DOI:
10.1039/D1RA04064B
(Paper)
RSC Adv., 2021,
11, 31226-31234
A green route to prepare metal-free phthalocyanine crystals with controllable structures by a simple solvothermal method
Received
25th May 2021
, Accepted 3rd September 2021
First published on 22nd September 2021
Abstract
Exploring the environmentally friendly and low-cost synthesis strategies of phthalocyanine (Pc) crystals in just one step is an absolute challenge. The solvothermal synthesis of phthalocyanine crystals shows the advantages of high-quality crystalline products, facile reaction and purification, and low cost. Nevertheless, only a few metal phthalocyanine crystals have been successfully synthesized via solvothermal reactions. In this study, we found that the crystalline β metal-free phthalocyanine needles could be directly prepared via the tetrapolymerization of phthalodinitrile catalyzed by DBU in solvothermal reactions. Similar to the preparation of β-phthalocyanine crystals, the α metal-free phthalocyanine crystals with the specific multiply-laminated structures can be obtained through solvothermal reactions assisted by DBN. SEM characterization showed that the individual β metal-free phthalocyanine has a well-defined quadrangular shape with smooth faces. However, the α metal-free phthalocyanine exhibits a distinctive undulating surface morphology. Both phthalocyanines showed satisfactory thermal stability (from room temperature to about 300 °C), excellent resistance to acid/alkali solution, and fast photoelectric response properties (order of magnitude of response time, 10−6 s) as tested by TG-DSC and TPV, respectively. It is noted that ethanol was used as the reaction medium and the resulting phthalocyanine crystals can be facilely purified using hot ethanol to dissolve the impurities adsorbed on the surfaces of phthalocyanine crystals. Compared to the traditional methods, no re-crystallization operation was carried out for our method. To the best of our knowledge, this is the first report on the solvothermal synthesis of metal-free phthalocyanine crystals with controllable crystal form adjusted by DBU/DBN in one step.
1. Introduction
Since phthalocyanine was found unexpectedly by Braun and Tchemiac in 1907,1 the special macrocycle structure, stable properties, and especially the coloring function as a pigment and dye have been considered intensively.2 Until now, more than 5000 kinds of phthalocyanine compounds and their derivatives have been synthesized. Furthermore, other than the traditional dyes, some novel technological applications of Pc-based compounds and materials have been explored extensively for their unique optical, electronic, catalytic, and various optoelectronic/photovoltaic properties.3–7 The synthesis methods for Pc-based materials have been gradually improved, especially the environmentally friendly, low-cost, and easily operable methods.
Solvothermal technology is an efficient synthesis method for most inorganic compounds. The particularly shaped crystalline structures can be achieved and the morphologies of products can be regulated largely due to the high reactivity of reactants and limited solubility of products in the reaction media under high-pressure conditions.8,9 Although great breakthroughs have been accomplished for the solvothermal synthesis of inorganic materials,10 for phthalocyanine and metal phthalocyanine complexes there has been limited solvothermal synthesis success and only a few phthalocyanine-based crystals have been reported. In 2001, McGaff's group presented the solvothermal synthesis of two nickel complexes with alkoxy-substituted phthalocyanine ligands by selecting nickel acetate tetrahydrate and 1,2-dicyanobenzene as raw materials, and alcohol as the solvent at 100 °C for at least 4 days.11 Xia reported the direct synthesis of needle-like copper phthalocyanine single-crystals with 1,3-diiminoisoindoline and Cu(AcO)2·2H2O in quinoline solvent at 270 °C for 8 h.12 After that, we reported the one-step solvothermal synthesis of needle-like manganese phthalocyanine,13 copper phthalocyanine,14 zinc phthalocyanine and cobalt phthalocyanine crystals,15 with absolute ethanol as the reaction medium. Recently, crystalline iron phthalocyanine was synthesized via the reaction between FeCl2·4H2O and 1,2-dimethylbenzene catalyzed by DBU.16 It is worth noting that the highly crystalline phthalocyanine can be directly formed and grown into big crystals with sizes even greater than 10 mm in solvothermal reactions mentioned above. These big metal phthalocyanine crystals are ideal candidates in the investigations of optoelectronic devices, field-effect transistors and so on.17–19 Accordingly, the quality of phthalocyanine products is closely related to the synthesis methods, which will largely determine their applications. Compared to the traditional methods of phthalocyanine complexes, solvothermal synthesis shows obvious advantages such as an environmentally friendly process, direct transformation into crystal products from reactants, and low purification cost without using concentrated H2SO4. Therefore, the syntheses of other unreported crystalline phthalocyanine materials deserve to be further explored by this method.
For the simplest metal-free phthalocyanine compound (C32H18N8, H2Pc), the known methods are as follows: (I) heating phthalonitrile (Phn) in a basic solvent with a high boiling point, such as quinolone or phyridine.20,21 (II) Heating or UV irradiation of Phn in the alcohol solvent with alkali metal alkoxide.22 In 1980, Ogawa and Shiraishi reported a metal-free catalyzed strategy in which the organic strong base DBU (or DBN) was used as the proton acceptor instead of alkali metal alkoxide to catalyze the formation of H2Pc from Phn in refluxed ethanol.23 Afterwards, the synthetic method based on DBU/DBN was improved continually, and various organic bases were designed as catalysts for the synthesis of Pc and its derivatives.24–26 Nevertheless, the phthalocyanine compounds were formed as precipitates in the solvent and phthalocyanine crystals could not be directly obtained via this method.
In this work, we present a one-step solvothermal route for the synthesis of high-quality metal-free phthalocyanine crystals. The well-defined quadrangular phthalocyanine crystals (β crystal form) could be prepared in ethanol with o-phthalodinitrile as the raw material catalyzed by DBU. These phthalocyanine crystals show regular and smooth surfaces and their maximum achieved length was 5 mm. However, the phthalocyanine crystals (α crystal form) obtained via DBN-catalyzed reactions showed multiply-laminated morphologies and small sizes. To ascertain the molecular structures, the growth behaviors and properties of the as-obtained crystals were determined, and characterization was performed via Fourier transform infrared (FT-IR) spectrometer, Raman spectrometer, scanning electronic microscopy (SEM) and X-ray powder diffraction (XRD). The thermal behavior was estimated via thermogravimetry-differential scanning calorimetry (TG-DSC) measurements.
