
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCationic polymer-grafted graphene oxide/CNT cathode-coating material for lithium–sulfur batteries†
Daun Jeonga,
Dong Gi Honga,
Jinsol Yooka,
Chan Yeong Koonga,
Soohyun Kimb,
Ki-Hyun Kimb,
Kwonnam Sohnb and
Jong-Chan Lee *a
*a
aSchool of Chemical and Biological Engineering, Institute of Chemical Processes, Seoul National University, Seoul 08826, Republic of Korea. E-mail: jongchan@snu.ac.kr
bLG Energy Solution, E5 Block, LG Science Park, 30, Magokjungang 10-ro, Gangseo-gu, Seoul 07796, Republic of Korea
First published on 21st July 2021
Abstract
A cathode-coating material composed of cationic polymer-grafted graphene oxide (CPGO) and carbon nanotube (CNT) was prepared, where the CPGO was synthesized by grafting quaternized 2-(dimethylamino)ethyl methacrylate (QDMAEMA) onto graphene oxide (GO) via atom transfer radical polymerization (ATRP). GO has good compatibility with carbon black, the main component of the cathode in lithium–sulfur (Li–S) batteries. Here, the cationic polymer having the QDMAEMA unit was intentionally grafted onto GO to decrease the shuttle effect by increasing the chemical adsorption of polysulfide (PS). In addition, when CNT was mixed with CPGO, the compatibility with carbon black was found to be further increased. The lithium–sulfur (Li–S) battery with a sulfur-deposited Super P® carbon black (S/C) cathode coated with a mixture of CPGO and CNT was found to have much improved cell performance compared to those coated without any coating material, with only CPGO, with the mixture of GO and CNT, and with the mixture of PQDMAEMA and CNT. For example, the Li–S battery with the cathode coated using the mixture of CPGO and CNT retained a discharge capacity of 744 mA h g−1 after 50 cycles at 0.2C-rate, while those of the Li–S batteries with bare S/C and CPGO-S/C cathodes were found to be much smaller, i.e., 488 mA h g−1 and 641 mA h g−1, respectively, under the same conditions. Therefore, the mixture of CPGO with CNT as the cathode-coating material showed a synergetic effect to enhance the cell performance of the Li–S battery system.
Introduction
With the advent of the era of electric vehicles, the development of next-generation lithium battery systems with high energy density for realizing long driving distances is the most highlighted issue.1–4 Since the traditional graphite anode-transition metal oxide cathode-based lithium-ion batteries have certain disadvantages for use in electric vehicles, including their low energy density (maximum energy density of 260 W h kg−1), which limits the driving distance, the demand for next-generation batteries with high energy density continues to grow.5,6 Among the various next-generation lithium battery systems, lithium–sulfur (Li–S) batteries are regarded as one of the most promising and practical candidates since they have a very high energy density (2600 W h kg−1) and sulfur is a naturally abundant resource.7Li–S batteries are mainly composed of a lithium metal anode (3860 mA h g−1, −3.04 V vs. standard hydrogen electrode) and sulfur-based cathode (1675 mA h g−1). This battery form has a unique multi-step discharge mechanism accompanying the phase transition of sulfur from a solid (S8) to solution (Li2Sx (x = 4–8)), and then to solid (Li2S) again.8,9 Since polysulfides (PSs) having the chemical structure of Li2Sx (x = 4–8) are soluble in liquid electrolyte, PSs formed during this multi-step phase transition process are dissolved into the liquid electrolyte and pass through the separator to ultimately reach the anode in an irreversible process.10 The PSs that reach the lithium metal anode are partially reduced to Li2S2 or Li2S, and then deposited on the anode surface, leading to the development of passivation layers. Since the deposited PS destabilizes the solid-electrolyte interface (SEI) layer, the discharge capacity and coulombic efficiency of Li–S batteries decay and the lithium dendrite growth is accelerated, which is known as the “shuttle effect”.10,11
To minimize the shuttle effect, various strategies have been studied, including the introduction of carbon host materials in the cathode,12 manipulation of the electrode architecture,13–16 development of organosulfur polymers,17–19 and the introduction of interlayers and separator coating materials.20–23 In particular, the introduction of a carbon host material, such as porous carbon, carbon nanotubes (CNTs), graphene, and carbon derivatives, has been most widely studied to solve the shuttle problem and much research has been performed in this area since Nazar and co-workers first utilized mesoporous carbon CMK-3 as the host material for molten sulfur.12,24–27 In addition, the carbon host material can also alleviate the other critical drawbacks of sulfur, such as its high intrinsic resistance (5 × 10−30 S cm−1) and volume expansion during the charge–discharge process.24,25 However, the capacity decay caused by the shuttle effect cannot be fully resolved by the carbon host material because of the poor affinity between the hydrophobic carbon and polar PS having an ionic characteristic.28,29
Herein, a cationic polymer-grafted graphene oxide (CPGO) was prepared and used as a coating material for the cathode in Li–S batteries to further mitigate the shuttle effect, wherein the CPGO was synthesized by polymerizing quaternized 2-(dimethylamino)ethyl methacrylate (QDMAEMA) through atom transfer radical polymerization (ATRP) using a GO-containing initiation moiety as a macroinitiator. To the best of our knowledge, this is the first report to use such a GO grafted by a cationic polymer as the coating material for Li–S battery systems. We intentionally grafted the cationic polymer on GO because polymers with cationic moieties introduced into Li–S batteries as the binder and coating materials in the cathode have been reported to alleviate the shuttle effect.30–33 We further found that the mixture of CPGO with CNT had a much improved coating ability on the cathode based on hydrophobic sulfur and Super P® carbon, resulting in a better battery performance of a Li–S battery fabricated by the cathode coated with the mixture over other Li–S batteries fabricated by the cathode coated with CPGO, a GO/CNT mixture, and a cationic polymer/CNT mixture.
Experimental
Materials
Graphene oxide (GO) was provided by Promico CO., Ltd (Korea). Carbon nanotubes (CNTs) were provided by LG Chem. (Korea). Sulfur, 2-(dimethylamino)ethyl methacrylate (DMAEMA), iodoethane, hydroquinone, copper(I) bromide, N,N,N′,N′′,N′′-pentamethyldiethylenetriamine, poly(acrylic acid) (Mv = 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), poly(vinyl alcohol) (Mw = 9000–10000, 80% hydrolyzed) were purchased from Aldrich and used as received. 2,2′-Azobis(isobutyronitrile) (AIBN) was purchased from Aldrich and recrystallized in ethanol before use. Carbon black, Super P® conductive, and 2-bromo-2-methylpropionyl bromide were purchased from Alfa Aesar and used as received. All the other reagents and solvents were obtained from reliable commercial sources and used as received.
