
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEncapsulation of BSA in hybrid PEG hydrogels: stability and controlled release†
Corine Tourné-Péteilh *a,
Maeva Barègea,
Mathieu Lionsa,
Jean Martinezb,
Jean-Marie Devoissellea,
Anne Aubert-Pouessela,
Gilles Subrab and
Ahmad Mehdia
*a,
Maeva Barègea,
Mathieu Lionsa,
Jean Martinezb,
Jean-Marie Devoissellea,
Anne Aubert-Pouessela,
Gilles Subrab and
Ahmad Mehdia
aICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: corine.tourne-peteilh@umontpellier.fr
bIBMM, Univ Montpellier, CNRS, ENSCM, Montpellier, France
First published on 17th September 2021
Abstract
Hybrid hydrogels based on silylated polyethylene glycol, Si-PEG, were evaluated as hybrid matrices able to trap, stabilize and release bovine serum albumin (BSA) in a controlled manner. Parameters of the inorganic condensation reaction leading to a siloxane (Si–O–Si) three dimensional network were carefully investigated, in particular the temperature, the surrounding hygrometry and the Si-PEG concentration. The resulting hydrogel structural features affected the stability, swelling, and mechanical properties of the network, leading to different protein release profiles. Elongated polymer assemblies were observed, the length of which ranged from 150 nm to over 5 μm. The length could be correlated to the Si–O–Si condensation rate from 60% (hydrogels obtained at 24 °C) to about 90% (xerogels obtained at 24 °C), respectively. Consequently, the controlled release of BSA could be achieved from hours to several weeks, with respect to the fibers' length and the condensation rate. The protein stability was evaluated by means of a thermal study. The main results gave insight into the biomolecule structure preservation during polymerisation, with ΔG < 0 for encapsulated BSA in any conditions, below the melting temperature (65 °C).
Introduction
Despite their growing interest as a new class of drugs, the delivery of therapeutic bio-macromolecules (i.e. peptides, proteins, nucleic acids and lipids) remains a challenge due to the peculiar instability of these fragile and complex compounds.1–3 The biological activities of such non-small bioactive molecules cover a wide range of biomedical applications from vaccination, chronic disease treatment, tissue engineering, to cancer therapy, accounting for a total market size of several million $.4,5Among the strategies developed to improve their bioavailability, the design of responsive hydrogels, which may encapsulate them stimulated a significant interest among scientists.6–12 Drug encapsulation in polymer hydrogels is one of the most suitable strategies to encapsulate and controlled release bioactive factors. Polymer hydrogels provide an aqueous microenvironment that meets physiological conditions expected to prevent the biomolecules' degradation or aggregation. However, polymer hydrogels designs suffer from little mechanical stability, resulting in poor sustained release properties. Chemical cross-linking between the polymer chains is investigated to reinforce polymer hydrogels' mechanical properties, using addition reactions, click-chemistry, or photo induced reactions, and limiting as far as possible the formation of non-desired side products.8,13–18
A new family of biocompatible hybrid functional hydrogels polymers, have emerged recently that can cross-link with stable chemical bonds, namely siloxane, Si–O–Si.19–22 It is defined by the coexistence of organic (e.g. polymers, peptides, amino acids, etc.) and inorganic parts (i.e. silsesquioxanes (SiO1.5) units). Inorganic sol–gel polymerization leads to hybrid hydrogels useful for the preparation of water-containing materials,20 allowing the inclusion of water soluble bioorganic molecules.19,22–24 Hydrolysis of alkoxysilane (e.g. triethoxysilane, –Si(OC2H5)3) can be triggered in acidic conditions, the subsequent condensation being obtained during neutralisation, giving rise to three-dimensional network siloxane bonds (Si–O–Si). Interestingly, both reactions can also be catalysed in physiological conditions22 allowing the preparation of bio inks and the 3D bio printing of living cells.21,25 It seemed obvious that the versatility of the sol–gel reaction and the ease to modulate the hydrogel network composition by adding other molecules or functions after silylation (e.g. fluorophores, drugs, bioactive peptides, amino acids, …) offer a large potential to obtain hybrid functional materials adapted to drug delivery.
Biomolecules encapsulation, especially proteins, remains a challenging task, due to the aim of biological activity conservation and, to the complex physico-chemistry properties of this molecular class. Protein or peptides incorporation in hydrogels could yield dynamic transformations in their secondary and tertiary conformations. These intramolecular modifications could translate changes in the polymer chains organisation inside the hydrogel (mesh size), and modify its intrinsic mechanical and chemical properties (stability, degradability).26–28 All of these parameters are expected to modify the hydrogels controlled release properties of the encapsulated proteins. Also, swelling properties of hydrogels is a way to protein controlled release. The water uptake of dried hydrogels usually leads to relaxation of the cross-linked polymer chains, that increases the apparent mesh size of the network, and then, drives the protein diffusion.29 Furthermore, additional cross-linking reactions, during the drying process, have been mentioned to increase elastic-refractive forces. These results could results in improving protein retention.
In this context, we investigated the design of hybrid materials for the entrapment and the release of proteins of interest. More precisely we propose to study how the alkoxysilane sol–gel condensation reaction, could be tuned to ensure the protein trapping efficiency, its stabilisation and the control of its release kinetics. As a proof of concept, we have used the BSA as a model, and hybrid bis-silylated polyethylene oxide (Si-PEG) as a building block for establishment of the hydrogel network. The sol–gel chemical cross-linking was done in different hydration and temperature conditions to study how these parameters could affect the hybrid polymer network for sustained release purpose, while keeping the protein stability as possible. After cross-linking, the hybrid material swelling response and the protein sustained release properties were studied. Structure to function relationships were examined regarding the condensation and the drying of the network versus the observed release properties. Sustained release properties were expected, in order to further design drug delivery systems able to release biomolecules over several weeks, improving treatment compliance of patients.