2. Experimental
2.1 Materials and reagents
1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU, 99%), 1,5-diazabicyclo[4.3.0]-5-nonene (DBN, 98%) and o-phthalodinitrile were purchased from Shanghai Aladdin Biochemical Technology Co. Ltd. Absolute alcohol was used as the reaction medium. A Teflon-lined autoclave (Nanjing Ruinik Technology Development Co. Ltd.) was used for the synthesis of H2Pc crystals.
2.2 Synthesis of metal-free phthalocyanine crystals
The schematic map of H2Pc synthesis is presented in Scheme 1. Here, 769 mg of powdered o-phthalodinitrile (6 mmol) was added to the bottom of a 28 mL Teflon-lined autoclave, followed by the successive injection of about 20 mL absolute ethanol and 50 μL DBU (or DBN). The autoclave was placed into a program-controlled oven with the temperature set at 180 °C for 6 h. After cooling to room temperature naturally, bright-purple crystalline needles were observed in the autoclave. These needles were removed and washed several times with hot ethanol via a Buchner funnel. Finally, the H2Pc crystals were dried at 100 °C in an oven overnight for characterization. The yields of H2Pc needles for different reaction intervals were 16% (3 h), 24% (6 h), 30% (12 h), and 31% (24 h), respectively.
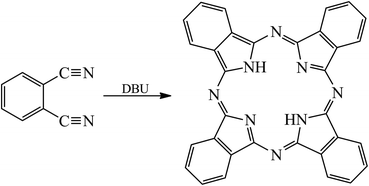 |
| | Scheme 1 The H2Pc macrocycle formation reaction. | |
2.3 Characterization of H2Pc crystals
The optical pictures of the as-obtained H2Pc crystals were taken using an Olympus SZX7 stereoscopic microscope. High-magnification morphological images were obtained using a scanning electron microscope (SEM, Nova NanoSEM 450). FT-IR spectroscopy was performed on a Thermo Nicolet Nexus FT-IR spectrometer using the standard KBr pellet method. Raman spectroscopy was performed on a Renishaw inVia Raman spectrometer with a continuous-wave laser at 532 nm. The laser power was set at 1% of 100 mW. X-ray diffraction (XRD) patterns of H2Pc crystals were investigated using a Bruker D8 ADVANCE powder X-ray diffractometer with Cu Kα radiation. To evaluate the thermal properties of the H2Pc crystals, thermogravimetry and differential scanning calorimetry (TG-DSC) measurements were performed using a Netzsch STA 409PC thermal analyzer in synthetic air at a heating rate of 10 °C min−1. Transient photovoltage (TPV) curve measurement was performed according to the following procedure. A sample chamber like a parallel-plate sandwich-like capacitor consisting of an ITO film, a mica spacer, and a layer of H2Pc crystals on the Cu foil substrate from top to bottom in sequence was used. An ultrafast laser pulse (λ = 355 nm and a pulse width of 4 ns) through a third-harmonic Nd:YAG laser (Quantel Brilliant Eazy: BRILEZ/IR-10) was employed as the light source. The transient surface photovoltage data were recorded on a 500 MHz digital oscilloscope (TDS 3054C, Tektronix).
3. Results and discussion
3.1 The mechanism of the H2Pc formation reaction
It is generally believed that the essential steps in the formation reactions of Pc are as follows.23,27 (I) The dissociation of ethanol into protons and ethoxy (CH3CH2O−) anions was activated by DBU (or DBN). (II) The nucleophilic attack by the CH3CH2O− anion towards the cyano-group of phthalodinitrile and the resulting generation of the 1-ethoxy-3-iminoisoindolenine intermediate. (III) The cyclization of four intermediates into a Pc macrocycle with a reduction of the double bond within it (Scheme 2). It is worth noting that the molecular structure of the solvent had a significant effect on the formation of the Pc compound. None of the Pc products were found in the DBU-catalyzed solvothermal reactions with non-protonic solvents, such as pyridine, tetrahydrofuran, and acetonitrile. Even for the different alcohol solvents (methanol, ethanol, and benzyl alcohol in our experiments), the shapes and yields of the Pc products were different. In consideration of the crystal quality, cost, and toxic properties, ethanol is the appropriate solvent for H2Pc synthesis.
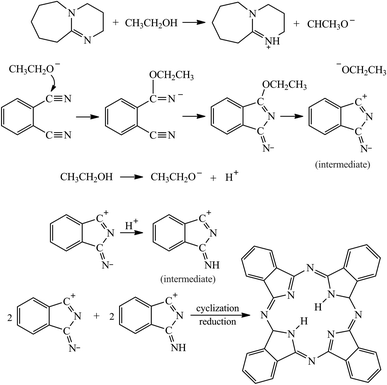 |
| | Scheme 2 The possible formation mechanism of H2Pc from o-phthalodinitrile, catalyzed by DBU. | |
Unlike the synthesis of MnPc, ZnPc and CoPc needles by using solvothermal methods,13,15 no promoter or activator was used. However, none of the H2Pc products in our experiments can be obtained without using DBU/DBN. We believe that the template effect of metal ions will promote their coordination with Pc macrocycles through a low activation energy path and result in the formation of MPc crystals in a moderate yield, at least. However, the cyclization of four phthalodinitrile molecules into a H2Pc macrocycle is attributed to the electrostatic attraction between intermediates, which is facilitated by ethoxy anions. Besides, the yield of H2Pc is proportional to the use of DBU/DBN, which also reflects the difference between MPc and H2Pc.
