
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysically crosslinked PVA/graphene-based materials/aloe vera hydrogel with antibacterial activity
Wildan Hanif a,
Andri Hardiansyah*b,
Ahmad Randyc and
Lia A. T. W. Asri*a
a,
Andri Hardiansyah*b,
Ahmad Randyc and
Lia A. T. W. Asri*a
aMaterials Science and Engineering Research Group, Faculty of Mechanical and Aerospace Engineering, Institut Teknologi Bandung, Jalan Ganesha 10, Bandung 40132, Indonesia. E-mail: lia.asri@material.itb.ac.id
bResearch Center for Physics, Indonesian Institute of Sciences (LIPI), Bld. 440-442 Kawasan Puspiptek, Serpong, South Tangerang 15314, Indonesia. E-mail: andri.hardiansyah@lipi.go.id
cResearch Center for Chemistry, Indonesian Institute of Sciences (LIPI), Bld. 452 Kawasan Puspiptek, Serpong, South Tangerang 15314, Indonesia
First published on 31st August 2021
Abstract
Burn is a major skin injury that occurs worldwide. For second-degree burns, special treatment should be given for creating a suitable wound healing environment. Hydrogel wound dressing as the primary care should possess extra properties that include antibacterial activity and cytocompatibility to enhance the treatment effectiveness. Additional therapy such as electrical stimulation can be applied as well promote wound healing. Herein, we used the tissue engineering concept to create a novel antibacterial and cytocompatible hydrogel made of polyvinyl alcohol (PVA), graphene-based material (GBM), and aloe vera extract (Av) through the freeze-thaw process. We prepared the PVA/GBM/Av hydrogel and examined its potential as a wound dressing. We found that it exhibited excellent hydrophilicity with a contact angle between 15 and 31 degrees and electrical conductivity within the range of 0.0102–0.0154 S m−1, which is comparable to that of the human skin tissue and possesses tensile strength up to 1.5 MPa with elongation of 405%. It also demonstrated good stability in phosphate buffer saline with a weight ratio of 73–80% after 14 days of immersion. We presented that the addition of graphene and graphene oxide (GO) inhibited the growth of Gram-positive Staphylococcus aureus ATCC 6538 with the lowest bacterial population observed in PVA/GO, which is 1.74 × 107 cfu mL−1 after 1 day incubation and 99.94% bacterial reduction. Furthermore, our PVA/GBM/Av showed no toxicity to 3T3 fibroblast cells after 48 h with viability up to 295% for PVA/GO/Av. In summary, our fabricated hydrogels have shown their potential as wound dressing with antibacterial and non-cytotoxic properties.
1 Introduction
Skin, as the largest organ in the human body that functions to protect other internal tissues and organs from the external environment, often experiences injuries and traumas including burns.1 The World Health Organization reported more than 180![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mortality case annually due to any causes of burns worldwide in 2018.2 Recently, there are many choices of treatment for burns including skin implantation, tissue-engineered skin substitute, and wound dressing covering.3–5 Further treatment such as giving external electrical stimulation to the wound bed can also be applied to the patient, depending on the severity of wounds.6 These treatments primarily aim at creating a suitable environment for wound healing.7
000 mortality case annually due to any causes of burns worldwide in 2018.2 Recently, there are many choices of treatment for burns including skin implantation, tissue-engineered skin substitute, and wound dressing covering.3–5 Further treatment such as giving external electrical stimulation to the wound bed can also be applied to the patient, depending on the severity of wounds.6 These treatments primarily aim at creating a suitable environment for wound healing.7
Advanced wound dressing has been employed to treat various wounds since it offers wide advantages compared to conventional wound dressing.8 Moreover, its superiority in healing over the conventional ones has been proved in clinical trials.9 Hydrogel wound dressing is a type of advanced wound dressing that is available in any size and shape, allows autocatalytic debridement, provides humidity that is useful in body liquid loss prevention, and is an excellent liquid absorbent.10,11 These properties make it ideal for burn treatment.10 Furthermore, hydrogel properties are tunable; therefore, it can be designed to have certain specific properties including antibacterial activity and good electrical conductivity.12,13 Antibacterial activity in wound dressings is required as bacterial colonies can prolong the healing time and make the wound site more serious.14 On the other hand, electrical conductivity comparable to that of the skin can trigger re-epithelization, which leads to wound closure and accelerates the wound healing time.15,16 For these reasons, researchers have developed electroconductive antibacterial hydrogels made of combined materials with the intention to investigate their effect on wound healing.17–19
Polyvinyl alcohol (PVA)-based hydrogels have been exploited in various biomedical applications including bone cartilage repair,20 skin scaffold,21 drug carrier,22 and wound dressing.23 Its ability to be synthesized through various crosslinking methods as well as ease of being combined with other materials that results in desired properties makes it preferable.24 Graphene-based materials (GBM) is one example of materials inserted in the PVA hydrogel. GBM has shown its potential to be applied in the biomedical field since graphene oxide (GO) was demonstrated for the first time as a drug vehicle in 2008.23 The purpose of combining PVA with GBM is commonly to enhance the mechanical properties of the hydrogel or to obtain the antibacterial effect.25,26 Despite its biocompatibility issue, however, it possesses great antibacterial activity, excellent electrical conductivity (except for GO, which is an insulator), and mechanical properties, which make it superior to other biomaterials.27 In addition, the involvement of GBM as an antibacterial agent could kill the multi-drug resistance bacteria and minimize the risk of the patient developing immunity to antibiotic drugs.28–30 Thus, the versatility of GBM, especially for biomedical application, still needs to be explored further.
The involvement of natural products and medicinal plants for designing biomaterials is somehow preferred as it could reduce the toxicity and has useful bioactivity to treat diseases.31,32 Aloe vera (Av) has long been believed to have wound healing efficacy.33 It contains many natural molecules including polysaccharides and glycoproteins, which are the two main sources of its biological activity.10 One of the Av molecule that plays the main role in wound healing, acemannan, is a type of polysaccharide that can impede bacterial growth and stimulate macrophage activity.10 Furthermore, the anti-inflammatory and antioxidant properties, which are useful to accelerate wound healing, have been discovered in Av.34,35 On the other hand, Av has also been reported to have good electrical conductivity.36
In this research, we developed PVA/GBM/Av hydrogel and investigated its potential as a wound dressing. PVA is responsible for the hydrogel architecture and to provide humidity. The GBM that were used herein were graphene (Gr) and GO with the aim to give anti-bacterial properties, electrical conductivity, and mechanical properties to the hydrogel. Since GO is widely known as an insulator material, relatively high temperature processing was chosen to transform it into reduced graphene oxide (rGO) in order to give better electrical conductivity properties.37 Lastly, the addition of Av aims to achieve bioactive properties of the hydrogel since it is well known for its efficacy in wound healing.10 To fabricate this hydrogel, we used the physical crosslinking method, freeze-thawing, to avoid the external chemical crosslinker agent that could affect the toxicity of the hydrogel.38 The hydrogel structure and morphology were characterized through scanning electron microscopy (SEM), Fourier transmission infrared (FTIR), and Raman spectroscopy. The hydrogel performances were tested through water contact angle measurement, electrical conductivity test, tensile test, degradation test, antibacterial assay, and cell cytotoxicity assay.
2 Experimental
2.1 Materials
Fully hydrolyzed PVA (99%, Mw = 146000–186000 g mol−1), GO (powder, 15–20 sheets, 4–10% edge-oxidized), and phosphate buffered saline (PBS, pH 7.4, sterile-filtered) were purchased from Sigma-Aldrich (USA). Gr powder was obtained from Strem Chemicals (USA). Av (powder, 200× concentrate) was purchased from the local company, Skin Dewi (Indonesia). All chemicals were used as received.
2.2 PVA/GBM/Av hydrogel preparation
Ten grams of PVA powder, 0.05 g GO, 0.05 g Av, and 100 mL of demineralized water, which was previously heated to 105 °C, were added to a beaker. The mixture was stirred at 105 °C until all the constituent materials were dispersed well. The solution was cooled down to room temperature and was homogenized after that using an ultrasonic bath for 30 min. This solution was cast by pouring 22.5 mL of the solution gently into a Petri dish with a diameter of 10 cm. Finally, the PVA/GO/Av solution was subjected to 3 cycles of successive freezing and thawing (freezing for 20 h at −15 °C and thawing for 4 h at 5 °C). The obtained hydrogels (average thickness = 1.821 ± 0.497 cm) were stored in a refrigerator at 5 °C before further use. All the hydrogels were synthesized using the same procedure, as explained above. The composition of the hydrogels was as follows: (1) PVA/Gr001/Av: 10 g PVA, 0.01 g Gr, and 0.05 g Av; (2) PVA/Gr005/Av: 10 g PVA, 0.05 g Gr, and 0.05 g Av.2.3 Characterization
All the samples were freeze-dried for 24 h prior to characterization with FTIR, Raman spectroscopy, and SEM. FTIR measurements were recorded with KBr pellets on a Prestige 21 Shimadzu. A sample shuttle measurement was performed to interleave the sample and scan the background. The spectra were measured at a resolution of 4 cm−1 with the number of scans of 40, at a wavelength of 4500 to 400 cm−1. Raman shifts were recorded by a Modular Raman Spectro Type iHR320 with an objective of 100×, grafting 1800 g mm−1, and 532 nm laser. The surface image was obtained using the SEM instrument Hitachi SU-3500, where the samples were coated with gold beforehand.2.4 Water contact angle measurement
The sample (n = 3) was cut to 1 cm × 3 cm dimensions and seated in the sample place of the measuring instrument. 1 μL demineralized water was dropped vertically on the sample surface. The image was taken using a Dino-Lite digital microscope premier AM3111/3113 series and captured right after the water droplet touched the surface. Afterward, the image was analyzed with the ImageJ software to measure the contact angle of water with the sample's surface.2.5 Conductivity test
The sample (n = 3) was cut to 1 cm × 3 cm dimensions. The test was conducted using a Keithley 2450 SourceMeter with the source mode of voltage sweep and a linear sweep type for 4-wire sensing. Voltage sweep was started from −4 V to 4 V with 401 number of steps. The distance between the probes was set to 1 cm. The conductivity value was calculated using the following formula.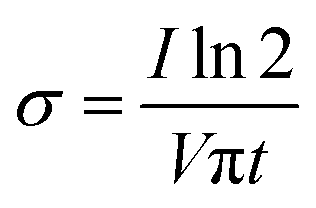
2.6 Tensile test
The samples of the three were cut into the designated type V geometry (refer to ASTM D638). The test was performed according to ASTM D638 at room temperature using a universal testing machine with a testing speed of 100 mm min−1. Each sample group has three samples to be tested, which was previously immersed in demineralized water and wiped by a tissue paper to remove excessive water on the hydrogel surface.2.7 Degradation test
The hydrogel (n = 3 per time point) was trimmed into 1 cm × 1 cm square-shaped and measured for its initial weight (Wi). Then, it was put into 2 mL pH 7.4 PBS solution. After a certain time point (1 d, 2 d, 3 d, 4 d, 5 d, 6 d, 7 d, and 14 d), each sample was washed with demineralized water prior to final weight (Wf) measurement to ensure that no salts were left within/on it. Weight ratio (WR) was calculated using the following formula.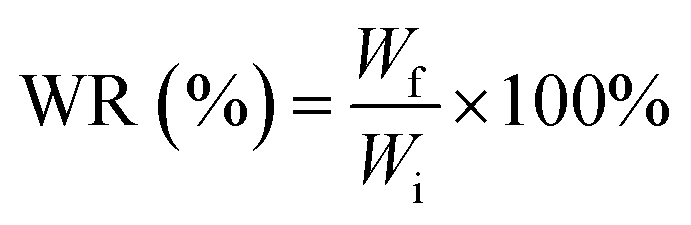
2.8 Antibacterial assay
:20) with MHB medium.To obtain the bacterial suspension with a desired population, total plate count (TPC) was conducted beforehand. The decimal dilution of the bacterial suspension (1:20) was carried out while 9 mL of 0.9% NaCl solution was pipetted into five different reaction tube. The first tube was used to dilute the bacterial suspension by 10−1 times by adding 1 mL of the bacterial suspension to the tube. Then, it was vortexed for homogenization. From the homogenized suspension in the first tube, 1 mL of the suspension was moved to the second tube to obtain 10−2 dilution. It was then homogenized via vortex and the steps were repeated until the fifth tube for 10−5 dilution. However, 1 mL of the resulting dilution in the fifth tube was removed after being homogenized.