000), poly(vinyl alcohol) (Mw = 9000–10000, 80% hydrolyzed) were purchased from Aldrich and used as received. 2,2′-Azobis(isobutyronitrile) (AIBN) was purchased from Aldrich and recrystallized in ethanol before use. Carbon black, Super P® conductive, and 2-bromo-2-methylpropionyl bromide were purchased from Alfa Aesar and used as received. All the other reagents and solvents were obtained from reliable commercial sources and used as received.
Synthesis of graphene oxide having bromo-initiation sites (GO-Br)
The graphene oxide macroinitiator having bromo-initiation sites was synthesized by the SN2 reaction of GO with 2-bromo-2-methylpropionyl bromide. GO (0.50 g) was dispersed in 100 mL of dimethylacetamide (DMAc) by sonication for 3 h and the resultant solution was added in to a 250 mL one-neck round-bottomed flask equipped with a magnetic stirring bar. Triethylamine (0.76 g, 7.5 mmol) and 2-bromo-2-methylpropionyl bromide (1.72 g, 7.5 mmol) were added to the solution and stirred for 24 h at room temperature under a nitrogen atmosphere. After the reaction, the crude product was filtered and washed with deionized water several times to remove the unreacted 2-bromo-2-methylpropionyl bromide and triethylamine. After being dried under vacuum conditions at 60 °C for 24 h, GO-Br was obtained.Synthesis of the quaternized 2-(dimethylamino)ethyl methacrylate (QDMAEMA)
DMAEMA (15.0 g, 0.095 mol), iodoethane (16.37 g, 0.105 mol), and hydroquinone (0.08 g, 0.73 mmol) were dissolved in 56 mL of acetonitrile and added to a 250 mL one-neck round-bottomed flask equipped with a magnetic stirring bar. After being stirred for 18 h at 45 °C, the crude product was precipitated in diethyl ether. After that, the crude product was filtered and washed with diethyl ether several times to remove unreacted DMAEMA and iodoethane. After being dried under vacuum conditions at room temperature for 24 h, a white powder was obtained with 90% yield. For convenience, quaternized 2-(dimethylamino)ethyl methacrylate is abbreviated as QDMAEMA. 1H NMR [400 MHz, [D6]DMSO, δ (ppm), TMS ref] of QDMAEMA: 4.52 (COO–CH2), 3.72 (CH2–CH2–O), 3.48 (CH2–CH2–N+(CH3)2), 1.91 (N+–CH2–CH3).Synthesis of poly(quaternized 2-(dimethylamino)ethyl methacrylate) (PQDMAEMA)
PQDMAEMA was synthesized by a free radical polymerization of QDMAEMA. QDMAEMA (2 g), and AIBN (0.02 g) were dissolved in 10 mL of acetone and the solution was deoxygenated three times by freeze–pump–thaw cycles. The resultant solution was heated at 80 °C for 16 h in an oil bath under nitrogen conditions. After the reaction, the crude product was precipitated in methylene chloride three times to remove any unreacted monomer. After filtering and being dried under vacuum at room temperature, PQDMAEMA was obtained as a powder in 70% yield. 1H NMR [400 MHz, [D6]DMSO, δ (ppm), TMS ref] of PQDMAEMA: 3.61 (COO–CH2), 3.24 (C–N+–(CH3)2(CH2CH3)), 1.35 (CH2–C–CH3).Synthesis of PQDMAEMA-grafted graphene oxide (CPGO)
The synthesized GO-Br (0.2 g) was dispersed in 15 mL of dimethylformamide (DMF) and added to a 100 mL Schlenk flask equipped with magnetic stirring bar. The solution was sonicated for 30 min and QDMAEMA (3.09 g, 9.72 mmol) was added to the solution. The solution was deoxygenated three times by freeze–pump–thaw cycles and copper(I) bromide (0.070 g, 0.486 mmol) was added and the mixture was deoxygenated again three times by freeze–pump–thaw cycles. N,N,N′,N′′,N′′-Pentamethyldiethylenetriamine (0.084 g, 0.486 mmol) was added to the mixture and deoxygenated once again by freeze–pump–thaw cycles and heated at 70 °C for 48 h in an oil bath under a nitrogen atmosphere (Scheme 1). After the reaction, the crude product was filtered and washed with methanol several times to remove copper catalyst and other reagents. After being dried under vacuum conditions at 60 °C for 24 h, a black powdery product was obtained. | ||
| Scheme 1 Synthesis of PQDMAEMA-grafted graphene oxide (CPGO). | ||
Preparation of CPGO-coated sulfur@Super P® carbon black (CPGO-S/C) powder for the cathode
Sulfur-deposited Super P® carbon black (S/C) powder was prepared by a melt diffusion of sulfur for Li–S batteries. Elemental sulfur and Super P® carbon black (7:3, w/w) were ground in a mortar and heated at 155 °C for 30 min under ambient conditions to obtain S/C powder. CPGO-coated S/C (CPGO-S/C) powder was prepared as follows. CPGO (3.0 mg) was dispersed in acetone (0.1 mg mL−1) by tip sonication for 45 min and the dispersed mixture was centrifuged at 3000 rpm for 5 min. After that, the supernatant was collected and mixed with S/C powder (0.3 g) dispersed in acetone solution (0.1 g mL−1) and stirred for 1 h. After filtration of the final mixture, the CPGO-S/C powder was obtained.
