
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterligand communication in a metal mediated LL′CT system – a case study†
Sara A. Dillea,
Kyle J. Colston a,
Stephen C. Ratvaskyb,
Jingzhi Pua and
Partha Basu*a
a,
Stephen C. Ratvaskyb,
Jingzhi Pua and
Partha Basu*a
aDepartment of Chemistry and Chemical Biology, Indiana University – Purdue University Indianapolis, Indianapolis, IN 46202, USA. E-mail: basup@iupui.edu
bDepartment of Chemistry and Biochemistry, Duquesne University, Pittsburgh, PA 15282, USA
First published on 12th July 2021
Abstract
A series of oxo-Mo(IV) complexes, [MoO(Dt2−)(Dt0)] (where Dt2− = benzene-1,2-dithiol (bdt), toluene-3,4-dithiol (tdt), quinoxaline-2,3-dithiol (qdt), or 3,6-dichloro-benzene-1,2-dithiol (bdtCl2); Dt0 = N,N′-dimethylpiperazine-2,3-dithione (Me2Dt0) or N,N′-diisopropylpiperazine-2,3-dithione (iPr2Dt0)), possessing a fully oxidized and a fully reduced dithiolene ligand have been synthesized and characterized. The assigned oxidation states of coordinated dithiolene ligands are supported with spectral and crystallographic data. The molecular structure of [MoO(tdt)(iPr2Dt0)] (6) demonstrates a large ligand fold angle of 62.6° along the S⋯S vector of the Dt0 ligand. The electronic structure of this system is probed by density functional theory (DFT) calculations. The HOMO is largely localized on the Dt2− ligand while virtual orbitals are mostly Mo and Dt0 in character. Modeling the electronic spectrum of 6 with time dependent (TD) DFT calculations attributes the intense low energy transition at ∼18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 to a ligand-to-ligand charge transfer (LL′CT). The electron density difference map (EDDM) for the low energy transition depicts the electron rich Dt2− ligand donating charge density to the redox-active orbitals of the electron deficient Dt0 ligand. Electronic communication between dithiolene ligands is facilitated by a Mo-monooxo center and distortion about its primary coordination sphere.
000 cm−1 to a ligand-to-ligand charge transfer (LL′CT). The electron density difference map (EDDM) for the low energy transition depicts the electron rich Dt2− ligand donating charge density to the redox-active orbitals of the electron deficient Dt0 ligand. Electronic communication between dithiolene ligands is facilitated by a Mo-monooxo center and distortion about its primary coordination sphere.
Introduction
Non-innocent ligands are known to play a vital role in biological systems across a wide variety of enzymes and metal centers. Ligand non-innocence occurs when there is strong interaction between redox-active ligand and metal-based orbitals.1 Non-innocent ligands can be simple reactive oxygen/nitrogen species (ROS/RNS) like superoxide (O2˙−), nitrosyl (NO˙) radicals or more complicated pterin and flavin systems.2 For example, the tyrosyl/tyrosinate redox pair plays an integral role in the catalytic cycle of the Cu cofactor containing galactose oxidase enzyme.3 Such systems have been modeled with biomimetic compounds to better understand their mechanism of action.4,5 Similar approaches have been utilized to model non-innocent dithiolene systems,6–8 however, only dithiolene ligands of like oxidation states have been utilized. A model system with dithiolene ligands in different redox states can be useful in understanding interligand communication and activity of molybdenum and tungsten containing enzymes.Oxidized dithiolene (i.e., dithione, Dt0) is an electron deficient ligand that can stabilize lower valent metal centers because of its π-accepting character.9–11 Fundamental understanding of this system is based on whether two redox active units can communicate, and how their interaction is manifested. The presence of both oxidized and reduced dithiolene provides the framework for a mixed-valence system in which the charge transfer can be defined. For example, the charge transfer transitions of a square planar d8 nickel compound as a class II mixed-valence system.10 Ligand-based mixed valent systems have also been reported in Ni-salen12 and M(III)-aminophenolate systems (M = Fe, Ru).13,14 Additionally, ligand-based donor–acceptor systems containing oxidized dithiolene have been observed in a structurally characterized oxo-Mo(IV) complexes.11,15
Herein we report the first example of an oxo-Mo dithiolene system where two chelating dithiolene units are of different redox states. We report the syntheses, structure, redox, and spectral properties of electronically asymmetric complexes [MoIVO(Dt2−)(Dt0)] (where Dt2− = benzene-1,2-dithiol (bdt), toluene-3,4-dithiol (tdt), quinoxaline-2,3-dithiol (qdt), or 3,6-dichloro-benzene-1,2-dithiol (bdtCl2); Dt0 = N,N′-dimethylpiperzine-2,3-dithione (Me2Dt0) or N,N′-diisopropylpiperzine-2,3-dithione (iPr2Dt0)) Chart 1. These complexes exhibit intense LL’CT bands where the electron rich Dt2− ligand serves to donate charge density to the electron poor Dt0 ligand. We report this system as a vehicle for understanding the inter-ligand communication between dithiolene units.