Afterwards, 1 mL from each of 10−3 dilution and 10−5 dilution tubes were added to a sterilized Petri dish. 20 mL of 50 °C MHA liquid was also poured into the Petri dish, which was mentioned earlier, followed by shaking the Petri dish in order to homogenize the mixture in it. This step was carried out twice for each. The suspension was incubated for 24 h at 37 °C, which then was counted for the number of bacterial colonies grown the next day. After reaching 1.6 × 10−6 cfu mL−1 of bacterial population, the antibacterial assay method of agar diffusion can be conducted.
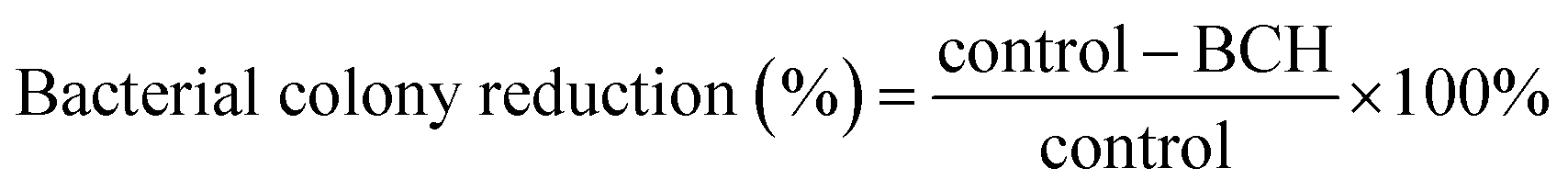
2.9 Cell cytotoxicity assay
3 Results and discussion
3.1 Preparation of the PVA/GBM/Av hydrogel
We successfully synthesized the PVA-based hydrogel with the combination of GBM and Av. We studied the influence of various concentrations of PVA in the resulting hydrogel and the freeze–thaw process as well. We prepared 4 groups of PVA with the concentration of 2 wt% (PVA2), 5 wt% (PVA5), 10 wt% (PVA10), and 12.5 wt% (PVA12.5). We examined the effect of the PVA concentrations through the observation of the PVA solution's physical appearance during the first two hours of freezing with 15 min gap per timepoint (Fig. 1A). The crystallite started to form during the freezing or nucleation of water molecules within the PVA solution. Nucleation corresponds to the process where the water molecule begins to freeze, leading to the phase separation of water and PVA. The crystallites were observed in PVA5, PVA10, and PVA12.5 at 30 min but were hardly seen in PVA2. In PVA5 and PVA10, crystallites started to develop from small white dots, which then propagated to its surroundings through the emerging of white branches. Nucleation in PVA12.5 was started by the white dot as well. But differently, these white dots did not spread to their surroundings by creating branches. They grew up following the pattern of their nuclei shapes; as shown by PVA12.5 at 45 min, the shape of the white area is circular with a thin transparent area as the boundary. In correlation to PVA concentration, PVA12.5 contains the most amount of the PVA chain, followed by PVA10, PVA5, and PVA2 consecutively. In case of PVA12.5 during early freezing, water molecules that begin to freeze were unable to move the PVA chains around them. It resulted in the localized round-shaped nucleation without propagation, causing noticeable phase separation as in PVA5. It happened due to the large number of PVA chains around them. In contrast, the PVA chains within PVA5 and PVA10 were forced by freezing water to get them separated from water; hence, phase separation occurred.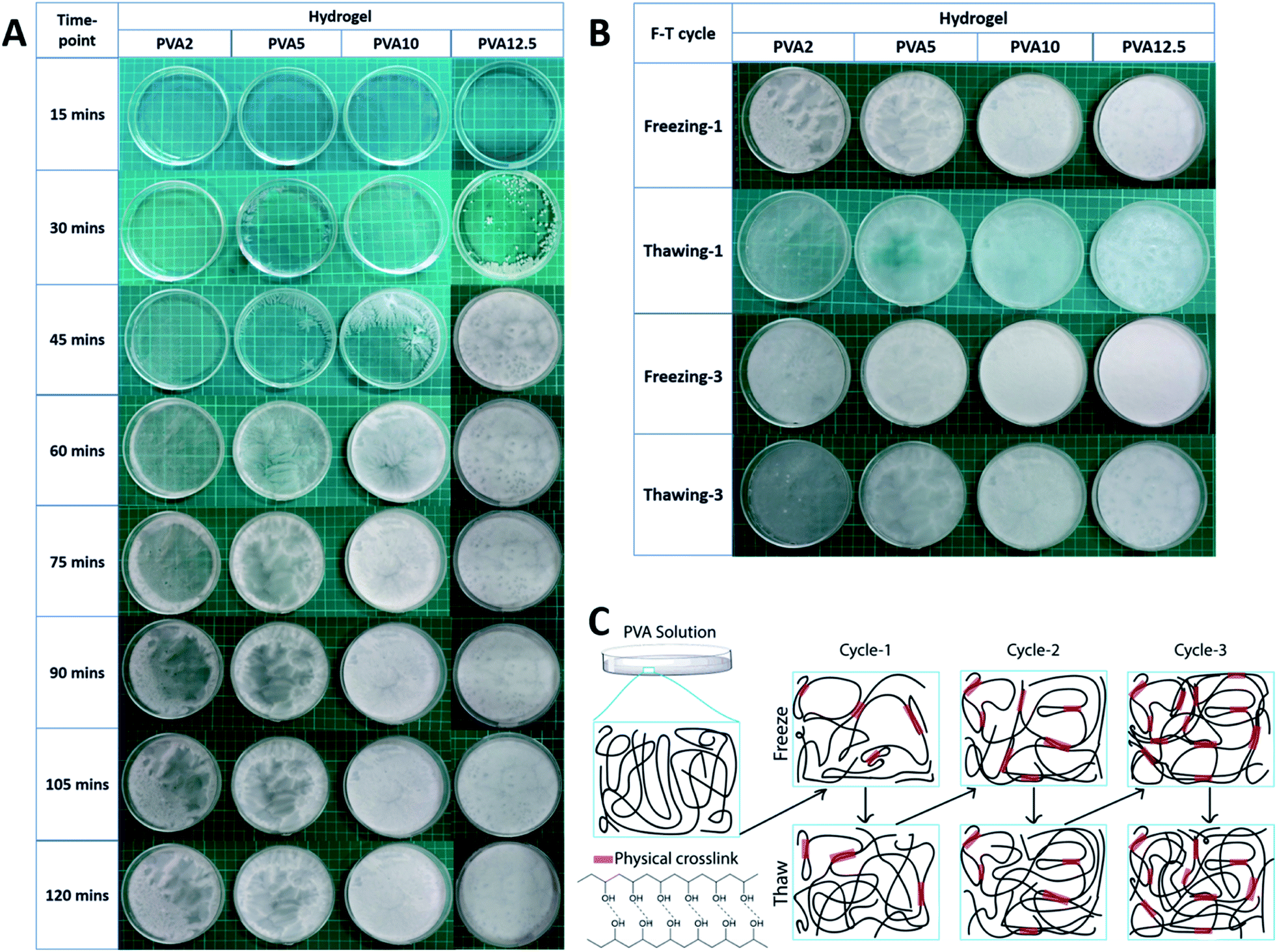 | ||
| Fig. 1 PVA concentration effect on the physical appearance of the hydrogel. Observed through (A) the first 2 hours of freezing of the first freeze–thaw cycle at the timepoint of 15 minutes and (B) after the first and third cycle of freeze-thawing. Phase separation occurred and was clearly observed in PVA2 and PVA5 while PVA10 and PVA12.5 were relatively more homogenous. (C) Physical crosslink (represented by red bar) formation mechanism of the PVA hydrogel via freeze-thawing. | ||
Surprisingly, PVA12.5 transformed to the frozen state completely after 45 min, followed by PVA10, PVA5, and PVA2 after 60 min, 75 min, and 105 min, respectively. After passing these timepoints, there was no significant change observed visually. It is clearly seen that there was a white region and a more transparent part in PVA2 and PVA5 after 120 min, which remained after 20 h. The high crosslinking degree resulted in a white region (crystalline region), the part formed by hydrogen bond interactions between the PVA chains.40 In contrast, the transparent region has less cross-linked PVA, showing less crystallinity and free water.41 These regions, which are distinguished by the localized PVA concentration, were caused by the phase separation phenomenon during freezing in freeze-thawing.42 This phenomenon occurred in PVA10 and PVA12.5 as well but the results of this are not clearly seen as in PVA2 and PVA5 due to the higher concentration of PVA.