Preparation of CPGO/CNT, GO/CNT, and PQDMAEMA/CNT-coated sulfur@Super P® carbon black (CPGO/CNT-S/C, GO/CNT-S/C, and PQDMAEMA/CNT-S/C) powders for the cathode
CPGO- and CNT-coated S/C (CPGO/CNT-S/C) powder was prepared as follows. CPGO (1.5 mg) and CNT (1.5 mg) were individually dispersed in acetone (0.1 mg mL−1) by tip sonication for 45 min and the dispersed mixtures were centrifuged at 3000 rpm for 5 min. After that, the supernatants were collected and mixed with S/C powder (3.0 g) dispersed in acetone solution (0.1 g mL−1) and stirred for 1 h. After filtration of the final mixture, the CPGO/CNT-S/C powder was obtained. S/C powder coated with GO and CNT (GO/CNT-S/C) and that coated with PQDMAEMA and CNT (PQDMAEMA/CNT-S/C) were also prepared in the same way.Characterization
1H NMR spectra were recorded on an Ascend™ 400 spectrometer (400 MHz) using [D6]DMSO (Cambridge Isotope Laboratories) as a solvent at room temperature, with TMS as a reference. Fourier transform infrared (FT-IR) spectra were recorded in the absorption mode on a Nicolet 6700 spectrophotometer with a resolution of 8 cm−1 in the vibrational frequency range from 600 to 4000 cm−1. Thermal gravimetric analysis (TGA) was conducted using a TA Instruments TGA Q-500 instrument under a nitrogen (N2) atmosphere. The samples were heated to 100 °C, isothermally for 10 min, and then heated to 700 °C at a heating rate of 10 °C min−1. The molecular weights (Mn, Mw) were analyzed by gel permeation chromatography (GPC) apparatus equipped with a quaternary pump and three columns: KF-806L, KD-806M, and GF-7M. The refractive index (RI) detector was calibrated using polystyrene standards. Field-emission scanning electron microscopy (FE-SEM) was performed on a JEOL JSM-6700F with an accelerating voltage of 10 kV. Transmission electron microscopy (TEM) was performed on a LIBRA 120 with an accelerating voltage of 120 kV. TEM specimens were prepared by drop casting a 0.1 wt% dispersion in acetone on the carbon-coated grid. The X-ray diffraction (XRD) spectra were obtained using Rigaku Smart Lab (Cu Kα) spectrometers. The surface compositions of the GO and CPGO were investigated by X-ray photoelectron spectroscopy (XPS, Kratos Inc., AXIS-HSi) using Al (1486.69 eV) as the radiation source. Survey scans were conducted, followed by a high-resolution scan in the C 1s, O 1s, N 1s, and I 3d regions, with a range of 0–1500 eV at an angle of 30°.Cell fabrication and electrochemical characterization
Cyclic voltammetry (CV) tests were performed using a WBSC3000 battery cycler (WonATech) with a cutoff voltage of 1.8–2.8 V (vs. Li/Li+) at 25 °C. The cell was assembled using a lithium metal anode, Celgard 2320 separator, and bare S/C or CPGO/CNT-S/C cathode with 40 μL of liquid electrolyte containing 1.0 M lithium bis(trifluoromethane)sulfonimide (LiTFSI), and 0.1 M lithium nitrate salt in DOL:DME (1:1 vol%) in a 2032 coin cell. For the charge/discharge tests of the Li–S batteries, bare S/C, CPGO-S/C, CPGO/CNT-S/C, GO/CNT-S/C, and PQDMAEMA/CNT-S/C powders were used as the cathode active material and dispersed in water with Super P (5 wt%), poly(vinyl alcohol) (0.5 wt%), and poly(acrylic acid) (10 wt%) using a Thinky mixer. The dispersed slurry was coated on an aluminum current collector and dried at 50 °C for 24 h. The loading amount of sulfur was 2.0–2.3 mg cm−1 in all the samples. The prepared cathode and lithium metal anode were assembled together in a 2032 coin cell with 40 μL of liquid electrolyte containing 1.0 M lithium bis(trifluoromethane)sulfonimide (LiTFSI), and 0.1 M lithium nitrate salt in DOL:DME (1:1 vol%) with Celgard 2320 as a separator. All the components were assembled in an argon-filled glove box (H2O < 0.5 ppm, O2 < 0.5 ppm). The charge/discharge tests of the Li–S batteries were performed with a WBSC3000 battery cycler (WonATech) with cutoff voltages of 1.8 = ∼2.8 V (vs. Li/Li+) at 25 °C. Electrochemical impedance spectroscopy (EIS) measurements were conducted by complex impedance spectroscopy from 10 °C to 80 °C with a Zahner Elektrik IM6 apparatus in the frequency range of 0.1 Hz to 1 MHz at an applied voltage of 10 mV.
Results and discussion
Synthesis and characterization of the cationic polymer-grafted graphene oxide (CPGO)
Cationic polymer-grafted graphene oxide (CPGO) was synthesized by the reaction of 2-bromo-2-methylpropionyl bromide with graphene oxide (GO) to introduce bromo-initiation sites, followed by atom transfer radical polymerization (ATRP) of the cationic monomer, QDMAEMA, as shown in Fig. 1. QDMAEMA was synthesized by the reaction between 2-(dimethylamino)ethyl methacrylate (DMAEMA) and iodoethane, where the chemical structure of QDMAEMA was confirmed by 1H NMR (Fig. S1†). Given the living characteristic of ATRP, the chain length of each polymer arm in CPGO should be very close.34 | ||
| Fig. 1 Synthetic routes for graphene oxide having bromo-initiation sites (GO-Br) and cationic polymer-grafted graphene oxide (CPGO). | ||
After grafting the cationic polymer on GO, the chemical composition was changed; whereby the contents of carbon, hydrogen, and nitrogen, increased as shown in the elemental analysis results (Table S1†). The successful introduction of the bromo-initiation sites and the grafting of the cationic polymer was also confirmed by the XPS study of GO, GO-Br, and CPGO. In the XPS spectrum of GO-Br, the characteristic peak of Br 3d could be newly observed by the introduction of bromo-initiation sites. In the XPS spectrum of CPGO, the characteristic peaks of the ester moiety from the C 1s and O 1s spectra became intense35 (Fig. S3†) and the characteristic peaks of the quaternized ammonium moiety and iodine newly appear, which could not be observed in GO and GO-Br (Fig. 2(e) and (f)).36,37 From the elemental analysis and XPS results, the amount of grafted cationic polymer was calculated using the content of nitrogen (3.1 and 3.2 wt% from the XPS and elemental analysis) and the molecular weight of the monomeric unit, and it was found to be about 70 wt% of CPGO. To investigate the molecular weight of the grafted cationic polymer of CPGO, the cationic polymer was detached from CPGO using a previously reported method,38 whereby the CPGO was treated with 1 M KOH/ethanol solution to hydrolyze the ester linkage between the grafted polymer and GO. After that, GPC analysis was carried out and the molecular weight of the grafted polymer obtained by GPC is shown in Fig. S4.† The molecular weight of the grafted polymer could be larger than the obtained one since the ester linkage in the grafted polymer can also be hydrolyzed by KOH solution.39 We believe that the polymers with different molecular weights can also affect the cell performance of Li–S batteries, with such work representing a different set of work for another possible publication. GO, GO-Br, CPGO, and PQDMAEMA have different thermal decomposition behaviors, as shown in Fig. 2(a), where the char yield of GO-Br was smaller than that of GO after the introduction of the bromo-initiation sites. Since the grafted cationic polymers were chemically bonded to hydroxyl group, the weight loss of CPGO below 230 °C was much less than that of GO (Fig. 2(a)).40–45 In addition, since the grafted cationic polymer was completely decomposed below 500 °C, a much larger weight loss was observed for CPGO below 500 °C and its char yield was also much less than that of GO. The grafted polymer of CPGO started to decompose at 230 °C and was completely decomposed below 500 °C in the TGA curve; thereby the amount of weight loss caused by the decomposition of the grafted polymer was about 65 wt%, which is quite close to the result obtained by the elemental analysis and XPS results. Such mismatched results from TGA and elemental analysis have been also reported by others before.46,47 We found that the ATRP condition at 70 °C for 48 h in DMF did not reduce GO, whereby the TGA curves of the pristine GO and the GO treated using the ATRP condition without the monomer were very close (Fig. S5†). Therefore, the CPGO should contain most of the oxygen functional groups that are very important characteristics of GO in coating applications.48 In the FT-IR spectra of GO, GO-Br, and CPGO in Fig. 2(b), the intensity of the peak in the range of 3100–3300 cm−1 assigned to the hydroxyl group in GO became very small with the introduction of bromo-initiation sites (GO-Br).49 After the grafting of the cationic polymer, the characteristic peak of the quaternized ammonium group at 1580 cm−1 newly appeared and the peaks corresponding to carbon–nitrogen and carbon–oxygen bonds in the grafted cationic polymer at 1220 and 1150 cm−1, respectively, became very intense.50 In the XRD patterns of GO and CPGO in Fig. S6,† the characteristic peak of GO (001) at 10° was not observed in CPGO, indicating that the amorphous structure was formed by grafting the cationic polymer.51 The TEM images and EDS mapping results also demonstrated the successful preparation of CPGO, where the characteristic elements of the grafted cationic polymer could be clearly observed in the EDS mapping images (Fig. S7†).