3.2 The molecular structure characterization of H2Pc
FT-IR and Raman spectroscopy are useful methods for verifying the molecular structures of organic compounds. The FT-IR spectrum of H2Pc needles is illustrated in Fig. 1a. The whole vibrational spectrum of H2Pc can be viewed as the combination of the vibrations of partial structures, such as macrocycles, isoindazole, benzene, C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) , N–H, and so on. The most intense peak at 1004 cm−1 is a characteristic vibrational peak of metal-free phthalocyanine.28 The weak absorption peak at 1041 cm−1 is considered as N–H in-plane bending. The intense and moderate absorption bands at 749, 733 and 717 cm−1 are assigned to N–H out-of-plane bending. The stretching of the N–H bond can be found at 3272 cm−1.29,30 The C–H stretching vibration is observed at 3048 cm−1.31 The Raman spectrum of H2Pc needles is recorded in Fig. 1b. The strong vibration bands in the range of 600 to 800 cm−1 are the characteristic vibrations of the Pc macrocycle. Specifically, the peaks at 679 and 792 cm−1 correspond to the breathing and deformation vibrations of the macrocycle, respectively. Besides, the stretching vibrations of the pyrrole parts within Pc can be observed in the three strong peaks at 1139, 1332, and 1533 cm−1, and their deformation vibration is located at 481 cm−1. The weak peak at 184 cm−1 could be evidence of the vibrational mode in which the four pyrroles move outward in phase. In addition, the three peaks at 1401, 1423, and 1611 cm−1 can be attributed to the stretching of benzene units in the Pc macrocycle. The weak peaks at 566 and 229 cm−1 can be assigned to the deformation of benzene and isoindole, respectively.32
, N–H, and so on. The most intense peak at 1004 cm−1 is a characteristic vibrational peak of metal-free phthalocyanine.28 The weak absorption peak at 1041 cm−1 is considered as N–H in-plane bending. The intense and moderate absorption bands at 749, 733 and 717 cm−1 are assigned to N–H out-of-plane bending. The stretching of the N–H bond can be found at 3272 cm−1.29,30 The C–H stretching vibration is observed at 3048 cm−1.31 The Raman spectrum of H2Pc needles is recorded in Fig. 1b. The strong vibration bands in the range of 600 to 800 cm−1 are the characteristic vibrations of the Pc macrocycle. Specifically, the peaks at 679 and 792 cm−1 correspond to the breathing and deformation vibrations of the macrocycle, respectively. Besides, the stretching vibrations of the pyrrole parts within Pc can be observed in the three strong peaks at 1139, 1332, and 1533 cm−1, and their deformation vibration is located at 481 cm−1. The weak peak at 184 cm−1 could be evidence of the vibrational mode in which the four pyrroles move outward in phase. In addition, the three peaks at 1401, 1423, and 1611 cm−1 can be attributed to the stretching of benzene units in the Pc macrocycle. The weak peaks at 566 and 229 cm−1 can be assigned to the deformation of benzene and isoindole, respectively.32
