
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe discovery of new phloroglucinol glycosides from Agrimonia pilosa and the mechanism of oxidative dearomatization of the methyl-substituted phloroglucinol derivatives†
Jia Zhang,
Ya-Nan Yang ,
Jian-Shuang Jiang,
Zi-Ming Feng,
Xiang Yuan,
Xu Zhang and
Pei-Cheng Zhang*
,
Jian-Shuang Jiang,
Zi-Ming Feng,
Xiang Yuan,
Xu Zhang and
Pei-Cheng Zhang*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing 100050, People's Republic of China. E-mail: pczhang@imm.ac.cn
First published on 24th June 2021
Abstract
Six methyl-substituted phloroglucinol glycosides (1–6) were isolated from Agrimonia pilosa, including four new compounds (1–3, 6). The aglycones (1a–4a) of 1–4 and their corresponding oxidized products (1c–4c) were also obtained from A. pilosa. The structures were determined by a series of spectroscopic analyses and chiral separation. Notably, the structures of aglycones 1a–4a were unstable and prone to oxidation spontaneously, to yield the dearomatized structures 1c–4c. The mechanism of oxidative dearomatization was disclosed as a free-radical chain reaction with 3O2 by the techniques of HPLC-HR-MS2, EPR spectra and DFT-calculation, and hydroperoxide was defined as the intermediate.
Introduction
Agrimonia pilosa is a plant in the Rosaceae family that is mainly distributed throughout East Asia. As a traditional Chinese medicine, the herbs of A. pilosa have been used to treat malaria, bleeding, dysentery and debility for many years.1 The chemical investigation of Agrimonia pilosa suggested the presence of phloroglucinols in various forms, such as the dimer agrimophol, with significant antituberculosis activity.2 During the phytochemical research on A. pilosa, six dimethyl-substituted phloroglucinol glycosides (1–6), four of their aglycones (1a–4a), and four pairs of dearomatized products (1c–4c) were isolated (Fig. 1). Compounds 1–3 and 6 are new phloroglucinol glycosides. However, there is an inescapable problem in which aglycones 1a–4a are readily oxidized by air to form the dearomatized products 1c–4c spontaneously.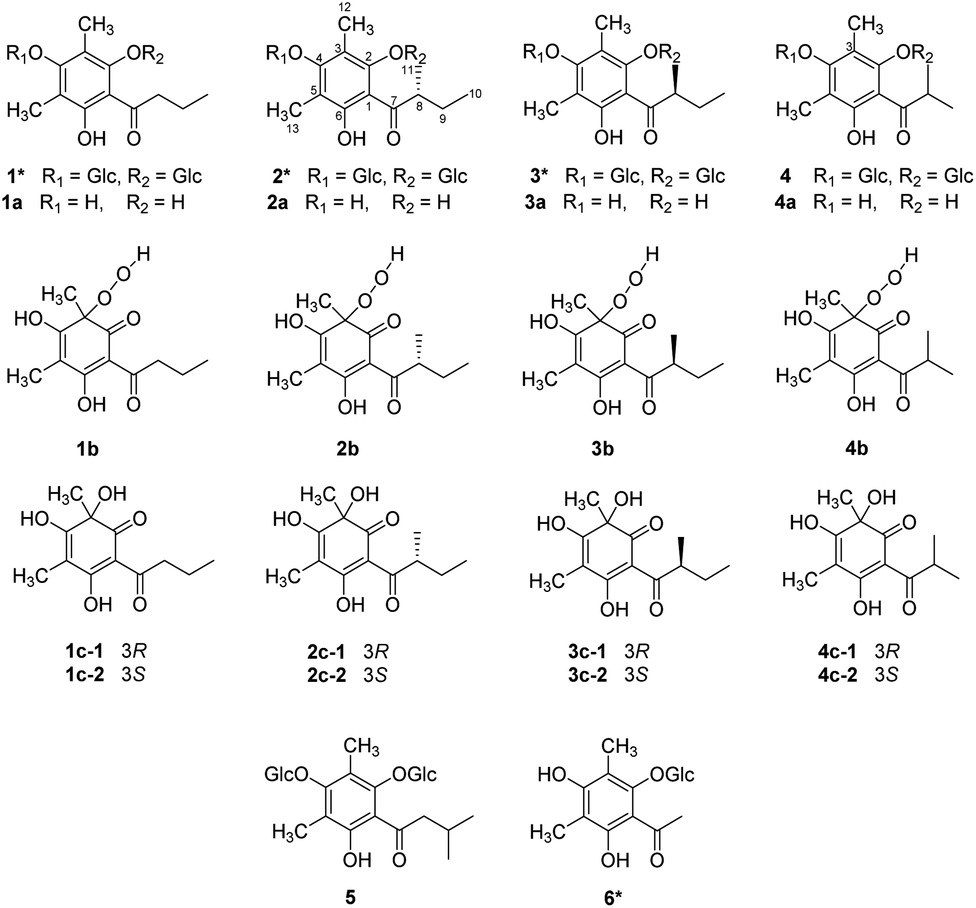 | ||
| Fig. 1 Chemical structures of compounds 1–6. | ||
Reference surveys revealed that the natural dearomatized phloroglucinol derivatives display remarkable pharmacological activities. α-Acids, isolated from hop (Humulus lupulus L.), showed antimetabolic activities,3 and hydroxysafflor yellow A (HSYA), isolated from Carthamus tinctorius,4 exhibited cardiovascular effects.5 Over the past few years, the dearomatization of isopentenyl- and C-glucosyl-substituted phloroglucinols has attracted much attentions