As expected, after the thawing of the first cycle (Fig. 1B), we can see that the less transparent area possesses a larger amount of water. Further, the freeze-thawing cycle creates a higher number of hydrogen bond as more phase separation occurred, which resulted in a more homogenous hydrogel (fully white), as shown after the completion of freezing in the third cycle. The final hydrogel shows that the most phase separation occurred in PVA5. The more or less appearance of phase separation results herein indicate that there still appear white and transparent regions that can be distinguished visually. Meanwhile, PVA2 exhibited little phase separation as well as PVA10 and PVA12.5 but due to the lower PVA concentration, it was more likely a water sheet with low mechanical strength.43 PVA10 and PVA12.5 are good in terms of the mechanical properties but PVA10 is much easier to dissolve with our method. This is the consequence of PVA chain segregation from water during freeze-thawing. By considering the final application of this hydrogel as the wound dressing and its fabrication process, we chose PVA10 as the most ideal concentration of PVA to synthesize the hydrogel for the wound dressing purpose.
In summary of the freeze-thawing method, Fig. 1C illustrates the individual process that occurs in each step of the freeze-thawing process. The PVA solution would automatically experience phase separation as it is the key point of physical crosslink formation.42 The water molecule that begins to freeze would push the surrounding PVA chains and make them closer to each other. This leads to the formation of the crystallite, which then acts as a crosslink through hydrogen bond interaction between hydrogen and oxygen in each PVA chain. On the other hand, during thawing, the chain will self-rearrange, move freely, and lose the weak interchain interaction. Further, the freeze-thawing cycle would increase the crosslink density as well as phase separation, which results in evenly distributed concentrated PVA.42
In order to make the PVA/GBM/Av hydrogel that is good in the mechanical properties, we synthesized the hydrogel from PVA with a concentration of 10 wt% and combined it with GBM, which are Gr and GO. GBM was added to increase the mechanical and antibacterial properties of the hydrogel. At the same time, GBM addition was expected to improve the hydrogel conductivity as well. We also added Av to serve as the additional bioactive agent for the anti-inflammation purpose since it contains acemannan, a natural anti-inflammatory compound.44 Fig. 2A shows all the hydrogel samples. Phase separation was observed only on PVA/Gr005 and PVA/Gr005/Av. Meanwhile, in PVA/Gr/Av and PVA/GO/Av, there is a noticeable small white circle, which is a sign of primary nucleation location of freezing during the freeze-thawing process. We proposed a structural model for PVA/Gr/Av and PVA/GO/Av, as shown in Fig. 2B and C, respectively. For Gr in PVA/Gr/Av, since it has neither oxygen nor hydrogen functional groups, it would not have any secondary interaction with PVA as well as other materials, while the Av in both hydrogels could have an interaction in the same manner as GO in PVA/GO/Av. They could form hydrogen bond interaction with the hydroxyl group possessed by PVA as all of them have oxygen functional groups. On the other hand, it is also possible for these materials to get trapped within the hydrogel network without any secondary interaction with PVA.
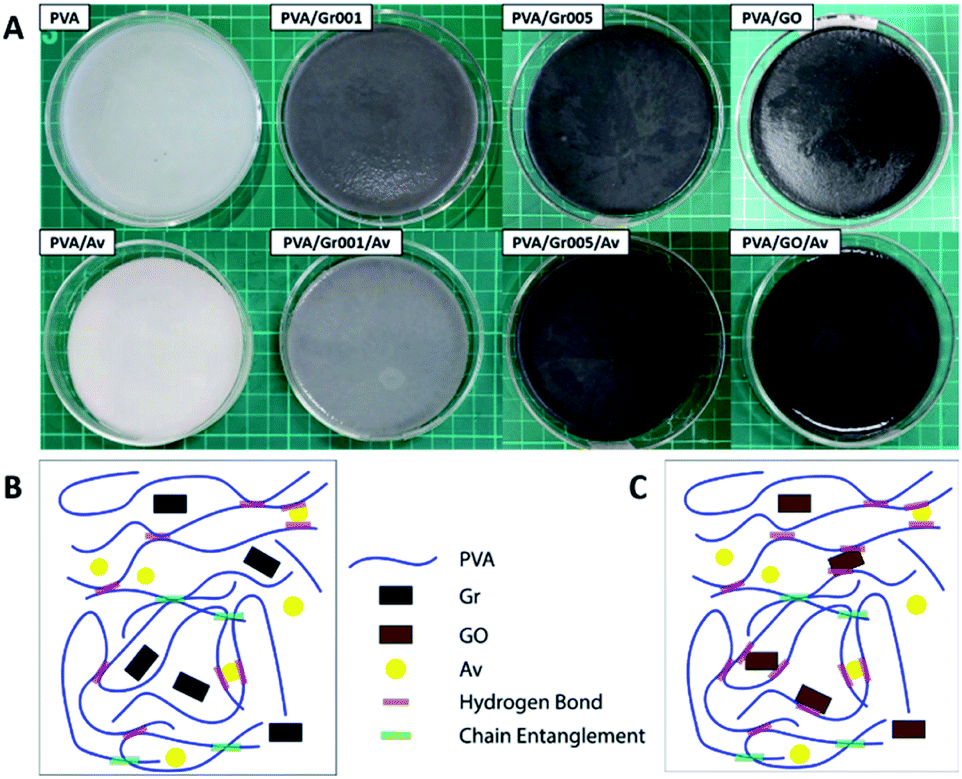 | ||
| Fig. 2 (A) All hydrogel samples with PVA concentration of 10 wt%. The proposed structure of the PVA/GBM/Av hydrogel: (B) PVA/Gr/Av and (C) PVA/GO/Av. | ||
3.2 Hydrogel characterization
The FTIR spectra (Fig. 3A) show the characteristic of PVA that is represented by a broad peak at 3000–3700 cm−1, which indicates the O–H stretching vibration.23,45 This peak also belongs to other materials including GO and Av that overlap with each other.46,47 C–H stretching vibration at 2939 cm−1 (PVA) and 2941 cm−1 (PVA/Av) are also presented. The peak at 1637 cm−1 (PVA), 1658 cm−1 (PVA/Av), 1629 cm−1 (PVA/Gr001 and PVA/Gr001/Av), and 1631 cm−1 (PVA/GO and PVA/GO/Av) are attributed to C–H bending vibration.23 The peak at 1093 cm−1 that represented –OH absorption appeared on all the hydrogels except for PVA/GO.48 The characteristic peaks of Av at 1436 cm−1 can be clearly seen on all the hydrogels with the addition of Av.49 Another peak of the chemical component of Av, which is at 1238 cm−1 (PVA/Av) and 1236 cm−1 (PVA/Gr001/Av), presented the response of C–O stretching vibrations of esters and phenolic groups.47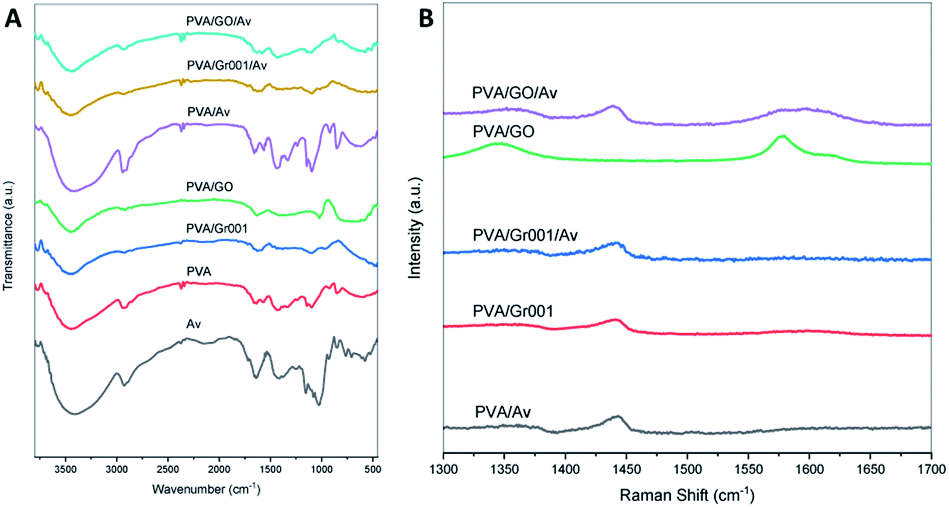 | ||
| Fig. 3 Characterization of the PVA/GBM/Av hydrogels: (A) FTIR spectra and (B) Raman shift of all the hydrogel samples. | ||
From Raman spectroscopy (Fig. 3B), we confirmed that GO added at the beginning of the preparation was reduced to rGO. However, we cannot determine whether it was partially or fully transformed through this characterization alone. For PVA/GO, the peak at 1577 cm−1 (G band) appeared with higher intensity compared to the D band, which was located at 1344 cm−1 (D band). After the deconvolution of the PVA/GO/Av curve, both the G band and D band were found at 1593 cm−1 and 1358 cm−1, respectively. The ID/IG ratio for both PVA/GO and PVA/GO/Av are 0.62 and 0.77, respectively. The higher intensity of the G band and the ID/IG ratio less than 1 indicate the more ordered structure in the graphene basal plane.37 The G band reflects the ordered structured of the graphene basal plane. Its appearance is due to the first order scattering of the E2g phonon from the sp2 carbon atoms. In contrast, the D band represents the defect at the edge and/or basal plane of graphene and its emergence is caused by the breathing mode of the k-point photons of A1g symmetry.25 Unlike GO, Gr commonly has the G band only as it comprised by the ordered structure of the basal plane without or with a little defect. It was found at 1590 cm−1 in PVA/Gr and PVA/Gr/Av. The small peak might be a consequence of the amount of Gr within the sample that is very small.