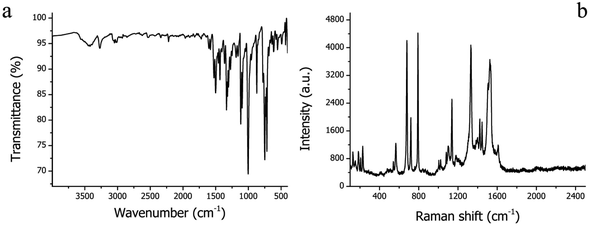 |
| | Fig. 1 (a) FT-IR and (b) Raman spectra of H2Pc crystals. | |
3.3 The surface morphologies and crystal structures of H2Pc
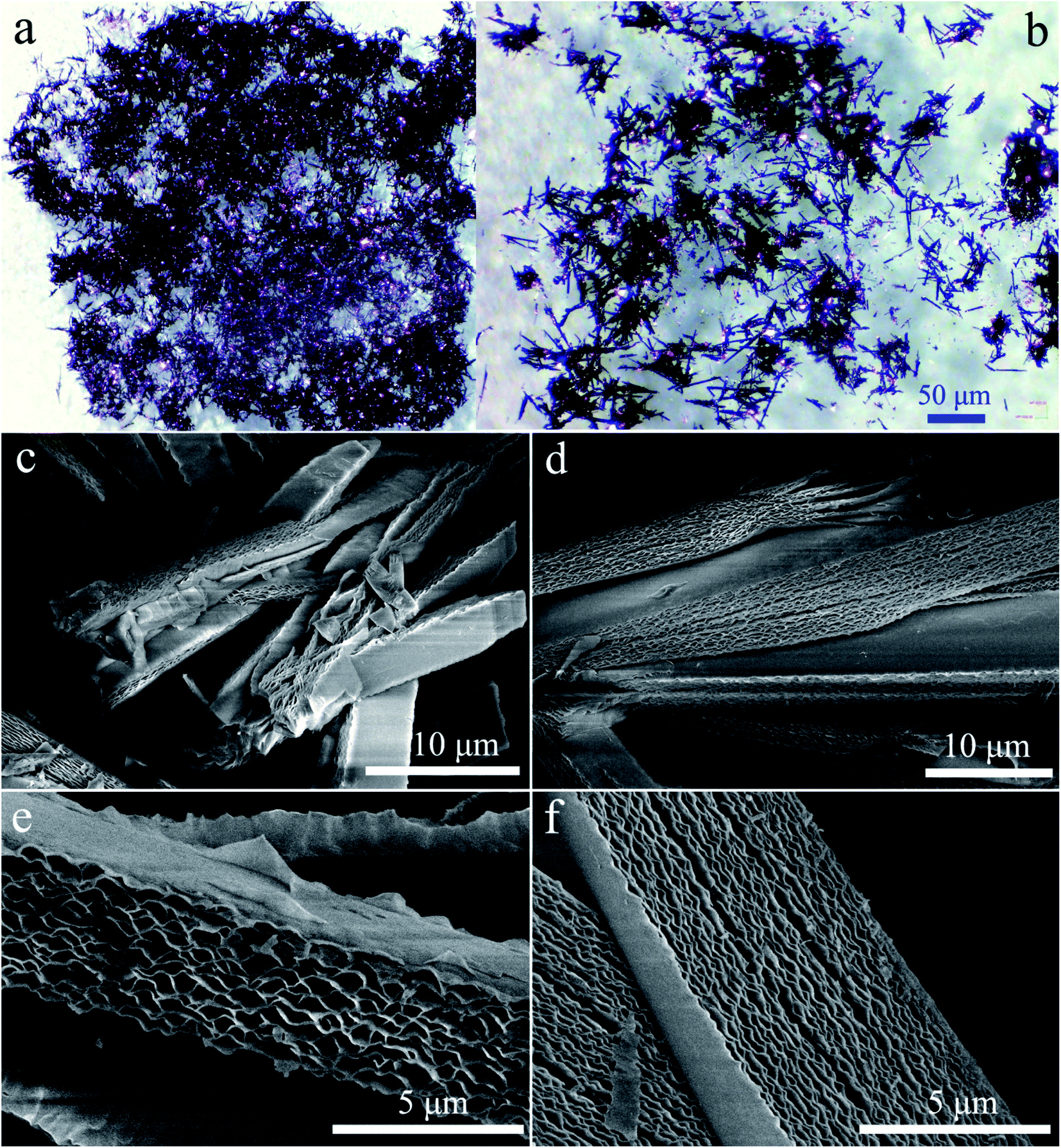
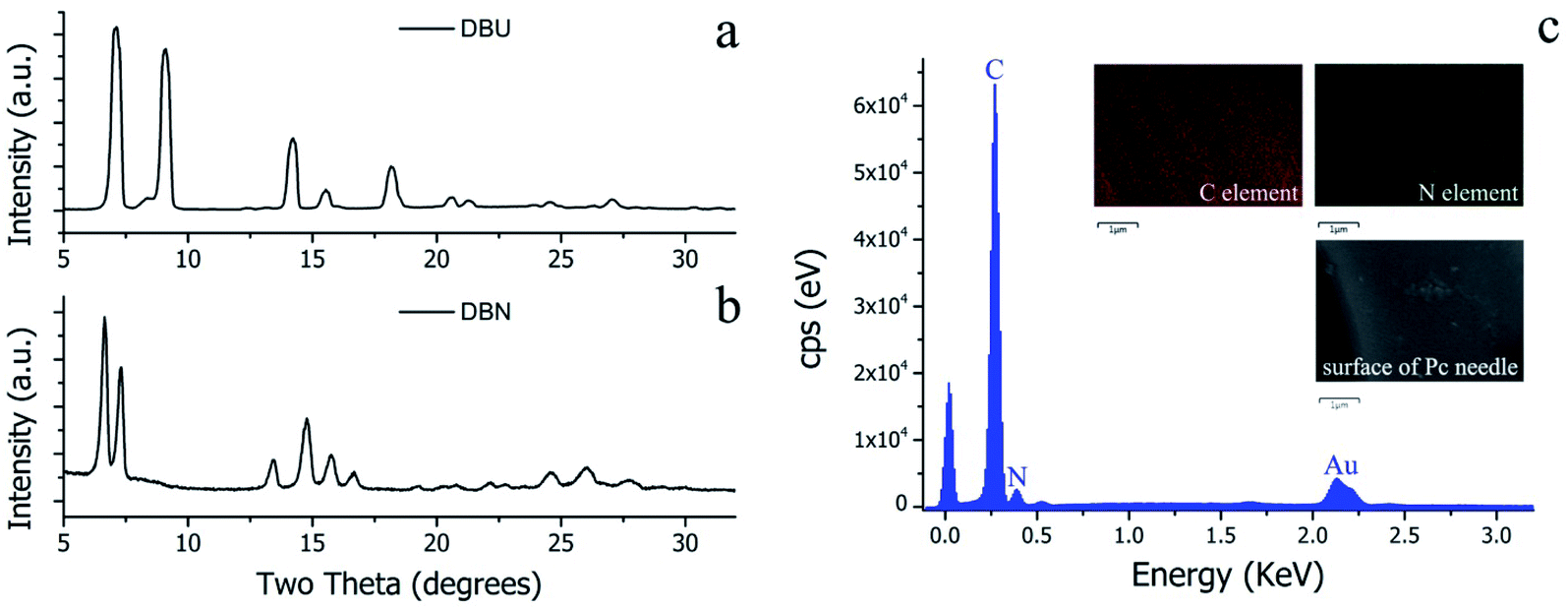
The geometric shapes of H2Pc crystals were firstly identified by using a stereoscopic microscope. From Fig. 2a, the H2Pc crystals prepared via DBU-catalyzed reactions show needle-like shapes with a high length-to-diameter ratio. Their maximum length was 5 mm. Moreover, these H2Pc needles were bright purple, which is difficult to distinguish from the color of ZnPc and CoPc needles prepared by our proposed method.15 The detailed surface morphologies of these H2Pc needles were further determined using SEM. Fig. 2b and c illuminate the well-defined quadrangular shapes with sharp, flat, or irregular end faces. According to careful observations on the prismatic planes (Fig. 2d and e), we found smooth and multilayered surfaces for these quadrangular crystals. Damaged surfaces of the quadrangular prisms were also found due to the weak mechanical strength of H2Pc molecular crystals. Based on the SEM results, a big and smooth surface was selected for the elemental scan measurement and EDX analyses. The EDX spectrum indicated that the weight percentages of C and N elements were 78.34 and 21.66%, and the atomic percentages of C and N elements were 80.83 and 19.17%, respectively. All these measured values are in accordance with the theoretical data. The elemental mapping images show the homogeneous distribution of the component elements of H2Pc needles (Fig. 4c). For comparison, the morphological analyses for the H2Pc crystals synthesized via DBN-catalyzed solvothermal reactions were performed. These crystals showed a smaller length of about 50 μm and were dark-purple under natural light (see Fig. 3a and b). To ascertain the possible reasons, the detailed SEM morphologies revealed that all these H2Pc crystals were composites of multiply-laminated structures on which no flat and smooth surfaces could be found (Fig. 3c and d). Furthermore, the high magnification SEM images showed that each plate in one laminated composite had wavy surfaces and many crevices were found between these wavy plates (Fig. 3e and f). The wavy morphologies are not beneficial for the parallel reflection of light, so the dark purple H2Pc crystals were observed.