 | ||
| Chart 1 | ||
Results and discussion
Syntheses and characterization
Analytically pure complexes 1–8 were synthesized via a ligand exchange between [MoOCl(Me2Dt0)2][PF6] or [MoOCl(iPr2Dt0)2][PF6] and corresponding Dt2− ligand. In the solid state, complexes are dark purple and their solutions in MeCN, CH2Cl2, THF, DMF, acetone, and ethyl acetate are varying shades of purple. The 1H NMR spectra of 1–8 exhibit resonances due to coordinated dithiolene ligands that are shifted downfield from uncoordinated dithiolene ligand. The solid-state IR spectra of 1–8 exhibit a strong thioamide C(=S)N stretching band (∼1500 cm−1) and a strong Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration (∼950 cm−1), which are consistent with vibrational frequencies reported previously for Mo-Dt0 complexes.9,11,15,16
O vibration (∼950 cm−1), which are consistent with vibrational frequencies reported previously for Mo-Dt0 complexes.9,11,15,16
Molecular structure
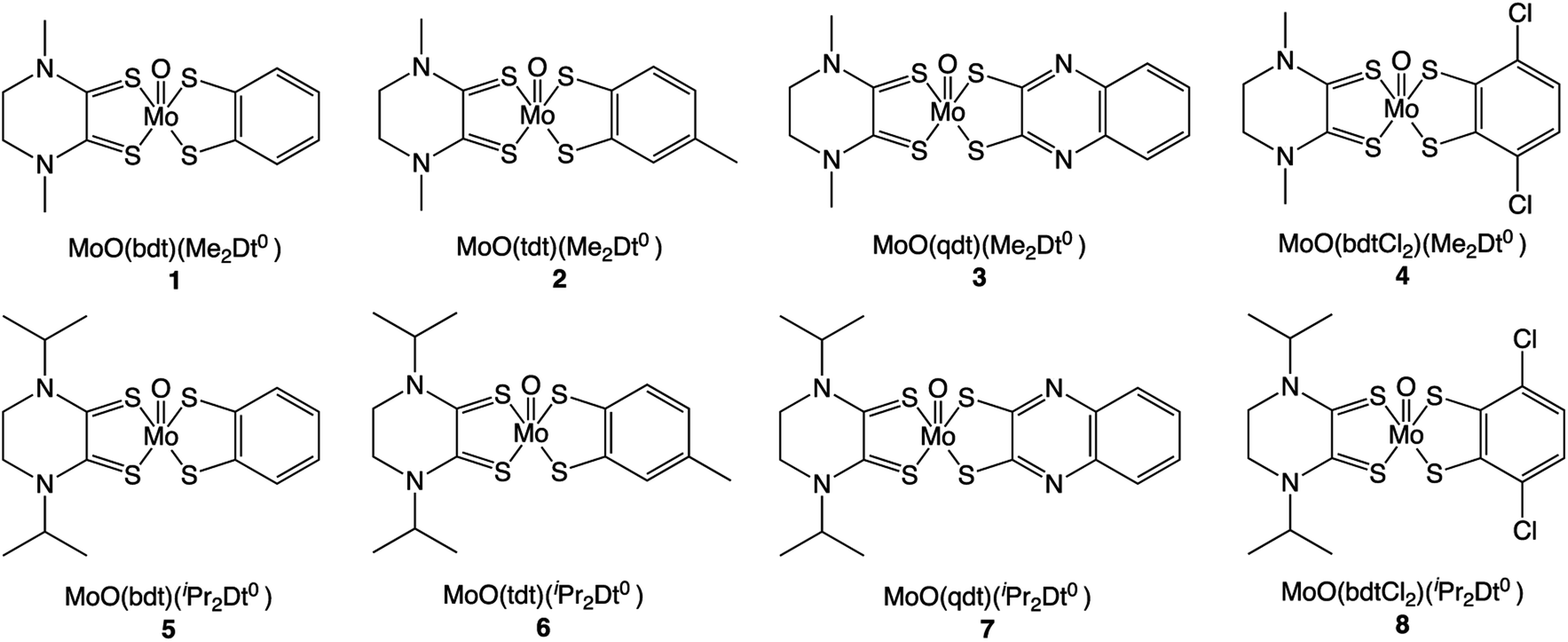
Single crystals of 6 were grown via vapor diffusion from MeCN/diethyl ether and the molecular structure was determined by X-ray crystallography (Fig. 1). The methyl substituent in the tdt fragment was disordered and was modeled for both orientations of the ring. Selected metric parameters are listed in Table 1. The molecular structure of 6 has provided insight into the redox state of the Mo atom. Structural descriptions of the putative oxidation states in known [MoO(Dt2−)2]2− complexes were used in understanding the redox states of the ligand and metal center in 6.2,17–20 | ||
| Fig. 1 Left: thermal ellipsoid plot (30%) of [MoO(tdt)(iPr2Dt0)]. Space group, Pbca; R1, 0.054; wR2, 0.100. Right: schematic demonstrating the drastic fold angle along the SS vector (defined by intersecting planes shown in grey) of the iPr2Dt0 ligand of 6. Crystallographic details are listed in Table S1.† | ||
| O1–Mo1 | 1.681 (2) | C4–C5a | 1.356(9) |
|---|---|---|---|
| a Average bond lengths reported due to disorder on tdt ligand. | |||
| Mo1–S3 | 2.3971 (8) | S3–Mo1–S4 | 84.87(3) |
| Mo1–S4 | 2.3714 (9) | O1–Mo1–S1a | 110.7 |
| S3–C8 | 1.724 (3) | O1–Mo1–S2a | 106.5 |
| S4–C9 | 1.717 (3) | S2–Mo1–S3a | 110.5 |
| C8–C9 | 1.475 (4) | S1–Mo1–S3a | 105.4 |
| C10–C11 | 1.493 (4) | S2–Mo1–S4a | 115.8 |
| Mo1–S1a | 2.374 (16) | S1–Mo1–S4a | 110.4 |
| Mo1–S2a | 2.356 (12) | S1–Mo1–S2a | 83.7 |
| S1–C1a | 1.765 (16) | O1–Mo1–S3 | 115.23(8) |
| S2–C2a | 1.776 (17) | O1–Mo1–S4 | 109.14(8) |
| C1–C2a | 1.382 (9) | ||
The redox state of the tdt and iPr2Dt0 ligands can be assigned based on the C–C bond length of their dithiolene moiety. The C4–C5 and C10–C11 bonds can be utilized as internal standards for CC and C–C bond, respectively, for a comparison. The length of the C1–C2 bond was consistent with the aromatic CC bond length (1.384 Å), which indicates that tdt remains reduced while coordinated. The C8–C9 bond length of 6 was consistent with the C10–C11 bond (1.493 Å) and longer than those reported for ene–dithiolate complexes.21–23 These bond distances support that the iPr2Dt0 remained fully oxidized in 6 in the solid state.