Fig. 4 shows the surface characteristic of each hydrogel, which was obtained through SEM. All hydrogels possess dense structure with porosities morphology but this porosity interconnectivity cannot be examined yet. The appearance of these porosities also confirmed that the freeze-thawing process was carried out successfully, as previously reported.41 The porosity formation in these hydrogels is the consequence of the repetition of the freeze-thawing cycle. In general, the porosity in the hydrogel for wound dressing would be useful as a way for water and active agents to move in and out from the hydrogel as well as the way for cellular mobilization.50 Thus, the porous morphology indirectly supports the antibacterial activity provided by the active agents through the release phenomenon.51 As an example, Rivero-Buceta et al. investigated that the porosities in the hydrogel could increase the ability of the antibacterial agent to kill the bacteria as it can improve the release of the bioactive agents to interact directly with the bacteria.52
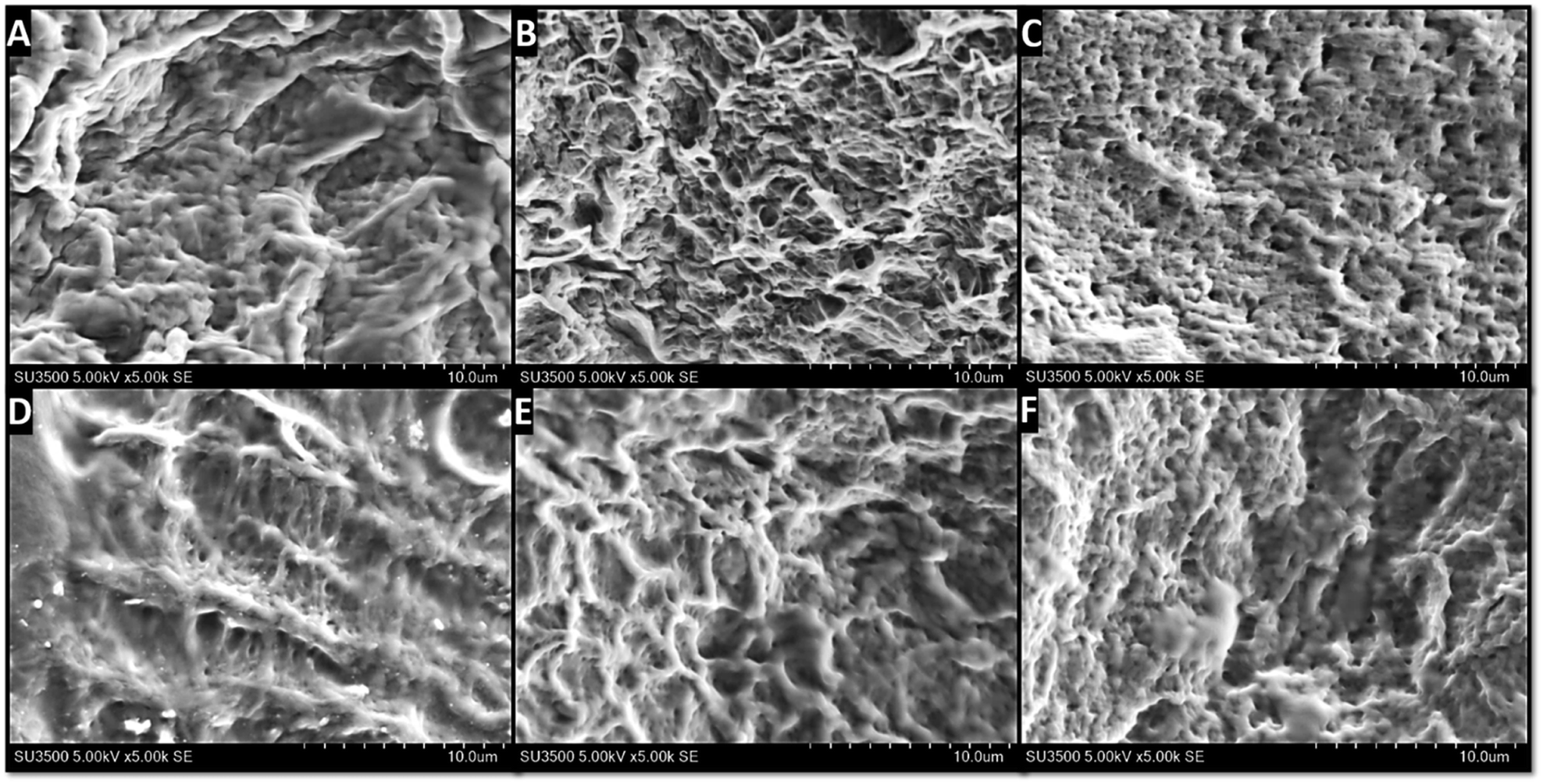 | ||
| Fig. 4 Surface characterization of the hydrogel through SEM imaging: (A) PVA; (B) PVA/Gr001; (C) PVA/GO; (D) PVA/Av; (E) PVA/Gr001/Av; (F) PVA/GO/Av. Porosities are observed in all the samples. | ||
3.3 Water contact angle measurement
The hydrogel should be hydrophilic as it is intended to be interactive with the human tissue at the wound site. All the constituent materials of our hydrogels are hydrophilic except for Gr, which is hydrophobic. GO is considered hydrophilic in general. However, the use of GO within our hydrogels is considered to be more hydrophobic as it is 10% edge-oxidized and is reduced to rGO due to heating during solution preparation. All the samples have a contact angle with water below 90 degrees (Table 1), which indicates that they are excellent in terms of the hydrophilicity. Nonetheless, we cannot conclude the effect of Gr or GO addition through this test since this property is also influenced by other factors including the surface roughness and the location of GBM in the hydrogel.53| Hydrogel | Water contact angle (degree) | Electrical conductivity (S m−1) |
|---|---|---|
| PVA | 30.79 ± 3.40 | 0.0132 ± 0.0 |
| PVA/Gr001 | 31.47 ± 0.30 | 0.0125 ± 0.00127 |
| PVA/Gr005 | 21.64 ± 2.07 | 0.0125 ± 0.00127 |
| PVA/GO | 27.39 ± 2.96 | 0.0110 ± 0.0 |
| PVA/Av | 29.39 ± 4.43 | 0.0154 ± 0.00221 |
| PVA/Gr001/Av | 30.56 ± 2.29 | 0.0117 ± 0.00127 |
| PVA/Gr005/Av | 14.95 ± 1.70 | 0.0102 ± 0.00127 |
| PVA/GO/Av | 28.97 ± 3.97 | 0.0154 ± 0.00221 |
3.4 Electrical conductivity
Our hydrogels are intended to mimic the electrical conductivity of the skin tissue, which is in the range of 0.026 S m−1 to 0.22 S m−1.54 Similar conductivity has been proven to promote and enhance the wound healing process.54,55 It is noteworthy that our hydrogels exhibited conductivity values that are close to that of natural dermis (0.22 S m−1) and epidermis (0.026 S m−1).17 We conducted the electrical conductivity test of the wet-state hydrogel through the 4-point probe method. The addition of Gr and GO to PVA decreased the conductivity of PVA slightly from 0.0132 S m−1 to 0.0125 S m−1 (for both PVA/Gr001 and PVA/Gr005) and 0.011 S m−1 (PVA/GO). However, after introducing Av into all the hydrogels, their conductivity increased except for PVA/Gr005/Av, which had decreased (Table 1). PVA/Av and PVA/GO/Av exhibit the highest conductivity with a value of 0.0154 S m−1. Even then, there is no significant difference between their conductivities.It is not clearly seen that the GBM affect the PVA hydrogel conductivity. Aycan et al. reported that the higher concentration of rGO within the polymeric network tends to agglomerate more easily than the lower one.56 This would lead to decreasing conductivity of the polymer since the rGO nanoparticles are not well-distributed in all the regions. This might be the reason that GBM addition, especially Gr, did not increase the conductivity significantly. Further, in case of GO, it did not improve the hydrogel conductivity even though Raman spectroscopy shows that it might have transformed to rGO. This result is in line with the previous study carried out by Slobodian et al., which showed that low temperature reduction reduces GO without a significance enhancement in the electrical conductivity.57 On the other hand, the addition of Av increased the conductivity value of all the samples except PVA/Gr. This result is considerably meaningful as the Av in our hydrogel is in its liquid/gel form and has been reported to have good electrical conductivity.36,58 Saberian et al. reported that the Av gel with the concentration of 0.5–2% exhibited electrical conductivity that is temperature-dependent, where from 30 °C to 60 °C, the conductivity value is about 0.0063–0.0236 S m−1.36 In addition, the Av gel has also been reported to be able to transmit current where it has been used for promoting the wound healing of the surgical wound model in Wistar rats by applying the Av gel and the microcurrent as electrical stimulation.59
In accordance with the electrical conductivity value effect on the wound healing process, our hydrogels should be able to reduce the wound healing time and fasten wound closure excellently as the conductivity of our hydrogels is comparable to the skin conductivity. As a comparison, He et al. demonstrated their hydrogel with conductivity within the range of 2.6 × 10−3–8.45 × 10−3 S m−1, which could lead to wound closure of the full thickness wound model in mice completely after 14 days.19 Another hydrogel made of functionalized chitosan and GO with 0.07 s–0.11 S m−1 range of conductivity has been proved as well by Zhang et al. to be able to significantly enhance the healing of a full thickness wound in mice with the closure of the wound site up to 98% after 14 days.60
3.5 Mechanical properties