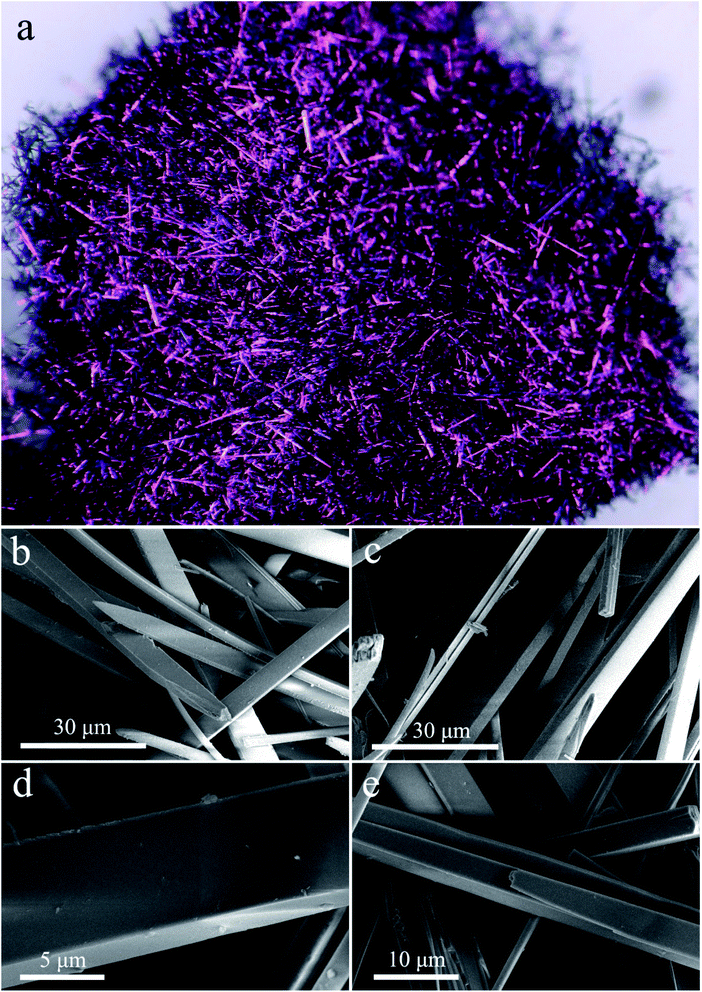 |
| | Fig. 2 (a) The optical image of H2Pc needle crystals. (b and c) Low-magnification SEM morphologies of H2Pc needles. (d–e) High-magnification SEM morphologies of H2Pc needles. All these H2Pc needles were obtained from DBU-catalyzed reactions. | |
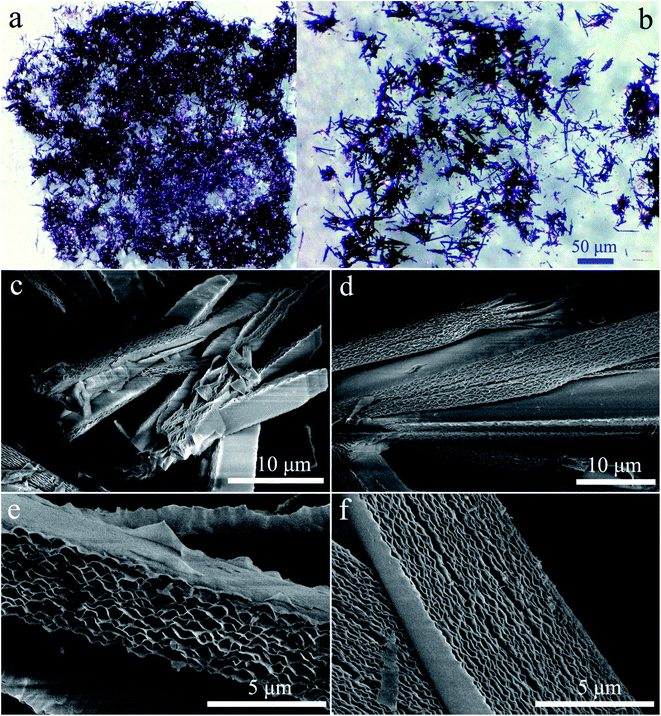 |
| | Fig. 3 (a and b) The optical images of small H2Pc crystals. (c and d) Low-magnification SEM morphologies of H2Pc crystals. (e and f) High-magnification SEM morphologies of H2Pc crystals. All the multiply-laminated H2Pc crystals were prepared via DBN-catalyzed reactions. | |
Although the molecular structures of DBU and DBN are similar, the surface morphologies of the resulting H2Pc crystals prepared using these organic bases in solvothermal reactions were quite distinctive. According to our previous work,13–15 the morphology of MPc crystals is closely related to its crystalline structure. Therefore, XRD investigation is a reasonable method for understanding the differences in crystalline structures. Fig. 4a and b present the X-ray diffraction patterns in the 2θ range of 5 to 32° for H2Pc crystals synthesized through DBU and DBN, respectively. The obvious difference between the two X-ray diffraction spectra is the position of peaks in the narrow range of 5–10°. Based on the reported crystallography data,33 the H2Pc needles via DBU synthesis can be identified as β-phthalocyanine (the matched JCPDS No. 37-1844). The detailed unit cell parameters of β-phthalocyanine are as follows: space group: P21/a (14), cell: a = 19.870(7) Å, b = 4.731(7) Å, c = 14.813(7) Å, α = 90°, β = 121.98(4)°, γ = 90°.33,34 The multiply-laminated H2Pc crystals via DBN synthesis were determined as α-phthalocyanine (the matched JCPDS No. 36-1882), and its detailed unit cell parameters are as follows. Space group: C2/n (15), cell: a = 26.121(4) Å, b = 3.7970(7) Å, c = 23.875(3) Å, α = 90°, β = 94.16(2)°, γ = 90°.33,35,36
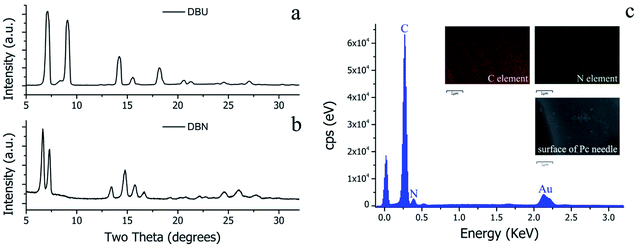 |
| | Fig. 4 (a) XRD patterns of H2Pc crystals from DBU-catalyzed reactions. The matched JCPDS card number is 37-1844. (b) XRD patterns of H2Pc crystals from DBN-catalyzed reactions. The matched JCPDS card number is 36-1882. (c) Energy-dispersive X-ray (EDX) spectra and elemental scan mapping images of a single H2Pc needle. The weight percentages (wt%) for C and N elements are 78.34 and 21.66%, and the atomic percentages (at%) for C and N elements are 80.83 and 19.17%, respectively. | |
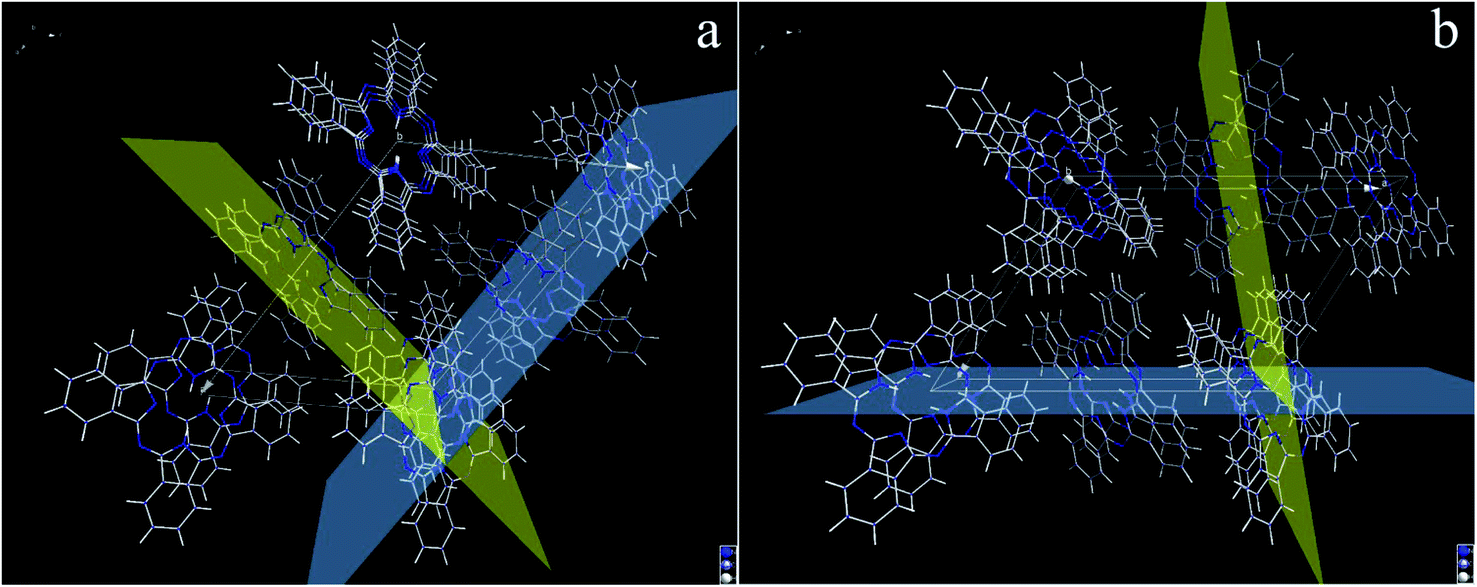
Herein, the two predominant crystal planes for β-phthalocyanine (Fig. 4a) can be indexed as (001) and (201(_)), which correspond to 2θ = 7.1° and 9.1°, respectively. The positional relationship between these two crystal planes is shown from different viewing directions in Fig. 5a and b. From the careful observations of the unit cell, the H2Pc macrocycles located in these two predominant planes keep the parallel positions so that strong π–π electronic interactions exist between them. Therefore, the formation of H2Pc needles with a high ratio of length to diameter can be explained through the continuous π–π stacking of H2Pc macrocycles during crystal growth.37,38
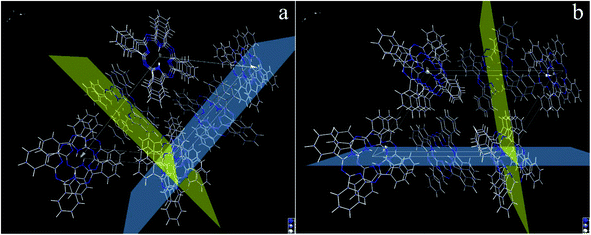 |
