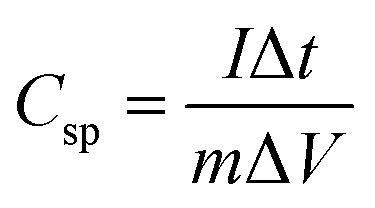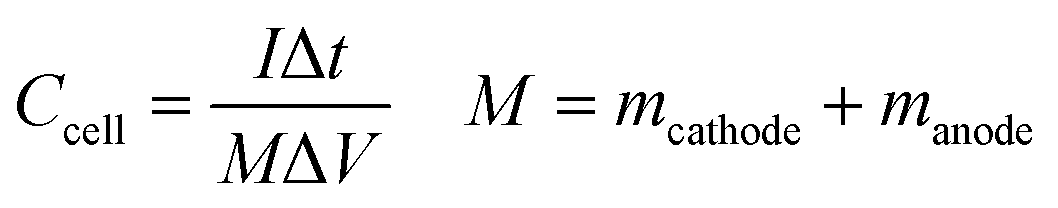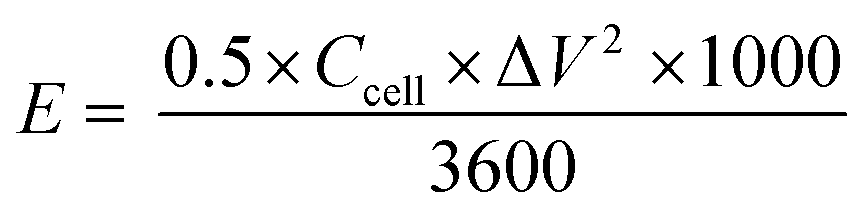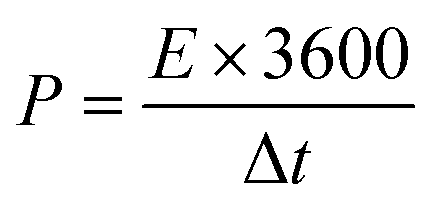DOI:
10.1039/D1RA04946A
(Paper)
RSC Adv., 2021,
11, 35828-35841
Ternary 3D reduced graphene oxide/Ni0.5Zn0.5Fe2O4/polyindole nanocomposite for supercapacitor electrode application†
Received
25th June 2021
, Accepted 21st October 2021
First published on 5th November 2021
Abstract
A facile two-step strategy has been reported for the preparation of a ternary 3D reduced graphene oxide/Ni0.5Zn0.5Fe2O4/polyindole nanocomposite (GNP) and this composite is applied as an electrode material for supercapacitor applications. Remarkably, Ni0.5Zn0.5Fe2O4 nanoparticles (NZF) decorated on reduced graphene oxide (GN2) are achieved by a facile hydrothermal method followed by coating with polyindole (PIN) through an in situ emulsion polymerization process. The structure, porosity, morphology, and thermal stability of the resulting ternary GNP hybrid material were characterized via X-ray diffraction (XRD), Raman spectroscopy, Brunauer–Emmett–Teller (BET) surface area measurements, transmission electron microscopy (TEM), and thermogravimetric analysis (TGA). This combination of hybrid material has a favorable mesoporous structure that enables high exposure of active sites for fast electron transport for supercapacitor applications. We demonstrate here that the ternary GNP hybrid electrode material is capable of delivering a favorable specific capacitance of ∼320 F g−1 at 0.3 A g−1 within the potential range from −0.1 to 1 V, with desirable rate stability and excellent cycling stability in the three-electrode system. Furthermore, an asymmetric supercapacitor (ASC) of a two-electrode configuration was fabricated using 3D RGO and GNP as the negative and positive electrodes, respectively. Such a device manifests a favourable Csp of 48.9 F g−1 at 0.5 A g−1 and retains stability of 84% even after 2000 cycles. This ASC device exhibits a significant energy density of 16.38 W h kg−1 at a power density of 1784 W kg−1. The synergistic effects of pseudo and double layer capacitive contributions from PIN and GN2 make this ternary GNP hybrid electrode material of great promise in supercapacitor applications.
Introduction
With the ever-increasing demand for renewable energy sources to fulfill future energy needs, electrochemical means for energy storage with high performance and environmentally friendly properties is of great significance.1,2 In this scenario, the development of supercapacitors (SCs) as a novel kind of energy storage device with high energy and power density has become one of the most targeted research areas.3 In line with the search for such novel electrode materials, recent research efforts have been focused on imparting modifications to the existing state-of-art materials, that are capable of meeting the real-time requirements for several applications.4,5 Conducting polymers have received unprecedented research interest and offer high pseudocapacitance through their rapid redox process and fast charge–discharge process.6 As a kind of conducting polymer, polyindole (PIN), a state-of-the-art pseudocapacitor material, is a suitable candidate for charge storage applications.7–10 PIN possessed the properties of both poly(para-phenylene) and PPy such as air-stable electrical conductance,11 slow hydrolytic degradation,11 high redox activity,12 high cycling stability and, excellent thermal stability as compared to PANI and PPy.13,14 Meanwhile, PIN is a non-planar, light-weight, very loosely compact, and arbitrarily oriented polymer with chain unevenness leading to low conductivity.15 To overcome these loopholes and to make it technically feasible, nanoparticle-induced moderations in PIN are carried out. Hence, heterogeneity increases the stereoregularity, provides better electrostatic orientation, and results in increased conducting properties.16 Mandira Majumder et al. reported the incorporation rare earth metal oxides in the PIN matrix as an effective way for improving the electrochemical and stability features of PIN.17 Xi Zhou et al. reported V2O5/polyindole (V2O5/PIN) decorated activated carbon cloth with a specific capacitance of 535.5 F g−1 at the current density of 1 A g−1.18
Recently binary transition metal oxides, especially spinel ferrites (MFe2O4, M = Mn, Co, Ni, Zn, or Mg) are of great interest as qualified pseudocapacitive electrode material.19–21 In comparison with the monometallic oxides, spinel ferrites are capable of triggering synergetic effects from both Fe and M ions, results in richer redox chemistry and provide the feasibility to tune the energy density.21,22 It has been reported that ZnFe2O4, NiFe2O4, Co3O4, and MnFe2O4 possess superior pseudocapacitance.23–26 As an important member of the mixed spinel ferrite family, Ni0.5Zn0.5Fe2O4 (NZF) exhibits promising potential to be a supercapacitor, due to good mechanical hardness, high thermal and chemical stability, easy synthesis, environmental friendliness, and high redox activity.27–29 Nevertheless, NZF has rarely been explored as a potential candidate for pseudocapacitors. D. K. Pawar et al. reported NZF thin film synthesized via chemical deposition method with a specific capacitance of 67 F g−1.27 A promising approach to enhance supercapacitative performance is designing NZF based hybrids. Taking into account that a binary hybrid system based on PIN and NZF can afford an enhanced electrochemical performance. However, the main shortcoming of such a pseudocapacitor electrode is poor cycling stability and unsatisfactory rate capability.22 Therefore it is significant to control the sizes and morphologies of PIN and NZF by dispersing them on high surface area support.
Among the carbonaceous materials with a porous structure, large surface area, and exceptional high conductivity, 3D graphene hydrogel/aerogel represents an attractive electrode material for supercapacitors.30,31 It has been certified that nanoparticles can be captured into the graphene network during the network formation in a single step process and prevent the nanomaterial from aggregation through balancing their high interface energy.32 Simultaneously, the presence of graphene can avoid any mechanical deformation in the redox process and sustain the basic structures of conducting polymer.33 The fabrication of hybrid material with graphene will result in the immobilization of active species to provide good electron transfer paths and thereby improve the stability of the entire hybrid.
In this context, we report a facile method to improve the performance of polyindole (PIN) via synthesis of a ternary 3D reduced graphene oxide/Ni0.5Zn0.5Fe2O4/polyindole (GNP) nanocomposite. A two-step synthesis method is involved in this design of GNP hybrid materials. Firstly, the binary 3D reduced graphene oxide/Ni0.5Zn0.5Fe2O4 (GN) hybrid is synthesized via a simple hydrothermal route. The nanoparticles homogeneously dispersed in the aqueous suspension of graphene oxide can be self-assembled together to form a network during reduction. Further, an in situ polymerization strategy was adopted for coating polyindole on the surface of binary GN hybrid material from ternary GNP hybrid. Such a ternary GNP hybrid is expected to tackle the drawbacks of each component, thereby leading to an enhanced specific capacitance and an excellent cycling life for supercapacitor applications.
Experimental section
Chemicals and materials
Natural graphite (99.5% pure, particle size < 50 mm) and Nafion (5% solution) were purchased from Alfa Aesar. Sodium nitrate (NaNO3), sulfuric acid (98% H2SO4), hydrochloric acid (HCl), potassium permanganate (KMnO4), hydrogen peroxide (30% H2O2) from Merck. Ferric chloride (FeCl3), iron nitrate (Fe(NO3)3·9H2O), zinc nitrate (Zn(NO3)2·6H2O), nickel nitrate (Ni(NO3)2·6H2O) with 99% of purity were obtained from Sigma Aldrich. Indole powder (monomer), ferric chloride (FeCl3), sodium dodecylbenzenesulfonate (SDBS), and citric acid were procured from Himedia – India. The ITO plates (1 cm × 1 cm × 1.5 mm) were used as the electrode substrate. Double distilled water and ethanol were used as a solvent for the synthesis.
Synthesis of Ni0.5Zn0.5Fe2O4 (NZF) nanoparticles
Ni0.5Zn0.5Fe2O4 nanoparticles have been synthesized by sol–gel auto combustion method.34 For the synthesis, nickel nitrate, zinc nitrate, and iron nitrate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:4 molar ratio) solutions were mixed thoroughly with citric acid (1:2.77 molar ratio with nitrates) to get a clear solution. A small amount of ammonia is added dropwise to the solution to adjust the pH value to 7. During this procedure, the solution was continuously stirred using a magnetic stirrer and kept at a temperature of 90 °C until gel forms. Then heated to 150 °C leads to the formation of nanopowders through a self-propagating combustion process. The loose powder was crushed well and calcined at 550 °C for 4 h to form the spinal phase.
:1:4 molar ratio) solutions were mixed thoroughly with citric acid (1:2.77 molar ratio with nitrates) to get a clear solution. A small amount of ammonia is added dropwise to the solution to adjust the pH value to 7. During this procedure, the solution was continuously stirred using a magnetic stirrer and kept at a temperature of 90 °C until gel forms. Then heated to 150 °C leads to the formation of nanopowders through a self-propagating combustion process. The loose powder was crushed well and calcined at 550 °C for 4 h to form the spinal phase.
Synthesis of PIN/10 Ni0.5Zn0.5Fe2O4 (PN10) nanocomposite
PIN/10 Ni0.5Zn0.5Fe2O4 nanocomposite (PN10) was synthesized via in situ polymerization of indole in an aqueous solution containing NZF nanofluid using FeCl3 as the oxidizing agent. 0.3 g NZF nanoparticles were mixed with 10 ml of SDBS solution and ultrasonicated for 10 min. Subsequently, the above mixture was gradually added into a 50 ml ethanol–water solution containing 3 g indole with SDBS (0.293 g) and sonicated for 30 minutes. Finally, 1.75 g of FeCl3 in 0.1 M HCl was added dropwise and the solution became to be a dark green color. The polymerization reaction was carried out for 24 h with continuous magnetic stirring. The precipitate finally obtained was washed by distilled water and ethanol several times before drying in an oven at 70 °C overnight to obtain PN10 nanopowders. Similarly, pure PIN powders were also synthesized via a simple chemical oxidative emulsion polymerization of indole in an aqueous solution of SDBS using FeCl3 as an oxidizing agent.35
Synthesis of graphite oxide (GO)
GO was synthesized from natural graphite powder according to the modified Hummers' method.36,37 Briefly, 1.0 g of graphite powder and 1.0 g NaNO3 was mixed with 96.0 ml conc. H2SO4 in an ice bath for 4 h. Under vigorous stirring, potassium permanganate (6.0 g) was added gradually to keep the temperature lower than 20 °C. Successively the ice bath was removed and the temperature of suspension was brought to 35–40 °C for about half an hour forming a thick brownish color paste. The pasty liquid was weakened with the slow addition of 200 ml of water and the solution was stirred for another 15 min. Finally, 20 ml of 30% H2O2 was added to the reaction mixture, resulting in a change of color from brown to brilliant yellow along with bubbling. The mixture was filtered and then washed with a 10% HCl aqueous solution (250 ml) by centrifugation to remove metal ions. This is followed by repeated washing with distilled water to remove the acid. The powder obtained was dried in a vacuum oven at 60 °C.
Synthesis of 3D reduced graphene oxide/Ni0.5Zn0.5Fe2O4 (GN) hydrogel
Binary 3D reduced graphene oxide/Ni0.5Zn0.5Fe2O4 (GN) nanocomposite hydrogel was synthesized via the hydrothermal method with GO and Ni0.5Zn0.5Fe2O4 (NZF) nanoparticles as the raw materials.38 For the synthesis, 2 mg ml−1 GO and different concentrations of NZF nanoparticles were uniformly dispersed in a solution of ethanol ad glycerol (ratio 3:1) in a 120 ml Teflon lined autoclave at 180 °C for 12 h. Two composites with the weight ratios of the raw material as GO to NZF nanoparticles, 1:1 (GN1) and 1:2 (GN2) were prepared. After the reaction was completed, the autoclave was allowed to cool naturally to room temperature in air and as-synthesized hydrogels were taken out with tweezers. Subsequently, the hydrogels were freeze-dried under a vacuum. Similarly, 3D RGO was also synthesized via the hydrothermal method in the absence of NZF nanoparticles.