Results and discussion
Texture and morphology of the hybrid hydrogels loaded with BSA
In this study, the BSA model protein was loaded using a sol–gel process, in hybrid hydrogels of bis-silylated polyethylene oxide (Si-PEG) (Fig. 1). The sol–gel process proceeded without fluorine catalyst.30 It was based on pre-maturation of the sol in a slightly acidic aqueous buffer (pH 4.9) at R.T. This step allowed the formation of small siloxane oligomer colloids, obtained by hydrolysis at moderate rate, followed by a first condensation reaction. These oligomers could be considered as the building blocks of the Si-PEG hydrogel, which ultimately cross-linked at pH 7.4, after the sol was neutralised, at a much faster condensation rate. The protein was introduced in the sol just after this neutralisation step, at a loading rate of 2.0 to 3.0% wt, respectively to PEG-Si gels dry polymer content. Then, hydrogel slabs were casted in a 96 wells plate, and the condensation reaction was conducted at different conditions of temperature (24 °C and 6 °C) and surrounding hygrometry (dry = 15–20% H2O, wet = 90% H2O, ambient = 30% H2O). A series of hydrogels was obtained, respectively named D24G, D6G, W24G, W6G, and A24G (Table 1). These experimental conditions, especially the water removal, were expected to modulate the final material's properties and the protein/material interactions. This should improved the material's controlled sustained release properties, while keeping the protein stability.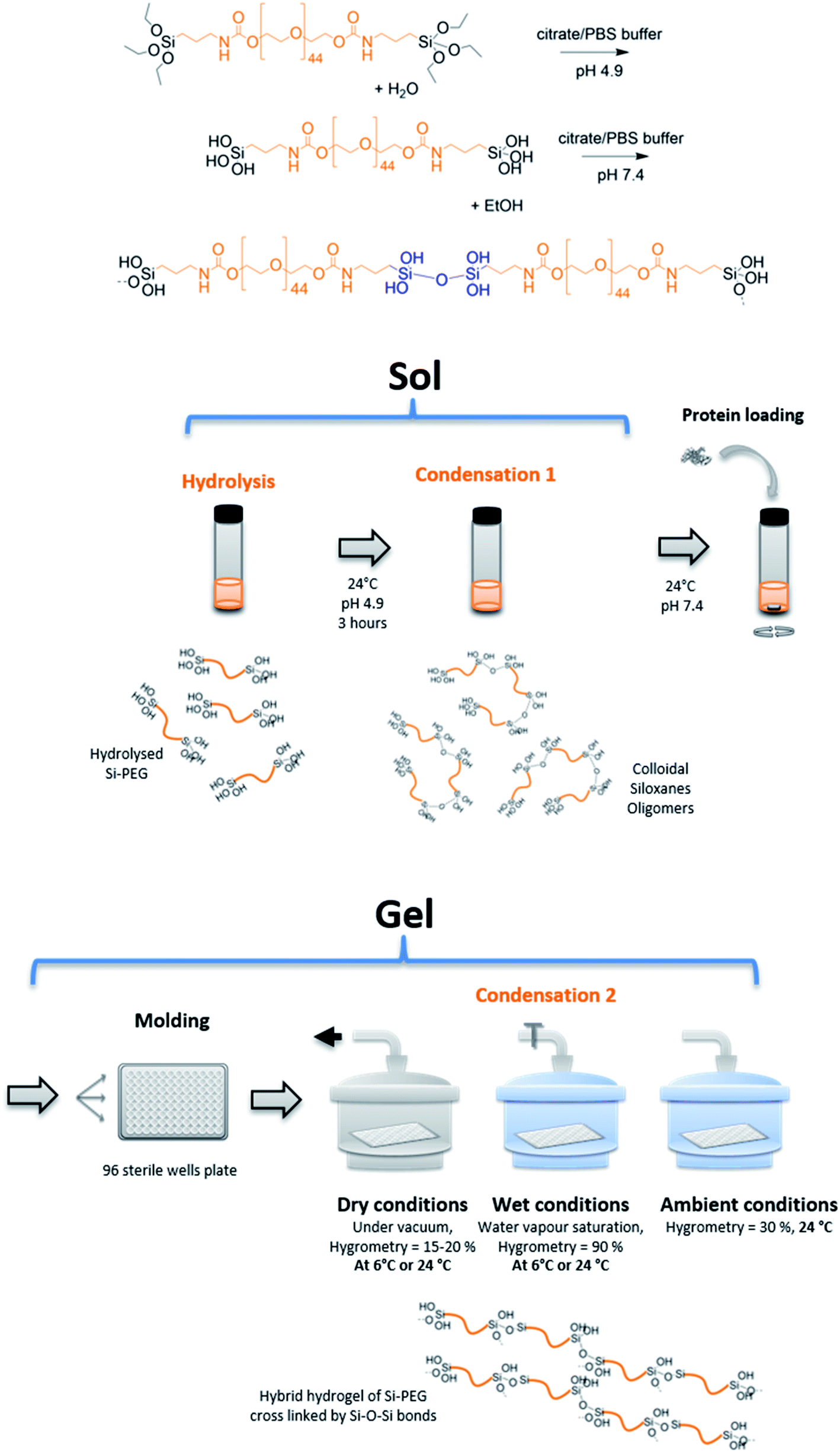 | ||
| Fig. 1 Principle of hybrid hydrogels cross-linking by sol–gel reaction and preparation. | ||
| Sample | T (°C) | HYGR (%) | τconda | RSiOHa (%) | QNM modulusb (MPa) | Structure networkb |
|---|---|---|---|---|---|---|
| a Determined on 29Si NMR spectra; [Si-PEG] = 30% wt.b Determined on PeakForce Quantitative Nanoscale Mechanical (QNM) analyses; [Si-PEG] = 20% wt/v. | ||||||
| D24G | 24 | 15 | 91.3 | 2 | 572 | Long range fibres lmoy > 5 μm |
| A24G | 24 | 30 | 60.3 | 6.6 | 117 | Web spy fibres lmoy = 150 nm |
| W24G | 24 | 90 | 74.3 | 18.2 | 20 | Spiky fibres lmoy = 125 nm |
| D6G | 6 | 15 | 87.7 | 8.5 | 75 | Long range fibres lmoy > 5 μm |
| W6G | 6 | 90 | 0 | 100 | 20 | Spiky fibres lmoy = 125 nm |
First, the condensation reaction efficiency was evaluated, by 29Si NMR, on Dry and Wet Gels (Fig. 2). Dry materials exhibited higher condensation rates, with τcond of 91.3% and 87.7%, obtained at 24 °C and 6 °C, respectively (Table 1). Then, for the wet materials obtained at 24 °C, τcond was 74.3% with surrounding hygrometry at 90% and 60.3% at ambient hygrometry. No condensation could be observed at 6 °C in wet conditions. In dry conditions, the formation of T-substructures was improved by the decrease of water content during the condensation reaction. The higher condensation rate was observed for D24G, as shown by the broad peaks corresponding to T3 (−65.0 ppm) and T2 (−56.0 ppm) species. D6G revealed some intermediate substructures as monomeric T0 (−49.5 ppm), dimeric T1 (−52.7 ppm), in addition to T2 (−58.2 ppm) and T3 (−66.6 ppm) entities. It is worth noting that residual silanol functions were still present as shown by the peak at −45.4 ppm. These indicated that at 6 °C, the condensation reaction was incomplete but effective due to the decrease of water content during the cross-linking reaction. In wet conditions, the condensation reaction was effective but incomplete, as evidenced by the presence of silanol signals at −38.8 ppm and −42.1 ppm for A24G and W24G. The corresponding peaks were sharp indicating high mobility of the species in the material. The incomplete condensation might be due to strong interactions between hydroxyl functions with the surrounding water, responsible for gaps between activated species and polymer chains.
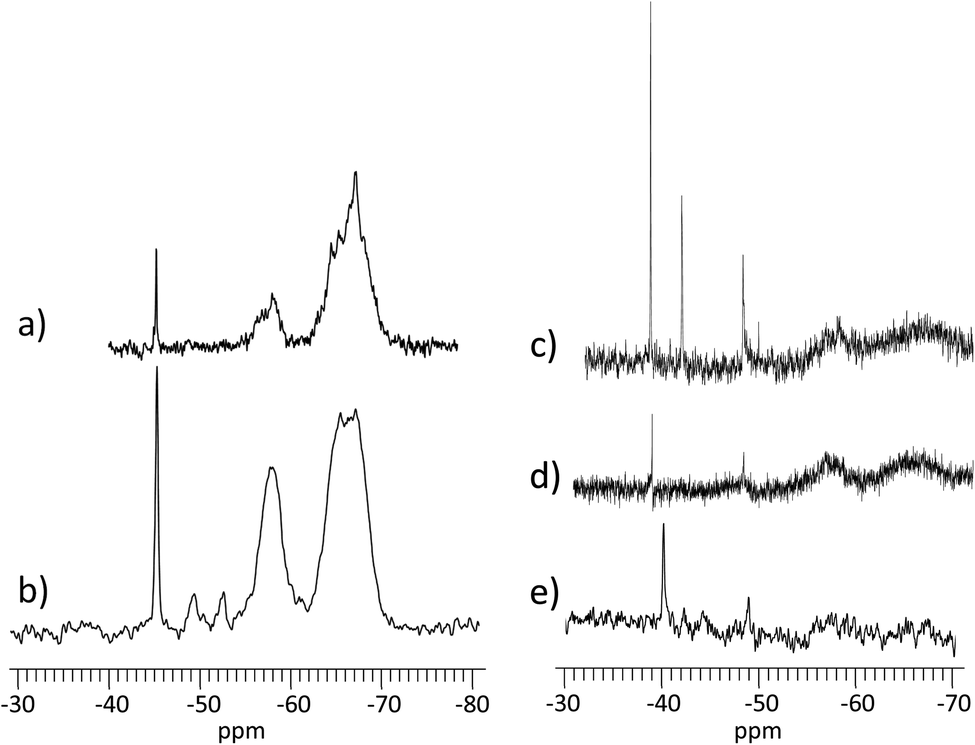 | ||
| Fig. 2 29Si NMR spectra on gels condensed in different conditions. (a) D24G; (b) D6G; (c) A24G; (d) W24G; (e) W6G. NB1: dry samples were analysed using MAS NMR solid-state; hydrated samples were analysed by liquid NMR. | ||
The structural organisation of the cross-linked hybrid hydrogels was obtained by Peak Force Quantitative Nanoscale Mechanical (QNM) analyses, allowing a quantitative mapping of the materials (Fig. 3 and ESI†). It gave a mean elastic modulus (Pa) by contact mode that was related to the materials topography. The clearest zones corresponded to the higher modulus values ascribed to the supramolecular assembly of Si-PEG entities loaded with the protein. All the materials exhibited fibrillary stacked structures. Longer lengths (>5 μm) were obtained in dry conditions, and shorter lengths in wet conditions in the range of 100 to 200 nm. These elongated structures were probably due to preferential Si–O–Si formation at distal ends of the Si-PEG chains. The formation of elongated structures could be confirmed by QELS measurements (see ESI†). They showed an increase of the mean aggregate sizes, produced by the condensation reaction between the Si-PEG polymer chains. The aggregation kinetics followed an exponential profile, which is characteristic of molecular assembly mechanisms leading to fibrillary structures starting oligomers and nuclei entities.31 Regarding the mechanical properties, for one temperature, the modulus values increased with the drying process (Table 1). This was correlated with the improvement of the fibre lengths and, to a lesser extent, with τcond increase. Here, we assumed that additional Si–O–Si bonds were formed between Si-PEG elongated chains that could explain the strengthening of the materials' structures obtained in dry conditions. We could notice that the fibres' density was highly packed in the material D6G.