The MoO bond length provides an insight into the oxidation state of the molybdenum center. A CCDC search24 of Mo-dithiolene complexes revealed the average MoIVO bond length to be 1.696 Å, which is 0.015 Å longer than the MoO bond in 6. Interestingly, the MoO bond length of 6 is closer the average MoVO bond length (1.680 Å). The shorter MoO distance in 6 suggests that Mo is donating electron density toward the electron-deficient dithione ligand which results in a shorter MoO bond that resembles a Mo(V) center.
Structures of electron-rich ene-1,2-dithiolate complexes and electron-deficient metal ions often exhibit a large folding of the dithiolene ligand along the S⋯S vector. Early works have provided a “bending” scheme for the bent-metallocene (Cp) dithiolene.25–27 In 6, the angle between the plane containing S3, C8, C9, and S4 and the plane containing Mo, S3 and S4 was 62.6°, such that the iPr2Dt0 ligand is bent towards the terminal oxo group along its S⋯S vector. The angle between the plane containing S1, C1, C2 and S2 and the plane containing Mo, S1, and S2 of the tdt ligand was folded 12.3° towards the oxo group.
Complex 6 is fundamentally different from complexes where the Mo-center is coordinated by two ene-1,2-dithiolate ligands. Both ligands are folded towards the terminal oxo-group, however, the dithione ligand has a fold angle that is 49° greater than the fully reduced ligand. [MoO(SPh)2(iPr2Dt0)] exhibited a fold of 70.47° along the S⋯S vector of iPr2Dt0, due to a strong pseudo Jahn–Teller (pJT) effect.15 We surmise that a similar distortion is operative in 6. The ∼8° smaller fold angle observed for 6 is a result of increased stability introduced by Dt2− chelation.
The five-coordinate Mo center in 6 exhibits a distorted square-pyramidal geometry as measured by the distortion parameter τ = (α − β/60) (where α = the largest basal angle and β = the second largest angle). The τ value for ideal square pyramidal geometry is 0.00.28–30 The τ value of 6 was determined to be 0.18, which indicates distortion from the ideal square pyramidal geometry. Such distortions have been observed in [MoIVO(Dt2−)2]2− complexes with τ values between 0.002–0.213.24 The distortion of 6 is a result of a pJT effect which also raises the Mo-center from the equatorial plane. The Mo-center was 0.84 Å above the equatorial plane created by the four sulfur donors, which is greater than the average value of MoIV-centers with a similar primary coordination sphere (0.74 Å).24 Raising the Mo-center above the sulfur plane stabilizes the dxy orbital to facilitate LL′CT. From the molecular structure of 6, it is evident that the chelating dithiolene ligands are present in differing oxidation states, and pJt effects produce distortion about the primary coordination sphere.