Synthesis of 3D reduced graphene oxide/Ni0.5Zn0.5Fe2O4/polyindole (GNP) nanocomposites
Ternary 3D reduced graphene oxide/Ni0.5Zn0.5Fe2O4/polyindole (GNP) nanocomposites are prepared via in situ polymerization of indole monomers in the presence of binary GN2. In a typical procedure, 0.5 g indole and sodium dodecylbenzenesulfonate (SDBS) in a molar ratio of 1:0.2 were mixed in 20 ml of ethanol and sonicated for 20 minutes. To this reaction mixture, 1 g of GN composite was added and further sonicated for another 20 minutes to a homogeneous solution. Subsequently, 20 ml of ferric chloride (1:2.57 molar ratios) were added to this mixture dropwise under vigorous stirring and after which, the resulting mixture was allowed to polymerize under stirring for 24 h at room temperature. Finally, the ternary GNP hybrid was filtered out and washed with deionized water and ethanol. Then dried under vacuum at 60 °C.
Characterizations
The material characterization such as phase identification, porosity, morphology, and chemical compositions of PIN and its composites were carried using powdered X-ray diffractometer (X'pert3 Powder with CuKα (λ = 1.5406 Å) radiation), Raman spectra were conducted on a laser micro-Raman spectrometer (JASCO NRS-4100), N2 adsorption/desorption isotherms were measured from BELSORP max apparatus and morphology study conducted by a transmission electron microscope using JEOL/JEM2100 with 200 kV accelerating voltage and lattice resolution of 0.14 nm. Thermogravimetric analysis (TGA) was performed on a STA 8000 TG-DTA analyzer. All cyclic voltammetry and charging–discharging measurements were performed using Zahner Zennium Pro electrochemical workstation attached with a personal computer and employing Thales XT electrochemical software and the impedance spectra were analyzed using semi-quantitative fittings program supplied by Zman Ver2.2 software.
Electrochemical characterization and fabrication of the asymmetric supercapacitor (ASC) device
A three-electrode cell consisted of the counter electrode (Pt foil 1 × 1 cm2), the reference electrode (Ag/AgCl electrode), and the working electrode (glassy carbon electrode) employed to determine the electrochemical performance of electrode materials. The fabrication of the working electrode was presented as follows. Firstly, the active material powder, Vulcan carbon powder, and Nafion solution with a weight ratio of 80:10:10 were mixed 50 μl isopropanol to form a homogeneous slurry and 5 μl coated on a glassy carbon electrode. Finally, the prepared electrodes were dried in a vacuum at 60 °C. The total weight of active material on the electrode is 0.2 mg. All the electrochemical tests were conducted at room temperature with a 1 M H2SO4 aqueous solution as the electrolyte. Also, an asymmetric supercapacitor (ASC) of the two-electrode configuration was fabricated with GNP as the positive electrode and 3D RGO as the negative electrode, respectively. In this ASC device, the electrodes were prepared by mixing 80 wt% of the active materials and 20 wt% conductive carbon paste powder in a 3:1 water/isopropanol mixture followed by the uniform coating of this slurry onto a 1 × 1 cm2 area of a clean ITO plate and drying in air overnight. A cellulose filter paper (Whatman filter paper 42) soaked in a 1 M H2SO4 aqueous solution was sandwiched between the two electrodes and used as the separator. The mass loading ratio of active material on positive (m+) and the negative electrode (m−) is confirmed by the equation, m+/m− = C−ΔV−/C+ΔV+. Here C−, C+, ΔV−, and ΔV+ represent the specific capacitances of the 3D RGO electrode and GNP electrode and the potential window of the 3D RGO electrode and GNP electrode, respectively. The area loading of the material on the cathode is 0.4 mg cm−2.
Specific capacitance values (C) of the materials were calculated from cyclic voltammograms using the equation,39
| |
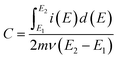 | (1) |
where
E1 and
E2 are the cut off potentials in cyclic voltammetry,
i(
E) is the instantaneous current,
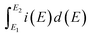
is the total voltammetric charge obtained by integration of the positive and negative sweeps in the cyclic voltammograms,
ν is the scan rate, and
m is the mass of the individual sample. When the charge–discharge method was used, the specific capacitance can be calculated according to the equation,
39| |
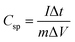 | (2) |
where
Csp is specific capacitance (F g
−1),
I and Δ
t are the discharge current and time, respectively, Δ
V is the potential range, and
m is the mass loading of electrode material on the GC electrode. Notably, the specific capacitance value of the ASC device can be expressed as,
| |
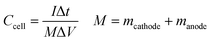 | (3) |
where
Ccell is the specific capacitance of the device and
M is the total mass of active materials deposited on both positive and negative electrodes.
The energy and power density of an ASC device can be derived by,
| |
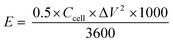 | (4) |
| |
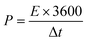 | (5) |
where
E (W h kg
−1) is the energy density,
P (W kg
−1) is the power density,
Ccell (F g
−1) is the specific capacitance of the device, Δ
V is the potential window, and Δ
t is the discharge time.
Results and discussion
Phase structure and morphology
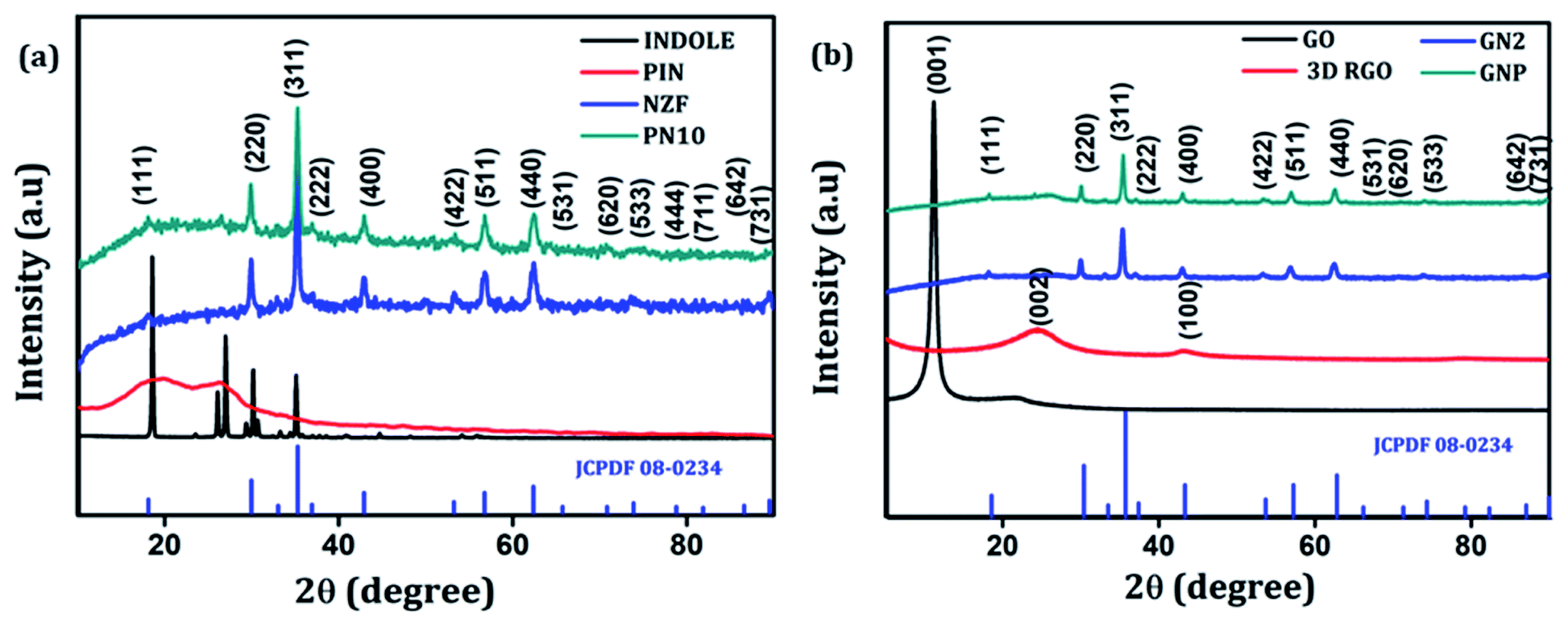
The crystallographic structure and phase purity of the as-prepared samples were examined by powder XRD analysis. The XRD diffraction patterns of indole, PIN, NZF, and PN10 are shown in Fig. 1(a), whereas GO, 3D RGO, GN2, and GNP composite diffraction patterns are shown in Fig. 1(b). It is evident from Fig. 1(a) that indole shows sharp crystalline peaks whereas PIN shows a broad peak at 18.8° and another one at 27.2°. The presence of broad peaks confirms the polymerization of indole and the diffraction peak at 27.2° indicates the presence of partial crystalline nature of polyindole.40 The XRD pattern of NZF gives characteristic diffraction planes at (111), (220), (311), (222), (400), (422), (511), (440), (531), (620), (533), (444), (711), (642), and (731) which can be indexed to the cubic spinel structure of NZF (JCPDF 08-0234). Diffraction patterns of PN10 displayed the signature peaks for NZF nanoparticles which suggest that nanoparticles have been successfully incorporated into the PIN matrix. In Fig. 1(b), pure GO shows a diffraction peak at 2θ = 11.1° attributed to the (001) reflection of the graphitic plane, corresponding to an inter-planar spacing of 7.91 Å.41 As a contrast, a broad (002) diffraction peak can be observed at 24.5° for 3D RGO with an interlayer spacing of 3.62 Å, which indicates restacking of GO under the hydrothermal conditions.42 The disappearance of the peak located at 11.1° in the diffraction pattern of 3D RGO can be assigned to the thorough reduction from GO to RGO. Such a broad peak in 3D RGO not only indicates the poor ordering of graphene sheets along their stacking direction but reflects that the framework of the 3D RGO is composed of few-layer stacked graphene sheets.43 Furthermore, it can be seen that almost all the diffraction peaks of GN2 are well-matched with the standard pattern of NZF nanoparticles (JCPDF 08-0234). However, the typical diffraction peak of graphene (002) is not observable in the XRD pattern for GN2 hydrogel. This shows that during the hydrothermal reaction, the crystal growth of NZF nanoparticles between the interlayers of graphene may destroy the regular layer stacking and lead to the exfoliation of the graphene sheet.19 For GNP nanocomposite, the diffraction peaks of NZF and broad peaks of the PIN can be observed, which suggests that NZF and PIN coexist in the prepared ternary nanocomposite.43
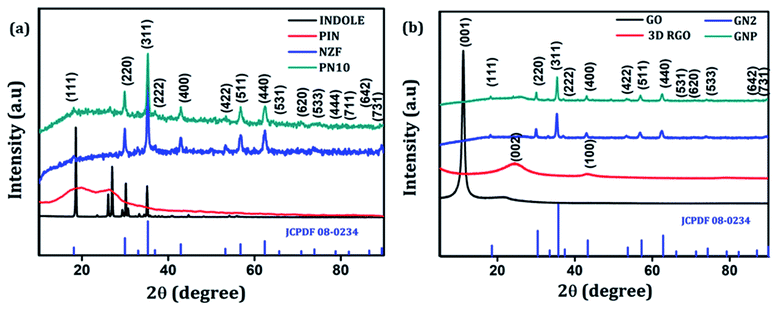 |
| | Fig. 1 (a) XRD patterns of indole, PIN, NZF, and PN10 and (b) XRD patterns of GO, 3D RGO, GN2 and GNP. | |
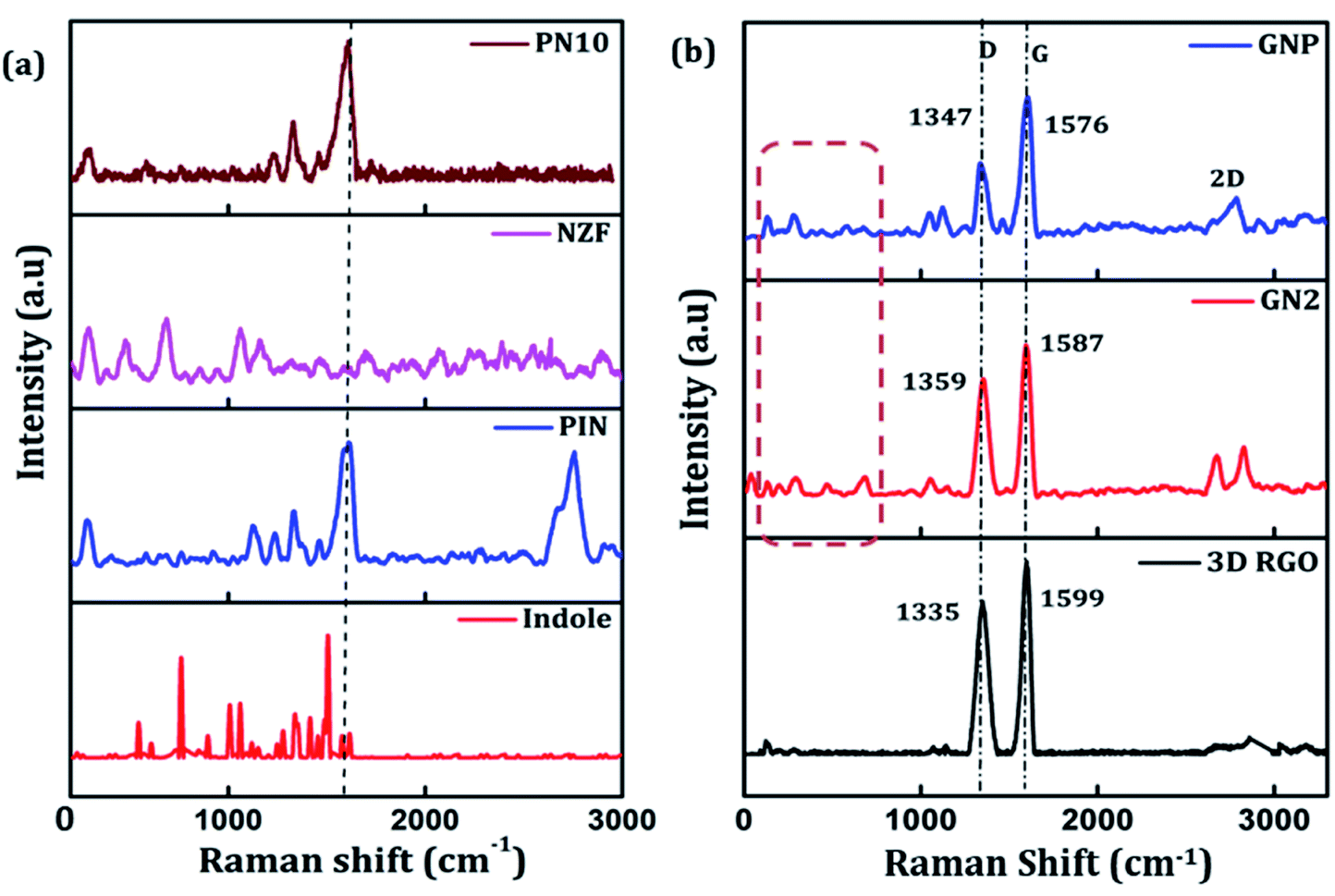