 | ||
| Fig. 3 PeakForce QNM® modulus images (bellow) recorded on gels condensed in different conditions. (a) D24G; (b) D6G; (c) W24G; (d) A24G; (e) W6G. | ||
The properties of these hybrid hydrogels and their interaction with the protein will be discussed in the following sections, highlighting the impact on the protein's stability and its release profile.
Evaluation of BSA stability
Stability of the loaded BSA was evaluated for each gel formulations in order to check that the loading process did affect the protein stability. It was done by following unfolded α-helix secondary structure emergence, which is ascribed to irreversible denaturation, in comparison with free BSA in solutions.32 Indeed, secondary structures of proteins are sensitive to the environment, such as pH, temperature, surrounding water, local hydrophobicity, which affect the intramolecular H bonds. Interactions with host materials, such as polymers, are also an important aspect of protein encapsulation issues.32–34 In the following, thermal melting study was conducted on the BSA loaded in the different hybrid hydrogels, to determine thermodynamic parameters that reflect protein stability. For that, circular dichroism spectra were recorded, and the negative ellipticity was used to evaluate the presence of α-helical secondary structure at a wavelength ≈ 220 nm, [θ]222 nm (deg cm2 mol−1). The decrease of this band indicates loss of the secondary structure leading to unfolded peptide chains.33,34 The ellipticity [θ]222 nm (deg cm2 mol−1) for each material was plotted as a function of temperature (see ESI†).First, the fraction of α-helix remaining, or folded, as a function of the temperature was represented in Fig. 4. It was obtained by normalisation of [θ]222 nm to [θ]222 nm,0°C, extrapolated to 0 °C, which corresponded to a full folded α-helix. Therefore, at 20 °C, all materials exhibited a high α-helix structure ratio close to 100%, like in aqueous solutions, including PEG solution (Table 2). This indicated there was no degradation that could be ascribed to the sol–gel process, and that could affect the structural integrity of the protein.
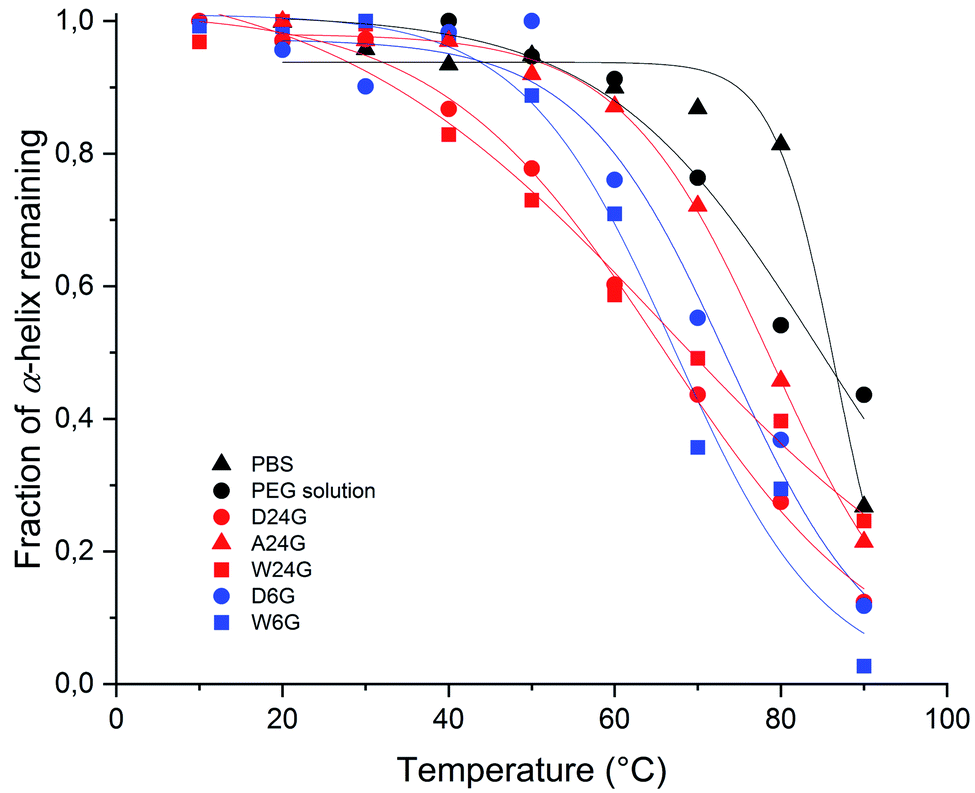 | ||
| Fig. 4 Evolution of fraction α-helix of loaded BSA remaining, or folded, in function of the temperature for each hybrid Si-PEG material. | ||
| Sample | α-Helix remaining at 20 °C (%) | Tm (°C) | R2 | Water contenta (% wt) |
|---|---|---|---|---|
| a Determined by TGA at the end of the maturation process from 25 °C to 200 °C; [Si-PEG] = 20% wt/v. | ||||
| H2O | 100 | 98 | 0.985 | — |
| PBS | 100 | 86.6 | 0.967 | — |
| PEG solution | 97 | 84.6 | 0.966 | — |
| D24G | 97 | 65.7 | 0.997 | 1.0 |
| A24G | 97 | 66.0 | 0.977 | 3.5 |
| W24G | 100 | 73.7 | 0.965 | 70.2 |
| D6G | 98 | 67.2 | 0.979 | 2.4 |
| W6G | 99 | 78.8 | 0.997 | 78.1 |
Then, the melting temperature, Tm (°C), was determined as the thermal unfolding transition, at equilibrium between two states of the secondary structure, where [α-helix]folded = [α-helix]unfolded.35,36 It gave indications on the protein propensity to rise either unfolded α-helix or to remain stable. The highest values of melting temperature were obtained for the wet materials, with Tm = 73.7 °C for W24G, and 78.8 °C for W6G (Table 2). This could be correlated to the water content, respectively 70.2 wt% and 78.1 wt%. Water might favour protein stabilisation by creating a layer hydration surrounding the peptides, like in aqueous solutions. Yet, it was noteworthy that BSA could be considered fully hydrated in any material. Actually, the amount of water reached at least 0.3–0.35 g g−1 of protein that is largely sufficient for protein full hydration.37
The free energy, ΔG (kcal mol−1) of α-helix unfolding was evaluated according to eqn (1):
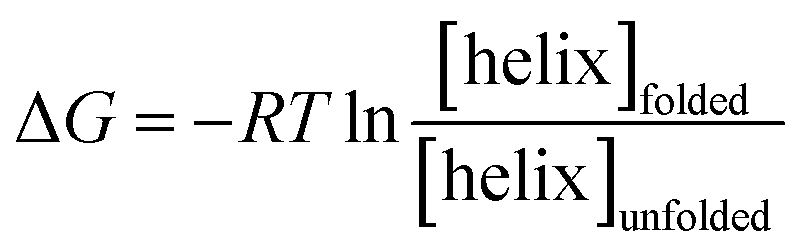 | (1) |
As we can see in Fig. 5, free energy values of BSA are negative, in all experimental conditions, up to about 60 °C (333 K), which is the BSA temperature denaturation. ΔG of the references in solution (in PBS or PEG solution) were ranged between −0.8 to −1.75 kcal mol−1. Free energy of BSA loaded in hydrogel A24G followed the same profile as in solution. This indicated that this material did not affect interactions inside the protein and reflected standard aqueous conditions. For temperatures lower than 40 °C (313 K), wet materials exhibited lower free energy values, ascribed to a highly folded α-helix stabilised by strong intra molecular interactions. This was also correlated with low interactions with the material that less affected the BSA structure.33,34,39