Redox chemistry
The redox reactivities of complexes 1–8 were investigated by cyclic voltammetry in MeCN solutions. Cyclic voltammograms for all complexes exhibited two, one-electron partially reversible redox couples due to the reduction of the Dt0 ligand. A representative cyclic voltammogram for 6 is shown in Fig. 2 and potentials are listed in Table 2. In addition, an irreversible metal centered oxidation couple was observed in 1–8. Plotting peak current against the square root of the scan rate for ligand-based couples produced a linear relationship, suggesting diffusion-controlled processes (Fig. S1†).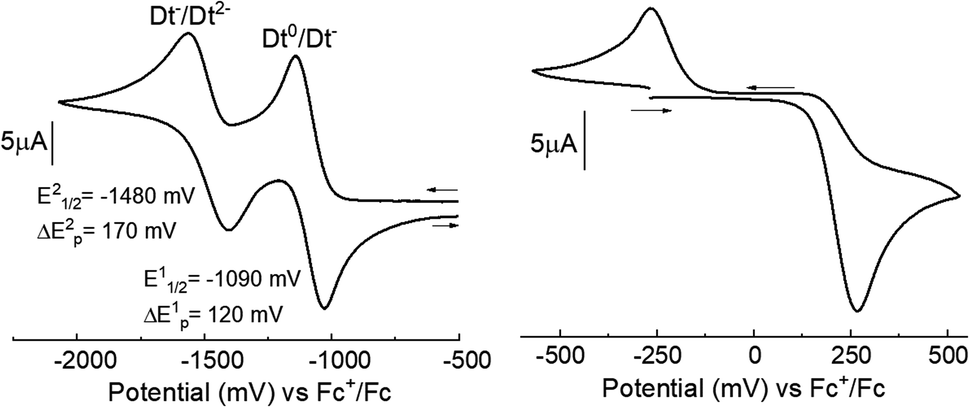 | ||
| Fig. 2 Cyclic voltammograms of 6 at a scan rate of 100 mV s−1. Experimental details are included with the physical methods of the ESI.† | ||
| Complex | E11/2(ΔE1p) (mV) | E21/2(ΔE2p) (mV) |
|---|---|---|
| [MoO(bdt)(Me2Dt0)] (1) | −1060(90) | −1370(100) |
| [MoO(tdt)(Me2Dt0)] (2) | −1070(70) | −1380(70) |
| [MoO(qdt)(Me2Dt0)] (3) | −950(90) | −1290(100) |
| [MoO(bdtCl2)(Me2Dt0)] (4) | −1000(90) | −1350(90) |
| [MoO(bdt)(iPr2Dt0)] (5) | −1060(100) | −1460(110) |
| [MoO(tdt)(iPr2Dt0)] (6) | −1090(120) | −1480(170) |
| [MoO(qdt)(iPr2Dt0)] (7) | −940(60) | −1400(120) |
| [MoO(bdtCl2)(iPr2Dt0)] (8) | −1000(110) | −1430(130) |
Two irreversible responses were observed at Epa 262 mV and Epc −284 mV for complex 6. The metal-based couples of previously reported complexes with the general formula: [MoIVO(Dt2−)2]2−, exhibit a reversible Mo(V)/Mo(IV) couple. For instance [MoO(bdt)2]2− exhibits a redox potential of −39 mV for the oxidation to the Mo(V) species.6 [MoO(tdt)2]2− and [MoO(mnt)2]2− exhibit redox potentials of −46 mV and 48 mV respectively for oxidation to Mo(V).6,22
Comparison between 6 and the previously reported complex Zn(mnt)(iPr2Dt0) and uncoordinated iPr2Dt0 ligand will help to better understand how coordination impacts ligand redox properties (Fig. S2 and S3†).31 The cyclic voltammogram of free iPr2Dt0 ligand exhibited two partially reversible reduction couples at −1887 mV (ΔEp = 134 mV) and −2088 mV (ΔEp = 141 mV), and [Zn(mnt)(iPr2Dt0)] produces two partially reversible reduction couples at −939 mV (ΔEp = 115 mV) and −1365 mV (ΔEp = 125 mV). For 6 and [Zn(mnt)(iPr2Dt0)], the ligand-based redox couples are shifted by 802 mV and 948 mV, respectively for the first couple and 604 mV and 723 mV for the second couple, respectively, compared to free iPr2Dt0. Therefore, coordination to a metal center facilitates reduction of the dithione ligand. The redox couples observed for 6 are 146 mV and 119 mV more negative when compared to those reported for [Zn(mnt)(iPr2Dt0)], suggesting that electron density is being donated to the Dt0 ligand by Mo. This trend is observed for all iPr2Dt0 complexes besides 7, which contains the electron withdrawing qdt ligand.
The electron-withdrawing and donating capabilities of both the Dt0 and Dt2− ligand impact the redox potentials. The addition of electron donating (tdt) or electron withdrawing (bdtCl2) resulted in relatively small (<60 mV) changes in redox potential, whereas the addition of another ring (qdt) resulted in 118 and 68 mV shifts of E11/2 and E21/2. The largest and most consistent shift (−98 mV) in Dt0 redox potential is observed for E21/2 between Me2Dt0 and iPr2Dt0 ligands. This suggests that the acceptor ligand is more sensitive to ligand substituents than the donor moiety of these complexes. The effects of Dt2− substituents indirectly influence the redox potentials of the Dt0 ligand through modulation of available Mo electron density. The influence of Dt2− on the redox couples of the Dt0 ligand show the sensitive nature of the electronic communication between ligands, which is mediated by the Mo center.