| | Fig. 5 (a and b) The unit cell of β phthalocyanine visualized from different views. The (001) and (201(_)) crystal planes in a unit cell of β phthalocyanine. The (001) plane is denoted as the blue plane and (201(_)) plane as the yellow plane. | |
It is known that H2Pc crystals are polymorphic, with three crystalline forms, i.e., α-form, β-form, and X-form. The current methods for the control of the H2Pc crystal form is crystal seed-induced transformation,39,40 vacuum sublimation,41 and thermal-induced transformation.42 Very recently, the ball-milling method was invented, by which the three crystal forms can be prepared directly and transformed in each other through regulating the ball-milling time, rotating speed, temperature and solvent.33 For our proposed solvothermal method, no special equipment, crystal seed materials, and complicated operations were needed. Compared with the reported methods, our method showed the advantages of simple synthesis, low cost of materials, and single experimental factor control.
3.4 The stability testing of H2Pc crystals
TG and DSC measurements were performed to observe the thermal stability of the resulting H2Pc crystals. Fig. 6a and b present the TG-DSC curves of H2Pc needles and multiply-laminated crystals in the temperature range of 40 to 1000 °C under synthetic air atmosphere, respectively. The H2Pc needles showed thermal stability before 330 °C, and then underwent the three successive stages of mass decrease until 700 °C, and the final weight loss was 100% after 700 °C. Interestingly, similar TG results were observed for multiply-laminated H2Pc crystals. Their detectable decrease in mass began at 280 °C and then fell into the stage of obvious weight loss from 280 °C to 725 °C, and finally achieved almost 100% weight loss after 725 °C. Some previous reports have proved that the β-phase of H2Pc crystals is the high-temperature phase that can be obtained from α-phase H2Pc through sublimation by heating above 475 K under the low pressure of nitrogen or helium gas.41,43 Although the phase transition from the a to β form cannot be precisely determined for no obvious changes in the TG-DSC curve near to 200 °C (Fig. 6b) under our measurement conditions, the TG curve with different gradients indicates the decomposition of the H2Pc skeleton to different degrees at the gradually incremental temperatures. Unlike the metal phthalocyanine, these H2Pc crystals can be completely decomposed due to their purely organic molecular structures. The resistance of H2Pc crystals to acid and alkali solution was also studied for stability estimation. A 6 mol L−1 HCl and 5 mol L−1 NaOH solution were prepared, respectively, to fully immerse H2Pc crystals at room temperature for at least 5 h. The SEM morphologies of the H2Pc samples were observed after immersion. For H2Pc needles, the prismatic planes and their edges remained unchanged either in HCl or NaOH solution (Fig. 7a and b). Similarly, no detectable dissolution or damage to the multiply-laminated structures was found after 5 h of immersion (Fig. 7c and d). All the phenomena indicated that the as-prepared H2Pc materials showed excellent acid/alkali resistance and have the potential for future application.