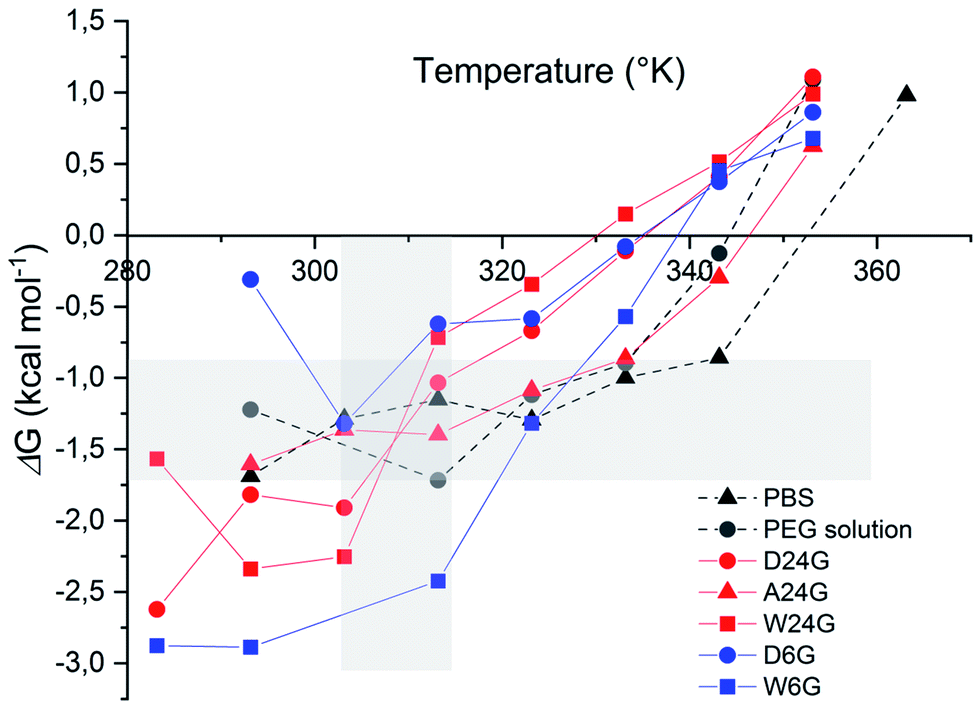 | ||
| Fig. 5 Free energy of the α-helix unfolding of BSA loaded in Si-PEG 20% wt/v materials. Grey area underlined reference free energy of BSA in PBS or PEG solution, and temperatures ranged between 30 °C and 40 °C. | ||
BSA in dry materials revealed higher ΔG values, but still in the negative range favourable to the α-helix stabilisation. This might be attributed to larger interactions of the protein with the material, due to lower water content and possibly a different distribution in the materials. Dry materials exhibiting higher τcond and long-range fibre structuration's, BSA was expected to be deeply trapped than in the wet materials.
Sustained release and responsive (swelling) properties
The protein release tests were performed in a simulated physiological medium PBS at 37 °C, for several weeks (Fig. 6 and Table 3). Each material condition and polymer density were evaluated. Swelling experiments were achieved in the same experimental conditions, at the same time. They were conducted up to material disruption. The swelling ratio, Qm, was calculated at different times of the kinetics. It was defined as the ratio between the weight, Ms, of material after it has taken water on the initial weight, Md.27,28 The swelling process reflects the materials capability to take water and, it is correlated to the intrinsic mechanical and deformable properties. The swellability of materials in case of drug delivery systems could be correlated to Fickian's diffusion to explain drug controlled release.40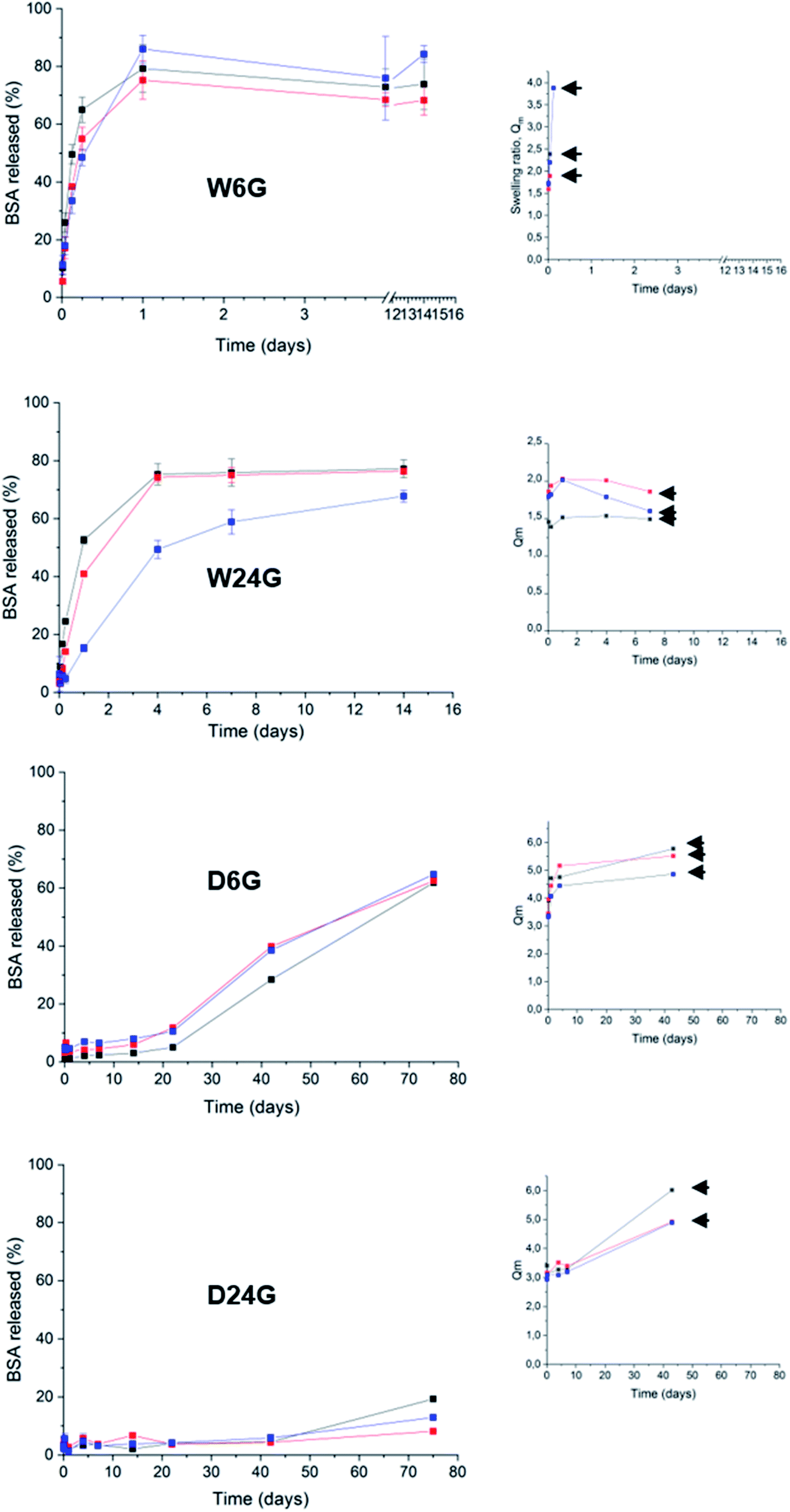 | ||
| Fig. 6 Release profiles of BSA in simulated physiological phosphate saline buffer at 150 mM pH 7.4 and 37 °C. On the right panel are kinetics of swelling ratio Qm, arrows were placed at the gel disruption points. Black traces corresponds to [PEG-Si] = 20% wt/v, red to [PEG-Si] = 25% wt/v and blue to [PEG-Si] = 30% wt/v. | ||
| Sample | 50% released | Release mechanism |
|---|---|---|
| D24G | >75 days | Swelling |
| Sustained release | ||
| A24G | <1 hour | Fick's diffusion |
| Burst effect | ||
| W24G | 1 to 5 days | Fick's diffusion |
| D6G | 60 days | Fick's diffusion |
| 2 regimes |
A first observation had shown that BSA release profiles were closely correlated to the way the materials were prepared. In fact, release kinetics could be gradually ranked from fast release or burst effect (50% BSA released for few hours) for the material W6G, to sustained release for several weeks (50% released for more than 75 days) for D24G. Especially, dry materials exhibited the best retention properties. This was consistent with both observations of (i) ΔG enhancement of BSA's α-helix substructures when it was trapped in the dry material, and (ii) the higher τcond values of the siloxane functions in the cross-linked Si-PEG network. Moreover, the dry materials exhibited better structural stability, as shown by the swelling plots (Fig. 6), where the gels' disruption occurred over 40 days. After disruption, gel's fragments of tens of microns in size, still remained in the aqueous medium for several extra days. Further, for this additional period, they behaved as small matrix systems, where the protein was dispersed, and continue to be released. Remarkably, water penetration inside the material D24G was slow, following a 0-order kinetics that could explain the particular sustained release of the protein, governed here by a swelling mechanism. Nevertheless, reduction of the gels size increased the contact surface with the aqueous medium, leading to a progressive acceleration of the protein release.