The hydrogel for wound dressing purposes must have sufficient mechanical properties, which includes tensile strength and modulus elasticity.61 We tested our hydrogels according to ASTM D638 with the results shown in Fig. 5 and Table 2. A previous study has reported that the addition of GBM to the PVA hydrogel increased the tensile strength of the PVA hydrogel.62,63 We found that GO addition to PVA increased the tensile strength of the hydrogel up to 1.5 MPa or about two times higher than PVA alone. Interestingly, this value is the highest among the other samples, even higher than that of PVA/Gr001 and PVA/Gr005. This is probably the result of the GBM interaction with the PVA hydrogel network. Even though Gr has higher tensile strength than GO, it interacts hydrophobically with PVA. It could lead to weak interaction with the PVA network and resulted in phase separation/agglomeration, as seen in Fig. 2A (PVA/Gr005 and PVA/Gr005/Av). The good interface between the PVA chains and GBM is the main factor behind the successfully enhanced mechanical properties of the PVA-GBM hydrogel.64 If it is not achieved, it would lead to the deterioration of its properties. Zhang et al. prepared freeze-thawed PVA hydrogel with the addition of 0.8 wt% GO and demonstrated that this hydrogel possesses a tensile strength of 3.48 MPa and breaking elongation up to 140%.62 Huang et al. found the tensile strength and elongation of the PVA hydrogel were enhanced, respectively, from 0.25 MPa to 0.543 MPa and from 350% to 405% with the addition of 0.05 wt% of GO.63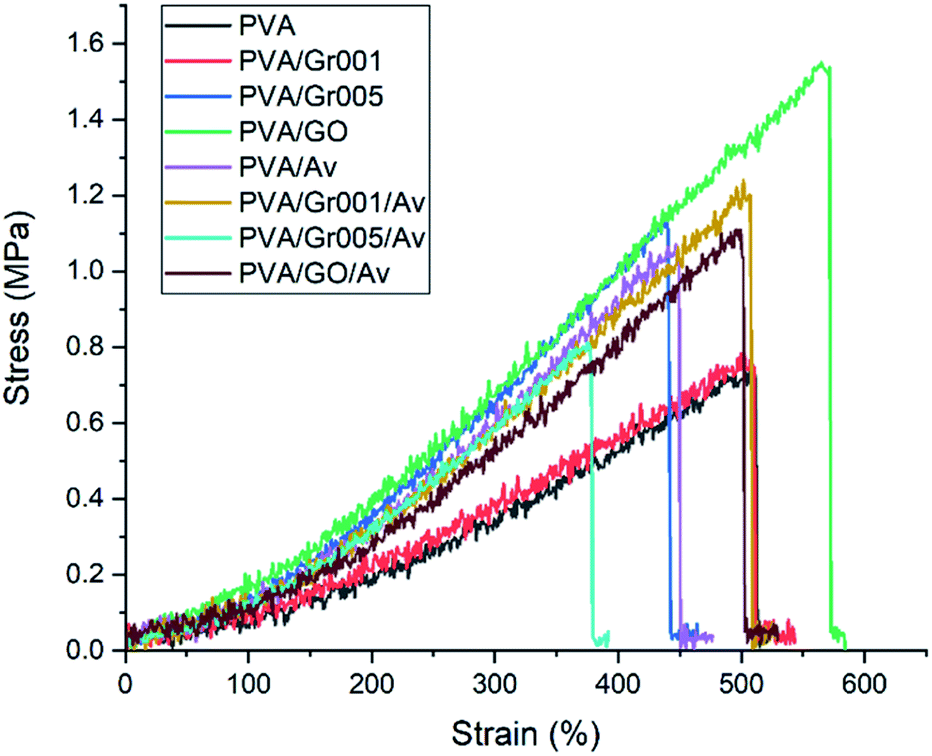 | ||
| Fig. 5 Stress–strain curve of all hydrogels. PVA/GO shows the highest tensile strength. | ||
| Hydrogel | Tensile strength (MPa) | Tensile strain (%) | Modulus elasticity (MPa) |
|---|---|---|---|
| PVA | 0.740 ± 0.030 | 459.42 ± 66.18 | 0.231 ± 0.049 |
| PVA/Gr001 | 0.820 ± 0.114 | 520.17 ± 11.41 | 0.228 ± 0.042 |
| PVA/Gr005 | 1.103 ± 0.220 | 436.45 ± 15.40 | 0.393 ± 0.072 |
| PVA/GO | 1.543 ± 0.040 | 508.68 ± 57.02 | 0.389 ± 0.033 |
| PVA/Av | 1.073 ± 0.015 | 438.31 ± 27.30 | 0.439 ± 0.025 |
| PVA/Gr001/Av | 1.213 ± 0.132 | 500.91 ± 17.00 | 0.341 ± 0.054 |
| PVA/Gr005/Av | 0.887 ± 0.150 | 397.57 ± 47.46 | 0.312 ± 0.021 |
| PVA/GO/Av | 1.123 ± 0.023 | 496.46 ± 30.34 | 0.331 ± 0.013 |
On the other hand, the incorporation of Av in this hydrogel system gives a different effect to each hydrogel. Both PVA/Av and PVA/Gr001/Av experienced increasing value of the tensile strength compared to PVA and PVA/Gr001. In contrast, the Av appearance in PVA/Gr005 and PVA/GO lowers this property; nonetheless, it is still higher than that of PVA alone. As Gr is hydrophobic, it tends to agglomerate, which leads to an inhomogeneous hydrogel and the higher Gr concentration elevates the chance of this to occur. Then, we expected that Av addition will not affect the hydrogel mechanical properties since it has never been reported to have the strengthening effect as a GBM.
In terms of the ability to strain, all the hydrogels demonstrated it excellently, which was represented by the tensile strain value of more than 400%. Besides, all the hydrogels also possess adequate elasticity with modulus elasticity within the range of 0.3–0.5 MPa. This property is very important to ensure that it could be adjustable, following the skin contour.18 Our hydrogels are considerably comparable to the skin mechanical properties, specifically, the tensile strength and elastic modulus.65,66 More importantly, the value of these properties in the hydrogel should be less or equal to the skin; thus, it can be applied effectively to the wound bed.
3.6 Degradation behavior
We evaluated the degradation behavior of all the hydrogels in PBS at the timepoint of 1 d, 2 d, 3 d, 4 d, 5 d, and 14 d. According to the clinical practice, hydrogel wound dressing in burn treatment is to be ideally changed after no more than 5 days of use.67 There is weight loss observed, which is represented by the decrease of WR from 100% to 85% in all the hydrogels after 1 day (Fig. 6), except for PVA, which gets increased. WR itself indicates the ratio of the weight of the sample at a particular timepoint to its initial weight. PVA/Av, PVA/Gr001/Av, and PVA/GO/Av are likely to decrease in the WR at each timepoint until 4 d. We assume that weight reduction is mainly caused by the dissolution of the non-crosslinked component to the water environment. Interestingly, their WR is somewhat higher after 5 d, then decrease again at 6 d, and increase slightly at 7 d (but less than WR at 5 d). Their WR after 6 d, 7 d, and 14 remained constant at about 80%.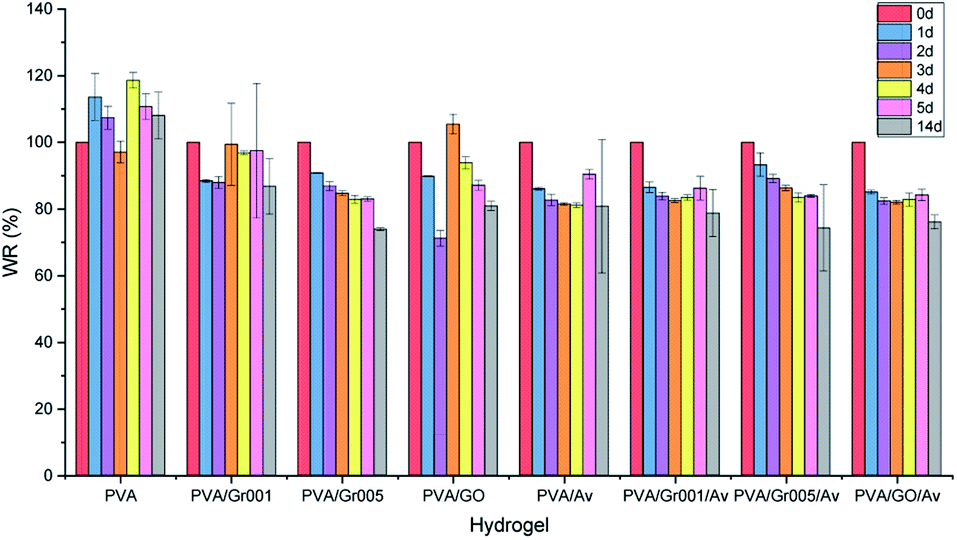 | ||
| Fig. 6 Weight ratio (WR) percentage profile of all the hydrogel groups, which were obtained by sample immersion in PBS for 1, 2, 3, 4, 5, and 14 days. | ||
Differently, PVA/Gr005 and PVA/Gr005/Av are two groups that experienced weight reduction from day to day with WR of PVA Gr/005 at 1 d and 14 d, consecutively, being 90.78% and 73.88%, while PVA/Gr005/Av are 93.26% and 74.28%, respectively, at the same time point. Same as PVA/Av, PVA/Gr001/Av, and PVA/GO/Av, these two also experienced a slight increase in the WR by 0.17% and 0.46%, consecutively, at 5 d. Despite this similar behavior, their WR keep getting lower until 14 d. However, from the degradation data profile of these two hydrogels and others, increasing the soaking time of all the hydrogels might not give further weight reduction.
There is an equilibrium point of the WR for these hydrogels where they absorb water and vice versa. This point appeared as the response of hydrogel in the salt environment. Still, WR will stay around the equilibrium point after reaching the stability. The same behavior of the PVA hydrogel has been reported by Hassan et al. where they demonstrated the swelling and dissolution of the freeze-thawed PVA hydrogel in deionized water.68 They confirmed that PVA gets degraded due to the dissolution of the not-strongly-attached PVA chains to the hydrogel network. As we employed the freeze-thawing process to create the physical crosslink between PVA, there are chances of PVA chains to not be involved in crystallite formation during freezing. Hence, it resulted in the dissolution of these chains in water. Then, after these weak-bonded chains have been fully dissolved, the hydrogel would remain in the equilibrium state, which have been mentioned previously.
We evaluated the degradation behavior of our hydrogels through their weight reduction. We suspect that the weight loss was primarily caused by the dissolution of PVA weakly attached to the hydrogel network. In this case, this PVA did not contribute well to crystallite formation. Hence, once it gets disrupted by the PBS solution, it would dissolve easily since no strong bonds connected it with the crosslinked PVA network. Secondly, we hypothesize that water released from the hydrogel is another cause of this phenomenon to occur since we used PBS solution as the medium of the test. Water released from the hydrogel network occurred due to hydrogen bond devastation that was caused by hydrogel–PBS solution interaction. Otherwise, the combination of these two causes worked synergistically to degrade the hydrogel.