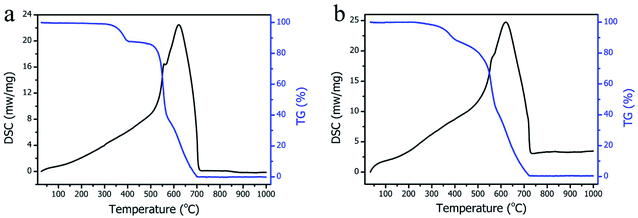 |
| | Fig. 6 TG-DSC curves of (a) H2Pc needles and (b) H2Pc multiply-laminated crystals. | |
 |
| | Fig. 7 The SEM morphologies of H2Pc needles after immersion in HCl solution (a), and in NaOH solution (b). The SEM morphologies of H2Pc crystals after immersion in HCl solution (c), and in NaOH solution (d). | |
3.5 Photoelectric properties
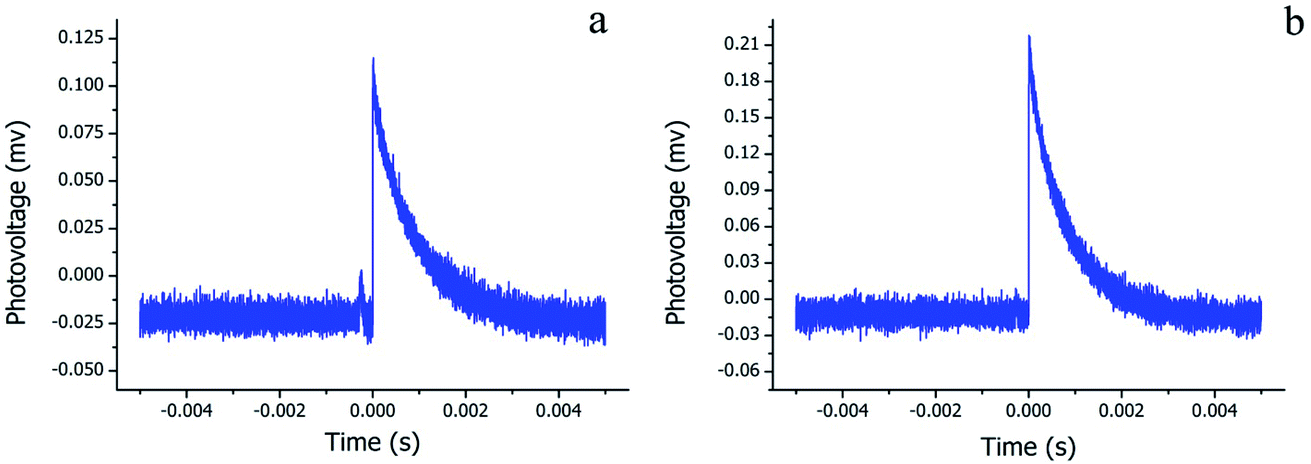
Since the Pc-based materials have the photoelectric properties of p-type semiconductors, TPV measurements were performed to assess the photoelectric performance of the H2Pc needles and multiply-laminated crystals. Fig. 8a and b show the TPV response curves of H2Pc samples with the recombination time as the variable and photovoltage as the function. The wavelength of the ultrafast laser was 355 nm and its pulse width was 4 ns. The fast and strong response of these H2Pc crystals under light irradiation indicated the high efficiency of the generation, separation, and flow of photogenerated electrons and holes. From detailed analyses, the maximum recombination times were 2 × 10−6 and 3 × 10−6 s for H2Pc needles (β-phase) and H2Pc multiply-laminated crystals (a-phase), respectively. The high photovoltage value (0.219 mv) of a-phase H2Pc means the effective separation of electrons and holes, largely for the specific multiply-laminated structures, which enhanced the separation efficiency. However, for the quadrangular H2Pc needles (β-phase), the electrons and holes were easily recombined within the bulk so that the low photovoltage value of 0.116 mv was measured for their condensed structures.
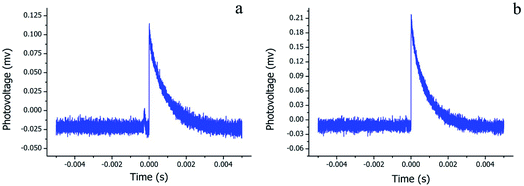 |
| | Fig. 8 Transient photovoltage curves of (a) H2Pc needles and (b) H2Pc multiply-laminated crystals. | |
4. Conclusions
In this work, an environmentally friendly and low-cost solvothermal method was proposed for the one-step synthesis of metal-free phthalocyanine crystals. The SEM and XRD characterizations show that the needle-like β-phthalocyanine crystals can be prepared via DBU-catalyzed reactions, while the multiply-laminated α-phthalocyanine crystals can be synthesized in DBN-catalyzed reactions. The distinguishable morphologies and different crystal structures of the H2Pc crystals resulted from the single experimental factor of the organic bases. Although the molecular structures of DBU and DBN are similar, the growth of H2Pc crystals can be well regulated through catalysis by these organic bases. Moreover, these H2Pc crystals showed excellent thermostability before 330 and 280 °C for the needles and multiply-laminated crystals, respectively. The fast and strong responses of these H2Pc crystals in TPV measurements illuminated that they can be utilized as suitable candidates for application in photoelectric devices.
Conflicts of interest
The authors declare that they have no conflicts of interest to the publication of this paper.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 51302241 and 21507107), Key Research & Develop and Promotion Projects of Henan Province in 2021 (No. 212102310525), Innovation and Entrepreneurship Training Program for University Students of Henan Province in 2020 (No. S202010480009), Training Plan of Young Core Teachers in Universities of Henan Province in 2018 (No. 2018GGJS142), and the Applied Basic Research Project of Education Department of Henan Province (21B430015).
References
- A. Braun and J. Tchemiac, Chem. Ber., 1907, 40, 2709–2714 CrossRef CAS.
- D. Wöhrle, G. Schnurpfeil, S. G. Makarov, A. Kazarin and O. N. Suvorova, Macroheterocycles, 2012, 5, 191–202 CrossRef.
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768–6816 CrossRef CAS PubMed.
- A. B. Sorokin, Chem. Rev., 2013, 113, 8152–8191 CrossRef CAS PubMed.
- X. S. Li, D. Y. Lee, J. D. Huang and J. Y. Yoon, Angew. Chem., Int. Ed., 2018, 57, 9885–9890 CrossRef CAS PubMed.