To describe more precisely the release mechanisms, the experimental profiles of BSA release were linearized using the semi-empirical model: Mt/M0 = Kt1/2, where the constant K encloses information on the diffusion coefficient and the material intrinsic properties.41 Here, we considered that diffusion takes place in a well-stirred finite volume. The concentration, Mt, in the surrounding system changes with time. The surface resistance may or may not be negligible.42 The linear deviation of Mt/M0 as a function of the square root of time, t1/2, generally known as the simplified Higuchi model, describes drug release as a diffusion process based on the Fick's law when linearity is observed.41 So that, the material D6G exhibited a release profile in two regimes both governed by Fick's diffusion (see ESI†). The first one was slow for 20 days, and released about 10% of protein. Then, a second regime increased the release rate, up to 60% within 50 days. Although fast water uptake at the beginning was observed, swelling did not affect solely the protein release. Maybe in D6G the degradation could be also responsible for BSA relief, due to lower mechanical properties, the modulus being of 75 MPa against 572 MPa for D24G.
Now regarding hybrid hydrogels obtained in wet conditions, faster release profiles driven by Fick's diffusion were observed. 50% of BSA were released after 3 hours from the W6G material. This was not surprising due to low interactions of the protein with the Si-PEG material, low mechanical properties and negligible condensation rate observed inside the material. It was confirmed by the swelling experiments, where the hydrogels were fully disrupted during this period leading to fast protein release. A better controlled was obtained on hydrogels W24G, which exhibited a Si–O–Si condensation rate of 74.3%. This value seemed to be significant to enhance the protein retention, but not enough to reach sustained release over weeks. This is due to lower mechanical properties that did not allow sufficient stability of the material, as shown by the swelling experiment. Indeed, the material got total disrupt at 7 days. Then, we can assume here that the release was driven by erosion/diffusion mechanisms in wet materials.
Proposed structure–function mechanism for entrapment and protein controlled release
The scheme below (Fig. 7) gives a representation of structure/function relationships of the materials obtained in different conditions of temperature and surrounding aqueous vapour. We demonstrated that these conditions clearly affected the Si-PEG polymer chains organisation during the cross-linking reaction. The condensation of hydrolysed alkoxy-siloxanes into stable siloxane Si–O–Si bonds drove the final material mechanical properties, the protein stability, and the controlled release. Both the formation of Si–O–Si bonds at neutral pH and water depletion favour higher condensation rates that produced elongated fibre Si-PEG polymer structures with probable intra-chains stable chemical cross-linking. This resulted in better resistance of the materials under the pressure of water penetration during the swelling event occurring in release experiments. In this way, the protein retention was enhanced and the sustained release could be reached for several weeks in the case of materials prepared at 24 °C and dry condensation conditions (D24G). Here, the swelling process mainly drove the release mechanism.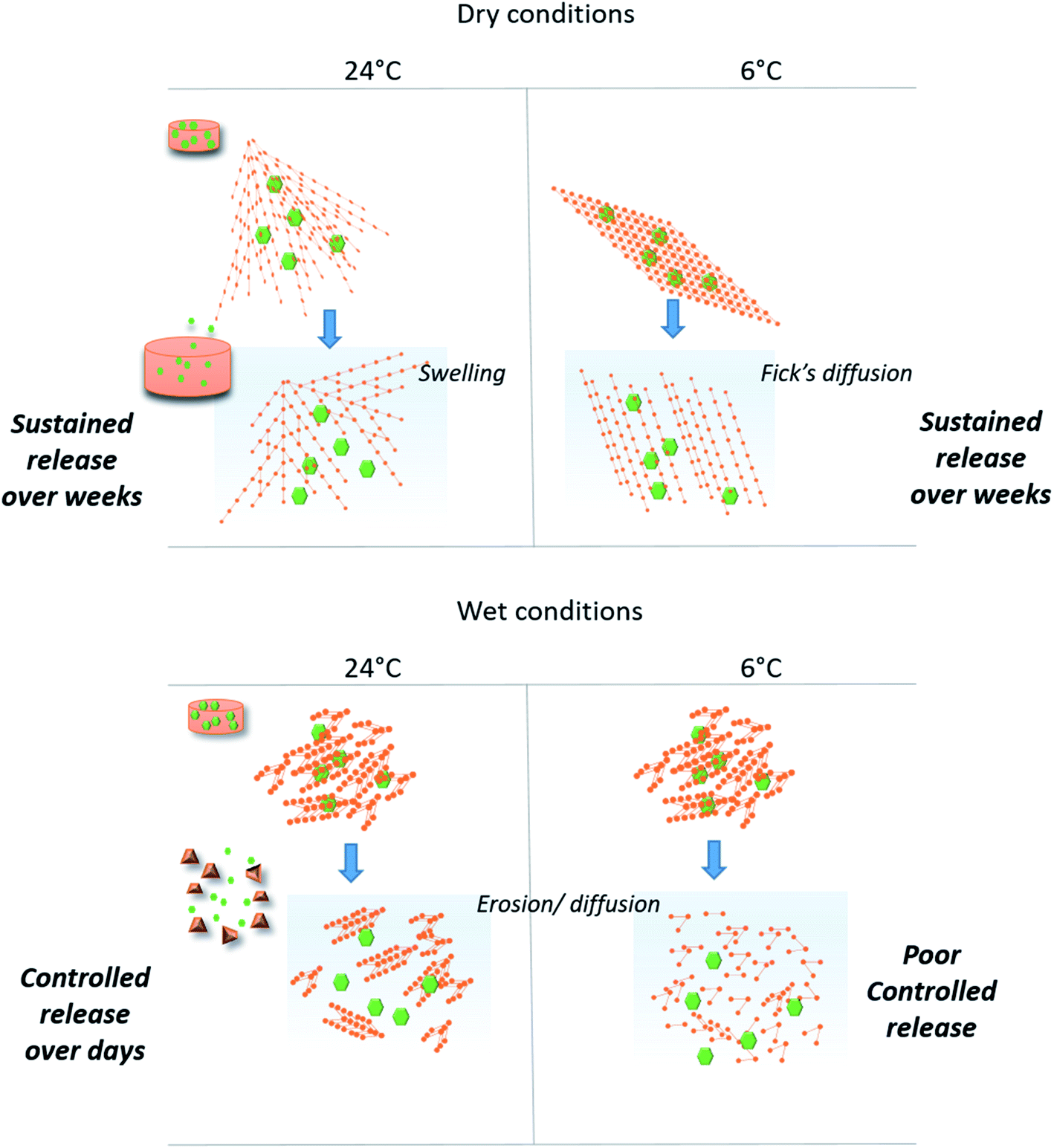 | ||
| Fig. 7 Scheme of hybrid materials' structure obtained in different conditions of Si–O–Si covalent links formation, and associated release BSA mechanisms. The synergic effects of both the condensation reaction and the drying process gave high mechanical stability of the chemical xerogels to swelling process, which promoted sustained release protein profiles over several weeks. Chemical hydrogels revealed faster release profiles from hours to several days, based on erosion/diffusion of the polymeric matrix. | ||
The synergic process of condensation and drying was observed at both ambient temperature and at 6 °C. Yet, at the lower temperature, the formation of Si–O–Si between the polymer chains was probably inhibited, that decreased the mechanical resistance during the swelling (D6G). In this case, diffusion was the main driving force that released the protein. The high retention properties observed for the 3 first weeks could be explained by high interactions between the protein and the material as indicated by the free energy ΔG values. Indeed, protein interactions may be in favour of the PEG chains when the water content decreased.