from synthetic chemists. Most dearomatizations were proceed by oxidation under alkaline environment. The oxidants include lead(II) acetate trihydrate,6 [bis(trifluoroacetoxy)iodo]benzene,7 tert-butylhydroperoxide,8 and oxygen.9 Meanwhile, sodium hydride,10 sodium bicarbonate,7 sodium hydroxide,8 and pyridine9 were utilized as alkaline. In 2019, our group employed ammonium chloride and ammonia buffer salt as alkaline to achieve the oxidative dearomatization of di-C-glucosyl-substituted phloroglucinols.11 While the mechanism of oxidative dearomatization on phloroglucinol by oxygen were barely studied, the phenomenon of auto oxidative dearomatization of 1a–4a inspired us to explore the in-depth mechanism. Consequently, a free-radical chain mechanism was determined by the techniques of HPLC-HR-MS2, EPR (electroparamagnetic resonance) spectra, and DFT calculations.
Results and discussion
The structures and absolute configurations of 2 and 3 were elucidated by extensive spectroscopic data (Tables S1 and S2†) and a chiral separation method. Of note is that 2 and 3 are a pair of epimers with the opposite chirality at C-8, sharing the same molecular formula of C25H38O14 by HR-ESI-MS. The 1H NMR spectrum of 2 exhibited two aliphatic methyl groups at δH 0.97 (3H, t, J = 7.5 Hz) and 1.00 (3H, d, J = 7.5 Hz), two aromatic methyl groups at δH 2.20 and 2.32 (each 3H, s), one methylene group at δH 1.42 and 1.91 (each 1H, m), one methine group at δH 3.74 (1H, overlapped), and two anomeric protons of the β-glucopyranosyl moiety at δH 4.43 and 4.67 (each 1H, J = 7.5 Hz). The 13C NMR spectrum of 2 displayed twenty-five carbon resonances, including the characteristic signals of two glucopyranosyl moieties and one phloroglucinol.The 1H–1H COSY correlations between H3-11 and H-8, H-8 and H2-9, and H2-9 and H3-10 implied the presence of the H3-11–H-8–H2-9–H3-10 proton spin system, corresponding to an α-methylbutyryl group. This was further verified by the HMBC correlations from H-8 to C-7/C-9/C-10/C-11, from H3-10 to C-8/C-9, and from H3-11 to C-7/C-8/C-9. The HMBC correlations from H-1′ to C-2 and from H-1′′ to C-4 placed two glucopyranosyl moieties at C-2 and C-4. In addition, HMBC correlations from H3-12 to C-2/C-3/C-4 and from H3-13 to C-4/C-5/C-6, suggested two aromatic methyl groups were located at C-3 and C-5, respectively (Fig. 2A). The configuration of two glucopyranosyl moieties were determined as β-D based on the 3J1′,2′, 3J1′′,2′′ values (7.5 Hz) and the retention time (20.39 min) by GC analyses (Fig. S2 and S3†). Hence, the structure of 2 was deduced as 3,5-dimethyl-α-methylbutyrylphloroglucinol-2,4-O-β-D-diglucopyranoside.