Raman spectroscopy can be employed to further determine the degree of graphitization and the crystalline structure of the prepared samples. Raman spectra of indole, PIN, NZF, and PN10 are shown in Fig. 2(a). The spectrum of PIN displayed characteristic Raman bands at 1220, 1333, 1390, 1460, and 1595 cm−1 corresponding to various C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC stretching vibrations.44,45 The strong bands at 1503 and 1145 cm−1 which attributed to modes (νN1C2 + δC2H) and (νC3H + νNC2) are intense for indole monomer and almost absent in PIN, conforming to the polymerization of indole monomers linked together via positions 2 and 3 of the pyrrole moiety.46 The diffraction pattern of NZF nanoparticles shows a high-frequency first-order mode A1g at 650–700 cm−1 and modes F2g(2) and F2g(3) at 450–550 cm−1, attributed to the symmetric stretching of oxygen atoms along with M–O bonds in the tetrahedral and octahedral coordination respectively.47 Compared to PIN, the characteristic C–N stretching band of PN10 nanocomposite was located at 1613 cm−1, and the frequency difference was measured to 18 cm−1. Thus the interaction of NZF nanoparticles with the PIN matrix can be visible from the Raman spectra of PN10 nanocomposites. Besides, the characteristic peaks of both PIN and NZF nanoparticles have appeared for the PN10 nanocomposite. This implied that the PN10 nanocomposite was successfully obtained. The intensity of peaks corresponding to NZF is greatly diminished, indicating the good wrapping of nanoparticles within the PIN matrix. The structure of the 3D RGO, GN2, and GNP nanocomposites was also characterized by Raman spectra and the results are shown in Fig. 2(b). For graphitic carbon-based materials, the G and D-bands and their overtones are the Raman characteristic bands.48 The Raman spectra of 3D RGO presents its D-band at 1335 cm−1 and G-band at 1599 cm−1. Furthermore, a weak and broad 2D peak was also observed at 2672 cm−1 along with a defect activated peak called D + G is which is visible near 2855 cm−1 in the spectrum.49 It is well known that the intensity ratio of D to G-band, ID/IG, was often used to characterize the degree of crystal disorder.50 The ID/IG ratio for 3D RGO was 0.86. The Raman spectrum for the prepared GN2 hydrogel shows D-band, G-band, 2D-band, and D + G band at 1359, 1587, 2674, and 2857 cm−1 respectively. It is seen that the NZF nanoparticles induce a blue shift in the D-band of GN2, suggesting the strong interaction between RGO and NZF. The ID/IG ratio for GN2 was 0.81, slightly lower than that of the 3D RGO. This suggests that GO is reduced, and defects have been reduced due to the interfacial interaction between the graphitic plane and NZF in the hybrid GN2 which affected the Raman peaks of graphene.38 There were very weak Raman bands for NZF nanoparticles in the spectrum of GN2 composite at the 200–600 cm−1 range. This observation can be explained as the graphene oxide and NZF nanoparticles were mixed in the 1:2 ratios, the volume of NZF nanoparticles was relatively small as compared to the volume of GO. Therefore, it is very difficult to find a part for NZF nanoparticles in the Raman spectrum of GN2 composite hydrogel. In the Raman spectra of GNP composite, the characteristic peaks of PIN are covered by the strong G and D-bands of graphene confirming the successful incorporation of GN2 hydrogel to the PIN matrix. When PIN comes in the field forming a GNP composite, there is a shift of D-band from 1359 to 1347 cm−1, showing a π–π interaction along with charge transfer from GN2 hydrogel to PIN.50
N and CC stretching vibrations.44,45 The strong bands at 1503 and 1145 cm−1 which attributed to modes (νN1C2 + δC2H) and (νC3H + νNC2) are intense for indole monomer and almost absent in PIN, conforming to the polymerization of indole monomers linked together via positions 2 and 3 of the pyrrole moiety.46 The diffraction pattern of NZF nanoparticles shows a high-frequency first-order mode A1g at 650–700 cm−1 and modes F2g(2) and F2g(3) at 450–550 cm−1, attributed to the symmetric stretching of oxygen atoms along with M–O bonds in the tetrahedral and octahedral coordination respectively.47 Compared to PIN, the characteristic C–N stretching band of PN10 nanocomposite was located at 1613 cm−1, and the frequency difference was measured to 18 cm−1. Thus the interaction of NZF nanoparticles with the PIN matrix can be visible from the Raman spectra of PN10 nanocomposites. Besides, the characteristic peaks of both PIN and NZF nanoparticles have appeared for the PN10 nanocomposite. This implied that the PN10 nanocomposite was successfully obtained. The intensity of peaks corresponding to NZF is greatly diminished, indicating the good wrapping of nanoparticles within the PIN matrix. The structure of the 3D RGO, GN2, and GNP nanocomposites was also characterized by Raman spectra and the results are shown in Fig. 2(b). For graphitic carbon-based materials, the G and D-bands and their overtones are the Raman characteristic bands.48 The Raman spectra of 3D RGO presents its D-band at 1335 cm−1 and G-band at 1599 cm−1. Furthermore, a weak and broad 2D peak was also observed at 2672 cm−1 along with a defect activated peak called D + G is which is visible near 2855 cm−1 in the spectrum.49 It is well known that the intensity ratio of D to G-band, ID/IG, was often used to characterize the degree of crystal disorder.50 The ID/IG ratio for 3D RGO was 0.86. The Raman spectrum for the prepared GN2 hydrogel shows D-band, G-band, 2D-band, and D + G band at 1359, 1587, 2674, and 2857 cm−1 respectively. It is seen that the NZF nanoparticles induce a blue shift in the D-band of GN2, suggesting the strong interaction between RGO and NZF. The ID/IG ratio for GN2 was 0.81, slightly lower than that of the 3D RGO. This suggests that GO is reduced, and defects have been reduced due to the interfacial interaction between the graphitic plane and NZF in the hybrid GN2 which affected the Raman peaks of graphene.38 There were very weak Raman bands for NZF nanoparticles in the spectrum of GN2 composite at the 200–600 cm−1 range. This observation can be explained as the graphene oxide and NZF nanoparticles were mixed in the 1:2 ratios, the volume of NZF nanoparticles was relatively small as compared to the volume of GO. Therefore, it is very difficult to find a part for NZF nanoparticles in the Raman spectrum of GN2 composite hydrogel. In the Raman spectra of GNP composite, the characteristic peaks of PIN are covered by the strong G and D-bands of graphene confirming the successful incorporation of GN2 hydrogel to the PIN matrix. When PIN comes in the field forming a GNP composite, there is a shift of D-band from 1359 to 1347 cm−1, showing a π–π interaction along with charge transfer from GN2 hydrogel to PIN.50
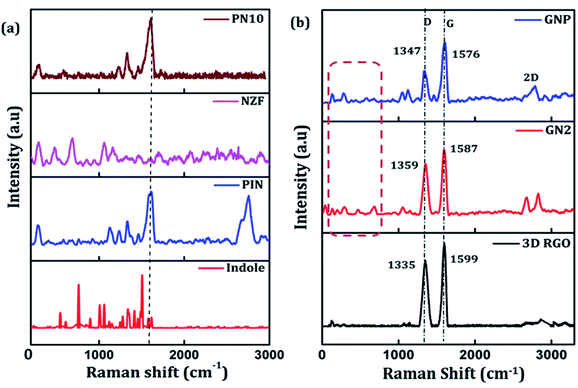 |
| | Fig. 2 (a) Raman spectra of indole, PIN, NZF, and PN10 and (b) Raman spectra of 3D RGO, GN2, and GNP composite. | |
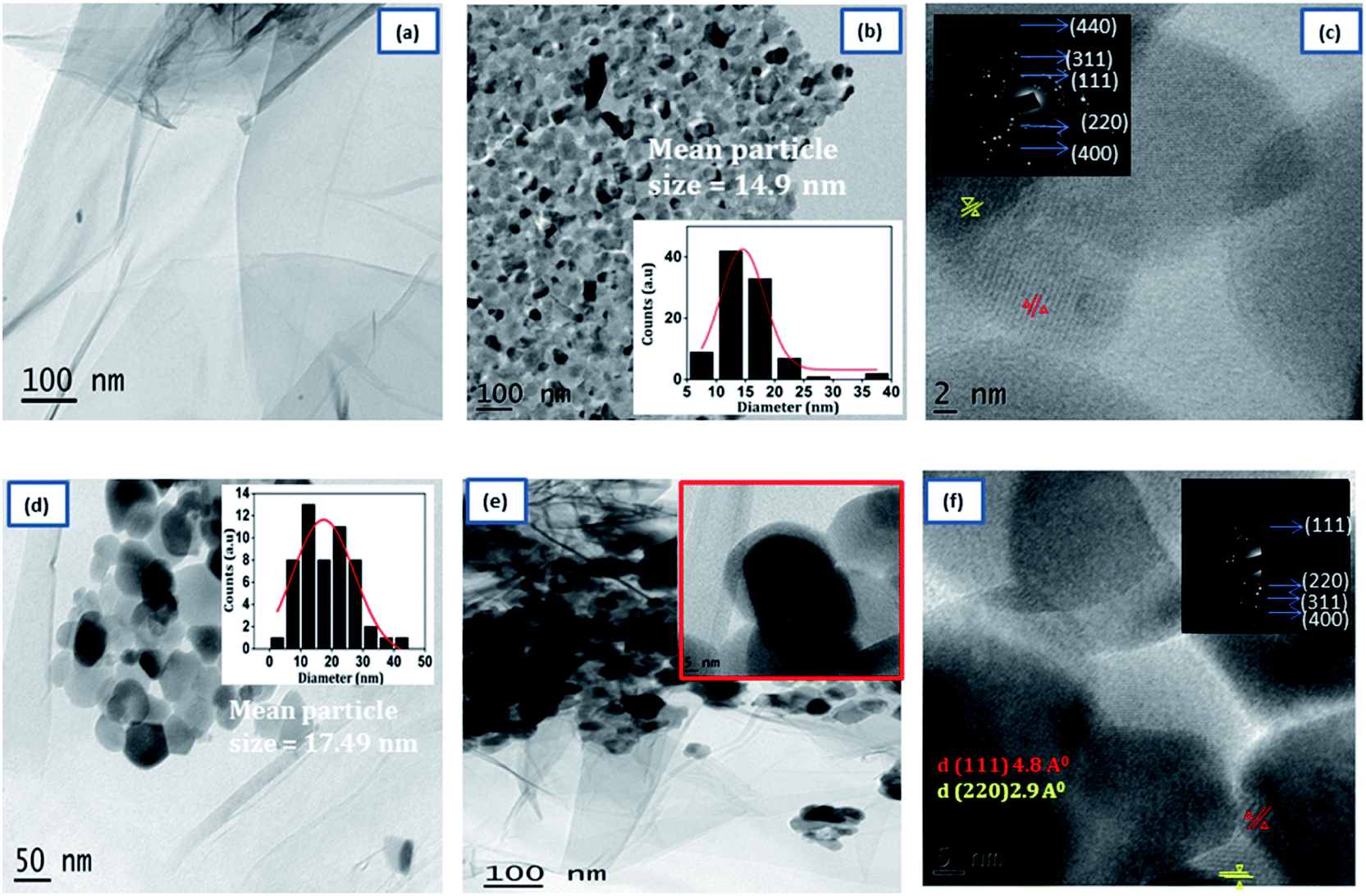
The detailed morphology of prepared samples was examined by TEM analysis were shown in Fig. 3. The TEM image of 3D RGO [Fig. 3(a)] shows that hydrogel consists of a large number of wrinkles and folds, capable of anchoring the NZF nanoparticles. As shown in Fig. 3(b), the pristine NZF nanoparticles are randomly aggregate with each other with an average particle size of about 14.9 nm, determined from the particle size distribution graph (shown in inset). In the high-resolution TEM graph of NZF nanoparticles [Fig. 3(c)] the atomic lattice fringes can be observed and the interplanar spacing was measured to be 4.8 Å, and 2.9 Å which were close to the {111} and {220} lattice planes of cubic spinel respectively. In Fig. 3(d), it can be intangibly noted that many NZF nanoparticles decorated on the wall of the graphene sheets having an average particle size of 17 nm and there are no apparent aggregates are observed. Fig. 3(e) clearly shows that the PIN is successfully integrated uniformly in the GNP hybrid system and it is evident that RGO hydrogel can act as good support for the embedment of organic and inorganic substances. An enlarged view is shown in the inset of Fig. 3(d). In the HRTEM image of PNG nanocomposite [Fig. 3(f)], the lattice between the adjacent fringes with an interplanar spacing of 4.8 Å and 2.9 Å can be identified, which reflects that the graphene hydrogel supported NZF nanoparticles were very stable during the in situ polymerization of indole monomer. Besides, the appearance of the bright arc in the selected area electron diffraction pattern (SAED) [inset Fig. 3(f)] corresponding to the XRD data of NZF nanoparticles and indicated that the spherical structures were formed by nanoparticles in highly preferred orientations.