In the absence of drying, the condensation reaction led to Si-PEG hybrid materials bearing short fibres in the range of 100–150 nm length with poor resistance to swelling (less than one hour). Moreover, the condensation reaction did not occur at 6 °C, resulting in poor controlled release (W6G). The profiles of protein release were controlled for one week by an erosion/diffusion mechanism in W24G.
The residual water content was not negligible, even in dry materials, providing good insight of the protein stability trapped inside the Si-PEG network, insuring a full protein hydration. We have evidenced that the protein stability was better in wet materials, because of higher unfolding temperature transition, and lower interactions with the polymer. Nevertheless, in dry materials, the protein melting temperatures were still below the protein temperature degradation of 60 °C, and above the storage and physiological temperatures. Finally, it was shown that interactions were stronger in dry materials than in wet materials, without any loss of the protein substructure.
Experimental
Materials
Polyethylene glycol, PEG, (PEG; Alfa Aesar; No. CAS 25322-68-3) of molecular weight 2000 g mol−1, was silylated triethoxy(3-isocyanatopropyl)silane, ICTPES (247.4 g mol−1, TCI Chemicals, No. CAS 24801-88-5), in presence of triethylamine (101.2 g mol−1, Sigma Aldrich, No. CAS 121-44-8).Buffered solutions were prepared by using monohydrated citric acid (210.14 g mol−1, C6H8O7·H2O, MERCK, 77-92-9), dehydrated Na2HPO4 (177.98 g mol−1, Sigma Aldrich, No. CAS 10028-24-7), KH2PO4 (136.09 g mol−1, Sigma Aldrich, No. CAS 7778-77-0).
The Bovine Serum Albumin (BSA) was purchased from SIGMA-ALDRICH (purity of 98%, 65 kDa), with an isoelectric point of 4.7 and an hydrodynamic diameter of 7 nm in water.
All the solutions were prepared with ultra pure Milli Q water.
Sigmacote® siliconizing reagent for glass from SIGMA-ALDRICH was used to prevent protein glass surfaces adhesion.
Silylation of PEG (Si-PEG)
Dehydrated PEG (20 g, 0.01 mol, 1 eq.) was reacted with ICTPES (7.42 mL, 0.03 mol, 3 eq.) in 65 mL of anhydrous THF, in the presence of triethylamine (11.12 mL, 0.08 mol, 8 eq.), for 72 h at 72 °C under stirring and argon flux. The bis-silylated hybrid Si-PEG, was obtained after THF evaporation, followed by four washings with pentane, and then, drying 5 h under vacuum.Gels preparation
Si-PEG hydrogels of 0.2 mL were prepared at 20, 25 and 30% wt in buffered solutions insuring the pH stability during the whole sol–gel process, including the hydrolysis and condensation reactions. No chemical catalyst was added, the reactions being solely controlled by adjusting pH. First, 1 mL of sol was hydrolysed in a buffered saline solution at pH 4.9 (0.1 M citric acid solution adjusted with a 0.2 M Na2HPO4 solution) for 3 hours then the pH was adjusted to exactly 7.4 with 4 M NaOH to initiate the condensation reaction. 50 μL of an aqueous solution of BSA (120 mg mL−1) was immediately added to the neutralised sol. The hybrid hydrogels were moulded in 96-well sterile plates to obtained cylindrical slabs. The 96-well plates were placed under vacuum to obtained a hygrometry of <15% (Dry Gels), or under water saturated atmosphere for hygrometry >90% (Wet Gels). Gels were left to stand under atmospheric conditions at 30% of hygrometry (Ambient Gels). Dry and wet conditions were performed at 24 °C and 6 °C. They were left to stand for 3 days in these conditions to complete the condensation reaction.29Si NMR
The 29Si NMR spectra were recorded at the end of the condensation reaction on gels containing 30% wt of Si-PEG, to ensure reliable 29Si detection. The PBS/citrate buffer was used with 20% v/v of D2O. The Gels and Wet Gels were analysed on a RMN BRUKER Avance III – 500 MHz using liquid or solid sequences. The condensation ratio τcond (%), was determined by integrating the peaks resonance for each siloxane T units (using DmFIT software43) and using eqn (2):
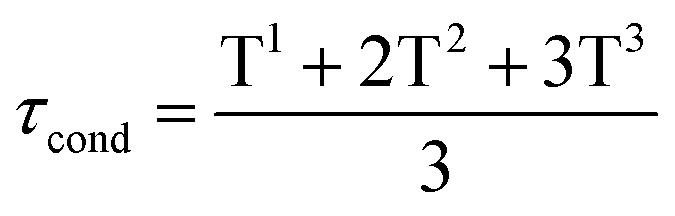 | (2) |
The Si–OH function remaining ratios, RSi–OH, were determined as the fraction of total integration bellow δ = −50 ppm.