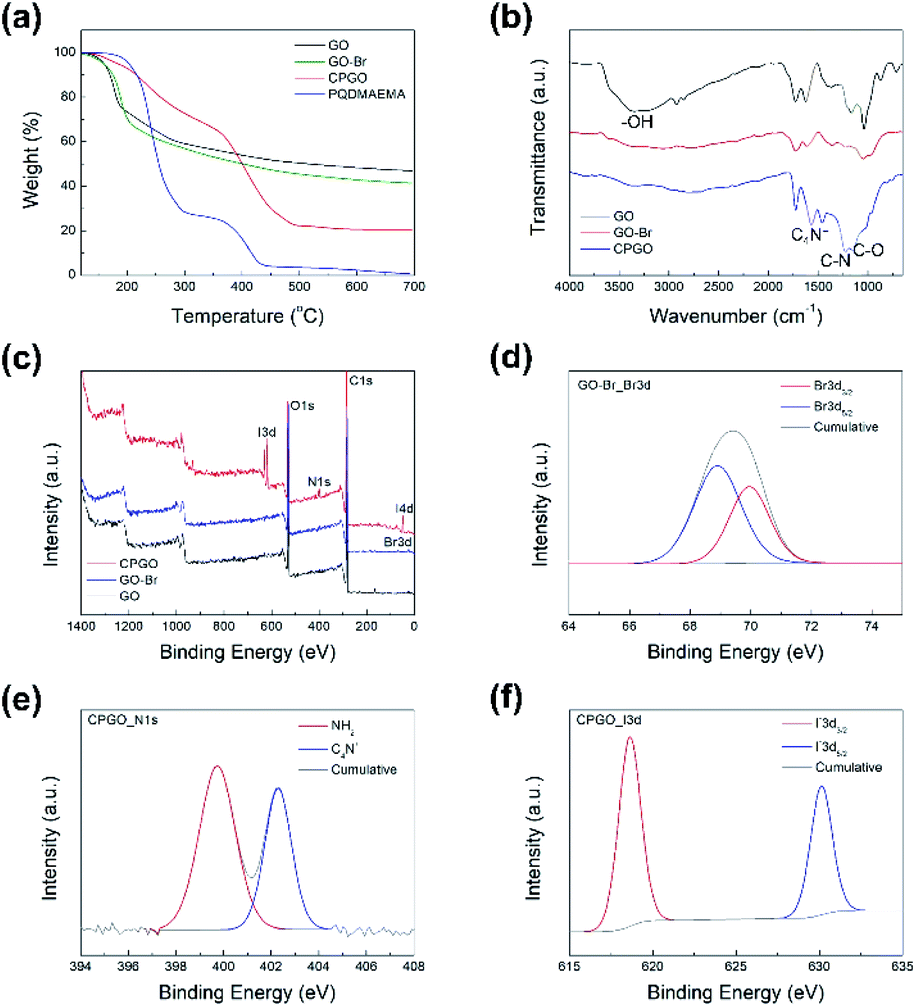 | ||
| Fig. 2 (a) TGA profiles of graphene oxide (GO), GO-Br, CPGO, and the linear cationic polymer (PQDMAEMA) where the PQDMAEMA was prepared intentionally from free radical polymerization (see Fig. S2† for the synthetic scheme and 1H NMR spectrum of PQDMAEMA), (b) FT-IR spectra of GO, GO having bromo-initiation sites (GO-Br), and CPGO, (c) wide-scan XPS spectra of GO, GO-Br, and CPGO, (d) Br 3d XPS spectrum of GO-Br, (e) N 1s XPS spectrum of CPGO, (f) I 3d XPS spectrum of CPGO. | ||
Preparation of the CPGO/CNT-coated S/C (CPGO/CNT-S/C) cathode
Fig. 3(a) shows the preparation method of the CPGO/CNT-coated S/C (CPGO/CNT-S/C) powder for Li–S batteries. Other powders coated with CPGO, GO/CNT, and PQDMAEMA/CNT were also prepared in the same way as described in Fig. 3(a). Fig. 3(b)–(d) show the TEM images of bare S/C, CPGO-S/C, and CPGO/CNT-S/C powders, respectively. After the coating of CPGO or CPGO/CNT on the S/C powder, the S/C powders were covered by CPGO (Fig. 3(c) and (d)). In the case of the CPGO/CNT-S/C powder, CNTs were further observed as expected where CNTs are known to increase the dispersion and conductivity of GO derivatives (Fig. 3(d)).52–54 The EDS mapping images in Fig. S8† also show that the S/C powders were covered by CPGO. | ||
| Fig. 3 (a) Preparation method of the CPGO/CNT-S/C powder. TEM images of: (b) sulfur-deposited Super P® carbon black (S/C), (c) CPGO-S/C, and (d) CPGO/CNT-S/C powders. SEM images of: (e) bare S/C, (f) CPGO-S/C, and (g) CPGO/CNT-S/C cathodes. | ||
Fig. 3(e)–(g) show the SEM images of the cathodes fabricated with bare S/C, CPGO-S/C, and CPGO/CNT-S/C powders, respectively. In all the samples, the weight ratio of S/C:conductive carbon:poly(acrylic acid) binder was 85:5:10, and the loading amount of sulfur was 2.0–2.3 mg cm−2. The S/C particles in Fig. 3(e) were found to be covered by CPGO and CPGO/CNT, as shown in Fig. 3(f) and (g), respectively, forming entangled nets enwrapping the S/C particles. Furthermore, the CPGO/CNT-S/C cathode showed that the powders were more uniformly coated by the better dispersion of the mixture of CPGO and CNT than CPGO alone. The cationic moiety in these cathode-coating materials is known to be able to capture anionic PS by strong ionic interaction.30,31 In addition, since the anionic PS can be diffused out from the S/C particles during battery cycling, a uniform coating is the key factor for the improvement of the cell performance.55,56 To demonstrate the synergetic effect of CPGO on the cell performance obtained by grafting of the cationic polymer onto GO, GO/CNT and PQDMAEMA/CNT-coated S/C (GO/CNT-S/C and PQDMAEMA/CNT-S/C) cathodes were also prepared to compare their cell performances with bare S/C, CPGO-S/C, and CPGO/CNT-S/C cathodes. Fig. S9† shows the SEM images of GO/CNT-S/C and PQDMAEMA/CNT-S/C cathodes, where the GO/CNT coating on S/C showed an entangled net-like morphology similar to that of the CPGO/CNT coating on S/C, while the PQDMAEMA/CNT coating on S/C showed aggregation because of the low compatibility between the hydrophilic cationic polymer and hydrophobic S/C (Fig. S9†).57 Therefore, the cationic polymer that can capture the anionic PS cannot be easily coated on hydrophobic S/C, while CPGO having the cationic polymer can be well-coated on S/C to form the entangled net structure shown in Fig. 3(g).