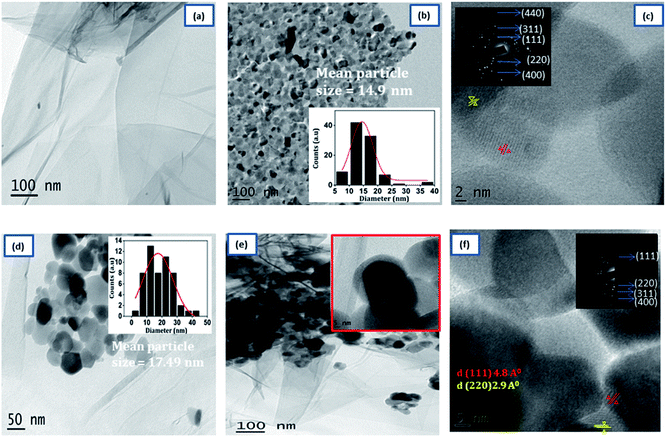 |
| | Fig. 3 Typical TEM images of (a) 3D RGO, (b) NZF, (d) GN2 (e) GNP. HRTEM images (c) NZF and (f) GNP composite. The inset in (b), and (d) shows the particle size distribution. | |
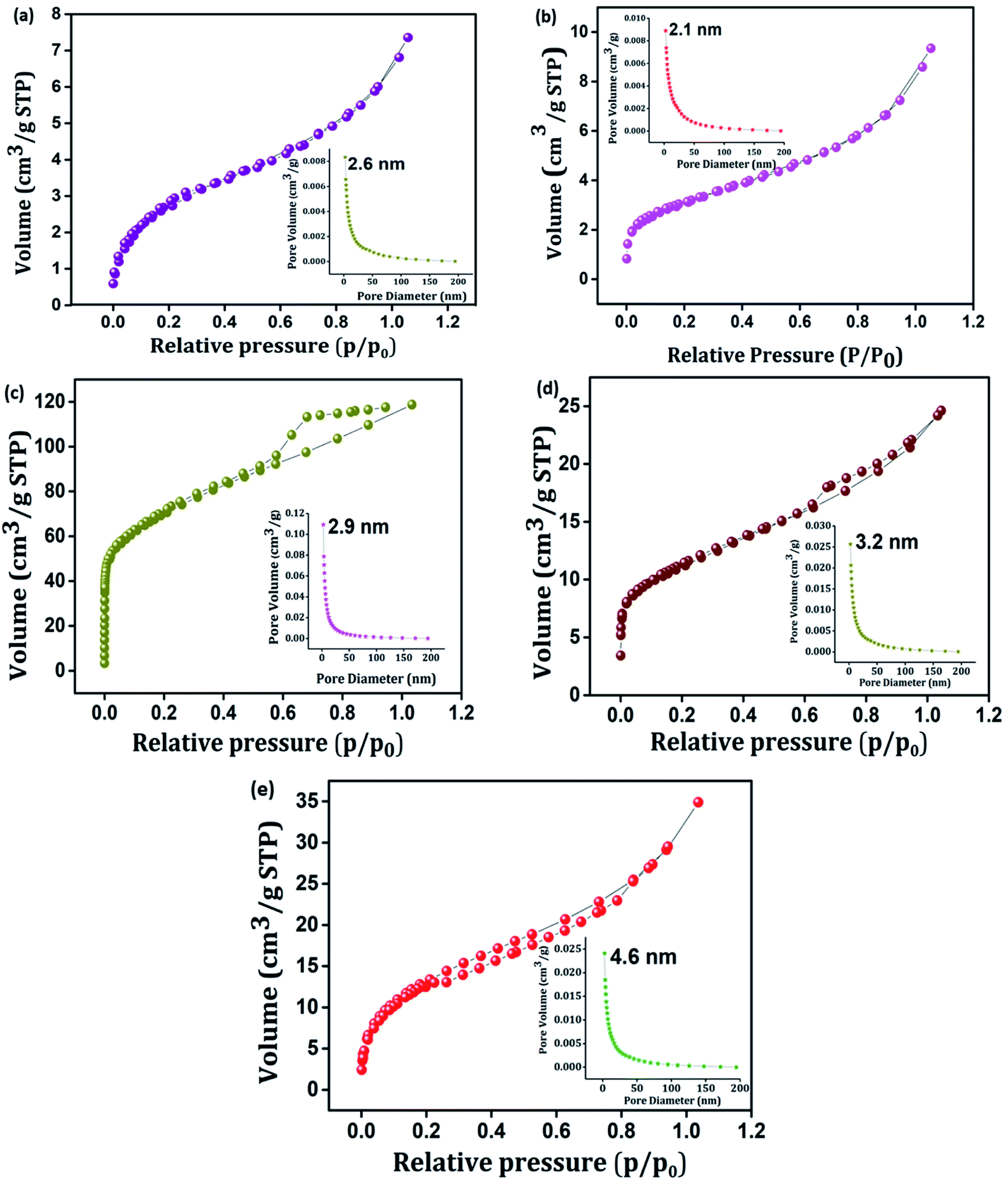
Porosity and the surface area play a significant role in determining the electrochemical properties of an active material.51 Typical nitrogen adsorption–desorption isotherms and corresponding pore size distribution plots of PIN, NZF, 3D RGO, GN2, and GNP composites were investigated and shown in Fig. 4(a)–(e). It is seen from Fig. 4 that, in contrast to PIN and NZF, the isotherms of 3D RGO, GN2, and GNP composites possess typical type IV characteristics with distinct H2, H2, and H3 hysteresis loops in the range between 0.3 and 0.7, respectively. This manifesting the typical mesoporous structure in the 3D framework of the composites.31,52 Further, according to the Barrett–Joyner–Halenda (BJH) method, the pore-size distribution plots (inset) of all electrode materials lie in the 2–50 nm range and clearly showed the mesoporosity. Specific surface area, cumulative pore volume, and average pore diameter of all the electrode materials are given in Table S1.† The high surface area leads to better faradaic redox reactions at surface sites and results in enhanced capacitive performance of the electrode materials.53 A favorable mesopore structure and improved surface area characteristics provide more diffusion and accession of electrolyte ions into the active sites of the GNP electrode materials and also may benefit from the high rate capability of the composite.19
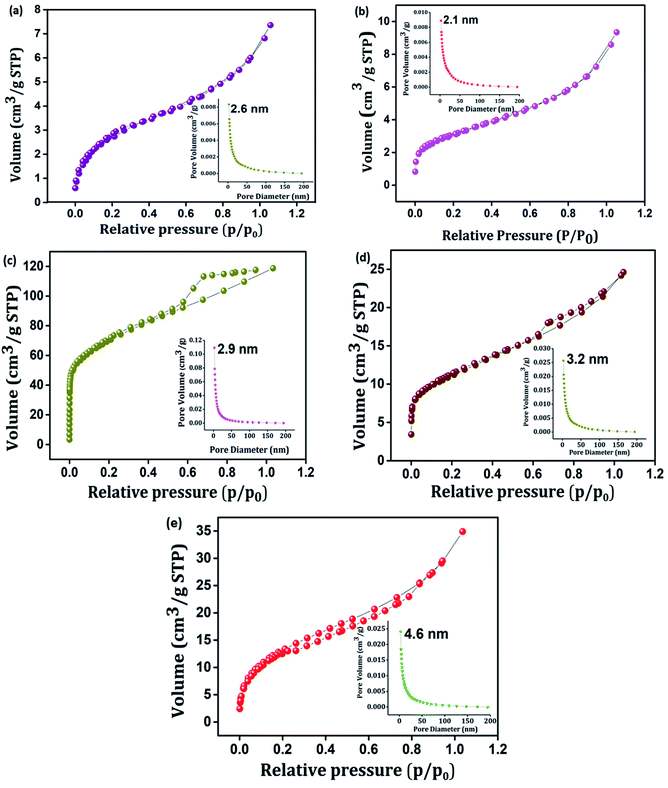 |
| | Fig. 4 N2 adsorption–desorption isotherms of (a) PIN, (b) NZF, (c) 3D RGO, (d) GN2, and (e) GNP. Inset shows BJH pore size distribution plot of (a) PIN, (b) NZF, (c) 3D RGO, (d) GN2 and (e) GNP. | |
Thermal stability
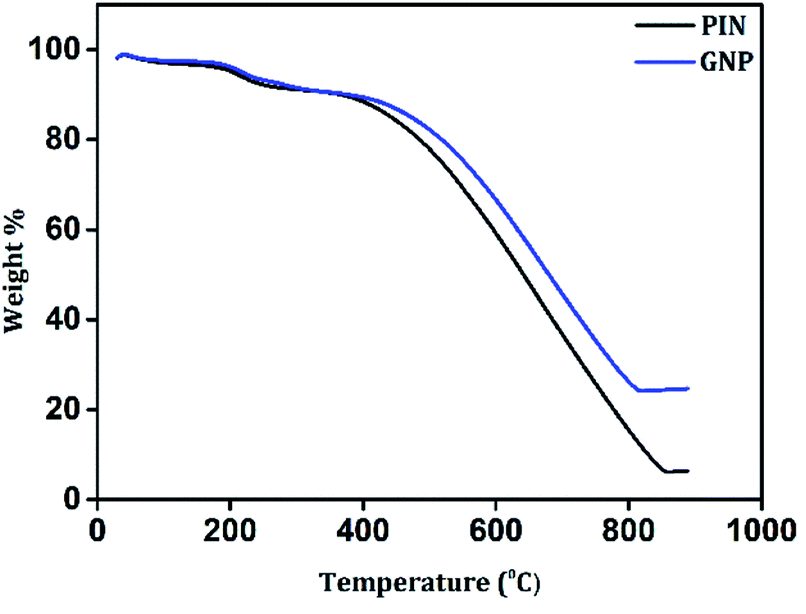
The impact of the incorporation of GN2 on the thermal stability of the PIN matrix was evaluated by TGA measurements under N2 atmosphere from 30 to 900 °C. As shown in Fig. 5, PIN and GNP composites possess a three-step degradation and exhibit similar weight loss features. The initial degradation below 100 °C led to weight losses of approximately 2% for both PIN and GNP, which is due to the removal of occluded moisture.54 Then, a weight loss of 7% was recorded for both PIN and GNP between 100–300 °C and was mainly attributed to the degradation of unreacted monomer, some oligomers, and doping anions from PIN structure.40 Upon increasing the temperature from 300 to 850 °C, maximum weight losses of 84 and 67% were recorded for PIN and GNP composites respectively, due to decomposition of the polymer skeleton.13,17 It is noteworthy that, the third step of degradation in PIN occurred at 360 °C, while GNP composite decomposes at a higher temperature around 415 °C, indicating higher thermal stability. At 850 °C, PIN retains only 6.5% of the initial weight, while GNP composite shows improved weight retention corresponding to 24.9%. Such a huge loss in weight for PIN reveals high thermal degradability of the PIN backbones while the formation of a stable layered structure with the incorporation of GN2 composite within PIN matrix limits the open exposure of the PIN backbone in GNP composite and hence, restricting its easy obliteration. The compositions of the constituent material in the GNP composite are also verified from the weight retention values so obtained from the thermograms. Accordingly, the content of GN2 is found to be 19.9% in the GNP composite. The enhancement of thermal stability of the GNP nanocomposite proposed a strong interfacial interaction between the PIN chain and GN2.