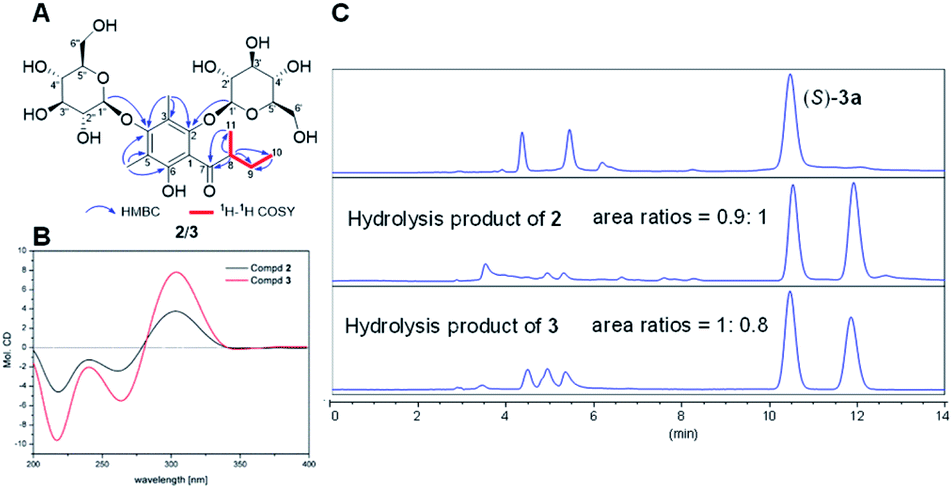 | ||
Fig. 2 (A) Key HMBC and COSY correlations of 2 and 3; (B) experimental ECD spectra of 2 and 3; (C) HPLC-DAD spectra of (S)-3a, hydrolysis products of 2 and 3 (HPLC condition: CHIRALPAK AD-H, 250 × 4.6 mm, n-hexane: IPA = 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, T = 40 °C, v = 1 mL min−1, λ = 280 nm). :10, T = 40 °C, v = 1 mL min−1, λ = 280 nm). | ||
The 1D and 2D NMR data of 3 were in good agreement with those of 2, except for the difference of the α-methylbutyryl group signals. Compared with 1H NMR spectrum of 2, the chemical shifts of H-9a, H-9b and H3-10 of 3 were shifted upfield by 0.38, 0.12, and 0.23 ppm, while the chemical shift of H3-11 of 3 was shifted downfield by 0.13 ppm. Detailed analysis of the NMR data revealed that the planar structure of 3 was identical with 2 but the opposite chirality at C-8, resulting in a pair of epimers.
Initially, the ECD spectra were expected to determine the absolute configuration of C-8, but the experimental ECD spectra of 2 and 3 were very similar, suggesting the ECD cotton effects were mainly affected by glucopyranosyl moieties (Fig. 2B). To determine the absolute configuration of C-8, (S)-aglycone 3a was synthesized from phloroglucinol through a series of chemical reactions (Section S3.6†), and compounds 2 and 3 were hydrolyzed by 2 M HCl at 60 °C (Section S3.4†). The products of the acidic hydrolysis were analyzed by a normal-phase chiral chromatographic column, and the unequal enantiomers 2a and 3a were monitored (Fig. 2C). This is due to the fact that carbonyl tautomerism occurred in the acidic hydrolysis of 2 and 3. However, the area ratios of the two peaks 0.9:1 in 2 and 1:0.8 in 3 suggested that the absolute configurations of C-8 in 2 and 3 were R and S, respectively.