Peak force quantitative nano-mechanical property mapping (PF-QNM)
Mechanical characterization was performed with a Multimode AFM instrument (from Bruker Corporation, USA) upgrade with Nanoscope V using PF-QNM imaging mode and a scanasyst-fluid (from Bruker) probe with a spring constant of the lever of 1.12 N m−1 and a tip radius of 150 nm. After a first step to calibrate sensitivity deflection and sync distance QNM on a sapphire sample from Bruker Instruments, these values were confirmed during measurement on a mix of polymers (PS-LDPE) from Bruker, providing an indentation modulus of around 400 MPa for LDPE phase. The applied maximum loading was set at 30 nN for all the measurements and the peak force amplitude was set at 300 nm. The obtained force versus separation curves were analysed in real time to obtain different mechanical properties: adhesion force, elastic modulus, deformation, energy dissipation. The well-known DMT model44 was used to fit retract curve to calculate indentation modulus (Poisson's coefficient set at 0). These values were then sent to different data channels that were shown as a set of different images simultaneously with the topography image.Release tests and swelling experiment
The casted hydrogels were soaked at 37 °C in a 150 mM physiological simulated saline buffer at 66 mM and pH 7.4 (9.073 g L−1 of KH2PO4 and 11.87 g L−1 of Na2HPO4·2H2O).The release tests were performed, in duplicates (n = 2), in 10 mL of physiological simulated buffer, with 0.05% m/v of azide, in SigmaCote® pre-treated containers to prevent protein surface adsorption. Samples of 0.150 mL (n = 2) were taken off at different times and, replaced by fresh medium. The protein was quantified with MicroBCA kit reactant (Thermofisher) following the microplate procedure (linear standard working range of 2–40 μg mL−1). Standard deviation of 2 kinetics was given as error bar on the BSA release curves.
Fick diffusion was validated when linearity was reached (with r2 ≥ 0.92 and Mt/M0 ≤ 0.6) for the function Mt/M0 = f(t1/2), where Mt is the concentration of solute released in the bulk for each sampling time, and M0 the solute concentration at infinite time (see ESI†).27
For the swelling experiment, the gels were weighted with precision, first as synthesised (Md) and, then at different times (Ms), up to disruption when occurring. The swelling ratio, Qm, was calculated as follows27,28 (eqn (3)):
| Qm = Ms/Md | (3) |
Thermal stability study and circular dichroism
The thermal stability studies of the loaded BSA were performed from 10 to 90 °C at fixed equilibrated temperatures.32 Spectra were recorded on a JASCO J-815 CD Spectrometer, in the 260 to 190 nm wavelengths range, at a scanning speed of 100 nm min−1 and a data pitch of 0.1 nm. Samples were placed in carefully cleaned quartz demountable cuvettes (Hellma QS type 106) with an optical path length of 0.1 mm. To prevent side dichroic interactions, PBS/citrate buffer was replaced by PBS at pH 7.4 prepared with a 66 mM KH2PO4 solution adjusted in pH with a 66 mM Na2HPO4 solution. Duplicates of CD at 222 nm were plotted in function of temperature, each time as possible (see ESI†).For references, BSA solutions were prepared in Milli Q water at a concentration of 4.4 μM, in PBS at pH 7.4 or in 20% wt/v PEG solution. CD spectra of the loaded BSA were done in hydrogels of Si-PEG at 20% wt/v in the different experimental conditions described above.
Thermal melting profiles were collected by monitoring the CD signal at 222 nm ([θ]222) as a function of temperature, using thermostatically controlled cuvette holder. Signal was recorded at temperature equilibrium. The curves of [θ]222 = f(T °C) could be fitted as follows35,36 (eqn (4)):
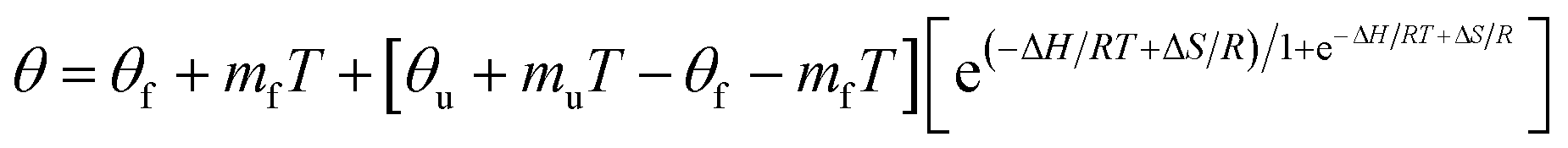 | (4) |
The melting temperature, Tm, was determined at the inflection point of the plots fraction of α-helix remaining = f(T °C),45 fitted by a Slogit function, using Origin 2020. Tm is thermal unfolding transition and corresponds to the equilibrium between folded and unfolded peptides.
The free energy, ΔG (kcal mol−1) was determined using the plots [θ]222 (unit deg cm2 dmol−1) = f(T °C), following eqn (5):38
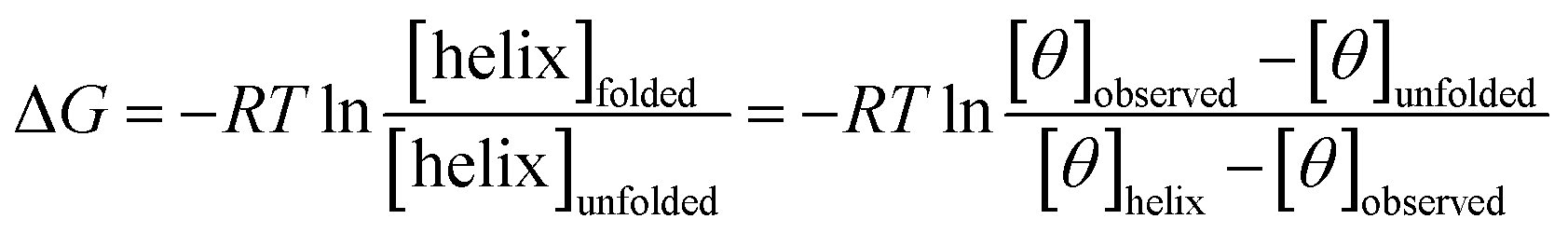 | (5) |
TG analysis
Thermic analyses to determine the water residual inside the gels were done at 10 °C min−1 under air flux from 25 to 500 °C using a Thermal Analyser (STA) 6000 (PerkinElmer).Conclusions
In this piece of work, we were able to adapt the mechanical properties of the hybrid hydrogels or xerogels, as a function of a desired protein delivery rate, from hours to several weeks. The release mechanisms were driven by several factors including the penetration of aqueous medium inside the hybrid materials resulting in the swelling, the protein diffusion, and the erosion of the material. As expected,46 the sol–gel proceeded chemo selectively towards amino acid side chains. This is a key advantage for therapeutic protein encapsulation during the loading process, as it avoids unwanted side reactions involving the biomolecule of interest.Beyond this proof of concept, the perspectives of designing tailored hybrid hydrogels for encapsulating proteins are as wide as the range of available silylated (bio) polymers and other hybrid biomolecules that may constitute the network. Besides the hybrid polymer nature, the cross-linking density, the hydrophilic/hydrophobic balance and even the isoelectric point of the gel could be chosen according to the protein to be delivered and the desired timeframe for delivery.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by Chimie Balard Cirimat Carnot Institute through the ANR program No. 16 CARN 0008-01. The materials characterisations were done on the facilities technology of the University of Montpellier, France. In particular, the authors thank Emmanuel Fernandez for solid NMR measurements in the Balard Plateforme d'Analyses et de Characterisation (PAC Balard), Karine Parra for liquid NMR measurements and Baptiste Legrand for CD measurements in the Laboratoire de Mesures Physiques (LMP), and, Michel Ramonda for QNM Peak Force analyses in the Centrale de Technologie en Micro et nanoelectronique (CTM).Notes and references
- S. D. Putney and P. A. Burke, Nat. Biotechnol., 1998, 16, 153–157 CrossRef CAS PubMed.