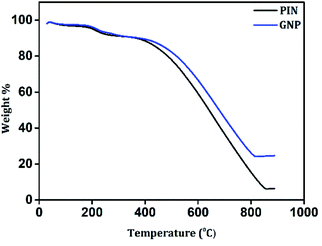 |
| | Fig. 5 TGA curves of PIN and GNP composite. | |
Electrochemical performance
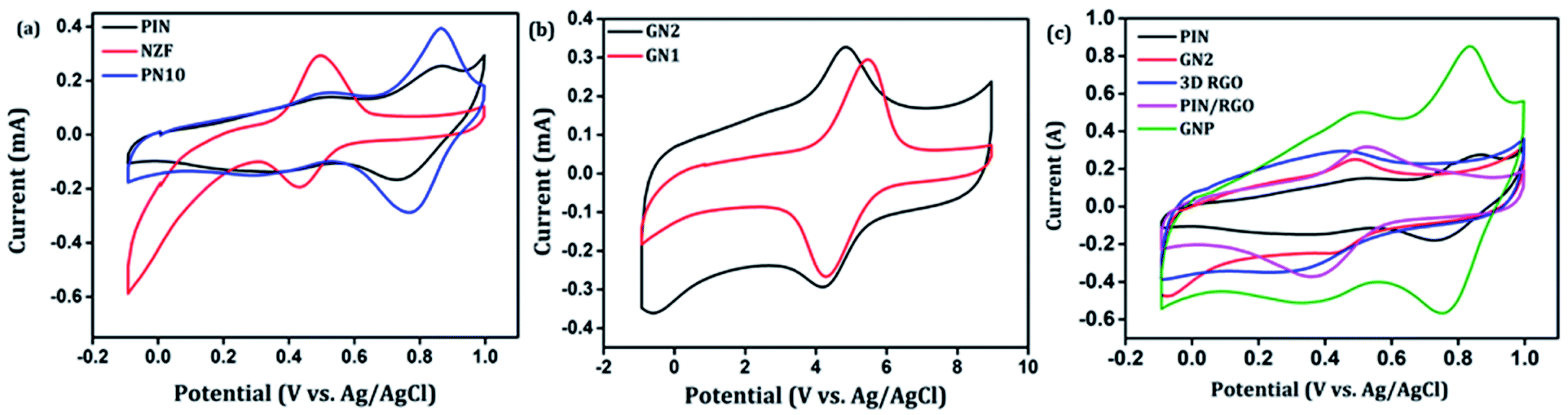
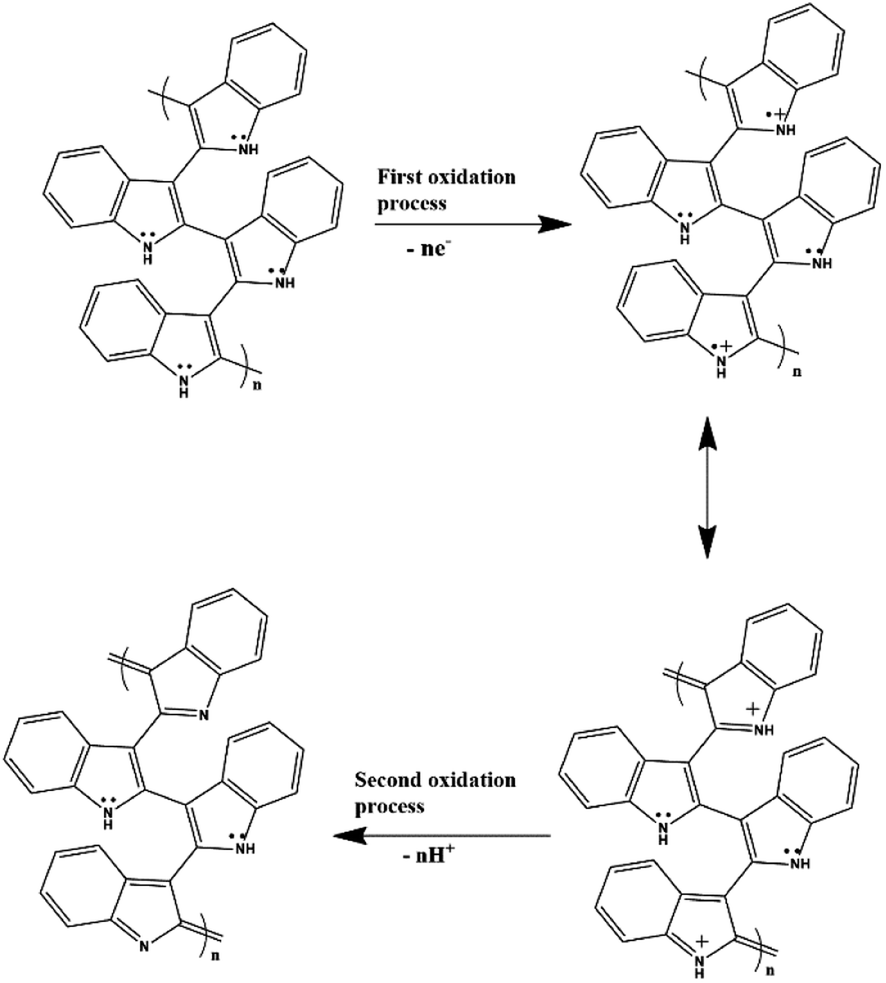
After confirming the incorporation of NZF nanoparticles and binary GN2 composite in PIN matrix, the advantages of these hybrid materials as active supercapacitor electrodes were explored using cyclic voltammetry (CV), galvanostatic charge–discharge measurements (GCD), and electrochemical impedance spectrum (EIS) in a three-electrode configuration in 1 M H2SO4. Fig. 6(a) shows the comparison of the CV response obtained for the PIN, NZF, and PN10 nanocomposites within the potential range of −0.1 to 1 V at the scan rate of 10 mV s−1. The PIN electrode gives two couple of anodic (at 0.48 and 0.86 V) and cathodic peaks (at 0.28 and 0.76 V) which indicate that the charge storage mechanism of the PIN electrode is through the process of electro-oxidation and electro-reduction. In the first redox process, the neutral species are converted to radical-cation whereas concomitant proton and electron exchange occurs at the most positive potentials during the second redox process.39,55,56 Scheme 1 represents the structural conversions observed for a PIN electrode in acidic solutions when the potential is changed from the fully reduced forms to the fully oxidized forms. The CV curve of NZF nanoparticle exhibit an anodic peak located at approximately 0.49 V and a corresponding cathodic peak potential at around 0.43 V, assigned to the reversible electrochemical reaction Ni2+/3+ and/or Fe3+/2+ revealing the presence of pseudocapacitive behavior of the electrode.57 The CV plot of PN10 nanocomposite combines the features of both PIN and NZF nanoparticles and exhibit two sets of oxidation and reduction peaks at 0.48, 0.86 V and 0.28 V, 0.76 V respectively. As known, the area under the CV loop is proportional to the specific capacitance of the electrodes.4 Thus the specific capacitances obtained for the PIN, NZF, and PN10 are ca. 52, 20, and 66 F g−1 respectively. This result demonstrates that the charge storage capability of the PN10 nanocomposite system is very slightly higher than that of PIN and NZF in terms of capacitance.
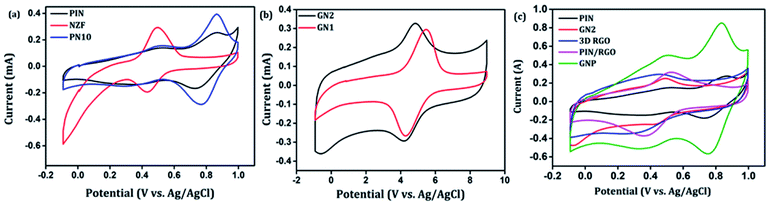 |
| | Fig. 6 Comparison of cyclic voltammograms of (a) PIN, NZF, and PN10 (b) GN1 and GN2 (c) PIN, GN2,3D RGO, PIN/RGO and GNP at the scan rate of 10 mV s−1. | |
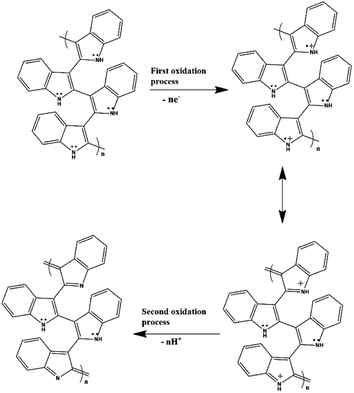 |
| | Scheme 1 Structural conversions are observed for a polyindole electrode in acidic solutions when the potential is changed from the fully reduced forms to the fully oxidized forms. | |
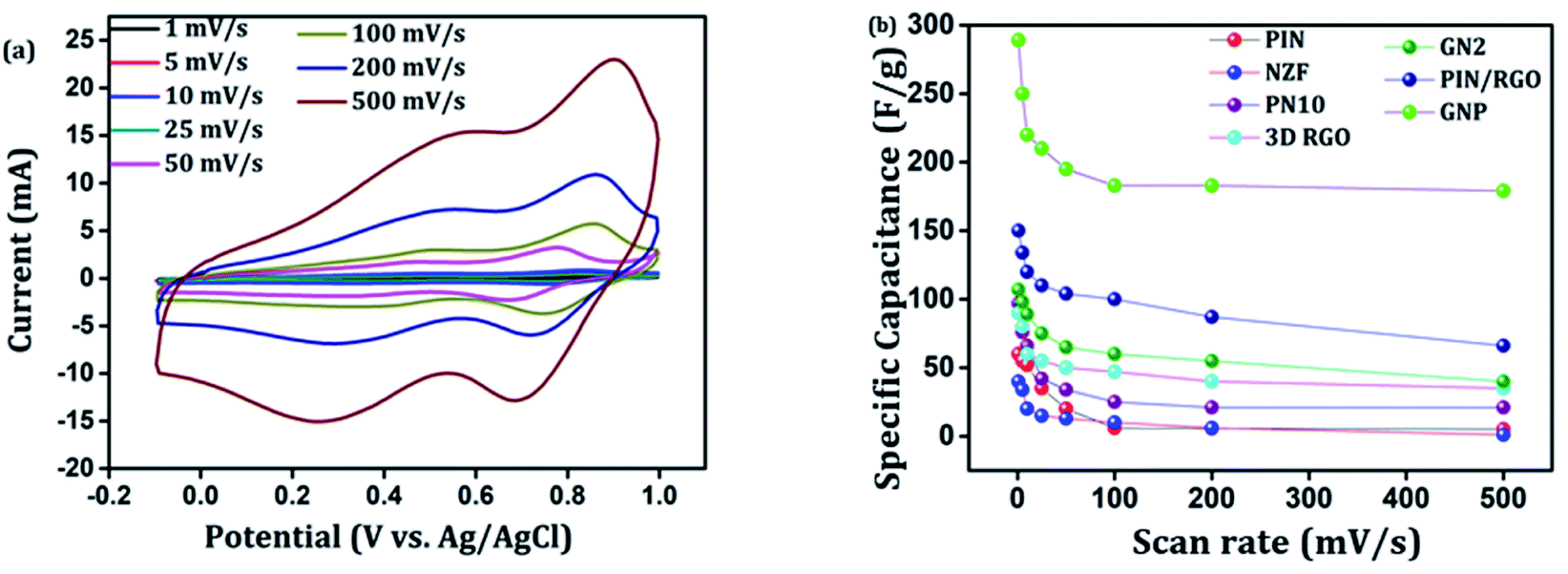
CV analysis of GN nanocomposites was also carried out and the results are presented in Fig. 6(b). The presence of 3D RGO can greatly enhance the electrical conductivity and control the nanostructure and morphology of NZF nanoparticles. Further, this leads to improved electrode/electrolyte contact areas and high rates of electrode reaction.58 The CV profile includes the comparison for GN1 and GN2 electrodes at the scan rate of 10 mV s−1 with varying concentrations of NZF nanoparticles in RGO hydrogel. It is notable from the CV study that, GN2 shows the highest capacitive property where the synergism between RGO hydrogel and NZF nanoparticle is maximum and hence, it was used for further study. Fig. 6(c) shows the comparison of the CV response obtained for the PIN, GN2, 3D RGO, PIN/RGO, and ternary GNP hybrid within the potential range of −0.1 to 1 V at the scan rate of 10 mV s−1. The specific capacitance obtained for the PIN, GN2, 3D RGO, PIN/RGO, and GNP is ca. 52, 75, 60, 99, and 220 F g−1, respectively. The CV profile of the GNP ternary hybrid combines the features of both PIN and GN2 hydrogel and there is an evident improvement in the specific capacitance of the PIN matrix (nearly 5 times) after the incorporation of GN hydrogel. The oxidation and reduction peaks of ternary GNP composites were at 0.50 V, 0.83 V, and 0.36 V, 0.74 V respectively. The shift of peaks concerning polyindole indicates the interaction between the functional group of 3D RGO on PIN, which affected the intrinsically electrochemical property of PIN. Combining with the BET results, a highly porous structure of GNP composite may benefit the electrolyte ion diffusion to active sites with less resistance and results in high capacitances.59,60 Besides, this π–π stacking, electrostatic and hydrogen-bonding interactions between the PIN molecular chains and NZF incorporated graphene nanosheets are also led to enhanced electrochemical performance.33,61,62 The effect of scan rate on the stability of different electrodes (PIN, NZF, PN10, 3D RGO, PIN/RGO, and GN2) was investigated within a wide scan range from 1 to 500 mV s−1 and shown in Fig. S1(a)–(f),† respectively. Relatively high rate capability performances of the ternary GNP electrode at all scan rates are depicted in Fig. 7(a). The CV profile of the electrode retained a similar shape at all scan rates due to its porous structure and was beneficial for the fast ion diffusion into the electrode.19 In addition to this, the variation of specific capacitance with a potential scan rate is shown in Fig. 7(b), which confirms that the rate capability of the GNP hybrid electrodes is also significantly improved. At a higher scan rate, electrolyte ions cannot make full use of the inner surface area by diffusing completely into the inner active sites of material.63,64 However, at a low scan rate electrolyte ions are completely diffused into the inner active sites of the material and make the redox transitions completely. As a result, specific capacitance decreases as the potential scan increases. The maximum specific capacitance obtained for GNP hybrid material is 289 F g−1 which is much higher than 60 F g−1 for the PIN at 1 mV s−1.
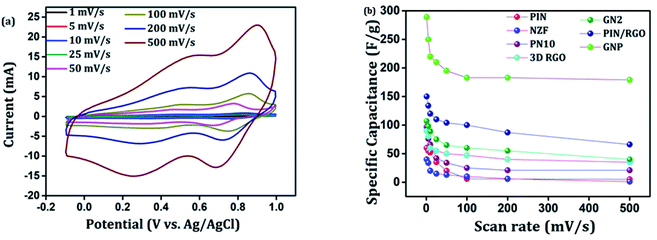 |
| | Fig. 7 (a) CV curves of ternary GNP nanocomposite at different scan rates, (b) variation of the specific capacitance with different scan rates for the PIN, NZF, PIN10, 3D RGO, PIN/RGO, GN2, and GNP composite. | |
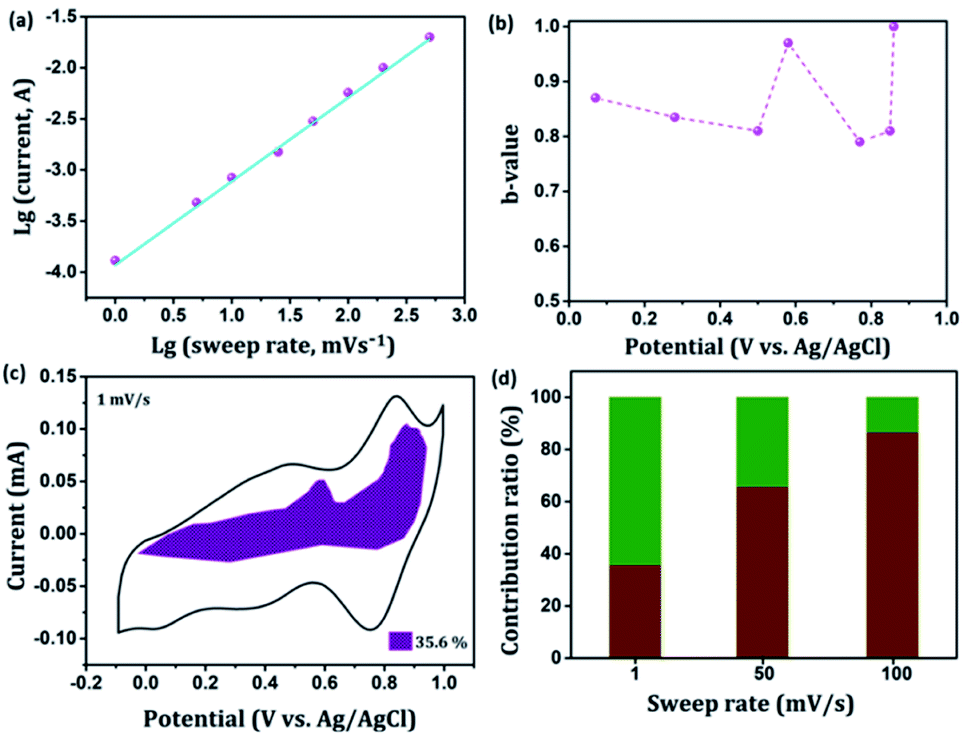
In general, the current (i) fits a relationship with the scan rate (ν) according to the power-law i = aνb, in which both a and b are constants. The b value can be employed to qualitatively evaluate the degree of capacitive effect with a value of 0.5 indicating ideal diffusion-controlled faradaic process, and a value of 1 suggesting surface redox reactions involving non diffusion-controlled processes.65 The parameter b is calculated from the slope of the linear plot of log(i) versus log(ν) (Fig. 8(a) and S2†). The b value at the cathodic peak regimes was estimated to be 0.81. When the potentials were far away from the cathodic peak potential, the b values were more close to 1 [Fig. 8(b)]. So it is evidenced that the total charge storage in the GNP electrode is a combination of a capacitance (or pseudocapacitance)-controlled process and a diffusion-controlled redox process. According to the power-law relationship, the ratio of capacitive (or pseudocapacitive) contribution can be quantified from another CV analysis by separating the current (i) into capacitive (k1v) effects (electrical double layer capacitance or redox pseudocapacitance) and intercalation process (k2v1/2) as discussed below,
Thus,
k1 and
k2 contents at various potentials can be determined by plotting
i/
v1/2 versus v1/2.
66 Fig. 8(c) shows the result of this analysis for the GNP electrode at 1 mV s
−1 with a capacitive (or pseudocapacitive) contribution of 35.6%. Besides, when the scan rate increases, the ratio of the capacitive (or pseudocapacitive) contribution gradually increases. A contribution value of 86% was obtained at a sweep rate of 100 mV s
−1 corresponds to charge storage due to surface reaction [
Fig. 8(d)].
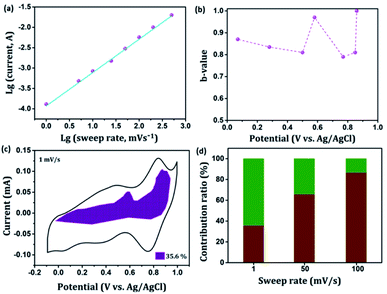 |
| | Fig. 8 Kinetics analysis of the electrochemical behavior in the GNP electrode. (a) The determination of the b-value at cathodic peak regimes. (b) The calculated b values at various potentials. (c) Separation of the pseudocapacitive and diffusion currents at a scan rate of 1 mV s−1. (d) Contribution ratios of capacitances from the surface-controlled pseudocapacitive reaction and diffusion-limited intercalation/deintercalation reaction at various sweep rates. | |
Further, galvanostatic charging–discharging (GCD) measurements were also taken in a stable window of −0.1 to 1 V which further demonstrates the electrical double layer capacitance (EDLC), pseudocapacitance, and internal resistance of the composites. Fig. 9(a) shows a comparison of the charge–discharge profiles of PIN, NZF, PN10, 3D RGO, PIN/RGO, GN2, and ternary GNP hybrid electrodes obtained at the current density of 0.5 A g−1. As can be manifested from Fig. 9(a), GNP ternary hybrid displays substantially prolonged discharging time as compared to other electrode materials. The specific capacitance calculated from the charge–discharge profile for the PIN, NZF, PN10, 3D RGO, PIN/RGO, GN2, and ternary GNP hybrid electrodes is 25.5, 10.4, 100, 85, 150, 97.1, and 258 F g−1 respectively. This result demonstrates that the charge storage capability of GNP hybrid material outperforms that of PIN and PN10 composite in terms of higher specific capacitance. It is also realized that the IR drop of the PIN is higher than the composite electrode, and shows an asymmetric behavior having to charge time significantly higher than the discharging time. This result indicating higher internal resistance of the PIN electrode than composites and is consistent with the high ESR obtained from the EIS analysis.67
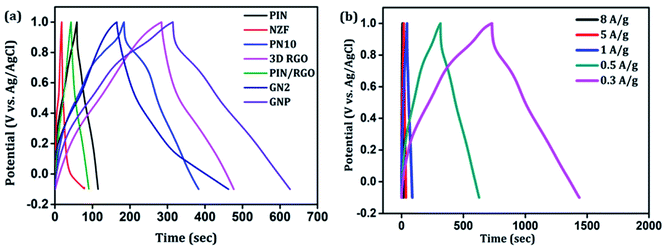 |
| | Fig. 9 (a) The charge–discharge profiles of PIN, NZF, PN10, 3D RGO, PIN/RGO, GN2, and ternary GNP hybrid electrodes obtained at the current density of 0.5 A g−1. (b) Galvanostatic charge–discharge curves of ternary GNP nanocomposite at different current densities. | |
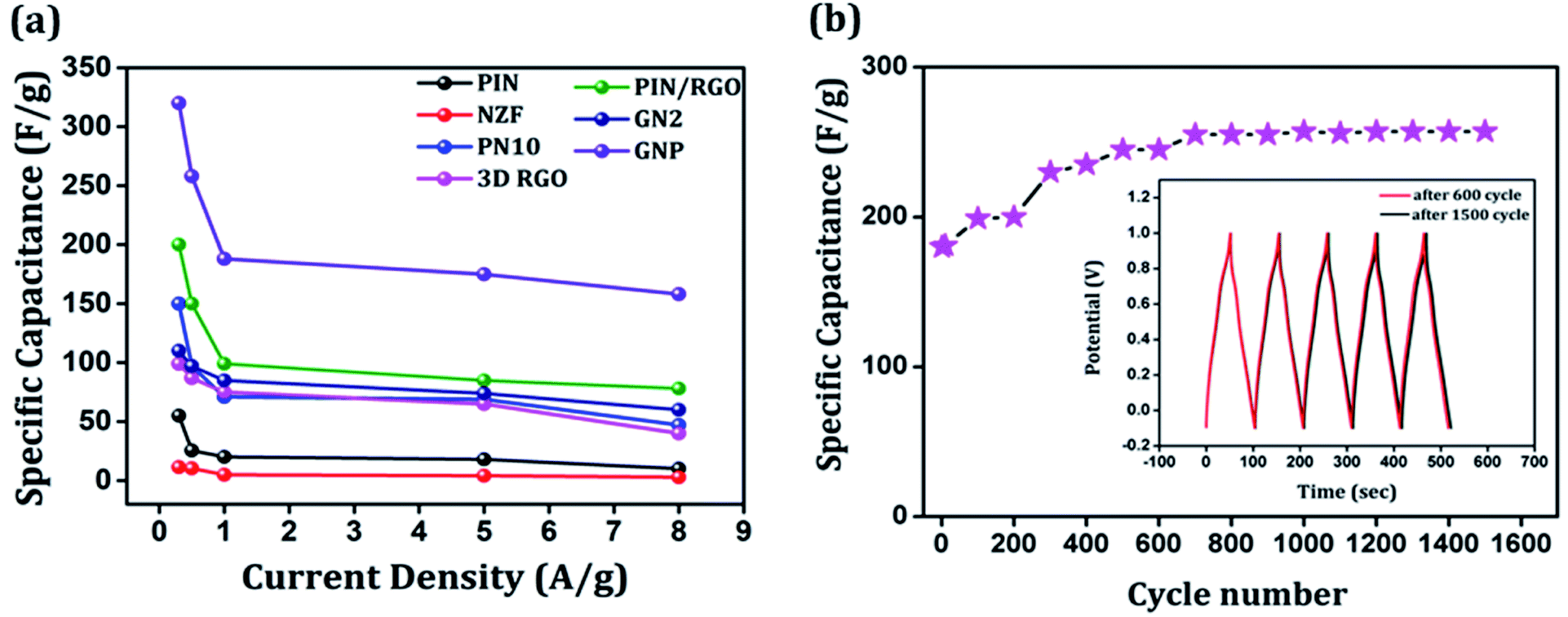
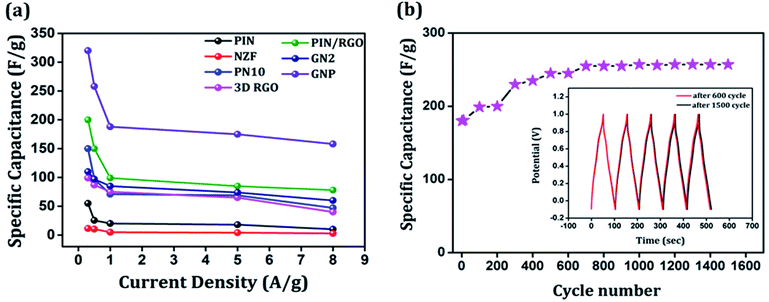
The galvanostatic charge–discharge curves of the GNP electrode at different current densities are shown in Fig. 9(b) confirms that the GNP electrode has better rate capability compared to other electrodes. Unlike the conventional triangular-shaped GCD curves, the GNP electrode shows a slight deviation from the triangular-shaped curve, which confirmed the presence of both the EDLC of 3D RGO and the pseudocapacitance of the PIN and NZF nanoparticles.68 As the current density decreased, the specific capacitance of the GNP electrode increased and reached a maximum value of 320 F g−1 at 0.3 A g−1, which was 5.8 and 2.1 times higher than the values observed for the PIN (55 F g−1) and PN10 (150 F g−1) composites, respectively. In addition to this, the retentions in the specific capacitance are calculated for the PIN, NZF, PN10, 3D RGO, PIN/RGO, GN2, and GNP electrodes, and shown in Fig. 10(a). The GNP electrode has a retention of 69.6% as the current density increases from 0.3 to 8 A g−1. Remarkably high activity of the GNP electrode compared to other electrodes is mainly attributed to the improved utilization of inner and outer active sites of the electrode material.4
 |
| | Fig. 10 (a) Variation of the specific capacitance with current density for the PIN, NZF, PN10, 3D RGO, PIN/RGO, GN2 and GNP, (b) cycling performance of GNP nanocomposite measured at a current density of 1 A g−1. Inset in (b) shows charge–discharge curves at the 600th cycle and 1500th cycle. | |
Cycling stability is a key parameter that assesses the performance of a supercapacitor. The cyclic stability of the GNP electrode was determined by conducting the charge–discharge tests at 1 A g−1 for 1500 cycles and shown in Fig. 10(b). As shown in the figure, the specific capacitance of the GNP electrode further increased to ∼257 F g−1 after 600 cycles and stays stable up to 1500 cycles. Such an enhancement in capacitances may be because of more complete intercalation and deintercalation of active species after some initial cycles.42,69 The cyclic stability of the GNP electrode is also evidenced in the inset of Fig. 10(b), which shows that charge–discharge curves after 600 and 1500 remain undistorted and essentially symmetric. This confirms that the as-synthesized GNP hybrid material not only possesses high specific capacitance and rate capability but also much improved cyclic stability.