Cell performance of Li–S batteries
Fig. 4(a) shows the cycle performance of the Li–S batteries assembled with bare S/C, CPGO-S/C, and CPGO/CNT-S/C cathodes at a 0.2C-rate. All the cells were assembled with a lithium metal anode and Celgard separator with 1 M LiTFSI and 0.1 M lithium nitrate in DOL/DME (1:1, v/v) liquid electrolyte. As shown in Fig. 4(a), the cycle performance of Li–S batteries was improved as the bare S/C cathode was coated with CPGO or CPGO/CNT because CPGO having the cationic polymer can capture the anionic PS. In addition, the cell assembled with the CPGO/CNT-S/C cathode showed better cycle performance than the cell with the CPGO-S/C cathode, and this can be attributed to the more uniform coating of CPGO/CNT on S/C than that of CPGO only on S/C.53,54 Consequently, the cell assembled with the CPGO/CNT-S/C cathode retained a discharge capacity of 744 mA h g−1 after 50 cycles, while the retained discharge capacity of the cells with bare S/C and CPGO-S/C cathodes were found to be smaller, such as 488 mA h g−1 and 641 mA h g−1 after 50 cycles, respectively. We also conducted cycle tests of the cells with GO/CNT-S/C and PQDMAEMA/CNT-S/C cathodes (Fig. S10†) to further verify the advantage of CPGO as the coating material. The cycle performance of the cells with bare S/C and GO/CNT-S/C cathodes was found to be close, while the cell with the PQDMAEMA/CNT-S/C cathode showed a poorer cycle performance than that of the cell with the bare S/C cathode. Although GO/CNT was found to be coated on S/C quite uniformly as CPGO/CNT, the cycle performance of the cell with the GO/CNT-S/C cathode was poorer than that of the cell with the CPGO/CNT-S/C cathode, because GO without any cationic moieties cannot well capture the anionic PS as CPGO with the cationic polymer. In the case of the cell with the PQDMAEMA/CNT-S/C cathode, the poorest cycle performance was observed because PQDMAEMA/CNT could not form a uniform coating due to the low compatibility between the hydrophilic PQDMAEMA and hydrophobic Super P carbon, even though the PQDMAEMA has a strong PS-capturing ability.30,31,57,58 Since the CPGO has a strong PS-capturing ability through the cationic polymer and a fair coating ability with Super P carbon by the GO moiety, the cycle performance of the cell with the CPGO-S/C cathode was found to be better than those of the cells with the GO/CNT-S/C and PQDMAEMA/CNT-S/C cathodes. The best cycle performance was observed from the cell with the CPGO/CNT-S/C cathode because of the advantageous feature of the mixture of CPGO and CNT having a cationic characteristic and much improved coating ability by the addition of CNTs.59
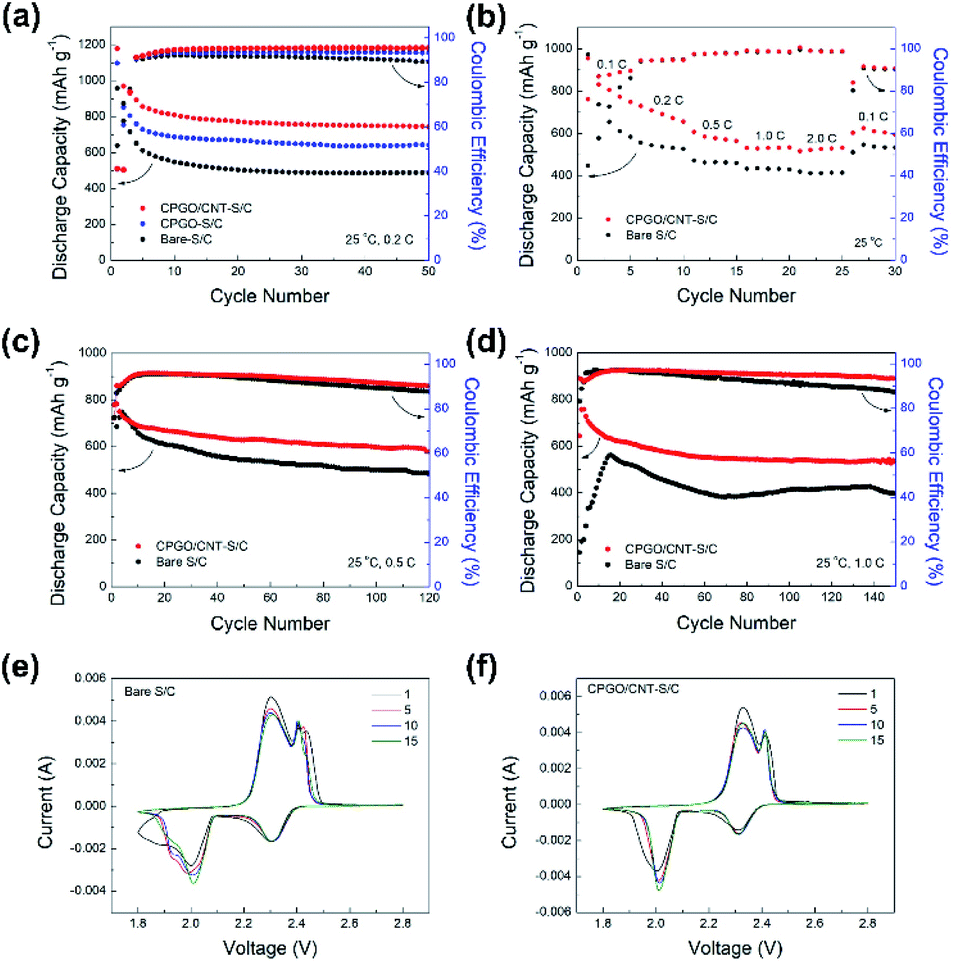 | ||
| Fig. 4 (a) Cycle performance of the cells assembled with bare S/C, CPGO-S/C, and CPGO/CNT-S/C cathodes with a C-rate of 0.2C at 25 °C. (b) Rate capability of the cells with bare S/C and CPGO/CNT-S/C cathodes at 25 °C, cycle performance of the cells assembled with bare S/C and CPGO/CNT-S/C cathodes with a C-rate of 0.5C (c) and 1C (d) at 25 °C. Cyclic voltammetry curves of the cells assembled with (e) bare S/C and (f) CPGO/CNT-S/C cathodes. | ||
Further long-term cycle tests at high current densities (0.5 and 1C) and rate capability tests of the cell with CPGO/CNT-S/C cathode were compared with those of the cell with the bare S/C cathode. As shown in Fig. 4(b), the cell with the CPGO/CNT-S/C cathode showed a better rate capability than the cell with the bare S/C cathode. In the long-term cycle tests at 0.5C and 1C in Fig. 4(c) and (d), the cell with the CPGO/CNT-S/C cathode exhibited a better cycle performance with a higher coulombic efficiency than the cell with the bare S/C cathode, where the discharge capacity values of the cell with the CPGO/CNT-S/C cathode were 579 mA h g−1 (after 120 cycles) and 533 mA h g−1 (after 150 cycles) at 0.5C and 1C, respectively. Also, as shown in the charge–discharge curves of the cells with the bare S/C and CPGO/CNT-S/C cathode at 0.5C (Fig. S11†), the cell with the CPGO/CNT-S/C cathode showed smaller overpotential values than the cell with the bare S/C cathode, which could be attributed to the small cell polarization due to the suppressed shuttle effect. Cyclic voltammetry (CV) tests were conducted to analyze the electrochemical redox reaction of Li–S batteries assembled with bare S/C and CPGO/CNT-S/C cathodes. From the CV curves in Fig. 4(e) and (f), typical two cathodic peaks of Li–S batteries at 2.3 V and 2.0 V are shown, corresponding to the reduction of elemental sulfur S8 to long chain PS (Li2Sx, x = 4–8), and then to the final discharge product, short chain PS (Li2S), respectively.60,61 The anodic peaks at 2.3 V and 2.4 V correspond to the reverse reaction of PS from the short chain to the long chain one, and then to elemental sulfur, respectively.60 In the CV curve of the cell with the CPGO/CNT-S/C cathode, the cathodic peak at around 2.0 V has a narrower shape without a shoulder peak than that of the cell with the bare S/C cathode. Also, the anodic peaks of the cell with the CPGO/CNT-S/C cathode at 2.3–2.4 V showed a smaller overpotential value of 0.16 V at the first cycle than the cell with the bare S/C cathode, which showed a value of 0.21 V, when the current value is 0.002 A. This result demonstrates that the cell polarization and diffusion of PS during cycling can be suppressed by the introduction of CPGO/CNT coating.60