DFT and excited state calculations
The electronic structure of 6 was explored using density functional level of theory (DFT). The orbital energy diagram and corresponding molecular orbitals are presented in Fig. 3 and C2 population analysis is presented in Table S2.† The HOMO and HOMO−1 orbitals are ∼85% tdt ligand in character; ∼44% of which is contributed by S atoms. There is an increased metal orbital contribution in the virtual orbitals. The LUMO is composed of ∼69% iPr2Dt0 ligand with significant (∼25%) Mo dxy participation.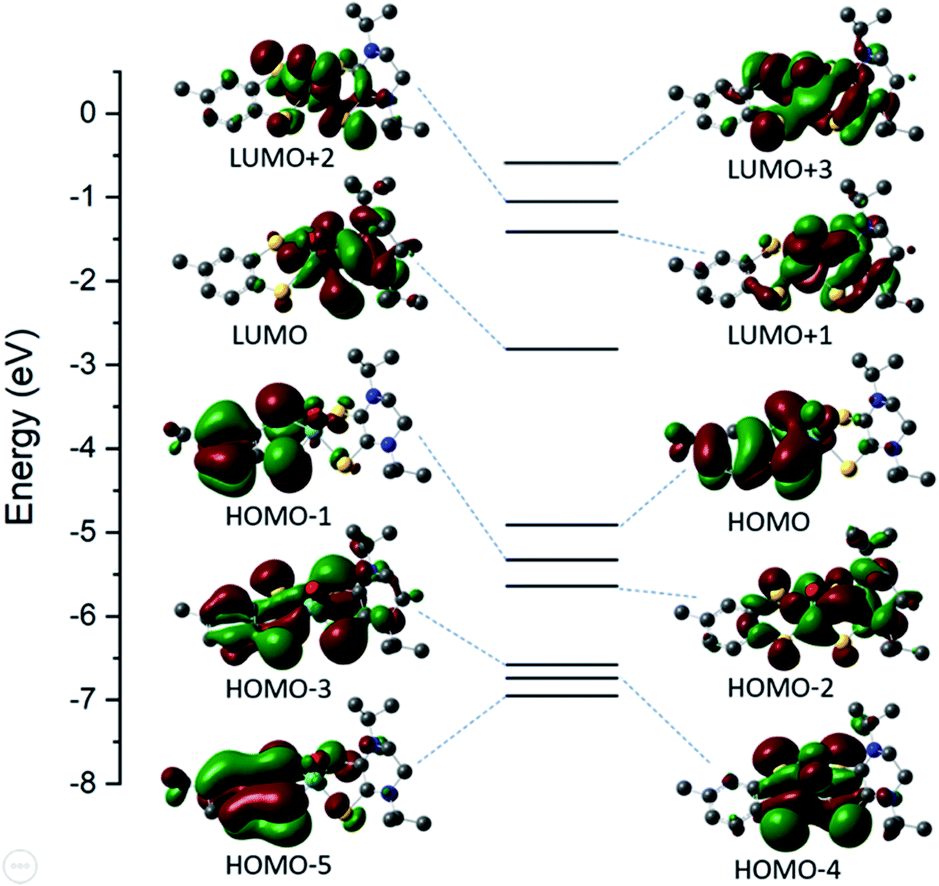 | ||
| Fig. 3 Energy diagram and corresponding molecular orbitals for 6. | ||
The electronic spectra for complexes 1–8 were recorded in MeCN (Fig. S4–S6†) and their low energy features are shown in Table 3. All complexes exhibit an intense low energy charge-transfer (CT) transition between 18903–18248 cm−1 and a higher energy transition between 2631524390 cm−1. The energy of the CT transition is affected by the solvent environment and complexes 1, 2, 4, 5, 6, and 8; all exhibit a positive solvatochromic effect (Fig. S7–S12†). Complexes 3 and 7 were found to only be stable in MeCN solutions which precluded collection of reliable spectral data in other organic solvents.
| Complex | λmax nm (ε, M−1 cm−1) | 2Hab (cm−1) |
|---|---|---|
| [MoO(bdt)(Me2Dt0)] (1) | 380 (1610), 532 (4400) | 4680 |
| [MoO(tdt)(Me2Dt0)] (2) | 380 (2320), 531 (6050) | 5360 |
| [MoO(qdt)(Me2Dt0)] (3) | 410 (6110), 548 (7450) | 6130 |
| [MoO(bdtCl2)(Me2Dt0)] (4) | 385 (2050), 531 (4460) | 4920 |
| [MoO(bdt)(iPr2Dt0)] (5) | 380 (2880), 529 (6900) | 6010 |
| [MoO(tdt)(iPr2Dt0)] (6) | 380 (2880), 533 (7500) | 6380 |
| [MoO(qdt)(iPr2Dt0)] (7) | 400 (8240), 543 (7070) | 5910 |
| [MoO(bdtCl2)(iPr2Dt0)] (8) | 390 (4070), 530 (9400) | 7540 |
Complexes 1–8 exhibit a markedly different electronic structure when compared to complexes with only Dt2− ligands.32 Electronically symmetric complexes exhibit a ligand-to-metal charge transfer band (LMCT) at 30487 cm−1 to 27472 cm−1.6,22,33,34 Complexes 1–8 exhibit an intense CT band that is red shifted by ∼6000 cm−1 from the LMCT observed in electronically symmetric complexes. The CT transition observed in complexes 1–8 is reminiscent of intense charge transfer transitions observed at ∼18000 cm−1 in [MoO(SPh)2(iPr2Dt0)], which was assigned to be a ligand–ligand charge transfer (LL′CT).15 The LL′CT band of [MoO(SPh)2(iPr2Dt0)] is observed at a higher energy to 6, due to the larger Dt0 fold angle. Larger fold angles misalign ligand-based donor and acceptor orbitals which increases the energy of the CT transitions.15
The low energy charge transfer process of 1–8 can be defined as a class II–III mixed-valence system (![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) 2Hab = 5900 cm−1).35 The Dt2− ligand serves as an electron donor while the Dt0 acts as an electron acceptor, forming a donor–acceptor system. This is similar to a previously reported non-innocent-dithiolene Mo system in which the Dt2− moiety serves as the electron donor in an intraligand charge transfer (ILCT) to quinoxaline π* orbitals.36,37 The electronic interaction is also consistent with the large fold angle observed in the solid state which facilitates increased orbital overlap between Dt0 and Mo moieties, allowing for more facile intramolecular electron transfer. The raised Mo-center observed in the crystallographic structure of 6 also helps to facilitate charge transfer by orienting the Mo dxy closer to the redox active orbitals of Dt0. This is supported by PCM-TDDFT calculations (Fig. 4) visualized with electron density difference maps (EDDMs).