- F. Madani, H. Hsein, V. Busignies and P. Tchoreloff, Pharm. Dev. Technol., 2020, 25, 133–148 CrossRef CAS PubMed.
- M. B. Reis, P. A. T. Pereira, G. F. Caetano, M. N. Leite, A. F. Galvão, F. W. G. Paula-Silva, M. A. C. Frade and L. H. Faccioli, PLoS One, 2017, 12, e0182381 CrossRef PubMed.
- D. Jain, S. S. Mahammad, P. P. Singh and R. Kodipyaka, Drug Dev. Ind. Pharm., 2019, 45, 1403–1420 CrossRef CAS PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- H. R. Culver, J. R. Clegg and N. A. Peppas, Acc. Chem. Res., 2017, 50, 170–178 CrossRef CAS PubMed.
- N. Oliva, J. Conde, K. Wang and N. Artzi, Acc. Chem. Res., 2017, 50, 669–679 CrossRef CAS PubMed.
- M. M. Pakulska, S. Miersch and M. S. Shoichet, Science, 2016, 351, aac4750 CrossRef PubMed.
- A. H. Nguyen, J. McKinney, T. Miller, T. Bongiorno and T. C. McDevitt, Acta Biomater., 2015, 13, 101–110 CrossRef CAS PubMed.
- Y. Tahara and K. Akiyoshi, Adv. Drug Delivery Rev., 2015, 95, 65–76 CrossRef CAS PubMed.
- M. K. Nguyen and E. Alsberg, Prog. Polym. Sci., 2014, 39, 1235–1265 CrossRef CAS.
- K. Ladewig, Expert Opin. Drug Delivery, 2011, 8, 1175–1188 CrossRef CAS.
- C.-C. Lin, A. Raza and H. Shih, Biomaterials, 2011, 32, 9685–9695 CrossRef CAS PubMed.
- R. A. A. Muzzarelli, Carbohydr. Polym., 2009, 77, 1–9 CrossRef CAS.
- H. Jiang, S. Qin, H. Dong, Q. Lei, X. Su, R. Zhuo and Z. Zhong, Soft Matter, 2015, 11, 6029–6036 RSC.
- G. N. Grover, J. Lam, T. H. Nguyen, T. Segura and H. D. Maynard, Biomacromolecules, 2012, 13, 3013–3017 CrossRef CAS PubMed.
- T. Loth, R. Hötzel, C. Kascholke, U. Anderegg, M. Schulz-Siegmund and M. C. Hacker, Biomacromolecules, 2014, 15, 2104–2118 CrossRef CAS PubMed.
- S. Kaga, S. Yapar, E. M. Gecici and R. Sanyal, Macromolecules, 2015, 48, 5106–5115 CrossRef CAS.
- C. Echalier, S. Jebors, G. Laconde, L. Brunel, P. Verdié, L. Causse, A. Bethry, B. Legrand, H. Van Den Berghe, X. Garric, D. Noël, J. Martinez, A. Mehdi and G. Subra, Mater. Today, 2017, 20, 59–66 CrossRef CAS.
- T. Montheil, C. Echalier, J. Martinez, G. Subra and A. Mehdi, J. Mater. Chem. B, 2018, 6, 3434–3448 RSC.
- C. Echalier, R. Levato, M. A. Mateos-Timoneda, O. Castaño, S. Déjean, X. Garric, C. Pinese, D. Noël, E. Engel, J. Martinez, A. Mehdi and G. Subra, RSC Adv., 2017, 7, 12231–12235 RSC.
- L. Valot, M. Maumus, T. Montheil, J. Martinez, D. Noël, A. Mehdi and G. Subra, ChemPlusChem, 2019, 84, 1720–1729 CrossRef CAS PubMed.
- C. Vinatier, D. Magne, A. Moreau, O. Gauthier, O. Malard, C. Vignes-Colombeix, G. Daculsi, P. Weiss and J. Guicheux, J. Biomed. Mater. Res., Part A, 2007, 80, 66–74 CrossRef CAS PubMed.
- N. Buchtová, G. Réthoré, C. Boyer, J. Guicheux, F. Rambaud, K. Vallé, P. Belleville, C. Sanchez, O. Chauvet, P. Weiss and J. Le Bideau, J. Mater. Sci.: Mater. Med., 2013, 24, 1875–1884 CrossRef PubMed.
- T. Montheil, M. Maumus, L. Valot, A. Lebrun, J. Martinez, M. Amblard, D. Noël, A. Mehdi and G. Subra, ACS Omega, 2020, 5, 2640–2647 CrossRef CAS PubMed.
- C. M. Kirschner and K. S. Anseth, Acta Mater., 2013, 61, 931–944 CrossRef CAS PubMed.
- S. P. Zustiak and J. B. Leach, Biotechnol. Bioeng., 2011, 108, 197–206 CrossRef CAS PubMed.
- S. P. Zustiak, H. Boukari and J. B. Leach, Soft Matter, 2010, 6, 3609 RSC.
- M. Betz, J. Hörmansperger, T. Fuchs and U. Kulozik, Soft Matter, 2012, 8, 2477 RSC.
- C. Tourne-Peteilh, B. Robin, M. Lions, J. Martinez, A. Mehdi, G. Subra and J.-M. Devoisselle, Chem. Commun., 2019, 55, 13112 RSC.
- T. Eichner and S. E. Radford, Mol. Cell, 2011, 43, 8–18 CrossRef CAS PubMed.
- T. Nishikawa, K. Akiyoshi and J. Sunamoto, J. Am. Chem. Soc., 1996, 118, 6110–6115 CrossRef CAS.
- J. Chen, X. Ma, Q. Dong, D. Song, D. Hargrove, S. R. Vora, A. W. K. Ma, X. Lu and Y. Lei, RSC Adv., 2016, 6, 56183–56192 RSC.
- S.-R. Hsieh, P. Reddy, C.-J. Chang, A. Kumar, W.-C. Wu and H.-Y. Lin, Polymers, 2016, 8, 238 CrossRef PubMed.
- E. K. O'Shea, R. Rutkowski and P. S. Kim, Cell, 1992, 68, 699–708 CrossRef.
- S. B. Kennedy, K. Littrell, P. Thiyagarajan, D. A. Tirrell and T. P. Russell, Macromolecules, 2005, 38, 7470–7475 CrossRef CAS.
- W. Wang, Int. J. Pharm., 2000, 203, 1–60 CrossRef CAS PubMed.
- C. M. Deber and S.-C. Li, Biopolymers, 1995, 37, 295–318 CrossRef CAS PubMed.
- K. Rajagopal, J. Wood, B. Tran, T. W. Patapoff and T. Nivaggioli, J. Pharm. Sci., 2013, 102, 2655–2666 CrossRef CAS PubMed.
- J. Siepmann, K. Podual, M. Sriwongjanya, N. A. Peppas and R. Bodmeier, J. Pharm. Sci., 1999, 88, 65–72 CrossRef CAS PubMed.
- P. Costa and J. M. Sousa Lobo, Eur. J. Pharm. Sci., 2001, 13, 123–133 CrossRef CAS PubMed.
- D. Y. Arifin, L. Y. Lee and C.-H. Wang, Adv. Drug Delivery Rev., 2006, 58, 1274–1325 CrossRef CAS PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- B. V. Derjaguin, V. M. Muller and Y. P. Toporov, J. Colloid Interface Sci., 1975, 53, 314–326 CrossRef CAS.
- M. Paul, V. Vieillard, E. Jaccoulet and A. Astier, Int. J. Pharm., 2012, 436, 282–290 CrossRef CAS PubMed.
- T. Montheil, C. Echalier, J. Martinez, A. Mehdi and G. Subra, in Peptide and Protein Engineering: From Concepts to Biotechnological Applications, ed. O. Iranzo and A. C. Roque, Springer US, New York, NY, 2020, pp. 69–92 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03547a |
| This journal is © The Royal Society of Chemistry 2021 |