SEM and EIS analyses before and after cycling
In order to further figure out the reason for the improvement of the cell performance by the CPGO/CNT coating, the morphology change of the lithium metal anode and cathode after cycling was examined by SEM. Fig. 5(a)–(c) show the surface morphology of the lithium metal anode dissembled from the cells with the bare S/C and CPGO/CNT-S/C cathodes before and after cycling. A very rough surface with many lithium dendrites was observed from the cell with the bare S/C cathode after cycling, while a much smoother surface with a few dendrites was observed from the lithium metal anode of the cell with the CPGO/CNT-S/C cathode. Since the CPGO/CNT coating can suppress the shuttle effect of the Li–S batteries, a stable SEI layer between the lithium metal anode and electrolyte can be developed and the lithium dendrite growth can be suppressed.62 | ||
| Fig. 5 Surface SEM images of (a) bare lithium metal before cycling, (b) lithium metal dissembled from the cell with bare S/C cathode, and (c) lithium metal dissembled from the cell with CPGO/CNT-S/C cathode after 120 cycles cycled at 0.5C. SEM images of the (d) bare S/C cathode and (e) CPGO/CNT-S/C cathode after 120 cycles cycled at 0.5C. | ||
The bare S/C and CPGO/CNT-S/C cathodes were also dissembled from the cells and the surface morphology change before and after cycling was observed by SEM. The SEM images of the bare S/C and CPGO/CNT-S/C cathodes before cycling are shown in Fig. 3(d) and (f). After the cycle tests, precipitated PSs on the surface of the Super P carbon particles could be observed because the PSs diffused out from the bare S/C cathode during cycling.63,64 As the elemental sulfur was reduced and PSs were formed, the volume of the sulfur expanded and the PSs diffused out from the Super P carbon. During the charge process, however, the PSs that have diffused out from the cathode cannot diffuse into the Super P carbon due to the poor compatibility between the hydrophobic carbon and anionic PSs. Consequently, the sulfur and PSs formed during the cycling precipitate on the S/C, thus forming precipitation layers as shown in Fig. 5(d).64,65 In the case of the CPGO/CNT-S/C cathode, however, the CPGO/CNT coating layer acts as a physical barrier to the volume expansion of sulfur and also CPGO having a cationic polymer can attract the dissolved anionic PSs, thus it showed much less precipitate on the surface, as shown in Fig. 5(e). In the EIS spectra, the impedance of the cell with the CPGO/CNT-S/C cathode was slightly smaller than the cell with the bare S/C cathode, possibly because CNT can decrease the impedance (Fig. S11†).66 After cycling, quite a smaller interfacial impedance was observed from the cell with the CPGO/CNT-S/C cathode due to the suppressed shuttle effect by the coating material.67 In addition, although the electrolyte resistance of both cells with the bare S/C and CPGO/CNT-S/C cathode was increased after cycling, because the PS increased the viscosity of the electrolyte, the resistance increment of the cell with the CPGO/CNT-S/C cathode was smaller than that of the cell with the bare S/C cathode due to the suppressed shuttle effect by the CPGO/CNT coating.68
We admit, however, that the performance of the Li–S battery system assembled with the CPGO/CNT-S/C cathode is not yet good enough to be used for practical applications. Furthermore, there are a lot of reports that show much better cell performance than ours.13–23 In this work, however, we mainly focused on the synthesis of the cationic polymer grafted on GO and the effect of CPGO as a coating material without the need for an optimization process for the cell fabrication. Still we clearly showed that the mixtures CPGO and CNTs were better coating material systems than CPGO alone, the mixture of GO and CNTs, and the mixture of cationic polymer (PQDMAEMA) and CNTs for the Li–S battery systems. Therefore, further works to optimize the fabrication procedure for the CPGO/CNT-S/C cathode, including investigating the change of the chemical structure and the molecular weight of the cationic polymer, the control of the grafting density, the change of the types of CNT, the ratio control between CPGO and CNT, and so on are under progress to try to improve the cell performance.