In case of PVA, PVA/Gr001, and PVA/GO, they seem more stable compared to other samples as they experienced swelling–deswelling at a higher point of WR. The hydrogels that experience swelling indicate that it possesses a lower degree of physical crosslinking. It causes the hydrogels to have more free volume and makes it easier to rearrange its chain during water uptake. Thus, it results in the swelling phenomenon of the corresponding hydrogels. However, we can also obviously see that these hydrogels also have decreased WR after swelling. This would happen because of the deeper unstable PVA chains being penetrated by the PBS solution and getting disrupted. At the end, these chains get dissolved in the PBS solution and result in the lower weight of the hydrogel. Despite the ability of the lower degree crosslinked PVA hydrogel for easier water absorption, it is actually unstable in terms of the weight. For this reason, we see that swelling and deswelling in these three hydrogels as well as in other hydrogels have occurred despite the number of changes in the weight. Fortunately, it is not a big matter in the case of wound dressing application since the number of PVA molecules that get dissolved is low and also the replacement with a new hydrogel dressing can be done anytime. In addition, a further test in the burns model in vivo is needed to see whether this behavior would happen in the same manner as the results of this test.
3.7 Antibacterial activity
The antibacterial property in wound dressing is important to reduce the probability of the wound getting infected by microbes such as the bacteria.69 The appearance of the bacterial colony at the wound site would prolong the inflammation stage in wound healing. Bacteria can also lead to serious pathological conditions such as infection and sepsis, which may lead to death.70 Our hydrogels were first evaluated through the agar diffusion method as the first screening method to see whether they have antibacterial activity through the emergence of the inhibition zone. Staphylococcus aureus ATCC 6538, Gram-positive bacteria, was employed in this test. As shown in Fig. 7, all the samples exhibited a slight inhibition zone except for PVA and PVA/Av. The inhibition zone indicates the reduction in the bacterial colony population within that area. From this, we can conclude that all the PVA/GBM and PVA/GBM/Av hydrogels have antibacterial activity. The formation of that zone is determined by several factors including the ability of the antimicrobial agent to diffuse into the surface of the hydrogel. The absence of the inhibition zone may not be generalized as the lack of antibacterial activity. Hence, another method to examine the antibacterial properties for materials that do not present this zone should be employed.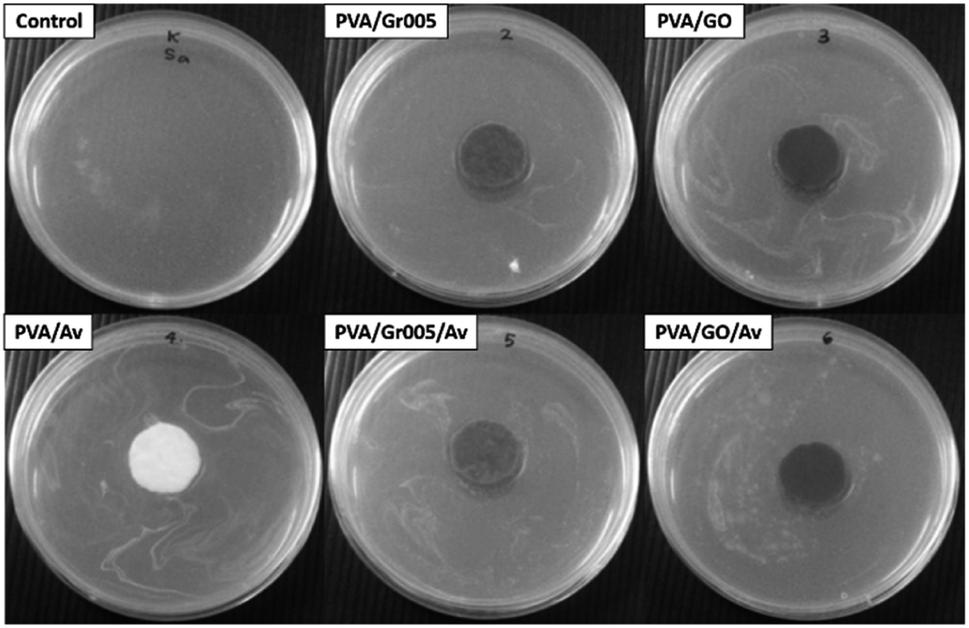 | ||
| Fig. 7 Antibacterial activity of the PVA/GBM/Av hydrogels, which was observed through the pour plate disc diffusion method. | ||
We used the TPC method to assess the effect of the antibacterial quantitatively. From this method, the antibacterial activity of PVA and PVA/Av can be measured as well. The initial bacterial colony population was 1.6 × 106 cfu mL−1. Table 3 presents the population number of the bacterial colonies and the bacterial reduction percentage with respect to our hydrogels. Compared to the PVA/Av hydrogel, the incorporation of GBM into the PVA hydrogel system could reduce and lower the growth of the bacterial colony significantly (p < 0.05), as confirmed through the TPC method. For the hydrogel without Av, PVA/GO had the lowest bacteria population number, which is 2.68 × 107 cfu mL−1. In addition, the interaction between GO and Av would increase the number of bacterial populations even though are still lower than that of PVA without GBM. Overall, this result might not satisfy our expectation since there were no hydrogels that are able to kill the bacterial colony completely. However, our hydrogels have shown the percentage of bacterial colony reduction up to 99.94% for PVA/GO, 99.64% for PVA/Gr005, 99.53% for PVA/GO/Av, and 99.34% for PVA/Gr005/Av. Nevertheless, from this evaluation, we can see that the potential of this hydrogel system to be developed further. As a comparison, the addition of Av alone without GBM to PVA reduced the bacterial colony only up to 95.88%. From here, we can see that GBM acts as the main contributor for enhancing the antibacterial activity in the PVA/GBM/Av hydrogel despite the ability of Av to kill the bacteria as well.71 The concentration of GBM might give another effect to this property as it is the biggest contributor of all the constituent materials. Besides, another factor from the bacteria in terms of its capability to grow under our hydrogels' physicochemical condition could influence the overall hydrogel's antibacterial activity as well.72
| Hydrogel | Bacterial colony population (cfu mL−1) | Bacterial reduction (%) |
|---|---|---|
| a Without hydrogel. | ||
| Controla | 4.8 × 1010 ± 0.00 | — |
| PVA/Gr005 | 3.18 × 108 ± 2.77 × 107 | 99.34 ± 0.058 |
| PVA/GO | 2.68 × 107 ± 2.28 × 106 | 99.94 ± 0.005 |
| PVA/Av | 1.98 × 109 ± 2.39 × 108 | 95.88 ± 0.497 |
| PVA/Gr005/Av | 1.74 × 108 ± 3.97 × 107 | 99.64 ± 0.083 |
| PVA/GO/Av | 2.28 × 108 ± 2.17 × 107 | 99.53 ± 0.045 |
GBM and Av were physically embedded in the PVA matrix and are able to leach out. We hypothesize that the antibacterial mechanism of PVA/GBM/Av is through the interaction of both GBM and Av in the hydrogel with the surrounding environment. It is definitely interesting to see the ability of the GBM to impede the growth and to kill both Gram-positive and Gram-negative bacteria73 as it has been reported previously to have excellent antibacterial properties.72 There are three different general mechanisms of GBM appearance that lead to bacteriostatic or/and bactericidal activity: knives-like edge-mediated cutting, oxidative stress, and bacterial membrane entrapment.74 The mechanism of edge cutting by GBM is started by direct contact between the GBM and the bacteria cell membrane.75 These edges at the molecular level are considered as sharp as knives and work in the same manner to cut the cell membrane mechanically, which then results in bacterial mortality.72 Differently, oxidative stress would lead to bacterial death or inactivation through a chemical pathway, which is either reactive oxygen species (ROS)-dependent or ROS-independent. ROS-dependent pathway oxidative stress occurs when GBM generates ROS, such as hydroxyl radicals, peroxide, singlet molecular oxygen, and superoxide anions, in a very large number, hence resulting in the accumulation of the ROS at the cellular level. This accumulated ROS would disrupt the bacterial body chemically, which eventually led to bacterial membrane degeneration, lipid peroxidation, protein inactivation, mitochondrial membrane depolarization and dysfunction, and cell necrosis.76 For the ROS-independent pathway, disruption comes from the cellular electronic transfer between rGO in specific with the bacteria and it is mostly induced by the conductive GBM such as Gr and rGO.75 Subsequently, for cell entrapment, GBM by its graphitic 2D layer might isolate the bacteria from the cellular environment.77 This event would block the bacteria to get the physicochemical conditions for living as well as the nutrients. Also, because of this, the bacteria cannot survive and die.