2Hab = 5900 cm−1).35 The Dt2− ligand serves as an electron donor while the Dt0 acts as an electron acceptor, forming a donor–acceptor system. This is similar to a previously reported non-innocent-dithiolene Mo system in which the Dt2− moiety serves as the electron donor in an intraligand charge transfer (ILCT) to quinoxaline π* orbitals.36,37 The electronic interaction is also consistent with the large fold angle observed in the solid state which facilitates increased orbital overlap between Dt0 and Mo moieties, allowing for more facile intramolecular electron transfer. The raised Mo-center observed in the crystallographic structure of 6 also helps to facilitate charge transfer by orienting the Mo dxy closer to the redox active orbitals of Dt0. This is supported by PCM-TDDFT calculations (Fig. 4) visualized with electron density difference maps (EDDMs).
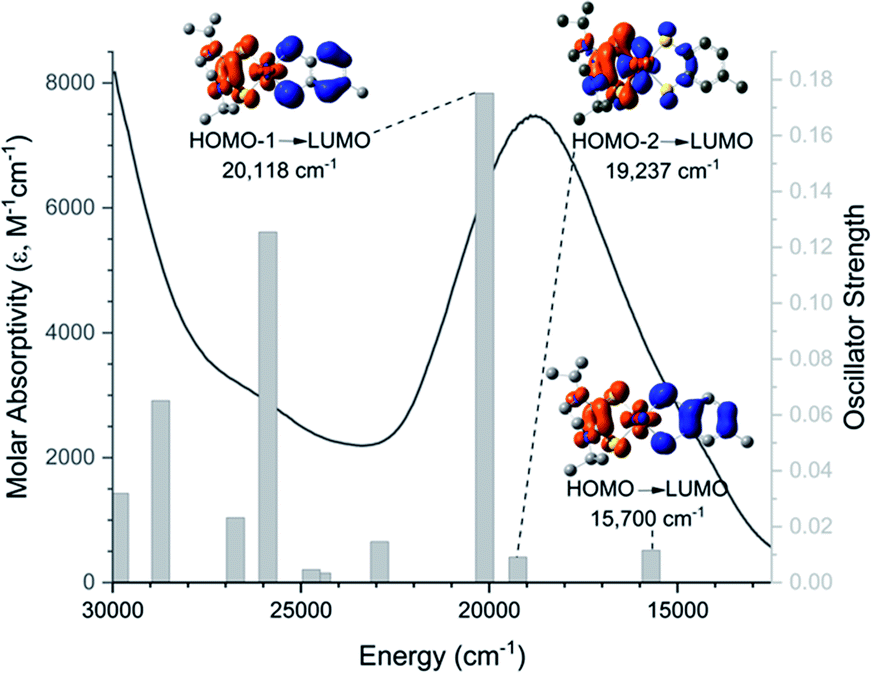 | ||
| Fig. 4 Calculated transitions (bars) imposed on experimental UV-Vis data of 6. PCM-TDDFT calculations were done using MeCN as the solvent to match experimental conditions. Transitions for the low energy band are paired with their corresponding electron density differential map (EDDM). Electron donating orbitals are blue and electron accepting orbitals are orange. | ||
Crystallographic data for a similar MoIV–Dt0 system shows the Dt0 fold angle to be sensitive to the electron donating and withdrawing properties of Dt0 substituents.38 Gas phase optimizations were performed on 2, 5, 7, and 8 to understand the intrinsic propensity of the Dt0 fold angle in relationship to dithiolene substituents. The Dt0 fold angle of 6 will be used as the reference since it is the only compound with a molecular structure. The substituents on the Dt2− ligand produced little change (<1°), whereas changing the substituent of the acceptor moiety resulted in a ∼5° difference in Dt0 fold angle (Table S3†). This is observed in the context of the electrochemical data, as the largest changes in redox potential are observed between corresponding Me2Dt0 and iPr2Dt0 complexes.
Correlations of the spectral data of 6 with the dipole moment of the different solvents the spectra were produce fits with R2 ∼ 0.90. The Kamlet–Taft model39,40 produces a better fit (R2 = 0.97) (eqn (1)) in which coefficients s, a and b are determined by regression analysis to be 1336; 23442; and 11962, respectively. The π* parameter describes the polarity and polarizability of the solvent, α is the hydrogen bond donation ability of the solvent, and β is the hydrogen bond acceptance ability of the solvent.39 Both α and β are stabilizing parameters that lower the energy of the electronic excited state, while π* plays a relatively small role in the solvatochromic shift.41 The ability for the solvent to provide hydrogen bonding to 6 could help to stabilize the electron deficient dithione ligand and facilitate LL′CT. The correlation coefficients for all complexes are found in the ESI (Tables S4 and S5†). Attempts to model solvatochromism with PCM-TD-DFT calculations were unsatisfactory due to limitations of implicit solvent methods.
| E (cm−1) = 25,891 + 1336 (π*) − 23,442(α) − 11,962(β) | (1) |
Complexes 1–8 are the first examples of oxo-Mo(IV) complex possessing two dithiolene ligands in different oxidation states. The interligand communication between dithiolene units is highlighted by the strong LL′CT observed in their absorption spectra and the relationship between the ligand structure and oxidation state. The electron deficient Dt0 ligand contains a large ligand fold angle to form a stabilizing interaction with the Mo dxy orbital. This is not observed for the Dt2− ligand because it is electron rich and does not need to fold towards the metal for stability.
Implications to dithiolene containing enzymes
The redox-active nature of dithiolene moieties suggests that their participation in biological processes can result in various dithiolene-based redox states. Indeed, a resonance Raman (rR) study on xanthine oxidase demonstrates that the dithiolene containing pterin cofactor participates in electron transfer processes.42 The coordination of multiple dithiolene units, such as molybdenum and tungsten containing enzymes, can produce dithiolene moieties of different oxidation states,43,44 not dissimilar to what is described in this work. There is also evidence of pterin ring redox-activity in YedY, a protein with a sulfite oxidase-like Mo biding domain,45 but the complex nature of the cofactor requires careful analysis of each redox-active unit to understand their impact on interligand communication. In the context of a biological system, the orientation and fold angle of dithiolene ligands could influence dithiolene oxidation state, interligand communication between dithiolene ligands, and partial charge of the Mo atom. Such a mechanism could be used to fine tune reactivity as different protein conformations could change dithiolene fold angles and influence cofactor reactivity. It is unlikely that fully oxidized dithiolene is fully realized in biological systems, however, the framework of a donor–acceptor system between dithiolene units is still applicable to understanding how dithiolene units electronically communicate and the role they play in a biological framework.Conclusions
Non-innocent ligands play a crucial role in a multitude of biological processes. Dithiolene ligands are just one type of non-innocent ligand which is known to exist in various molybdenum and tungsten containing enzyme. Interligand communication between dithiolene ligands has been reported using the first examples of electronically asymmetric monooxo-Mo(IV) dithiolene complexes. Complexes 1–8 have been fully characterized, and the molecular structure of 6 confirms the presence of both discrete oxidized and reduced dithiolene ligands bound to a MoIV-oxo center. Theoretical calculations and experimental results support interligand communication as an intense charge transfer band at ∼529 nm which has been assigned as a LL′CT (Dt2− π → Dt0 π*). Calculating the Hab of 6 indicates these complexes to be class II–III mixed valence systems. It has been proposed that the redox states of the dithiolene ligands coordinated to the molybdenum cofactor (Moco), of the DMSO reductase family of enzymes, are different,46,47 and the impact of such a system is highlighted in his work. The fold angle of the fully oxidized dithiolene ligand observed in the crystal structure is much larger than that of the fully reduced ligand. Dithiolene fold angle in the context of a biological system could influence ligand oxidation state and result in changes at the Mo atom that influence the reactivity of the cofactor.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the School of Science, IUPUI, is gratefully acknowledged. We acknowledge the National Institutes of Health (GM061555 and GM139064) for partial support. This research was supported in part by Lilly Endowment, Inc., through its support for Indiana University Pervasive Technology Institute, and in part by the Indiana METACyt Initiative. The Indiana MTACyt Initiative at IU was also supported in part by Lilly Endowment, Inc.Notes and references
- R. Mukherjee, Inorg. Chem., 2020, 59, 12961–12977 CrossRef CAS PubMed.
- W. Kaim and B. Schwederski, Coord. Chem. Rev., 2010, 254, 1580–1588 CrossRef CAS.
- M. M. Whittaker and J. W. Whittaker, Biophys. J., 1993, 64, 762–772 CrossRef CAS PubMed.
- H. Oshita, T. Suzuki, K. Kawashima, H. Abe, F. Tani, S. Mori, T. Yajima and Y. Shimazaki, Chem.–Eur. J., 2019, 25, 7649–7658 CrossRef CAS PubMed.
- Y. Wang, J. L. DuBois, B. Hedman, K. O. Hodgson and T. D. P. Stack, Science, 1998, 279, 537–540 CrossRef CAS PubMed.
- J. P. Donahue, C. R. Goldsmith, U. Nadiminti and R. H. Holm, J. Am. Chem. Soc., 1998, 120, 12869–12881 CrossRef CAS.
- G. Moula, M. Bose and S. Sarkar, New J. Chem., 2019, 43, 8332–8340 RSC.
- H. Sugimoto, M. Sato, K. Asano, T. Suzuki, T. Ogura and S. Itoh, Inorg. Chim. Acta, 2019, 485, 42–48 CrossRef CAS.
- V. N. Nemykin, J. G. Olsen, E. Perera and P. Basu, Inorg. Chem., 2006, 45, 3557–3568 CrossRef CAS PubMed.
- B. Mogesa, E. Perera, H. M. Rhoda, J. K. Gibson, J. Oomens, G. Berden, M. J. van Stipdonk, V. N. Nemykin and P. Basu, Inorg. Chem., 2015, 54, 7703–7716 CrossRef CAS PubMed.
- E. Perera and P. Basu, Dalton Trans., 2009, 5023–5028 RSC.
- F. Thomas, Dalton Trans., 2016, 45, 10866–10877 RSC.
- A. Saha, A. Rajput, P. Gupta and R. Mukherjee, Dalton Trans., 2020, 49, 15355–15375 RSC.
- A. Rajput, A. K. Sharma, S. K. Barman, D. Koley, M. Steinert and R. Mukherjee, Inorg. Chem., 2014, 53, 36–48 CrossRef CAS PubMed.