Conclusions
In this study, we presented the preparation of cationic polymer-grafted graphene oxide (CPGO) and its application as a cathode-coating material for Li–S batteries. It was found that CPGO having the cationic polymer could be quite uniformly coated on S/C powder, resulting in an improvement of the cell performance because the grafted cationic polymer on CPGO can capture the anionic PSs. In addition, we found that the uniformity of the coating on S/C powder could be further improved by mixing CNT with CPGO. As a result, compared to the cell performance of the cells with the bare S/C (without any coating), CPGO-S/C, cationic polymer/CNT-S/C, or GO/CNT-S/C cathodes, the cell with the CPGO/CNT-S/C cathode showed the best discharge capacity values of 599 and 538 mA h g−1 after 100 cycles at 0.5 and 1C, respectively. The improved cycle performance of Li–S batteries with the CPGO/CNT-S/C cathode can be ascribed to the fast electrochemical kinetics achieved by the suppression of both the shuttle effect and volume change of sulfur during cycling by the coating of the cathode with the mixture of CPGO and CNT, where CPGO contained a cationic polymer and CNT could improve the compatibility with the hydrophobic carbon material in the cathode. We hope this study provides a direction for the cathode-coating strategies of Li–S batteries with S/C cathodes, which have high potential for next-generation energy storage systems.Author contributions
Daun Jeong: investigation, formal analysis, methodology, writing – original draft. Dong Gi Hong: investigation, writing – review and editing. Jinsol Yook: methodology, writing – review and editing. Chan Yeong Koong: writing – review & editing. Soohyun Kim: funding acquisition. Ki-Hyun Kim: funding acquisition. Kwonnam Sohn: funding acquisition. Jong-Chan Lee: supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the LG Energy Solution and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF-2018R1A5A1024127 and NRF-2020R1A2C2008114).References
- M. A. Hannan, M. M. Hoque, A. Mohamed and A. Ayob, Renewable Sustainable Energy Rev., 2017, 69, 771–789 CrossRef.
- M. A. Hannan, M. S. H. Lipu, A. Hussain and A. Mohamed, Renewable Sustainable Energy Rev., 2017, 78, 834–854 CrossRef.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- B. Zhu, X. Wang, P. Yao, J. Li and J. Zhu, Chem. Sci., 2019, 10, 7132–7148 RSC.
- M. Rana, S. A. Ahad, M. Li, B. Luo, L. Wang, I. Gentle and R. Knibbe, Energy Storage Materials, 2019, 18, 289–310 CrossRef.
- A. Manthiram, Y. Fu, S. H. Chung, C. Zu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- X. Zhang, K. Chen, Z. Sun, G. Hu, R. Xiao, H.-M. Cheng and F. Li, Energy Environ. Sci., 2020, 13, 1076–1095 RSC.
- D. Liu, C. Zhang, G. Zhou, W. Lv, G. Ling, L. Zhi and Q. H. Yang, Adv. Sci., 2018, 5, 1700270 CrossRef PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- X. Yang, Y. Chen, M. Wang, H. Zhang, X. Li and H. Zhang, Adv. Funct. Mater., 2016, 26, 8427–8434 CrossRef CAS.
- J. Ma, Z. Fang, Y. Yan, Z. Yang, L. Gu, Y.-S. Hu, H. Li, Z. Wang and X. Huang, Adv. Energy Mater., 2015, 5, 1500046 CrossRef.
- Z. Li, J. Zhang and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 12886–12890 CrossRef CAS PubMed.
- L. Ma, S. Wei, H. L. Zhuang, K. E. Hendrickson, R. G. Hennig and L. A. Archer, J. Mater. Chem. A, 2015, 3, 19857–19866 RSC.
- H. Kim, J. Lee, H. Ahn, O. Kim and M. J. Park, Nat. Commun., 2015, 6, 7278 CrossRef CAS PubMed.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, A. Somogyi, P. Theato, M. E. Mackay, Y. E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- S. Wei, L. Ma, K. E. Hendrickson, Z. Tu and L. A. Archer, J. Am. Chem. Soc., 2015, 137, 12143–12152 CrossRef CAS PubMed.
- H. M. Kim, H.-H. Sun, I. Belharouak, A. Manthiram and Y.-K. Sun, ACS Energy Lett., 2016, 1, 136–141 CrossRef CAS.
- G. Zhou, L. Li, D. W. Wang, X. Y. Shan, S. Pei, F. Li and H. M. Cheng, Adv. Mater., 2015, 27, 641–647 CrossRef CAS PubMed.
- J. Balach, H. K. Singh, S. Gomoll, T. Jaumann, M. Klose, S. Oswald, M. Richter, J. Eckert and L. Giebeler, ACS Appl. Mater. Interfaces, 2016, 8, 14586–14595 CrossRef CAS PubMed.
- G. Li, F. Lu, X. Dou, X. Wang, D. Luo, H. Sun, A. Yu and Z. Chen, J. Am. Chem. Soc., 2020, 142, 3583–3592 CrossRef CAS PubMed.
- J. Liang, Z.-H. Sun, F. Li and H.-M. Cheng, Energy Storage Materials, 2016, 2, 76–106 CrossRef.
- A. Fu, C. Wang, F. Pei, J. Cui, X. Fang and N. Zheng, Small, 2019, 15, e1804786 CrossRef PubMed.
- X.-X. Peng, Y.-Q. Lu, L.-L. Zhou, T. Sheng, S.-Y. Shen, H.-G. Liao, L. Huang, J.-T. Li and S.-G. Sun, Nano Energy, 2017, 32, 503–510 CrossRef CAS.
- M. Zheng, Y. Chi, Q. Hu, H. Tang, X. Jiang, L. Zhang, S. Zhang, H. Pang and Q. Xu, J. Mater. Chem. A, 2019, 7, 17204–17241 RSC.
- H. J. Peng, G. Zhang, X. Chen, Z. W. Zhang, W. T. Xu, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12990–12995 CrossRef CAS PubMed.
- X. Tao, J. Wang, C. Liu, H. Wang, H. Yao, G. Zheng, Z. W. Seh, Q. Cai, W. Li, G. Zhou, C. Zu and Y. Cui, Nat. Commun., 2016, 7, 11203 CrossRef CAS PubMed.
- H. Wang, M. Ling, Y. Bai, S. Chen, Y. Yuan, G. Liu, C. Wu and F. Wu, J. Mater. Chem. A, 2018, 6, 6959–6966 RSC.
- X. F. Liu, H. Chen, R. Wang, S. Q. Zang and T. C. W. Mak, Small, 2020, 16, e2002932 CrossRef PubMed.