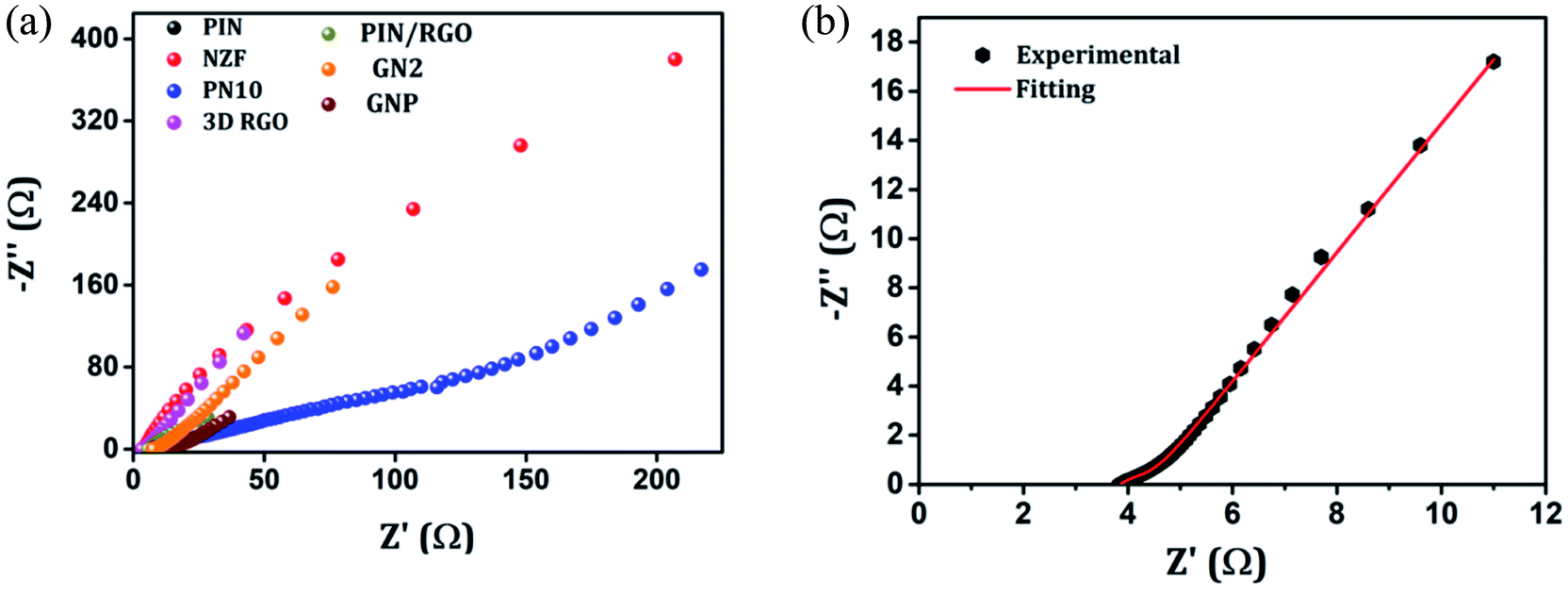
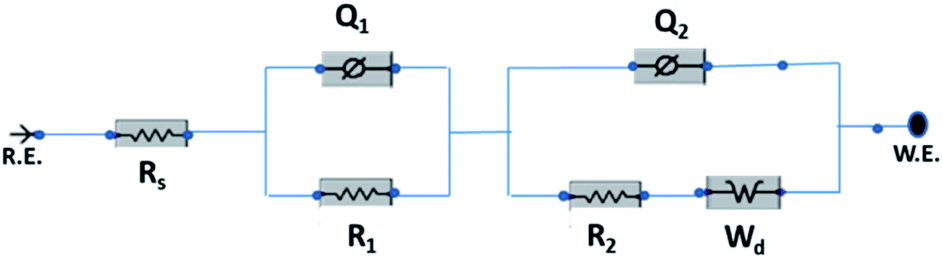
The electrochemical evaluated by the EIS establishes the kinetic feature of ion diffusion.70 The Nyquist plots obtained for all the seven electrode materials in a frequency range of 0.1 Hz to 100 kHz with a potential amplitude of 10 mV are shown in Fig. 11. The Nyquist plots behavior of electrode materials was further of the EIS consists of three parts.71,72 The first part is at the higher frequency region; the x-intercept of the starting curve represents the effective series resistance Rs (ESR) which is the sum of solution resistance, intrinsic resistance of the active material, and contact resistance of the active material to the current collector substrate. Here all seven electrodes exhibit an ESR value ≤ 10 Ω. The second part, the semicircle at the mid-frequency region ascribed to the charge transfer resistance (Rct) due to the faradaic reaction and double layer capacitance (Cdl) at the electrode–electrolyte interface and is equal to the diameter of the semicircle.67,73 The third part is the vertical spike at the low-frequency region, which is a result of the diffusion resistance or Warburg resistance (Wd) to the ionic transport at the electrode–electrolyte interface. The enhanced Warburg region indicates a larger variation in ion diffusion path length, which consecutively increases the obstruction of ion movement. To understand the behavior of the as-prepared samples, we proposed an equivalent circuit model (Fig. 12) to fit the experimental data as shown in Fig. 11(b) and S3.† The high reliability of the fitted results can be verified by the chi-square value on the order of 1 × 10−4. The interpreted data are summarized in Table S2.† In the equivalent circuit, the term Rs and Wd signifies the solution resistance and Warburg resistance, respectively. Q1 and Q2 are the constant phase elements of capacitance relative to the inner layer and diffusion layer, respectively. R1 represents the pore electrolyte and intermediate layer resistance and R2 is the resistance of the barrier layer.74 The GNP electrode shows low values of RS (3.98 Ω), R1 (7.68 Ω), and R2 (0.419), these obtained low values are of great importance as they affect the power and energy performance, but also reduce undesired heat dissipation throughout the charge–discharge processes.75 It is observed that the Rs, R1, and R2 values are reduced for GN2 composite and further reduced for GNP ternary composite compared to pure PIN electrode. It is ascribed to the enhanced electrical conductivity acquired through compositing with GN2 and PIN. The effect is reflected in the CV and GCD curves of the respective electrode and is explained in the previous section. The GNP ternary nanocomposite also attained lesser Wd values, suggesting a short diffusive path of the electrolyte ions within the nanocomposite.
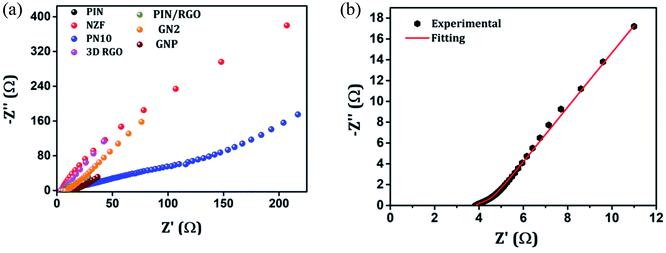 |
| | Fig. 11 (a) Nyquist plots of the EIS in the frequency range of 0.1 Hz to 100 kHz for the PIN, NZF, PN10, 3D RGO, PIN/RGO, GN2 and GNP. (b) Nyquist plots of GNP after fitting with an equivalent circuit. | |
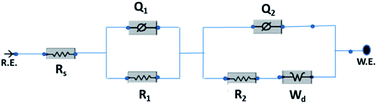 |
| | Fig. 12 Equivalent circuit used for fitting the Nyquist plots. | |
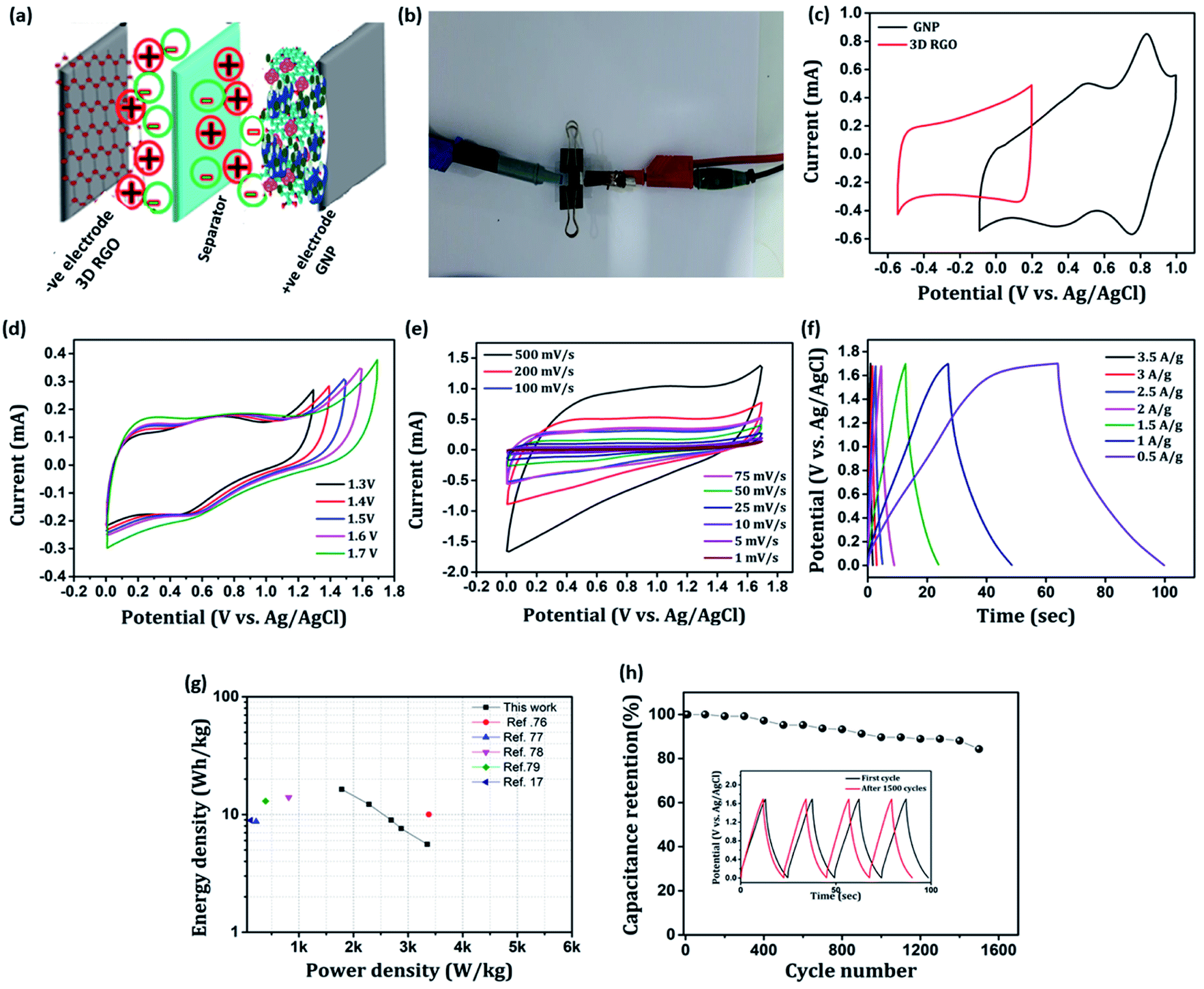
Towards practicability check, we fabricated an asymmetric device configuration (GNP∥3D RGO) by sandwiching a Whatman filter paper 42 soaked in 1 M H2SO4 aq. solution (the electrolyte) between the positive (GNP nanocomposite) and negative (3D RGO) electrodes, and the results are shown in Fig. 13. Fig. 13(a) and (b) shows schematic representation and the photograph of as-fabricated ASC device. The stable working potential window of the ASC device is confirmed by recording the cyclic voltammogram of both positive and negative electrodes at the possible potential range as shown in Fig. 13(c). Apparently, the maximum working potential limit of the ASC device can be tested up to 1.7 V to achieve better electrochemical performance as shown in Fig. 13(d). Typical cyclic voltammograms recorded for ASC at different scan rates (1–500 mV s−1) are presented in Fig. 13(e). It is found that, as scan rate increases, the redox behavior declines, which reveals that only double-layer capacitance maintains the rate capability. The charging and discharging processes of the ASC device are further evaluated by employing the GCD technique for various current densities (0.5–3.5 A g−1), shown in Fig. 13(f). The calculated maximum specific capacitance value of the ASC at a current density of 0.5 A g−1 is 48.9 F g−1. The estimated energy density, E (W h kg−1), and power density, P (W kg−1) of the ASC device is presented as a Ragone plot as shown in Fig. 13(g). The ASC device could deliver a maximum energy density of 16.38 W h kg−1 at the power density of 1784 W kg−1. These results are compatible with some recently reported devices of PIN nanocomposites.17,76–79 The cyclic stability of the ASC device is investigated by employing the GCD techniques between 0 and 1.7 V at an applied current density of 1 A g−1 for 1500 cycles. The typical capacitive retention plot is presented in Fig. 13(h). The results reveal that the ASC retains 84% of its original capacitance after 1500 cycles. This result demonstrates the practical application of the GNP∥3D RGO ASC device.
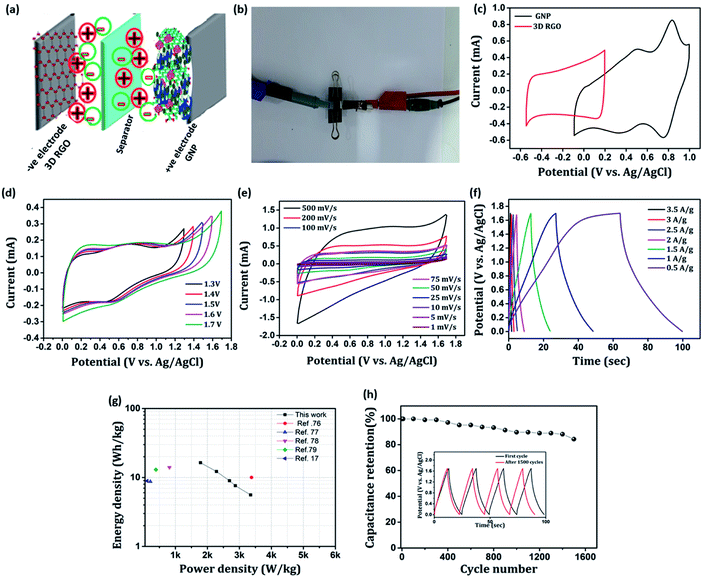 |
| | Fig. 13 (a) Schematic representation of the as-fabricated asymmetric supercapacitor, (b) snapshot of the as-fabricated asymmetric supercapacitor in the two electrode configuration, (c) CV profiles of 3D RGO and GNP electrodes at 10 mV s−1 scan rate, (d) CV profiles of 3D RGO∥GNP devices at 50 mV s−1, (e) CV recorded for ASC at different scan rates, (f) GCD measurements for ASC at different current densities, (g) Ragone plot derived from GCD techniques for ASC device, (h) cyclic stability of the ASC device. Inset in (h) shows charge–discharge curve at first and after 1500 cycles. | |
Conclusions
Ni0.5Zn0.5Fe2O4 nanoparticles (NZF) decorated on reduced graphene oxide (GN2) are achieved by a facile hydrothermal method followed by coating with polyindole (PIN) through in situ emulsion polymerization process to form a ternary GNP hybrid material is reported. The structure, porosity, morphology, and thermal stability of the resulting ternary GNP hybrid material were characterized via X-ray diffraction (XRD), Raman spectroscopy, Brunauer–Emmett–Teller (BET) surface area measurements, transmission electron microscopy (TEM), and thermogravimetric analysis (TGA). The fabricated ternary GNP electrode material exhibits a mesoporous structure and almost 5 times improvement in the specific capacitance and excellent cyclic stability compared to PIN electrode material. The enhancement in the performance is attributed to the presence of highly conducting GN hydrogel which reduces the electrical resistance of the composite and results in increased utilization of the active material. This facile strategy using conducting polymer, PIN via making a ternary composite with 3D reduced graphene oxide/Ni0.5Zn0.5Fe2O4 (GN) hydrogel is remarkably improved the performance of the final supercapacitor electrode. Hence this simple strategy can offer great promise for the next energy storage generation based on conducting polymer nanocomposites. To confirm the practical application of this ternary composite electrode, GNP∥3D RGO asymmetric supercapacitor devices are assembled. It is found that the ASC device can perform over an extended potential range of 1.7 V and exhibits a specific capacitance of 48.9 F g−1 at 0.5 A g−1. This ASC device exhibits a significant energy density of 16.38 W h kg−1 at the power density of 1784 W kg−1. In addition, the ASC device can retain stability of 84% even after 2000 cycles. These findings on the GNP ternary composite suggest that this composite can be used as an efficient positive electrode material for supercapacitor devices.
Conflicts of interest
“There are no conflicts to declare”.
Acknowledgements
Anjitha. T. is grateful to CSIR for her CSIR-JRF fellowship. The authors are thankful to Central Instrumentation Facility, University of Calicut for various measurements.
Notes and references
- P. Simon and Y. Gogotsi, Nat. Mater., 2020, 19, 1151–1163 CrossRef CAS PubMed.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301 RSC.
- F. Li, J. Liu, Y. Ma, Z. Shang, Q.-a. Huang and X. Huang, J. Mater. Chem. A, 2019, 7, 15378–15386 RSC.
- B. K. Balan, H. D. Chaudhari, U. K. Kharul and S. Kurungot, RSC Adv., 2013, 3, 2428–2436 RSC.
- S. Zhang and N. Pan, Adv. Energy Mater., 2015, 5, 1401401 CrossRef.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- M. Tebyetekerwa, S. Yang, S. Peng, Z. Xu, W. Shao, D. Pan, S. Ramakrishna and M. Zhu, Electrochim. Acta, 2017, 247, 400–409 CrossRef CAS.
- R. Oraon, A. De Adhikari, S. K. Tiwari, S. Bhattacharyya and G. C. Nayak, RSC Adv., 2016, 6, 64271–64284 RSC.
- R. P. Raj, P. Ragupathy and S. Mohan, J. Mater. Chem. A, 2015, 3, 24338–24348 RSC.
- H. Mudila, S. Rana, M. Zaidi and S. Alam, Fullerenes, Nanotubes, Carbon Nanostruct., 2015, 23, 20–26 CrossRef CAS.
- P. Pandey and R. Prakash, J. Electrochem. Soc., 1998, 145, 999 CrossRef CAS.
- K. Dhanalakshmi and R. Saraswathi, J. Mater. Sci., 2001, 36, 4107–4115 CrossRef CAS.
- P. S. Abthagir, K. Dhanalakshmi and R. Saraswathi, Synth. Met., 1998, 93, 1–7 CrossRef CAS.
- D. Billaud, E. Maarouf and E. Hannecart, Synth. Met., 1995, 69, 571–572 CrossRef CAS.
- H. Mudila, P. Prasher, M. Kumar, A. Kumar, M. Zaidi and A. Kumar, Mater. Renewable Sustainable Energy, 2019, 8, 9 CrossRef.
- W. Zhou and J. Xu, Polym. Rev., 2017, 57, 248–275 CrossRef CAS.
- M. Majumder, R. B. Choudhary, A. K. Thakur, C. S. Rout and G. Gupta, New J. Chem., 2018, 42, 5295–5308 RSC.
- X. Zhou, Q. Chen, A. Wang, J. Xu, S. Wu and J. Shen, ACS Appl. Mater. Interfaces, 2016, 8, 3776–3783 CrossRef CAS PubMed.
- L. Li, H. Bi, S. Gai, F. He, P. Gao, Y. Dai, X. Zhang, D. Yang, M. Zhang and P. Yang, Sci. Rep., 2017, 7, 43116 CrossRef CAS PubMed.
- S. Majid, Electrochim. Acta, 2014, 138, 1–8 CrossRef.
- M. S. Javed, C. Zhang, L. Chen, Y. Xi and C. Hu, J. Mater. Chem. A, 2016, 4, 8851–8859 RSC.
- P. Xiong, C. Hu, Y. Fan, W. Zhang, J. Zhu and X. Wang, J. Power Sources, 2014, 266, 384–392 CrossRef CAS.
- S.-Y. Wang, K.-C. Ho, S.-L. Kuo and N.-L. Wu, J. Electrochem. Soc., 2005, 153, A75 CrossRef.
- Z. Wang, X. Zhang, Y. Li, Z. Liu and Z. Hao, J. Mater. Chem. A, 2013, 1, 6393–6399 RSC.
- H. Liu, X. Gou, Y. Wang, X. Du, C. Quan and T. Qi, J. Nanomater., 2015, 2015, 1–9 Search PubMed.
- S.-L. Kuo and N.-L. Wu, J. Power Sources, 2006, 162, 1437–1443 CrossRef CAS.
- D. Pawar, J. Shaikh, B. Pawar, S. Pawar, P. Patil and S. Kolekar, J. Porous Mater., 2012, 19, 649–655 CrossRef CAS.
- D. Pawar, S. Pawar, P. Patil and S. Kolekar, J. Alloys Compd., 2011, 509, 3587–3591 CrossRef CAS.
- F. Meng, M. Yang, L. Zhao, Y. Zhang, X. Shang, P. Jin and W. Zhang, Ceram. Int., 2017, 43, 15959–15964 CrossRef CAS.
- X.-L. Wu and A.-W. Xu, J. Mater. Chem. A, 2014, 2, 4852–4864 RSC.
- Y. Xu, Z. Lin, X. Huang, Y. Liu, Y. Huang and X. Duan, ACS Nano, 2013, 7, 4042–4049 CrossRef CAS PubMed.
- W. Chen, S. Li, C. Chen and L. Yan, Adv. Mater., 2011, 23, 5679–5683 CrossRef CAS PubMed.
- P. Xiong, H. Huang and X. Wang, J. Power Sources, 2014, 245, 937–946 CrossRef CAS.
- A. Džunuzović, N. Ilić, M. V. Petrović, J. Bobić, B. Stojadinović, Z. Dohčević-Mitrović and B. Stojanović, J. Magn. Magn. Mater., 2015, 374, 245–251 CrossRef.
- K. Phasuksom and A. Sirivat, Synth. Met., 2016, 219, 142–153 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- Y. Ding, Q. Liao, S. Liu, H. Guo, Y. Sun, G. Zhang and Y. Zhang, Sci. Rep., 2016, 6, 32381 CrossRef CAS PubMed.
- X. Ma, W. Zhou, D. Mo, Z. Wang and J. Xu, Synth. Met., 2015, 203, 98–106 CrossRef CAS.
- T. Anjitha, T. Anilkumar, G. Mathew and M. Ramesan, Polym. Compos., 2019, 40, 2802–2811 CrossRef CAS.
- M. Tokarczyk, G. Kowalski, A. Witowski, R. Koziński, K. Librant, M. Aksienionek, L. Lipińska and P. Ciepielewski, Acta Phys. Pol., A, 2014, 126, 1190–1194 CrossRef.
- X.-C. Dong, H. Xu, X.-W. Wang, Y.-X. Huang, M. B. Chan-Park, H. Zhang, L.-H. Wang, W. Huang and P. Chen, ACS Nano, 2012, 6, 3206–3213 CrossRef CAS PubMed.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- A. Kumar and R. Prakash, J. Nanosci. Nanotechnol., 2014, 14, 2501–2506 CrossRef CAS PubMed.
- I. Marriam, W. Yuanhao and M. Tebyetekerwa, Energy Storage Materials, 2020, 336–359 CrossRef.
- H. Talbi, J. Ghanbaja, D. Billaud and B. Humbert, Polymer, 1997, 38, 2099–2106 CrossRef CAS.
- Z. Ž. Lazarević, A. N. Milutinović, Č. D. Jovalekić, V. N. Ivanovski, N. Daneu, I. Mađarević and N. Ž. Romčević, Mater. Res. Bull., 2015, 63, 239–247 CrossRef.
- L. Shahriary and A. A. Athawale, Int. J. Renewable Energy Environ. Eng., 2014, 2, 58–63 Search PubMed.
- D. C. Elias, R. R. Nair, T. Mohiuddin, S. Morozov, P. Blake, M. Halsall, A. C. Ferrari, D. Boukhvalov, M. Katsnelson and A. Geim, Science, 2009, 323, 610–613 CrossRef CAS PubMed.
- K. Singh, A. Ohlan, V. H. Pham, R. Balasubramaniyan, S. Varshney, J. Jang, S. H. Hur, W. M. Choi, M. Kumar and S. Dhawan, Nanoscale, 2013, 5, 2411–2420 RSC.
- Y. Yan, T. Kuila, N. H. Kim, S. H. Lee and J. H. Lee, Carbon, 2015, 85, 60–71 CrossRef CAS.
- F. J. Sotomayor, K. A. Cychosz and M. Thommes, Acc. Mater. Surf. Res., 2018, 3, 36–37 Search PubMed.
- P. Forouzandeh, V. Kumaravel and S. C. Pillai, Catalysts, 2020, 10, 969 CrossRef CAS.
- R. Wang, W. Zhou, K. Lin, F. Jiang, Z. Wang, J. Xu, Y. Zhang, A. Liang, G. Nie and X. Duan, Electrochim. Acta, 2020, 349, 136410 CrossRef CAS.
- H. Talbi, D. Billaud, G. Louarn and A. Pron, Spectrochim. Acta, Part A, 2001, 57, 423–433 CrossRef CAS.
- H. Talbi and D. Billaud, Synth. Met., 1998, 93, 105–110 CrossRef CAS.
- A. Ghasemi, M. Kheirmand and H. Heli, Russ. J. Electrochem., 2019, 55, 206–214 CrossRef CAS.
- C. Cai, J. Fu, C. Zhang, C. Wang, R. Sun, S. Guo, F. Zhang, M. Wang, Y. Liu and J. Chen, RSC Adv., 2020, 10, 29090–29099 RSC.
- S. Ramesh, H. Yadav, C. Bathula, S. Shinde, A. Sivasamy, H.-S. Kim, H. S. Kim and J.-H. Kim, J. Mater. Res. Technol., 2020, 9, 11464–11475 CrossRef CAS.
- Q. Zhou, D. Zhu, X. Ma, J. Xu, W. Zhou and F. Zhao, RSC Adv., 2016, 6, 29840–29847 RSC.
- M. Zhong, Y. Song, Y. Li, C. Ma, X. Zhai, J. Shi, Q. Guo and L. Liu, J. Power Sources, 2012, 217, 6–12 CrossRef CAS.
- P. Li, Z. Jin, L. Peng, F. Zhao, D. Xiao, Y. Jin and G. Yu, Adv. Mater., 2018, 30, 1800124 CrossRef PubMed.
- D. Zhao, Q. Zhang, W. Chen, X. Yi, S. Liu, Q. Wang, Y. Liu, J. Li, X. Li and H. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 13213–13222 CrossRef CAS PubMed.
- H. Vijeth, S. Ashokkumar, L. Yesappa, M. Niranjana, M. Vandana and H. Devendrappa, J. Mater. Sci.: Mater. Electron., 2019, 30, 7471–7484 CrossRef CAS.
- F. Yu, Z. Liu, R. Zhou, D. Tan, H. Wang and F. Wang, Mater. Horiz., 2018, 5, 529–535 RSC.
- M. Sathiya, A. Prakash, K. Ramesha, J. M. Tarascon and A. K. Shukla, J. Am. Chem. Soc., 2011, 133, 16291–16299 CrossRef CAS PubMed.
- P. K. Kalambate, R. A. Dar, S. P. Karna and A. K. Srivastava, J. Power Sources, 2015, 276, 262–270 CrossRef CAS.
- Y. Eeu, H. Lim, Y. Lim, S. Zakarya and N. Huang, J. Nanomater., 2013, 2013, 1–6 CrossRef.
- X. Lu, D. Zheng, T. Zhai, Z. Liu, Y. Huang, S. Xie and Y. Tong, Energy Environ. Sci., 2011, 4, 2915–2921 RSC.
- T. Purkait, G. Singh, D. Kumar, M. Singh and R. S. Dey, Sci. Rep., 2018, 8, 1–13 CAS.
- P. M. Shafi, V. Ganesh and A. C. Bose, ACS Appl. Energy Mater., 2018, 1, 2802–2812 CrossRef CAS.
- A. Gupta, S. Sardana, J. Dalal, S. Lather, A. S. Maan, R. Tripathi, R. Punia, K. Singh and A. Ohlan, ACS Appl. Energy Mater., 2020, 3, 6434–6446 CrossRef CAS.
- X. Pan, Y. Zhao, G. Ren and Z. Fan, Chem. Commun., 2013, 49, 3943–3945 RSC.
- P. Anjana, S. S. Kumar and R. Rakhi, J. Mater. Sci.: Mater. Electron., 2021, 1–9 Search PubMed.
- B. Pandit, B. R. Sankapal and P. M. Koinkar, Sci. Rep., 2019, 9, 1–13 CAS.
- E. Azizi, J. Arjomandi, A. Salimi and J. Y. Lee, Polymer, 2020, 195, 122429 CrossRef CAS.
- M. Majumder, R. B. Choudhary, S. P. Koiry, A. K. Thakur and U. Kumar, Electrochim. Acta, 2017, 248, 98–111 CrossRef CAS.
- B. Purty, R. Choudhary, R. Kandulna and R. Singh, AIP Conf Proc, 2018, 1953, 030178 CrossRef.
- H. Mudila, S. Rana and M. G. Zaidi, Adv. Mater. Lett., 2017, 8, 269–275 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04946a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Pradeepan Periyat
a and
Pradeepan Periyat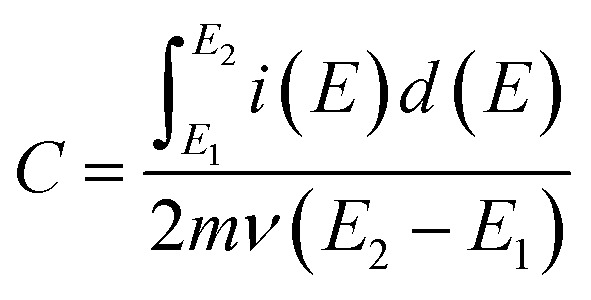
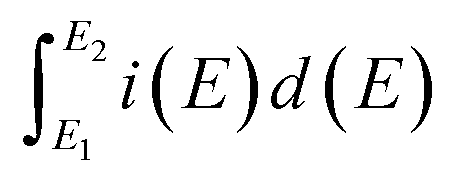 is the total voltammetric charge obtained by integration of the positive and negative sweeps in the cyclic voltammograms, ν is the scan rate, and m is the mass of the individual sample. When the charge–discharge method was used, the specific capacitance can be calculated according to the equation,39
is the total voltammetric charge obtained by integration of the positive and negative sweeps in the cyclic voltammograms, ν is the scan rate, and m is the mass of the individual sample. When the charge–discharge method was used, the specific capacitance can be calculated according to the equation,39