- B. D. Zheng, Q. X. He, X. S. Li, Y. Yoon and J. D. Huang, Coord. Chem. Rev., 2021, 426, 213548 CrossRef CAS.
- G. de la Torre, C. G. Claessens and T. Torres, Chem. Commun., 2007, 20, 2000–2015 RSC.
- G. Demazeau, J. Mater. Sci., 2008, 43, 2104–2114 CrossRef CAS.
- D. R. Modeshia and R. I. Walton, Chem. Soc. Rev., 2010, 39, 4303–4325 RSC.
- D. Sud and G. Kaur, Polyhedron, 2021, 193, 114897 CrossRef CAS.
- C. D. Molek, J. A. Halfen, J. C. Loe and R. W. Mcgaff, Chem. Commun., 2001, 24, 2644–2645 RSC.
- D. C. Xia, S. K. Yu, R. S. Shen, C. Y. Ma, C. H. Cheng, D. M. Ji, Z. Q. Fan, X. Wang and G. T. Du, Dyes Pigm., 2008, 78, 84–88 CrossRef CAS.
- D. P. Li, S. X. Ge, G. F. Sun, Q. He, B. J. Huang, G. Z. Tian, W. Y. Lu, G. B. Li, Y. L. Chen, S. N. An and Z. Zheng, Dyes Pigm., 2015, 113, 200–204 CrossRef CAS.
- S. X. Ge, Y. G. Zhang, B. J. Huang, S. P. Huang, W. W. T, Y. Lei, Q. He, G. L. Tu, Q. Q. Qin, S. S. Niu, M. T. Li, D. P. Li and Z. Zheng, Mater. Lett., 2016, 163, 61–64 CrossRef CAS.
- D. P. Li, S. X. Ge, T. C. Yuan, J. J. Gong, B. J. Huang, W. W. Tie and W. W. He, CrystEngComm, 2018, 20, 2749 RSC.
- T. T. Liu, F. Y. Zhang, L. X. Ruan, J. W. Tong, G. W. Qin and X. M. Zhang, Mater. Lett., 2019, 237, 319–322 CrossRef CAS.
- K. Xiao, R. J. Li, J. Tao, E. A. Payzant, I. N. Ivanov, A. A. Puretzky, W. P. Hu and D. B. Geohegan, Adv. Funct. Mater., 2009, 19, 3776–3780 CrossRef CAS.
- O. A. Melville, B. H. Lessard and T. P. Bender, ACS Appl. Mater. Interfaces, 2015, 7, 13105–13118 CrossRef CAS PubMed.
- Q. X. Tang, H. X. Li, M. He, W. P. Hu, C. M. Liu, K. Q. Chen, C. Wang, Y. Q. Liu and D. B. Zhu, Adv. Mater., 2006, 18, 65–68 CrossRef CAS.
- I. M. Heilbron, F. Irving and R. P. Linstead, US Pat., 2153620, April 1939 Search PubMed.
- I. M. Heilbron, F. Irving, R. P. Linstead and J. F. Thorpe, British Pat., 410814, May 1934 Search PubMed.
- P. A. Barrett, C. E. Dent and R. P. Linstead, J. Chem. Soc., 1936, 1719 RSC.
- H. Tomoda, S. Saito, S. Ogawa and S. Shiraishi, Chem. Lett., 1980, 9, 1277–1280 CrossRef.
- D. Wöhrle, G. Schnurpfeil and G. Knothe, Dyes Pigm., 1992, 18, 91–102 CrossRef.
- B. I. Kharisov, U. O. Mendez and J. Rivera de la Rosa, Russ. J. Coord. Chem., 2006, 32, 617–631 CrossRef CAS.
- T. Koczorowski, J. Ber, T. Sokolnicki, A. Teubert, W. Szczolko and T. Goslinski, Dyes Pigm., 2020, 178, 108370 CrossRef CAS.
- W. Zheng, C. Z. Wan, J. X. Zhang, C. H. Li and X. Z. You, Tetrahedron Lett., 2015, 56, 4459–4462 CrossRef CAS.
- X. X. Zhang, M. Bao, N. Pan, Y. X. Zhang and J. Z. Jiang, Chin. J. Chem., 2004, 22, 325–332 CrossRef CAS.
- M. P. Sammes, J. Chem. Soc., Perkin Trans. 2, 1972, 160–162 RSC.
- A. W. Snow, J. R. Griffith and N. P. Marullo, Macromolecules, 1984, 17, 1614–1624 CrossRef CAS.
- H. F. Shurvell and L. Pinzuti, Can. J. Chem., 1966, 44, 125–136 CrossRef CAS.
- R. Aroca and R. O. Loutfy, J. Raman Spectrosc., 1982, 12, 262–265 CrossRef CAS.
- X. L. Li, Y. X. Feng, C. Y. Li, H. H. Han, X. Q. Hu, Y. N. Ma and Y. H. Yang, Green Process. Synth., 2021, 10, 95–100 CrossRef.
- S. Matsumoto, K. Matsuhama and J. Mizuguchi, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 131–133 CrossRef.
- J. Janczak, Pol. J. Chem., 2000, 74, 157 CAS.
- J. Janczak and R. Kubiak, J. Alloys Compd., 1992, 190, 121–124 CrossRef CAS.
- S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430–3434 CrossRef CAS PubMed.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- R. W. Radler Jr and N. Y. Penfield, US Pat., 3594163, 1971 Search PubMed.
- P. J. Brach and H. A. Six, US Pat., 3657272, 1971 Search PubMed.
- N. M. Amar, R. D. Gould and A. M. Saleh, Curr. Appl. Phys., 2002, 2, 455–460 CrossRef.
- B. Y. Pu, X. G. Li, S. R. Wang, Y. Xiao, Z. Q. Li and Y. F. Chen, Chem. Ind. Eng., 2017, 34, 1–7 CAS.
- M. Füstöss-Wégner, Thermochim. Acta, 1978, 23, 93–102 CrossRef.
Footnote |
| † Peng Zhang and Dapeng Li contributed equally to this work |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Peng Zhang†ab,
Suxiang Ge*a,
Guofu Suna,
Qin Hea,
Wenjun Faa,
Yun Lic and
Juntao Mab
*a,
Peng Zhang†ab,
Suxiang Ge*a,
Guofu Suna,
Qin Hea,
Wenjun Faa,
Yun Lic and
Juntao Mab