The structures of 1 and 6 were also identified as new compounds (Section S1†), 4 and 5 were elucidated as kunzeaphlogin F and D, respectively.12
Apart from the phloroglucinol glucosides, a series of dimethyl-substituted phloroglucinol derivatives 1a–4a and 1c–4c were also isolated (Section S2†). During the separation of 1a–4a, it's interesting to find that when the mixture of 1a–4a was stored at methanol under room temperature, new peaks 1b–4b, 1c and 4c were monitored by HPLC analyses after eight days (Fig. 3A). As time went on, peaks 1b–4b were disappeared and peaks 1c–4c were increased a lot after 6 months (Fig. 3A).
 | ||
| Fig. 3 (A) HPLC-DAD spectra of 1a–4a; (B) HPLC-DAD spectra of 0.05 M (S)-aglycone 3a. (HPLC condition: 0–16 min, 35–70% MeCN–H2O, T = 40 °C, v = 1 mL min−1, λ = 254 nm). | ||
The structures of 1c–4c have been elucidated as the dearomatized products of 1a–4a (Fig. 1), but 1b–4b can't be isolated purely due to their poor stability. Therefore, high performance liquid chromatography (HPLC)-high resolution tandem mass spectrometry (HR-MS2) was applied for elucidating the structures of 1b–4b (Section S4†). For example, 4b were diagnosed as hydroperoxide by the high resolution quasi-molecular ions m/z 257.1018 and main fragment ions m/z 224.1041, 205.0863, 197.0809, 181.0496 (Scheme 1). Subsequently, 0.05 M synthesized (S)-aglycone 3a was stored at oxygen-saturated methanol under room temperature and monitored by HPLC every two days. The results indicated that 3a first rapidly transformed into 3b and then 3b transformed into 3c gradually (Fig. 3B). Those findings support the conclusion that dimethyl-substituted phloroglucinol derivatives can transform into the dearomatized structures spontaneously, and the hydroperoxides acted as the intermediate.
 | ||
| Scheme 1 The main fragmentation pathway of 4b. | ||
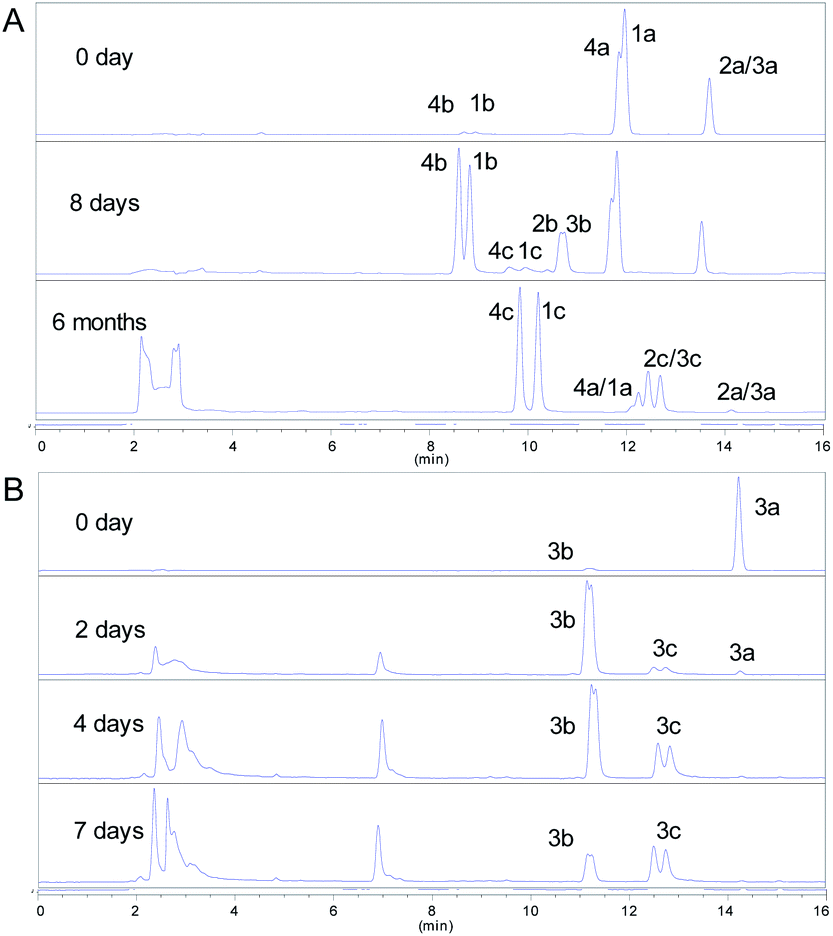
