This journal is © The Royal Society of Chemistry 2021 |