- J. Yang, B. Mogesa, P. Basu and M. L. Kirk, Inorg. Chem., 2016, 55, 785–793 CrossRef CAS PubMed.
- R. P. Mtei, E. Perera, B. Mogesa, B. Stein, P. Basu and M. L. Kirk, Eur. J. Inorg. Chem., 2011, 2011, 5467–5470 CrossRef CAS PubMed.
- P. J. Chirik, Inorg. Chem., 2011, 50, 9737–9740 CrossRef CAS PubMed.
- G. Skara, B. Pinter, P. Geerlings and F. De Proft, Chem. Sci., 2015, 6, 4109–4117 RSC.
- R. Eisenberg and H. B. Gray, Inorg. Chem., 2011, 50, 9741–9751 CrossRef CAS PubMed.
- P. Basu, A. Nigam, B. Mogesa, S. Denti and V. N. Nemykin, Inorg. Chim. Acta, 2010, 363, 2857–2864 CrossRef CAS PubMed.
- S. Boyde, S. R. Ellis, C. D. Garner and W. Clegg, J. Chem. Soc., Chem. Commun., 1986, 1541–1543 RSC.
- H. Oku, N. Ueyama, A. Nakamura, Y. Kai and N. Kanehisa, Chem. Lett., 1994, 607–610 CrossRef CAS.
- P. P. Samuel, S. Horn, A. Doering, K. G. V. Havelius, S. Reschke, S. Leimkuehler, M. Haumann and C. Schulzke, Eur. J. Inorg. Chem., 2011, 2011, 4387–4399 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed , The Cambridge Structural Database, accessed April 28, 2021..
- J. W. Lauher and R. Hoffmann, J. Am. Chem. Soc., 1976, 98, 1729–1742 CrossRef CAS.
- H. K. Joshi, J. J. A. Cooney, F. E. Inscore, N. E. Gruhn, D. L. Lichtenberger and J. H. Enemark, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3719–3724 CrossRef CAS PubMed.
- N. J. Wiebelhaus, M. A. Cranswick, E. L. Klein, L. T. Lockett, D. L. Lichtenberger and J. H. Enemark, Inorg. Chem., 2011, 50, 11021–11031 CrossRef CAS PubMed.
- D. S. Marlin, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2001, 40, 7003–7008 CrossRef CAS PubMed.
- M. Marchivie, P. Guionneau, J. F. Letard and D. Chasseau, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 25–28 CrossRef PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- S. C. Ratvasky, B. Mogesa, M. J. van Stipdonk and P. Basu, Polyhedron, 2016, 114, 370–377 CrossRef CAS PubMed.
- K. Ray, T. Petrenko, K. Wieghardt and F. Neese, Dalton Trans., 2007, 1552–1566 RSC.
- H. Sugimoto, M. Tarumizu, K. Tanaka, H. Miyake and H. Tsukube, Dalton Trans., 2005, 3558–3565 RSC.
- E. S. Davies, R. L. Beddoes, D. Collison, A. Dinsmore, A. Docrat, J. A. Joule, C. R. Wilson and C. D. Garner, J. Chem. Soc., Dalton Trans., 1997, 3985–3996 RSC.
- K. D. Demadis, C. M. Hartshorn and T. J. Meyer, Chem. Rev., 2001, 101, 2655–2685 CrossRef CAS PubMed.
- K. G. Matz, R. P. Mtei, B. Leung, S. J. N. Burgmayer and M. L. Kirk, J. Am. Chem. Soc., 2010, 132, 7830–7831 CrossRef CAS PubMed.
- K. G. Matz, R. P. Mtei, R. Rothstein, M. L. Kirk and S. J. N. Burgmayer, Inorg. Chem., 2011, 50, 9804–9815 CrossRef CAS PubMed.
- S. A. Dille, K. J. Colston, B. Mogesa, J. Cassell, E. Perera, M. Zeller and P. Basu, Eur. J. Inorg. Chem., 2021, 2021, 914–922 CrossRef CAS.
- M. J. Kamlet, J. L. Abboud and R. W. Taft, J. Am. Chem. Soc., 1977, 99, 6027–6038 CrossRef CAS.
- M. J. Kamlet and R. W. Taft, J. Am. Chem. Soc., 1976, 98, 377–383 CrossRef CAS.
- W. J. Cheong and P. W. Carr, Anal. Chem., 1988, 60, 820–826 CrossRef CAS.
- C. Dong, J. Yang, S. Leimkuehler and M. L. Kirk, Inorg. Chem., 2014, 53, 7077–7079 CrossRef CAS PubMed.
- P. Basu and S. J. N. Burgmayer, Coord. Chem. Rev., 2011, 255, 1016–1038 CrossRef CAS PubMed.
- P. Basu and S. J. N. Burgmayer, J. Biol. Inorg Chem., 2015, 20, 373–383 CrossRef CAS PubMed.
- C. C. Lee, N. S. Sickerman, Y. Hu and M. W. Ribbe, ChemBioChem, 2016, 17, 453–455 CrossRef CAS PubMed.
- R. A. Rothery, B. Stein, M. Solomonson, M. L. Kirk and J. H. Weiner, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14773–14778 CrossRef CAS PubMed.
- R. Hille, J. Hall and P. Basu, Chem. Rev., 2014, 114, 3963–4038 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2021145. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra04716g |
| This journal is © The Royal Society of Chemistry 2021 |