- M. J. Lacey, F. Jeschull, K. Edstrom and D. Brandell, Chem. Commun., 2013, 49, 8531–8533 RSC.
- S. S. Zhang, J. Electrochem. Soc., 2012, 159, A1226–A1229 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 3154–3160 CrossRef CAS.
- K. Dave, K. H. Park and M. Dhayal, RSC Adv., 2015, 5, 95657–95665 RSC.
- T. Susi, T. Pichler and P. Ayala, Beilstein J. Nanotechnol., 2015, 6, 177–192 CrossRef PubMed.
- T. Li, Y. Hu, C. A. Morrison, W. Wu, H. Han and N. Robertson, Sustainable Energy Fuels, 2017, 1, 308–316 RSC.
- S. Rahimi-Razin, V. Haddadi-Asl, M. Salami-Kalajahi, F. Behboodi-Sadabad and H. Roghani-Mamaqani, Int. J. Chem. Kinet., 2012, 44, 555–569 CrossRef CAS.
- P. van de Wetering, N. J. Zuidam, M. J. van Steenbergen, O. A. G. J. van der Houwen, W. J. M. Underberg and W. E. Hennink, Macromolecules, 1998, 31, 8063–8068 CrossRef CAS.
- G. Gonçalves, P. A. A. P. Marques, A. Barros-Timmons, I. Bdkin, M. K. Singh, N. Emami and J. Grácio, J. Mater. Chem., 2010, 20, 9927–9934 RSC.
- Z. Sekhavat Pour and M. Ghaemy, Compos. Sci. Technol., 2016, 136, 145–157 CrossRef CAS.
- E. S. Goda, B. S. Singu, S. E. Hong and K. R. Yoon, Mater. Chem. Phys., 2020, 254, 123465 CrossRef CAS.
- Y. Yang, J. Wang, J. Zhang, J. Liu, X. Yang and H. Zhao, Langmuir, 2009, 25, 11808–11814 CrossRef CAS PubMed.
- Y. Deng, J. Z. Zhang, Y. Li, J. Hu, D. Yang and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4451–4458 CrossRef CAS.
- H. Roghani-Mamaqani, RSC Adv., 2015, 5, 53357–53368 RSC.
- H. Lee, J. Han, K. Kim, J. Kim, E. Kim, H. Shin and J.-C. Lee, J. Ind. Eng. Chem., 2019, 74, 223–232 CrossRef CAS.
- B. Wang, D. Yang, J. Z. Zhang, C. Xi and J. Hu, J. Phys. Chem. C, 2011, 115, 24636–24641 CrossRef CAS.
- Y. N. Singhbabu, B. Sivakumar, J. K. Singh, H. Bapari, A. K. Pramanick and R. K. Sahu, Nanoscale, 2015, 7, 8035–8047 RSC.
- Z. Çiplak, N. Yildiz and A. Çalimli, Fullerenes, Nanotubes, Carbon Nanostruct., 2014, 23, 361–370 CrossRef.
- K. H. Jung, H. J. Kim, M. H. Kim, S. Kong, H. Seo and J. C. Lee, Macromol. Mater. Eng., 2020, 305, 2000348 CrossRef CAS.
- A. Jabbar, G. Yasin, W. Q. Khan, M. Y. Anwar, R. M. Korai, M. N. Nizam and G. Muhyodin, RSC Adv., 2017, 7, 31100–31109 RSC.
- Y. Cheng, S. Lu, H. Zhang, C. V. Varanasi and J. Liu, Nano Lett., 2012, 12, 4206–4211 CrossRef CAS PubMed.
- Y. Li, T. Yang, T. Yu, L. Zheng and K. Liao, J. Mater. Chem., 2011, 21, 10844–10851 RSC.
- H. J. Kim, M.-Y. Lim, K. H. Jung, D.-G. Kim and J.-C. Lee, J. Mater. Chem. A, 2015, 3, 6798–6809 RSC.
- H. Kim, J. T. Lee, D.-C. Lee, A. Magasinski, W.-i. Cho and G. Yushin, Adv. Energy Mater., 2013, 3, 1308–1315 CrossRef CAS.
- M. Yu, A. Wang, F. Tian, H. Song, Y. Wang, C. Li, J. D. Hong and G. Shi, Nanoscale, 2015, 7, 5292–5298 RSC.
- J.-H. Lee, S.-B. Wee, M.-S. Kwon, H.-H. Kim, J.-M. Choi, M. S. Song, H. B. Park, H. Kim and U. Paik, J. Power Sources, 2011, 196, 6449–6455 CrossRef CAS.
- J. Yu, X. Li, Y. Shu, L. Ma, X. Zhang and Y. Ding, Electrochim. Acta, 2019, 293, 458–465 CrossRef CAS.
- Y. Hua, F. Li, Y. Liu, G.-W. Huang, H.-M. Xiao, Y.-Q. Li, N. Hu and S.-Y. Fu, Compos. Sci. Technol., 2017, 149, 294–304 CrossRef CAS.
- M. Li, J. Zhou, J. Zhou, C. Guo, Y. Han, Y. Zhu, G. Wang and Y. Qian, Mater. Res. Bull., 2017, 96, 509–515 CrossRef CAS.
- C. Zhao, C. Yu, M. Zhang, J. Yang, S. Liu, M. Li, X. Han, Y. Dong and J. Qiu, J. Mater. Chem. A, 2015, 3, 21842–21848 RSC.
- X. Zhou, X. Luo, H. Wang, J. Yang, H. Xu, M. Jia and J. Tang, J. Mater. Sci., 2019, 54, 9622–9631 CrossRef CAS.
- R. Ye, J. Bell, D. Patino, K. Ahmed, M. Ozkan and C. S. Ozkan, Sci. Rep., 2017, 7, 17264 CrossRef PubMed.
- J. Yan, X. Liu and B. Li, Adv. Sci., 2016, 3, 1600101 CrossRef PubMed.
- Y. Chen, N. Liu, H. Shao, W. Wang, M. Gao, C. Li, H. Zhang, A. Wang and Y. Huang, J. Mater. Chem. A, 2015, 3, 15235–15240 RSC.
- Y. Rangom, X. S. Tang and L. F. Nazar, ACS Nano, 2015, 9, 7248–7255 CrossRef CAS PubMed.
- H. Qu, J. Ju, B. Chen, N. Xue, H. Du, X. Han, J. Zhang, G. Xu, Z. Yu, X. Wang and G. Cui, J. Mater. Chem. A, 2018, 6, 23720–23729 RSC.
- Z. Deng, Z. Zhang, Y. Lai, J. Liu, J. Li and Y. Liu, J. Electrochem. Soc., 2013, 160, A553–A558 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03744g |
| This journal is © The Royal Society of Chemistry 2021 |