In the case of our hydrogels, the effect of GBM on the antibacterial activity was probably caused the sharp edges of the GBM that are able to damage the bacterial cell membrane and oxidative stress. The same result was confirmed as well by Surudzic et al., where they hypothesized that the same mechanisms occurred in their PVA/Gr hydrogel.25 Nevertheless, Av should have given the antibacterial effect synergistically with GBM. The antibacterial activity provided by Av is mainly due to the interaction between the active compounds in Av, such as anthraquinones, pyrocatechol, cinnamic acid, p-coumaric acid, and ascorbic acid.71,78 Anthraquinones play a role as tetracycline, obstructing the ribosomal A site, which leads to bacterial protein synthesis inhibition.79 Pyrocatechol has a phenolic group that is able to increase hydroxylation, leading to protein denaturation and ends up in the disruption of the cellular membrane.80 Cinnamic acid hinders both glucose uptake and ATP energy production in the bacterial cells, while p-coumaric acid and ascorbic acid similarly interrupt the enzymatic activity in the bacteria.80
3.8 Cell viability
Hydrogel as a wound dressing should be biocompatible as it would lay on the wound bed and get in contact with the surrounding tissue directly. As the preliminary way to investigate this property in our hydrogels, we carried out the cell viability assay using alamarBlue resazurin staining. We used 3T3 fibroblast cells as the cell model since the fibroblasts have a crucial role in wound healing such as to generate the protein for new extracellular matrix formation.81 The results were then collected after 1 h and 48 h. Fig. 8 shows the results of this test and confirmed that all the hydrogels exhibited no toxicity toward the cells, compared to the group treated with 5% DMSO to induce toxicity. All the hydrogels maintained the number of viable cells as the control after 1 h in about 100% except for PVA/GO, which has 40% higher cell viability than the control. After 48 h, PVA/GO/Av showed the highest percentage, which is 295% (40–50% higher than that of the others) after 2 days. The cell viability percentage in all the hydrogels experienced escalation, indicating that the cells are able to proliferate. This result also implies that the combination of GBM with PVA and Av could generate a fully non-cytotoxic material. Since PVA and Av are well-known as biocompatible materials, it is likely they were helpful in reducing the cytotoxicity in the GBM that is dose-dependent.18,82 However, an additional in vivo test should be eventually employed to have better representation of the PVA/GBM/Av biocompatibility.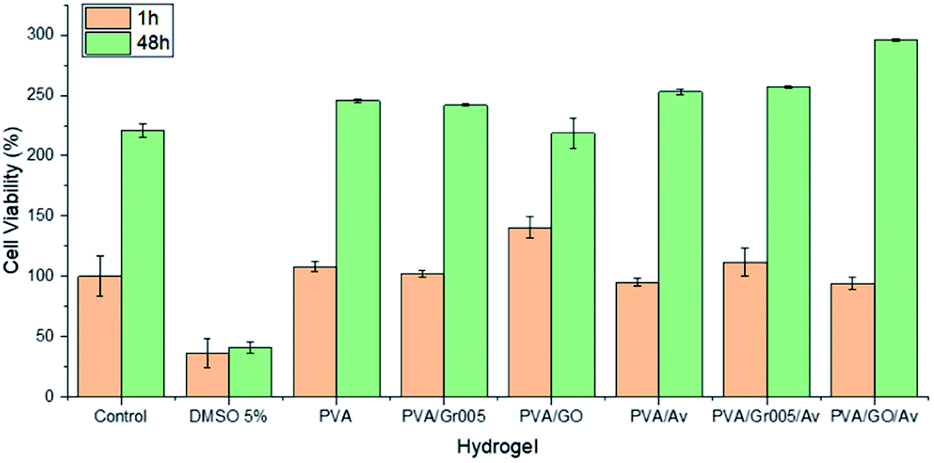 | ||
| Fig. 8 Viability of the fibroblast 3T3 cells on the PVA/GBM/Av hydrogels. The control and DMSO 5% are the negative and positive controls, respectively. | ||
4 Conclusions
We successfully fabricated the PVA/GBM/Av hydrogel via physical crosslink formation, i.e., freeze-thawing. The PVA concentration plays a remarkable role in determining the physical appearance of the hydrogel, which was caused by phase separation that occurred during the freeze-thawing process. The addition of GBM and Av to the PVA hydrogel was confirmed through FTIR and Raman spectroscopy. Through Raman spectroscopy as well, the GO added was confirmed to have transformed to rGO. The hydrogels have porosities, as observed through SEM, where these porosities would be useful as the pathway for the active agents, cells, and water mobilization at the wound site. The potential of the PVA/GBM/Av hydrogel for wound dressing application have been demonstrated through some tests, which aimed to determine whether our hydrogels fulfil the requirement of the wound dressing. Our hydrogels exhibited excellent hydrophilicity with a water contact angle less than 90 degrees, electrical conductivity that is comparable to that of the human skin tissue, and possess similar mechanical properties as that of the skin tissue. This PVA/GBM/Av hydrogel is stable in PBS with a weight ratio of about 73–80% after 14 days of immersion. The presence of GBM in the hydrogel would inhibit the growth of Gram-positive Staphylococcus aureus ATCC 6538 with the lowest bacterial population observed in PVA/GO (1.74 × 107 cfu mL−1) after 1 day incubation, which indicates 99.94% of bacterial reduction. Lastly, our PVA/GBM/Av hydrogels are cytocompatible with 3T3 fibroblast cells, with a viability up to 295% after 48 h for PVA/GO/Av. Despite these results that have satisfied the wound dressing requirement, further investigation is still needed to evaluate the performance of our hydrogels more comprehensively.Author contributions
Wildan Hanif: conceptualization, investigation, methodology, validation, writing original draft; Andri Hardiansyah: conceptualization, validation, supervision, writing–review and editing; Ahmad Randy: investigation, methodology; Lia A. T. W. Asri: conceptualization, validation, project administration, supervision, writing–review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Research, Community Service and Innovation Grant Program-ITB 2021 (P2MI-ITB). We would like to thank Low Dimensional Functional Material Laboratory at Research Center for Physics, Indonesian Institute of Sciences for all supports in materials and facilities. We also thank to ELSA LIPI system for the characterization at Indonesian Institute of Sciences.References
- Y. Wang, J. Beekman, J. Hew, S. Jackson, A. C. Issler-Fisher, R. Parungao, S. S. Lajevardi, Z. Li and P. K. Maitz, Adv. Drug Delivery Rev., 2018, 123, 3–17 CrossRef CAS PubMed.
- WHO, https://www.who.int/en/news-room/fact-sheets/detail/burns, accessed 10 June 2020.
- C. Hall, C. Hardin, C. J. Corkins, A. Z. Jiwani, F. John, A. Carlsson and R. Chan, Comprehensive Physiology, 2018, 8, 371–405 Search PubMed.
- S. M. Watt and J. M. Pleat, Adv. Drug Delivery Rev., 2018, 123, 82–106 CrossRef CAS PubMed.
- Z. Li and P. Maitz, Burns & Trauma, 2018, 6, 13 Search PubMed.
- R. Balint, N. J. Cassidy and S. H. Cartmell, Tissue Eng., Part B, 2013, 19, 48–57 CrossRef CAS.
- M. A. M. Jahromi, P. S. Zangabad, S. M. M. Basri, K. S. Zangabad, A. Ghamarypour, A. R. Aref, M. Karimi and M. R. Hamblin, Adv. Drug Delivery Rev., 2018, 123, 33–64 CrossRef.
- S. Dhivya, V. V. Padma and E. Santhini, BioMedicine, 2015, 5, 24–28 CrossRef PubMed.
- K. Heyer, M. Augustin, K. Protz, K. Herberger, C. Spehr and S. J. Rustenbach, Dermatology, 2013, 226, 172–184 CrossRef CAS PubMed.
- J. Boateng and O. Catanzano, J. Pharm. Sci., 2015, 104, 3653–3680 CrossRef CAS PubMed.
- E. A. Kamoun, E. S. Kenawy and X. Chen, J. Adv. Res., 2017, 8, 217–233 CrossRef CAS PubMed.
- R. Balint, N. J. Cassidy and S. H. Cartmell, Acta Biomater., 2014, 10, 2341–2353 CrossRef CAS PubMed.
- S. Li, S. Dong, W. Xu, S. Tu, L. Yan, C. Zhao, J. Ding and X. Chen, Adv. Sci., 2018, 5, 1700527 CrossRef PubMed.
- D. Simoes, S. P. Miguel, M. P. Ribeiro, P. Coutinho, A. G. Mendonca and I. J. Correia, Eur. J. Pharm. Biopharm., 2018, 127, 130–141 CrossRef CAS PubMed.
- M. Zhao, B. Song, J. Pu, T. Wada, B. Reid, G. Tai, F. Wang, A. Guo, P. Walczysko, Y. Gu, T. Sasaki, A. Suzuki, J. V. Forrester, H. R. Bourne, P. N. Devreotes, C. D. McCaig and J. M. Penninger, Nature, 2006, 442, 457–460 CrossRef CAS PubMed.
- Y. Liang, H. Tian, J. Liu, Y. Lv, Y. Wang, J. Zhang and Y. Huang, Bioelectrochemistry, 2020, 135, 107578 CrossRef CAS PubMed.
- X. Zhao, H. Wu, B. Guo, R. Dong, Y. Qiu and P. X. Ma, Biomaterials, 2017, 122, 34–47 CrossRef CAS PubMed.
- Y. Zhao, Z. Li, S. Song, K. Yang, H. Liu, Z. Yang, J. Wang, B. Yang and Q. Lin, Adv. Funct. Mater., 2019, 29, 1901474 CrossRef.
- J. He, M. Shi, Y. Liang and B. Guo, Chem. Eng. J., 2020, 394, 124888 CrossRef CAS.
- L. Peng, Y. Zhou, W. Lu, W. Zhu, Y. Li, K. Chen, G. Zhang, J. Xu, Z. Deng and D. Wang, BMC Musculoskeletal Disord., 2019, 20, 257 CrossRef.
- M. Bahadoran, A. Shamloo and Y. D. Nokoorani, Sci. Rep., 2020, 10, 7342 CrossRef CAS PubMed.
- X. Zhou, C. Hou, T.-L. Chang, Q. Zhang and J. F. Liang, Colloids Surf., B, 2020, 187, 110645 CrossRef CAS PubMed.
- Y. Li, J. Wang, Y. Yang, J. Shi, H. Zhang, X. Yao, W. Chen and X. Zhang, Mater. Sci. Eng., C, 2021, 118, 111447 CrossRef CAS PubMed.
- A. Kumar and S. S. Han, Int. J. Polym. Mater. Polym. Biomater., 2017, 66, 159–182 CrossRef CAS.
- R. Surudzic, A. Jankovic, M. Mitric, I. Matic, Z. D. Juranic, L. Zivkovic, V. Miskovic-Stankovic, K. Y. Rhee, S. J. Park and D. Hui, J. Ind. Eng. Chem., 2016, 34, 250–257 CrossRef CAS.
- R. Lima-Sousa, D. d. Melo-Diogo, C. G. Alves, C. S. Cabral, S. P. Miguel, A. G. Mendonca and I. J. Correia, Mater. Sci. Eng., C, 2020, 117, 111294 CrossRef CAS PubMed.
- J. Yi, G. Choe, J. Park and J. Y. Lee, Polym. J., 2020, 52, 823–837 CrossRef CAS.