Some mechanisms of oxidative dearomatization, such as Diels–Alder reactions, [2 + 2] cycloaddition, or free-radical reaction, have been introduced before,13 and singlet oxygen was often applied as the oxygen donor. But when we add TEMP (2,2,6,6-tetramethylpiperidine) to the reaction system of 4a for detecting singlet oxygen, the EPR spectra didn't showed the signal of the adduct of TEMP and singlet oxygen (Fig. S6†), indicating oxygen participating in the reaction was the triple state (3O2) instead of singlet state (1O2). To further explore the mechanism, DMPO (5,5-dimethyl-1-pyrroline N-oxide) was employed as a spin trapper and monitored by HPLC, the oxidative dearomatization was prevented (Fig. S7 and S8†). Obviously, the oxidative dearomatization of 4a involves a radical intermediate. Furthermore, the characteristic signal of the adduct of alkyl radical (R˙) and DMPO in the EPR spectra (Fig. 4) suggested that the oxidative dearomatization of 4a is a free-radical reaction. Based on the above HPLC and EPR results, the free-radical mechanism for 4a were proposed as Scheme 2. Firstly, 4a lose a hydrogen atom and give the radical a1. Because a1 and a2 were a pair of resonance structures, a1 can transform into a2. Then, radical a2 react with oxygen to give the peroxy radical a3, followed by the hydrogen atom transfer from 4a, giving the intermediate 4b. In the next step, the hydroperoxide 4b transform into the oxygen free-radical a4 and hydroxyl radical (˙OH) through the homolysis of peroxy bond. Finally, the oxygen radical a4 captures hydrogen atom of 4a to gain the dearomatized product 4c. As the chain carrier, radical a2 was considered as the alkyl radical trapped by DMPO.
 | ||
| Fig. 4 Electroparamagnetic resonance (EPR) spectra of 4a (room temperature in oxygen-saturated acetonitrile). The concentrations of DMPO and 4a were 500 and 50 mmol L−1, respectively. | ||
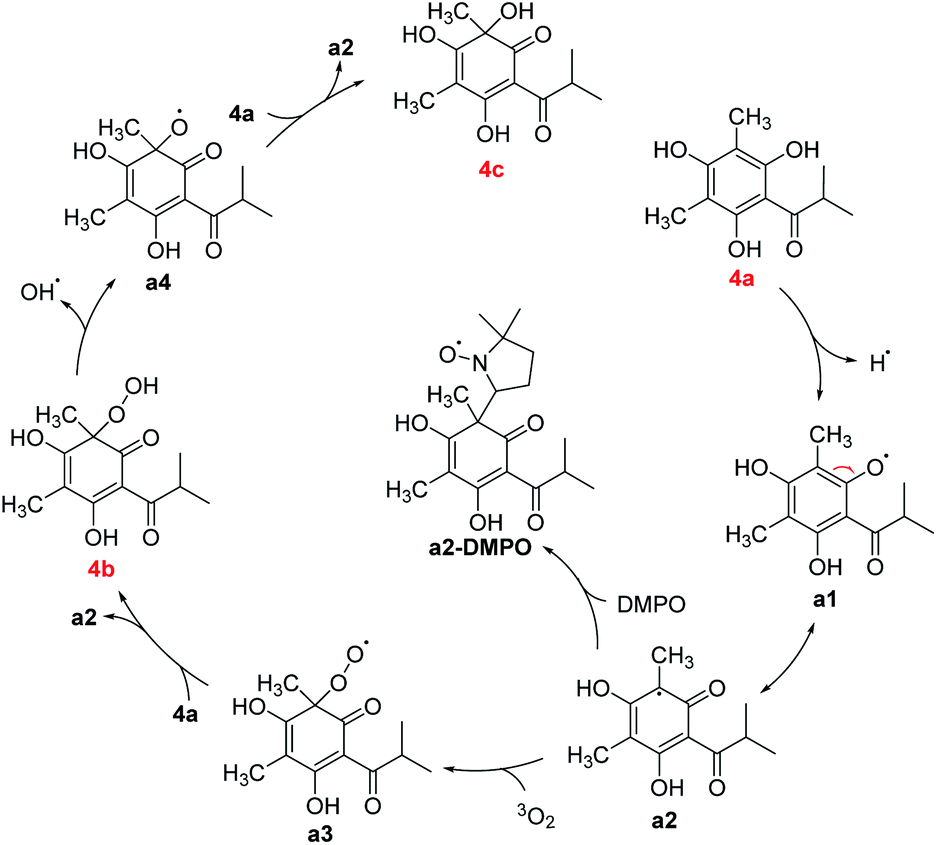 | ||
| Scheme 2 Proposed mechanism of oxidative dearomatization of 4a. | ||
Density functional theory (DFT) research was performed to better understand the mechanisms, the Gibbs free energy profile was established as shown in Fig. 5. The transition states of the reactions of a2 and 3O2, a3 and 4a, a4 and 4a were defined as TS-1–3, respectively. Firstly, the Gibbs free energy barrier for the electrophilic addition of radical a2 to 3O2 was 17.7 kcal mol −1, and the relative energy of a3 was 6.7 kcal mol−1. Likewise, when the radical peroxy radical captures the hydrogen atom of 4a, exhibiting a similar relative energy barrier 17.0 kcal mol −1, but the relative energy of product was −26.6 kcal mol −1, leading the production of the intermediate 4b. In fact, the rate of generating 4b was faster than 4c, but it still needs two days to achieve, which accord with the calculated energy barriers (17.7 and 17.0 kcal mol −1). There is no transition state during the homolysis of O–O bond, proved by the potential energy surface scanning (Fig. S12†). The BDE of O–O bond was calculated as 45.3 kcal mol −1, indicating the cleavage of O–O bond was the rate-limiting step, which was also correspond to the slow generation of 4c from 4b. Besides, the process from a4 to 4c was barrier-less, the relative energy of TS-3 and the product 4c were −10.2 and −47.3 kcal mol −1. Generally, the auto oxidative dearomatization pathway of dimethyl-substituted phloroglucinol derivatives were kinetically and thermodynamically accomplished.
 | ||
| Fig. 5 Gibbs free energy profiles for the free-radical chain reaction of 4a and the structures of transition states TS-1–3. | ||
When the 2-hydroxyl of the phloroglucinol derivatives get glycosylation or esterification, such as 1–7 (Fig. 6), the structures couldn't transform into the hydroperoxides or dearomatized structures. Structurally, two free phenolic hydroxyls at C-2 and C-6 were essential for the auto oxidative dearomatization, one hydroxyl formed a strong hydrogen bond with the carbonyl, and another hydroxyl was regarded as the hydrogen atom donor attributed to the radicals.
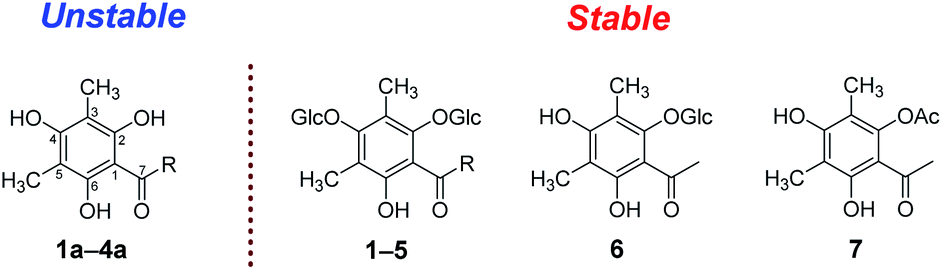 | ||
| Fig. 6 Stability of the substituted phloroglucinol derivatives (R = n-propyl, isopropyl, isobutyl). | ||
Conclusions
As a research hot spot, studies of phloroglucinol derivatives have been ongoing for many years. In present study, fortunately, six phloroglucinol glucosides (1–6), together with four aglycones (1a–4a) and four pairs of dearomatized products (1c–4c) were isolated from A. pilosa. Moreover, this is the first report on the instability and structural variability of dimethyl-substituted phloroglucinol derivatives. We found that dimethyl-substituted phloroglucinol derivatives were readily oxidized to their dearomatized structures, and the intermediates were determined as hydroperoxides by HPLC-HR-MS2. With the aid of EPR and DFT calculations, the mechanism was clarified as a free-radical chain reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was financially supported by the Chinese Academy of Medical Sciences (CAMS) Initiative for Innovative Medicine (No. 2019-I2M-1-005).Notes and references
- (a) H. Kato, W. Li, M. Koike, Y. H. Wang and K. Koike, Phenolic glycosides from Agrimonia pilosa, Phytochemistry, 2010, 71, 1925–1929 CrossRef CAS PubMed; (b) H. W. Kim, J. Park, K. B. Kang, T. B. Kim, W. K. Oh, J. Kim and S. H. Sung, Acylphloroglucinolated catechin and phenylethyl isocoumarin derivatives from Agrimonia pilosa, J. Nat. Prod., 2016, 79, 2376–2383 CrossRef CAS PubMed; (c) D. H. Nguyen, U. M. Seo, B. T. Zhao, D. D. Le, S. H. Seong, J. S. Choi, B. S. Min and M. H. Woo, Ellagitannin and flavonoid constituents from Agrimonia pilosa Ledeb. with their protein tyrosine phosphatase and acetylcholinesterase inhibitory activities, Bioorg. Chem., 2017, 72, 293–300 CrossRef CAS PubMed.
- (a) N. Zhao, M. N. Sun, K. Burns-Huang, X. J. Jiang, Y. Ling, C. Darby, S. Ehrt, G. Liu and C. Nathan, Identification of Rv3852 as an agrimophol-binding protein in Mycobacterium tuberculosis, PLoS One, 2015, 10, e0126211 CrossRef PubMed; (b) J. Wu, R. Mu, M. N. Sun, N. Zhao, M. M. Pan, H. S. Li, Y. Dong, Z. G. Sun, J. Bai, M. W. Hu, C. F. Nathan, B. Javid and G. Liu, Derivatives of natural product agrimophol as disruptors of intrabacterial pH homeostasis in Mycobacterium tuberculosis, ACS Infect. Dis., 2019, 5, 1087–1104 CrossRef CAS PubMed.
- M. Van Cleemput, K. Cattoor, K. De Bosscher, G. Haegeman, D. De Keukeleire and A. Heyerick, Hop (Humulus lupulus)-derived bitter acids as multipotent bioactive compounds, J. Nat. Prod., 2009, 72, 1220–1230 CrossRef CAS PubMed.
- Z. M. Feng, J. He, J. S. Jiang, Z. Chen, Y. N. Yang and P. C. Zhang, NMR solution structure study of the representative component hydroxysafflor yellow A and other quinochalcone C-glycosides from Carthamus tinctorius, J. Nat. Prod., 2013, 76, 270–274 CrossRef CAS PubMed.
- (a) X. Bai, W. X. Wang, R. J. Fu, S. J. Yue, H. Gao, Y. Y. Chen and Y. P. Tang, Therapeutic potential of hydroxysafflor yellow A on cardio-cerebrovascular diseases, Front. Pharmacol., 2020, 11, 01265 CrossRef CAS PubMed; (b) Y. Sun, D. P. Xu, Z. Qin, P. Y. Wang, B. H. Hu, J. G. Yu, Y. Zhao, B. Cai, Y. L. Chen, M. Lu, J. G. Liu and X. Liu, Protective cerebrovascular effects of hydroxysafflor yellow A (HSYA) on ischemic stroke, Eur. J. Pharmacol., 2018, 818, 604–609 CrossRef CAS.
- M. R. Cann, A. M. Davis and P. V. R. Shannon, The synthesis of some novel deoxyhumulone analogues. Observations on the air-oxidation of 2′,4′,6′-trihydroxy-3′-isopentyl-5′-(3-methylbut-2-enyl)isovalerophenone and its corresponding humulone derivatives, J. Chem. Soc., Perkin Trans. 1, 1984, 1413–1421 RSC.
- T. Hayashi, K. Ohmori and K. Suzuki, Synthetic study on carthamin: problem and solution for oxidative dearomatization approach to quinol C-glycoside, Synlett, 2016, 27, 2345–2351 CrossRef CAS.
- E. Collins and P. V. R. Shannon, Dimethylallylation products of phloroacetophenone; a convenient one-stage synthesis of deoxyhumulones, J. Chem. Soc., Perkin Trans. 1, 1973, 419–424 RSC.
- (a) S. Sato, T. Nojiri and J. I. Onodera, Studies on the synthesis of safflomin-A, a yellow pigment in safflower petals: oxidation of 3-C-β-D-glucopyranosyl-5-methylphloroacetophenone, Carbohydr. Res., 2005, 340, 389–393 CrossRef CAS PubMed; (b) T. Suzuki, M. Ishida, T. Kumazawa and S. Sato, Oxidation of 3,5-di-C-(per-O-acetylglucopyranosyl) phloroacetophenone in the synthesis of hydroxysafflor yellow A, Carbohydr. Res., 2017, 448, 52–56 CrossRef CAS PubMed.
- (a) S. Sato, T. Kumazawa, H. Watanabe, K. Takayanagi, S. Matsuba, J. I. Onodera, H. Obara and K. Furuhata, Synthesis of carthamin acetate, the red pigment in safflower petals, Chem. Lett., 2001, 30, 1318–1319 CrossRef; (b) S. Sato, H. Obara, T. Kumazawa, J. I. Onodera and K. Furuhata, Synthesis of (+),(−)-model compounds and absolute configuration of carthamin; a red pigment in the flower petals of safflower, Chem. Lett., 1996, 25, 833–834 CrossRef; (c) H. Obara, S. Namai and Y. Machida, Synthesis of 2-[[3-hydroxy-S-[3-(4-hydroxyphenyl)-1-oxo-2-propenyl]-3-methyl-2,4,6-trioxocyclohex-1-yl]methylene]-4-hydroxy-6-[3-(4-hydroxyphenyl)-1-oxo-2-prophenyl]-4-methyl-1,3,5-trioxocyclohexane, an analog of carthamin, Chem. Lett., 1986, 15, 495–496 CrossRef.
- W. Gao, Z. Chen, Y. N. Yang, J. S. Jiang, Z. M. Feng, X. Zhang, X. Yuan and P. C. Zhang, Base-catalyzed oxidative dearomatization of multisubstituted phloroglucinols: an easy access to C-glucosyl 3,5,6-trihydroxycyclohexa-2,4-dienone derivatives, Carbohydr. Res., 2019, 484, 107756 CrossRef CAS PubMed.
- N. Kasajima, H. Ito, T. Hatano and T. Yoshida, Phloroglucinol diglycosides accompanying hydrolyzable tannins from Kunzea ambigua, Phytochemistry, 2008, 69, 3080–3086 CrossRef CAS PubMed.
- (a) A. A. Ghogare and A. Greer, Using singlet oxygen to synthesize natural products and drugs, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS; (b) Q. J. Song, T. Niu and H. J. Wang, Theoretical study of the reaction of 2,4-dichlorophenol with 1O2, J. Mol. Struct.: THEOCHEM, 2008, 861, 27–32 CrossRef CAS; (c) S. Barradas, G. Hernandez-Torres, A. Urbano and M. C. Carreno, Total synthesis of natural p-quinol cochinchinenone, Org. Lett., 2012, 14, 5952–5955 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structural elucidations of 1 and 6, general experimental procedure, HPLC-MS2 analysis, DFT calculation details, UV, IR, NMR, and HRESIMS spectra. See DOI: 10.1039/d1ra03588f |
| This journal is © The Royal Society of Chemistry 2021 |