- M. Yousefi, M. Dadashpour, M. Hejazi, M. Hasanzadeh, B. Behman, M. d. l. Guardia, N. Shadjou and A. Mokhtarzadeh, Mater. Sci. Eng., C, 2017, 74, 568–581 CrossRef CAS PubMed.
- X. Wu, S. Tan, Y. Xing, Q. Pu, M. Wu and J. X. Zhao, Colloids Surf., B, 2017, 157, 1–9 CrossRef CAS PubMed.
- L. Bi, X. Wang, X. Cao, L. Liu, C. Bai, Q. Zheng, J. Choo and L. Chen, Talanta, 2020, 220, 121397 CrossRef CAS PubMed.
- N. Ibrahim, S. K. Wong, I. N. Mohamed, N. Mohamed, K. Y. Chin, S. I. Nirwana and A. N. Shuid, Int. J. Environ. Res. Public Health, 2018, 15, 2360 CrossRef CAS PubMed.
- R. A. K. Wardhani, L. A. T. W. Asri, H. Rachmawati, K. Khairurrijal and B. S. Purwasasmita, Int. J. Nanomed., 2020, 15, 6433–6449 CrossRef CAS.
- M. D. Boudreau and F. A. Beland, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2006, 24, 103–154 CrossRef CAS PubMed.
- M. Hes, K. Dziedzic, D. Gorecka, A. Jedrusek-Golinska and E. Gujska, Plant Foods Hum. Nutr., 2019, 74, 255–265 CrossRef PubMed.
- A. E. Stoica, C. Chircov and A. M. Grumezescu, Materials, 2020, 13, 2853 CrossRef CAS PubMed.
- H. Saberian, Z. H. Esfahani and A. Banakar, Iran. J. Chem. Chem. Eng., 2018, 37, 157–165 CAS.
- S. Chakraborty, T. Ponrasu, S. Chandel, M. Dixit and V. Muthuvijayan, R. Soc. Open Sci., 2018, 5, 172017 CrossRef CAS PubMed.
- G. Paradossi, F. Cavalieri and E. Chiessi, J. Mater. Sci.: Mater. Med., 2003, 14, 687–691 CrossRef CAS.
- M. Othman, H. S. Loh, C. Wiart, T. J. Khoo, K. H. Lim and K. N. Ting, J. Microbiol. Methods, 2011, 84, 161–166 CrossRef CAS.
- Y. Hou, C. Chen, K. Liu, Y. Tu, L. Zhang and Y. Li, RSC Adv., 2015, 5, 24023–24030 RSC.
- T. Nakano and T. Nakaoki, Polym. J., 2011, 43, 875–880 CrossRef CAS.
- J. L. Holloway, A. M. Lowman and G. R. Palmese, Soft Matter, 2013, 9, 826–833 RSC.
- S. Butylina, S. Geng and K. Oksman, Eur. Polym. J., 2016, 81, 386–396 CrossRef CAS.
- R. Kumar, A. K. Singh, A. Gupta, A. Bishayee and A. K. Pandey, Phytomedicine, 2019, 60, 152996 CrossRef CAS PubMed.
- P. Tiamduangtawan, C. Kamkaew, S. Kuntowatchara, E. Wimolmala and K. Saenboonruang, Radiat. Phys. Chem., 2020, 177, 109164 CrossRef CAS.
- X. Xiao, G. Wu, H. Zhou, K. Qian and J. Hu, Polymers, 2017, 9, 259 CrossRef PubMed.
- P. Jithendra, A. M. Rajam, T. Kalaivani, A. B. Mandal and C. Rose, ACS Appl. Mater. Interfaces, 2013, 5, 7291–7298 CrossRef CAS.
- X. Wu, W. Li, K. Chen, D. Zhang, L. Xu and X. Yang, Mater. Today Commun., 2019, 21, 100702 CrossRef CAS.
- D. A. Gyles, A. D. P. Junior, L. D. Castro, A. S. Brigida, M. L. N. Lamarao, W. L. R. Barbosa, J. O. S. Junior and R. M. Ribeiro-Costa, Polymers, 2020, 12, 690 CrossRef CAS PubMed.
- S. Zou, Z. Wei, Y. Hu, Z. Tong and C. Wang, Polym. Chem., 2014, 5, 4227–4234 RSC.
- C.-W. Peng, H.-Y. Lin, H.-W. Wang and W.-W. Wu, Mater. Sci. Eng., C, 2012, 32, 2491–2500 CrossRef CAS.
- V. Rivero-Buceta, M. R. Aguilar, A. M. Hernandez-Arriaga, F. G. Blanco, A. Rojas, M. Tortajada, R. A. Ramirez-Jimenez, B. Vazquez-Lasa and A. Prieto, Int. J. Biol. Macromol., 2020, 162, 1869–1879 CrossRef CAS.
- P. Zhang, C. Zhao, T. Zhao, M. Liu and L. Jiang, Adv. Sci., 2019, 6, 1900996 CrossRef CAS PubMed.
- J. Qu, X. Zhao, Y. Liang, Y. Xu, P. X. Ma and B. Guo, Chem. Eng. J., 2019, 362, 548–560 CrossRef CAS.
- Y. Liang, B. Chen, M. Li, J. He, Z. Yin and B. Guo, Biomacromolecules, 2020, 21, 1841–1852 CrossRef CAS.
- D. Aycan, B. Selmi, E. Kelel, T. Yildirim and N. Alemdar, Eur. Polym. J., 2019, 121, 109308 CrossRef CAS.
- O. M. Slobodian, P. M. Lytvyn, A. S. Nikolenko, V. M. Naseka, O. Y. Khyzhun, A. V. Vasin, S. V. Sevostianov and A. N. Nazarov, Nanoscale Res. Lett., 2018, 13, 138 CrossRef PubMed.
- F. Azmi, D. Sispriatna, K. Ikhsan, M. Masrura, S. S. Azzahra, Mahidin and M. D. Supardan, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 334, 012053 CrossRef.
- F. A. S. Mendonca, J. R. P. Junior, M. A. M. Esquisatto, J. S. Mendonca, C. C. Franchini and G. M. T. dos Santos, Acta Cir. Bras., 2009, 24, 150–155 CrossRef PubMed.
- B. Zhang, J. He, M. Shi, Y. Liang and B. Guo, Chem. Eng. J., 2020, 400, 125994 CrossRef CAS.
- A. Oryan, E. Alemzadeh and A. Moshiri, J. Wound Care, 2017, 26, 5–19 CrossRef CAS PubMed.
- L. Zhang, Z. Wang, C. Xu, Y. Li, J. Gao, W. Wang and Y. Liu, J. Mater. Chem., 2011, 21, 10399–10406 RSC.
- Y. Huang, M. Zhang and W. Ruan, J. Mater. Chem. A, 2014, 2, 10508–10515 RSC.
- J. Wang, X. Wang, C. Xu, M. Zhang and X. Shang, Polym. Int., 2011, 60, 816–822 CrossRef CAS.
- M. F. Griffin, B. C. Leung, Y. Premakumar, M. Szarko and P. E. Butler, Journal of Otolaryngology - Head & Neck Surgery, 2017, 46, 33 Search PubMed.
- M. Pawlaczyk, M. Lelonkiewicz and M. Wieczorowski, Postepy Dermatol Alergol, 2013, 30, 302–306 CrossRef PubMed.
- B. Atiyeh, J. P. Barret, H. Dahai, F. Duteille, A. Fowler, S. Enoch, E. Greenfield, A. Magnette, H. Rode and X. Zhao-fan, International Best Practice Guidelines: Effective skin and wound management of non-complex burns, Wounds International, London, 2014 Search PubMed.
- C. M. Hassan and N. A. Peppas, Macromolecules, 2000, 33, 2472–2479 CrossRef CAS.
- Z. Fan, B. Liu, J. Wang, S. Zhang, Q. Lin, P. Gong, L. Ma and S. Yang, Adv. Funct. Mater., 2014, 24, 3933–3943 CrossRef CAS.
- M. G. Jeschke, M. E. van Baar, M. A. Choudhry, K. K. Chung, N. S. Gibran and S. Logsetty, Nature Reviews Disease Primers, 2020, 6, 11 CrossRef PubMed.
- S. Jain, N. Rathod, R. Nagi, J. Sur, A. Laheji, N. Gupta, P. Agrawal and S. Prasad, J. Clin. Diagn. Res., 2016, 10, ZC41–ZC44 CAS.
- H. Mohammed, A. Kumar, E. Bekyarova, Y. Al-Hadeethi, X. Zhang, M. Chen, M. S. Ansari, A. Cochis and L. Rimondini, Frontiers in Bioengineering and Biotechnology, 2020, 8, 465 CrossRef.
- D. Lin, T. Qin, Y. Wang, X. Sun and L. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 1320–1329 CrossRef CAS.
- A. Hardiansyah, M.-C. Yang, H.-L. Liao, Y.-W. Cheng, F. Destyorini, Y. Irmawati, C.-M. Liu, M.-C. Yung, C.-C. Hsu and T.-Y. Liu, Nanomaterials, 2020, 10, 674 CrossRef CAS PubMed.
- M. D. Rojas-Andrade, G. Chata, D. Rauholiman, J. Liu, C. Saltikov and S. Chen, Nanoscale, 2017, 9, 994–1006 RSC.
- X. Zeng, G. Wang, Y. Liu and X. Zhang, Environ. Sci.: Nano, 2017, 4, 2248–2266 RSC.
- M. Y. Xia, Y. Xie, C. H. Yu, G. Y. Chen, Y. H. Li, T. Zhang and Q. Peng, J. Controlled Release, 2019, 307, 16–31 CrossRef CAS.
- P. P. Athiban, B. J. Borthakur, S. Ganesan and B. Swathika, J. Conservative Dent., 2012, 15, 246–248 CrossRef CAS.
- M. H. Radha and N. P. Laxmipriya, Journal of Traditional and Complementary Medicine, 2015, 5, 21–26 CrossRef.
- R. Lawrence, P. Tripathi and E. Jeyakumar, Braz. J. Microbiol., 2009, 40, 906–915 CrossRef CAS.
- A. Stunova and L. Vistejnova, Cytokine Growth Factor Rev., 2018, 39, 137–150 CrossRef CAS.
- K. H. Liao, Y. S. Lin, C. W. Macosko and C. L. Haynes, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |