
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent trends in dehydroxylative trifluoro-methylation, -methoxylation, -methylthiolation, and -methylselenylation of alcohols
Yan Caoa,
Roya Ahmadi
 *b,
Mohammad Reza Poor Heravi
c,
Alibek Issakhovde,
Abdol Ghaffar Ebadi
f and
Esmail Vessally
c
*b,
Mohammad Reza Poor Heravi
c,
Alibek Issakhovde,
Abdol Ghaffar Ebadi
f and
Esmail Vessally
c
aSchool of Mechatronic Engineering, Xi'an Technological University, Xi'an, 710021, China
bDepartment of Chemistry, College of Basic Sciences, Yadegar-e-Imam Khomeini (RAH) Shahre Rey Branch, Islamic Azad University, Tehran, Iran. E-mail: roya_ahmadi_chem@yahoo.com
cDepartment of Chemistry, Payame Noor University, P.O. Box 19395-3697, Tehran, Iran
dDepartment of Mathematical and Computer Modelling, al-Farabi Kazakh National University, 050040, Almaty, Kazakhstan
eDepartment of Mathematics and Cybernetics, Kazakh British Technical University, 050000, Almaty, Kazakhstan
fDepartment of Agriculture, Jouybar Branch, Islamic Azad University, Jouybar, Iran
First published on 13th December 2021
Abstract
Owing to the prevalence of hydroxyl groups on molecules, much attention has been paid to the synthesis of functionalized organic compounds by dehydroxylative functionalization of parent alcohols. In this context, dehydroxylative trifluoromethylation, trifluoromethoxylation, trifluoromethylthiolation, and trifluoromethylselenylation of readily available alcohols have recently emerged as intriguing protocols for the single-step construction of diverse structures bearing C–CF3, C–OCF3, C–SCF3, and C–SeCF3 bonds, respectively. This Mini-Review aims to summarize the major progress and advances in this appealing research area with special emphasis on the mechanistic features of the reaction pathways.
 Roya Ahmadi | Dr Roya Ahmadi began her bachelor's degree in pure chemistry in 1992 at Shahid Beheshti University in Tehran, completed her master's degree in inorganic chemistry at Kharazmi University of Tehran (Teacher Training) in 2000, and received her PhD in mineral chemistry from Islamic Azad University, Tehran Science and Research branch in 2007. In the same year, she worked as an assistant professor at Islamic Azad University, Imam Khomeini's Yadegar branch, and in 2014, was promoted to associate professor in the field of experimental and computational chemistry. Over the years, she has published more than 115 articles in chemistry journals and 163 papers in national and international conferences as posters and lectures. Additionally, she has provided guidance and counseling to more than 75 graduate students, and master's and doctoral degree recipients. |
 Mohammad Reza Poor Heravi | Mohammad Reza Poor Heravi was born in Seiyahkal-Lahijan, Guilan, Iran in 1965. He received a B.Sc. degree in pure Chemistry from Tabriz University, Iran, in 1987, a M.Sc. degree in Organic Chemistry from Tabriz University, Iran in 1990 under the supervision of Professor A. Shahrisa, and a PhD degree from Isfahan University, Iran, under the supervision of Professor H. Loghmanim. He was a visiting researcher in 2006 at The Sheffield University, UK, with Professor Richard F. W. Jackson. He became a science faculty member of Payame Noor University in 1990, and Associate Professor in 2012. His research interests include applications of solvent-free conditions, ionic liquids, fluorination reactions, designing fluorinating reagents, ultrasound irradiation in organic synthesis, and the study of methodology in organic chemistry. |
 Abdol Ghaffar Ebadi | Dr Abdol Ghaffar Ebadi completed his doctoral degree in Environmental Biotechnology (Algology) from Tajik Academy of Sciences. Currently, he is a researcher in TAS in Tajikistan and a faculty member at the Islamic Azad University of Jouybar in Mazandaran. Dr Ebadi has published more than 400 scientific papers in qualified international journals and attended more than 50 international conferences. He has collaborated with many research project teams around world such as those in China, Malaysia, and Thailand. His interests are environmental biotechnology, biochemistry, and gene pathways in the phytoremediation processes. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in pure chemistry from the University of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is employed at Payame Noor University as a Professor of organic chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis, and spectral studies of organic compounds. |
1. Introduction
The incorporation of fluorinated moieties and particularly the trifluoromethyl (CF3), trifluoromethoxy (OCF3), trifluoromethylthio (SCF3), and trifluoromethylseleno (SeCF3) groups into organic molecules such as pharmaceuticals and agrochemicals can often substantially improve their physical, chemical, and biological properties because of the electronic properties, unique size, lipophilicity, and metabolic stability of these groups.1,2 There are different examples of commercially available human and veterinary drugs that contain a CF3, OCF3, or SCF3 moiety in their structures (Scheme 1).3 However, there are no analogous CF3Se-containing drugs, which is likely due to the limited synthetic options for their preparation.4 Compounds containing the titled functionalities are versatile synthetic intermediates and can function as suitable building blocks for the preparation of many valuable functional materials.5 Considering the widespread biological activities and synthetic usefulness of CF3/OCF3/SCF3/SeCF3-containing compounds, it has been of great synthetic interest to develop new, efficient, and practical methods for the introduction of these privileged moieties into organic molecules.6–9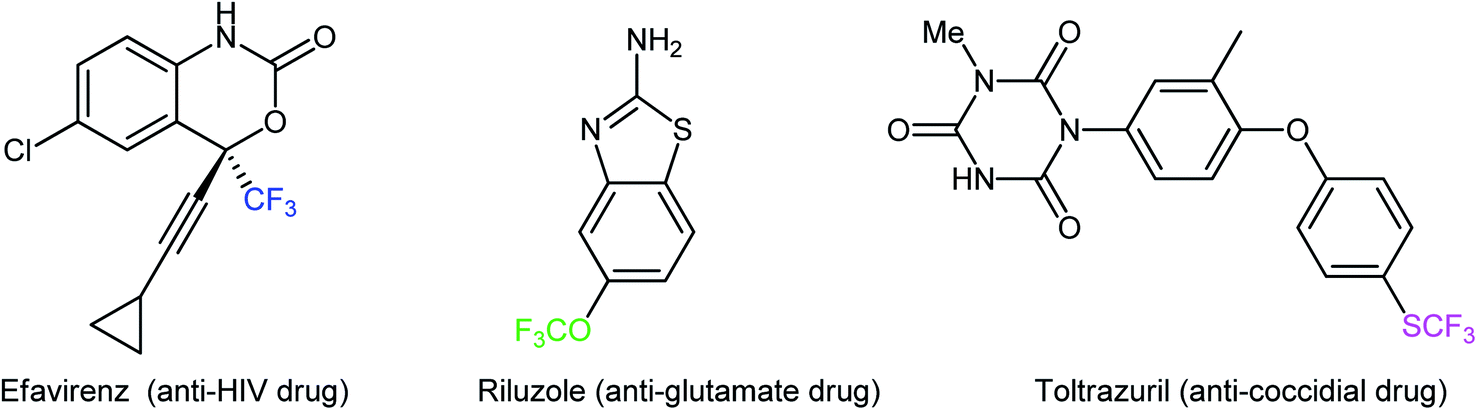 | ||
| Scheme 1 Selected examples of CF3/OCF3/SCF3-based drugs. | ||
In recent years, direct dehydroxylative functionalization of alcohols has become one of the hottest research topics in organic chemistry because it is a powerful and general strategy for the construction of various valuable functionalized organic compounds from inexpensive and abundantly available alcohols without isolation of intermediates.10 In this regard, dehydroxylative trifluoro-methylation, -methoxylation, -methylthiolation, and -methylselenylation of alcohols have captured the imagination of the organic chemical community and have become promising synthetic methods for constructing C–CF3, C–OCF3, C–SCF3, and C–SeCF3 bonds, respectively. These synthetic processes are advantageous because the starting materials possess high selectivity and stability, are abundant and inexpensive, with low toxicity, and there is no need for isolation of intermediates. In continuation of our interest in organofluorine chemistry11 and modern organic synthesis,12–18 in this Mini-Review, we will highlight the most important advances and progress in the arena of dehydroxylative trifluoro-methylation, -methoxylation, -methylthiolation, and -methylselenylation of alcohols (Scheme 2), with a particular emphasis on the mechanistic aspects of the reaction pathways.
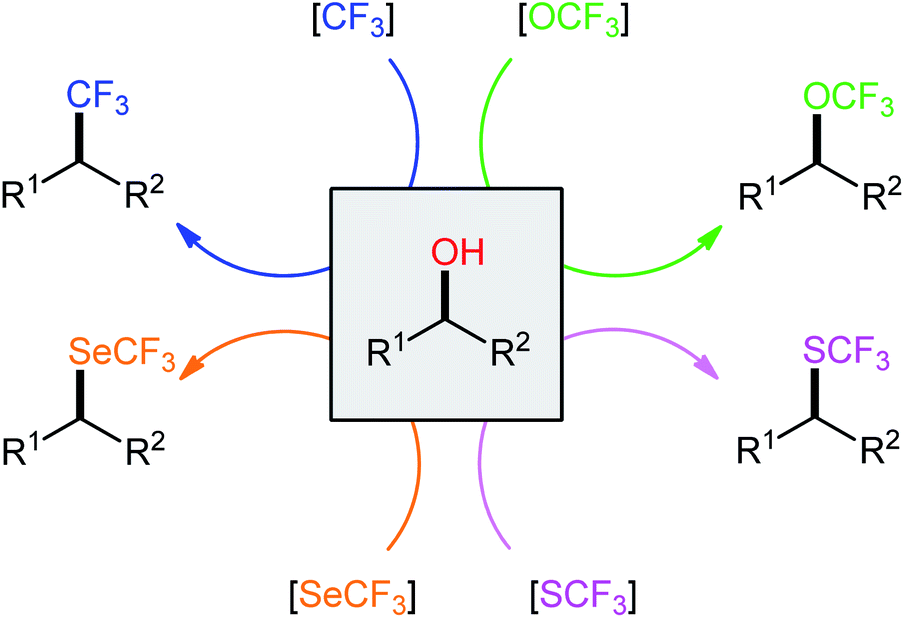 | ||
| Scheme 2 Direct dehydroxylative trifluoro-methylation, -methoxylation, -methylthiolation, and -methylselenylation of alcohols. | ||
2. Dehydroxylative trifluoromethylation
After their seminal work on the conversion of allylic alcohols to trifluoromethanes through a two-step esterification/decarboxylative trifluoromethylation procedure,19 Altman and colleagues developed bench-stable phenyl bromodifluoroacetate (PhO2CCF2Br) as a new nucleophilic trifluoromethylation reagent, which can directly convert alcohols to trifluoromethanes in a single operation without isolation of intermediates.20 They showed that treatment of various allylic alcohols 1 with over-stoichiometric amounts of phenyl bromodifluoroacetate 2 in the presence of the CuI/DMEDA/HO2CCF2Br combination as a catalytic system in DMF at room temperature afforded the corresponding deoxytrifluoromethylated products 3 in good to high yields (Scheme 3a). The substrate scope was evaluated on nine terminally substituted allylic alcohols, which proved that various linear cinnamyl alcohols and endocyclic alkenols were compatible with this reaction. Moreover, this strategy was successfully applied for the direct deoxytrifluoromethylation of a series of propargylic alcohols 4 (by replacement of the DMEDA ligand with Phen) and (hetero)benzylic alcohols 6 (by employing MeO2CCF2Br reagent instead of HO2CCF2Br and using over-stoichiometric amounts of KI) (Scheme 3b and c). Of note, propargylic alcohols underwent rearrangement and resulted in allenylic structures. Furthermore, the synthetic utility of this procedure was highlighted by gram-scale synthesis of (4,4,4-trifluorobut-1-en-1-yl)benzene (75% yield on a 20 mmol scale). Finally, the synthetic potentiality of this strategy was showcased by preparing a fluorinated analogue of tebufenpyrad, an acaricide. Regrettably, the authors did not investigate the applicability of secondary and tertiary alcohols in their methodology. On the basis of several control experiments, a plausible mechanism was proposed by the authors for this trifluoromethylation protocol involving the following key steps (Scheme 4): (i) transesterification between phenyl bromodifluoroacetate 2 and the starting alcohol 1 to generate alkyl bromodifluoroacetate intermediate A; (ii) formation of the active Cu–CF3 species B via interaction of the LnCuI precatalyst with HO2CCF2Br; (iii) direct nucleophilic trifluoromethylation of intermediate A with Cu–CF3 species B to give the desired product 3 and carboxylate-coordinated [Cu] complex C; and (iv) CO2 liberation and halogen exchange to recover the active Cu–CF3 species B and completion of the catalytic cycle.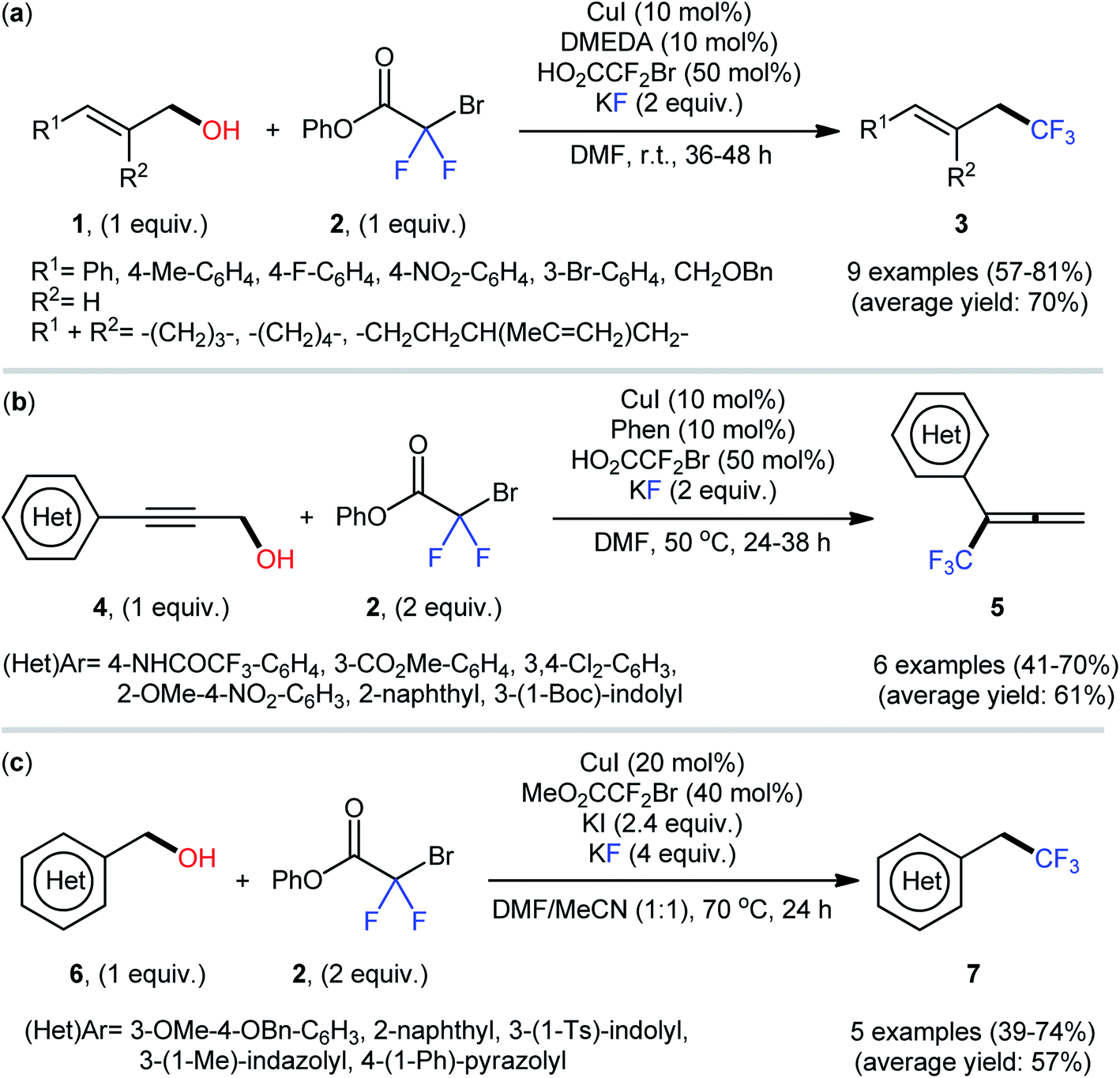 | ||
| Scheme 3 Cu-catalyzed deoxytrifluoromethylation of (a) allylic alcohols 1; (b) propargylic alcohols 4; and (c) (hetero)benzylic alcohols 6 with phenyl bromodifluoroacetate 2 developed by Altman. | ||
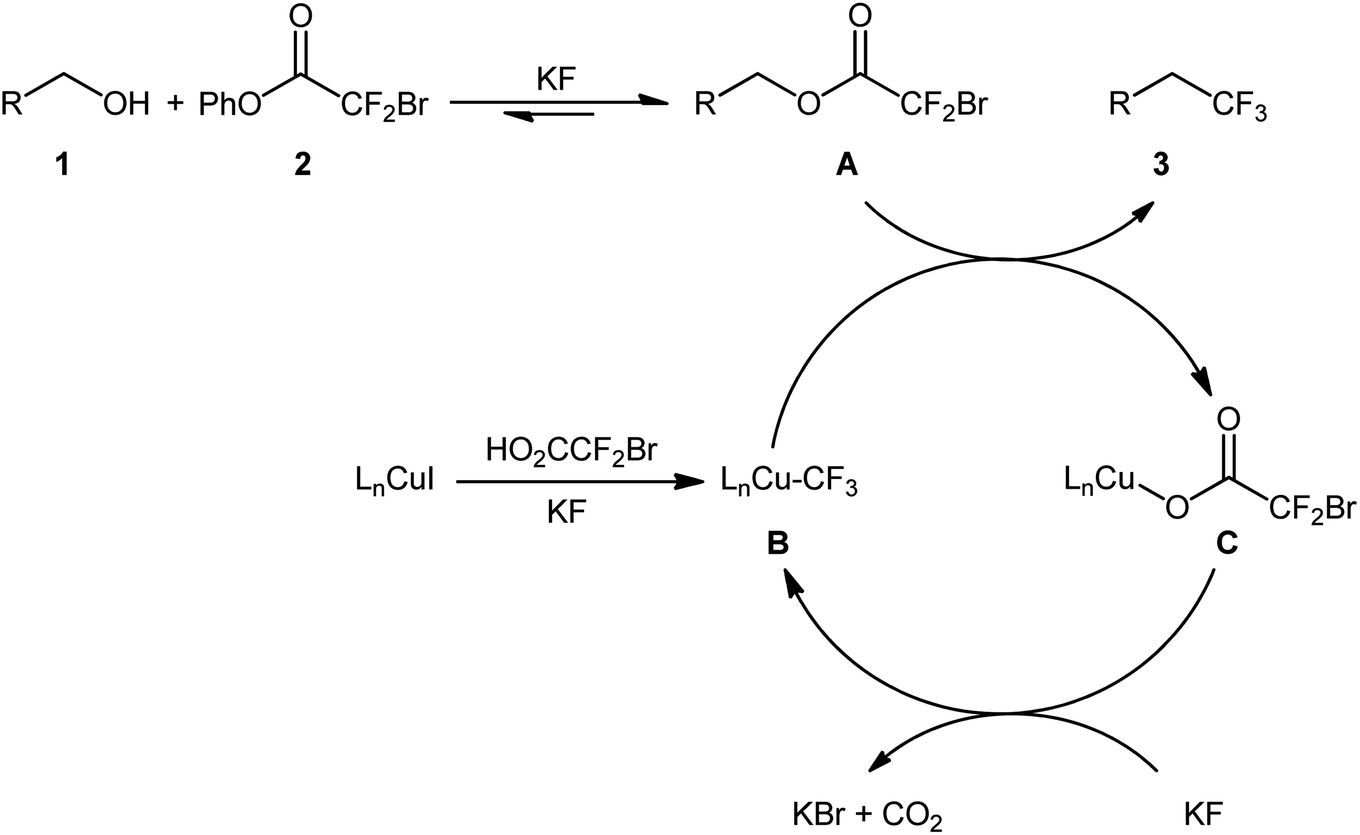 | ||
| Scheme 4 The plausible mechanism for the reactions in Scheme 2. | ||
In another report, this research group also developed [1,1′-biphenyl]-4-yl-2-bromo-2,2-difluoroacetate (BBDFA) as an efficient trifluoromethylating reagent for Cu-catalyzed dehydroxylative trifluoromethylation of alcohols.21 The reagent was synthetized on a 100 g scale in 93% yield via chlorination of commercially available bromodifluoroacetic acid with oxalyl chloride in the presence of a catalytic amount of DMF, followed by esterification of the resulting acid chloride with 4-phenylphenol, and it showed good thermal stability and high ability for trifluoromethylation of examined alcohols. However, the usefulness of this reagent was only demonstrated by deoxytrifluoromethylation of cinnamyl alcohol and 2-naphthalenemethanol, without any substrate scope exploration.
Drawing inspiration from these works, very recently, the Wu group, in collaboration with the Xiao group, described an interesting Cu(0)-catalyzed dehydroxylative trifluoromethylation of a library of (hetero)benzylic alcohols 8 with Chen's reagent (methyl fluorosulfonyldifluoroacetate; 9) in the presence of the Ph3P/ICH2CH2I system, which acted as the activator of the hydroxyl group.22 The reactions were performed in DMF at 80 °C, tolerated a series of synthetically useful functionalities (e.g., –Br, –I, –CO2Me, –CN, –NO2), and provided the desired (2,2,2-trifluoroethyl)arenes 10 in modest to good yields (Scheme 5a).
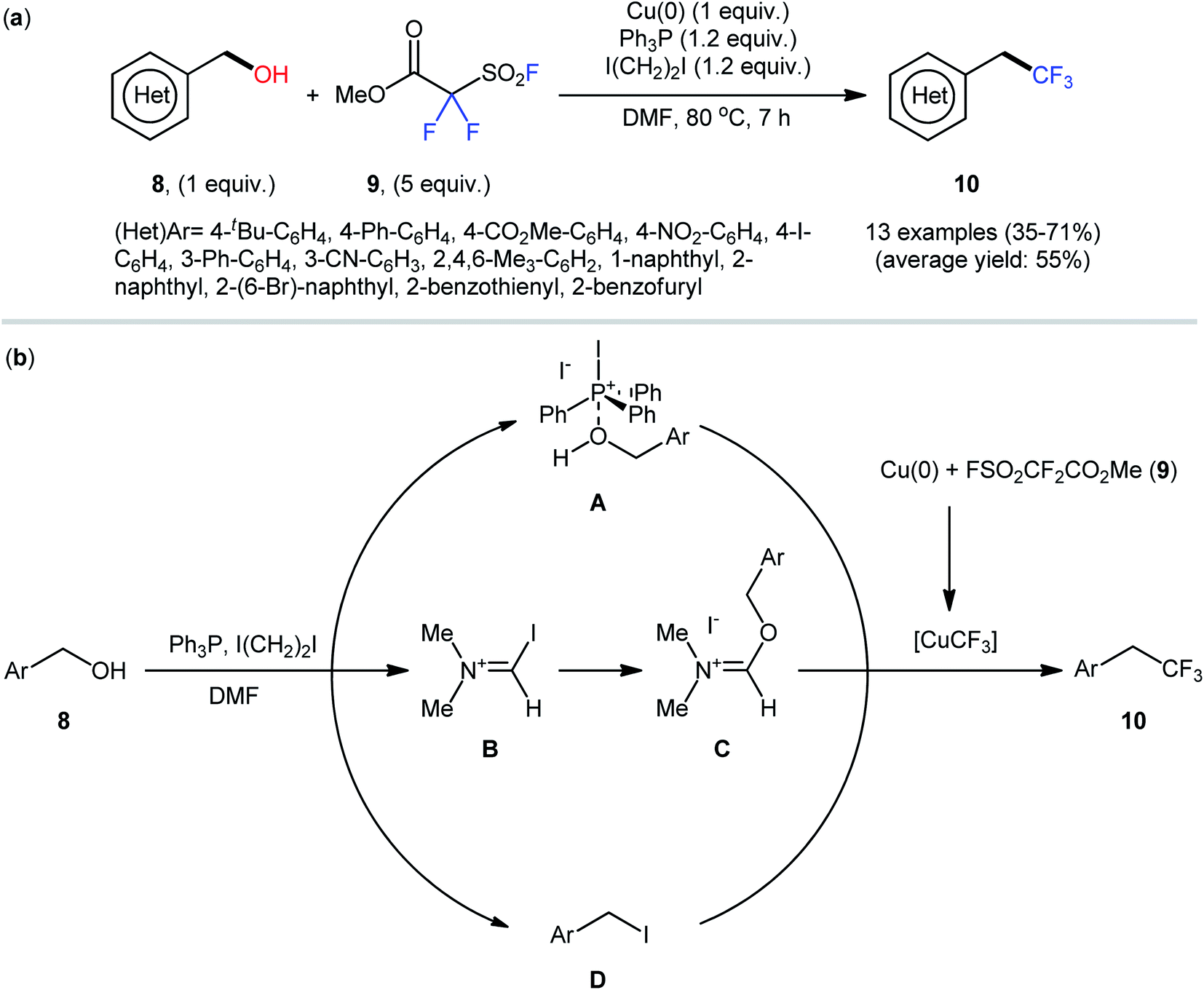 | ||
| Scheme 5 (a) Wu-Xiao's synthesis of (2,2,2-trifluoroethyl)arenes 10. (b) The proposed pathways for the formation of (2,2,2-trifluoroethyl)arenes 10. | ||
Regarding the influence of electronic effects of substituent groups in the phenyl ring periphery of benzylic alcohols, electron-rich substrates afforded higher yields compared to electron-poor ones. It is noteworthy that although aryl bromides remained intact under these conditions, undesired trifluoromethylation of C–I bonds was observed in the case of aryl iodides. Regrettably, alkyl alcohols did not exhibit reactivity under standard reaction conditions. It was noteworthy that under relatively similar conditions, dehydroxylative difluoromethylation and trifluoromethylthiolation of a diverse range of aliphatic alcohols with TMSCF2H and AgSCF3, respectively, also effectively proceeded to afford the corresponding difluoromethylated and trifluoromethylthiolated compounds in reasonable yields. While the detailed mechanistic picture remains unclear, the authors speculated that phosphonium A, iminium C, or (hetero)benzyl halide D might be key intermediates for this trifluoromethylation reaction (Scheme 5b).
3. Dehydroxylative trifluoromethoxylation
At the outset of 2018, Tang and colleagues discovered that a combination of nucleophilic trifluoromethoxylating reagent 12 (trifluoromethyl tosylate), quaternary ammonium salt (tetramethylammonium bromide; TMABr), and fluoride source (CsF) enabled direct dehydroxylative trifluoromethoxylation of various primary and secondary alcohols 11, giving the corresponding alkyl trifluoromethyl ethers 13 in moderate to high yields, ranging from 40% to 87% (Scheme 6).23 The synthetic broad scope of the protocol was established using a library of various alkyl, benzyl, allyl, and propargyl alcohols. Significantly, the authors showcased the potentiality of this process by dehydroxytrifluoromethoxylation of complex bioactive molecules such as cholane-3,12,24-triol (a natural product) and pleuromutilin (an antibacterial drug). The reaction demonstrated a high degree of chemoselectivity, and in the presence of a secondary or tertiary alcohol, the reaction preferentially took place at the position of a primary alcohol.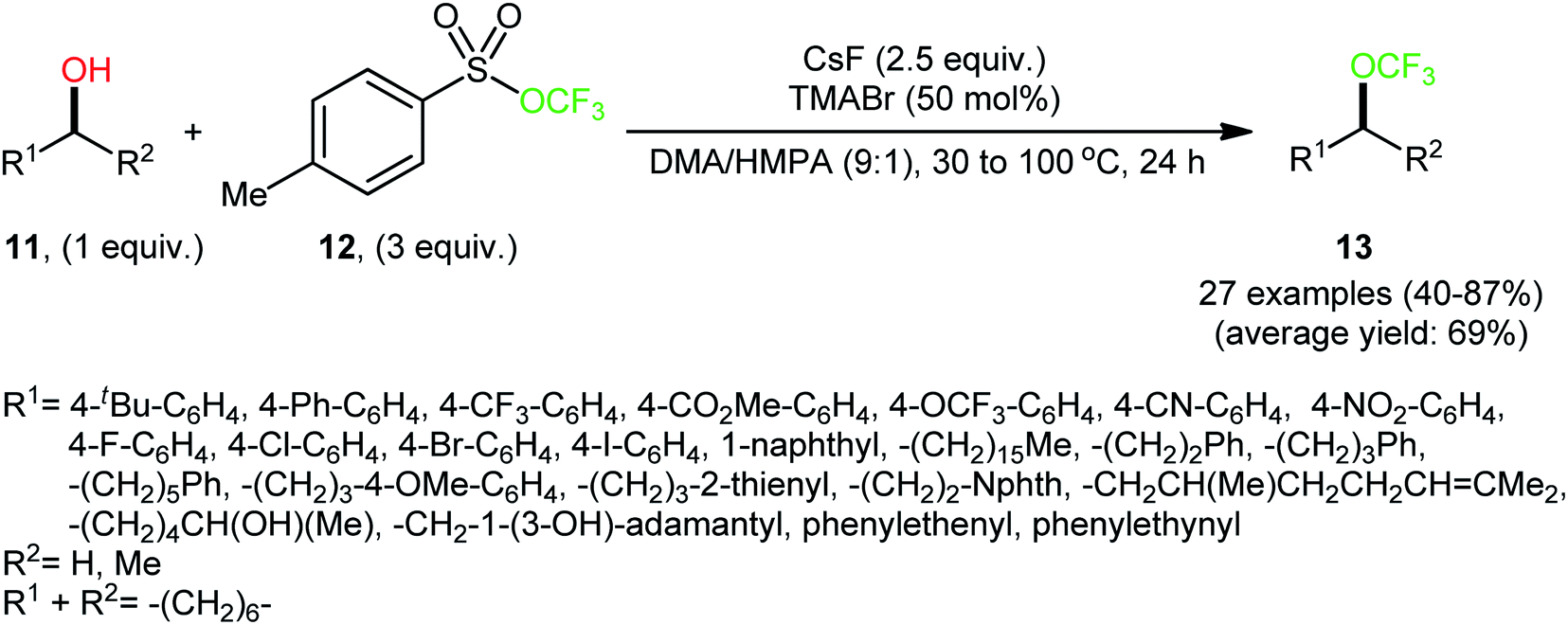 | ||
| Scheme 6 Metal-free direct dehydroxytrifluoromethoxylation of alcohols 11 with trifluoromethyl tosylate 12. | ||
Based on mechanistic studies (isotope labelling and 19F NMR experiments), the author proposed a plausible mechanistic pathway for the above transformation, as depicted in Scheme 7.24 The reaction begins with the release of trifluoromethoxide anion (CF3O−) from reagent 12 under the action of a fluoride salt. Subsequently, CF3O− undergoes decomposition to produce carbonic difluoride A which, upon esterification with alcohol 11, generates alkyl fluoroformate intermediate B. Finally, nucleophilic substitution reaction of activated species B with in situ-generated CF3O− affords the final product 13. Of note, the studies indicated that the presence of TMABr in the reaction mixture is crucial for improving the nucleophilicity of OCF3 anion, while in the absence of any quaternary ammonium salt, inferior results in terms of product yield were observed.
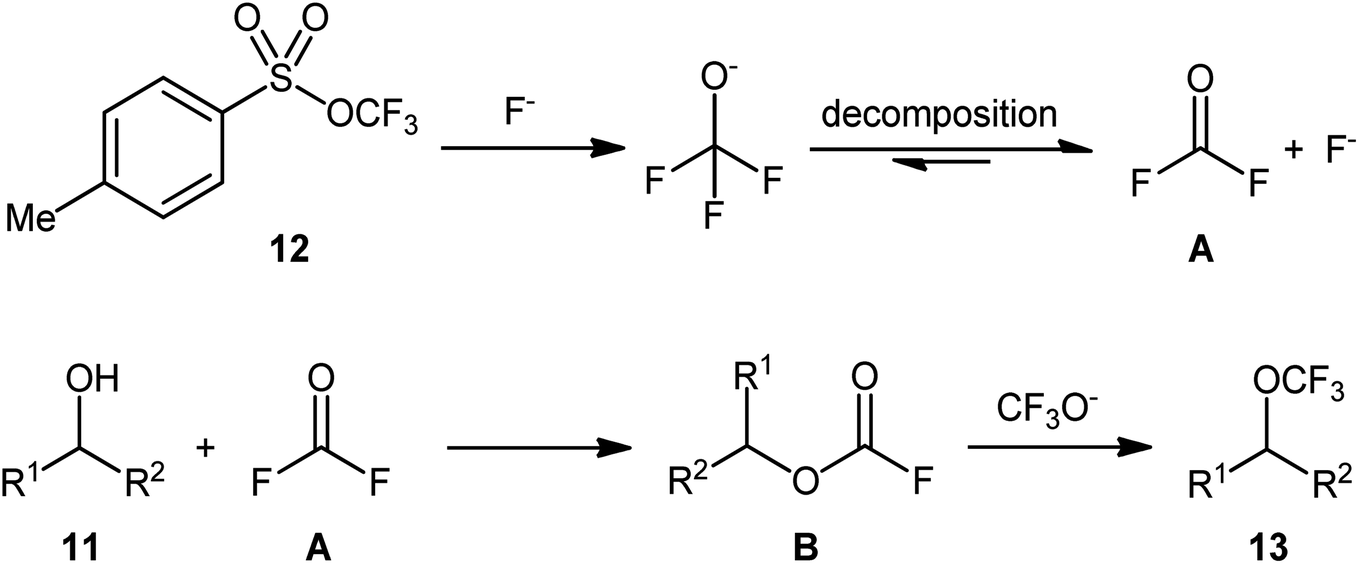 | ||
| Scheme 7 Mechanism proposed to explain the formation of alkyl trifluoromethyl ethers 13. | ||
In an attempt to further demonstrate the strength of this novel and interesting alkyl trifluoromethyl ether synthesis, Lin and Xiao along with their co-workers documented an elegant Ph3P/ICH2CH2I-promoted trifluoromethoxylation of aliphatic alcohols using AgOCF3 as a nucleophilic trifluoromethoxylating reagent, which allowed very rapid access to the corresponding dehydroxytrifluoromethoxylated products.25 Through exploration and optimization of this dehydroxylative functionalization, the authors identified that the reaction rate is highly dependent on the nature of solvent. Among several solvents tested (e.g., DMSO, DMF, NMP, and toluene), DMF was found to be the most effective. Furthermore, the outcome of this transformation was also dramatically dependent on the reaction temperature. The best results were obtained by performing the process at 80 °C. A higher or lower temperature resulted in lower yields. With these optimized reaction conditions, 25 trifluoromethyl ethers 15 were synthesized in 16–76% yields from the corresponding aryl/benzyl/allyl/propargyl alcohols 14 (Scheme 8). Notably, a diverse range of functional groups such as fluoro, chloro, bromo, iodo, cyano, nitro, ester, and ether functionalities were demonstrated to be well-tolerated by this protocol. However, the major drawback of this synthetic protocol was its very low efficiency for functionalization of alkyl alcohols. Intriguingly, the authors nicely solved this problem by replacing Ph3P with Ph2PCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 and performing the process at 100 °C. However, the only reported case of a secondary alkyl alcohol led to a mediocre yield.
CH2 and performing the process at 100 °C. However, the only reported case of a secondary alkyl alcohol led to a mediocre yield.
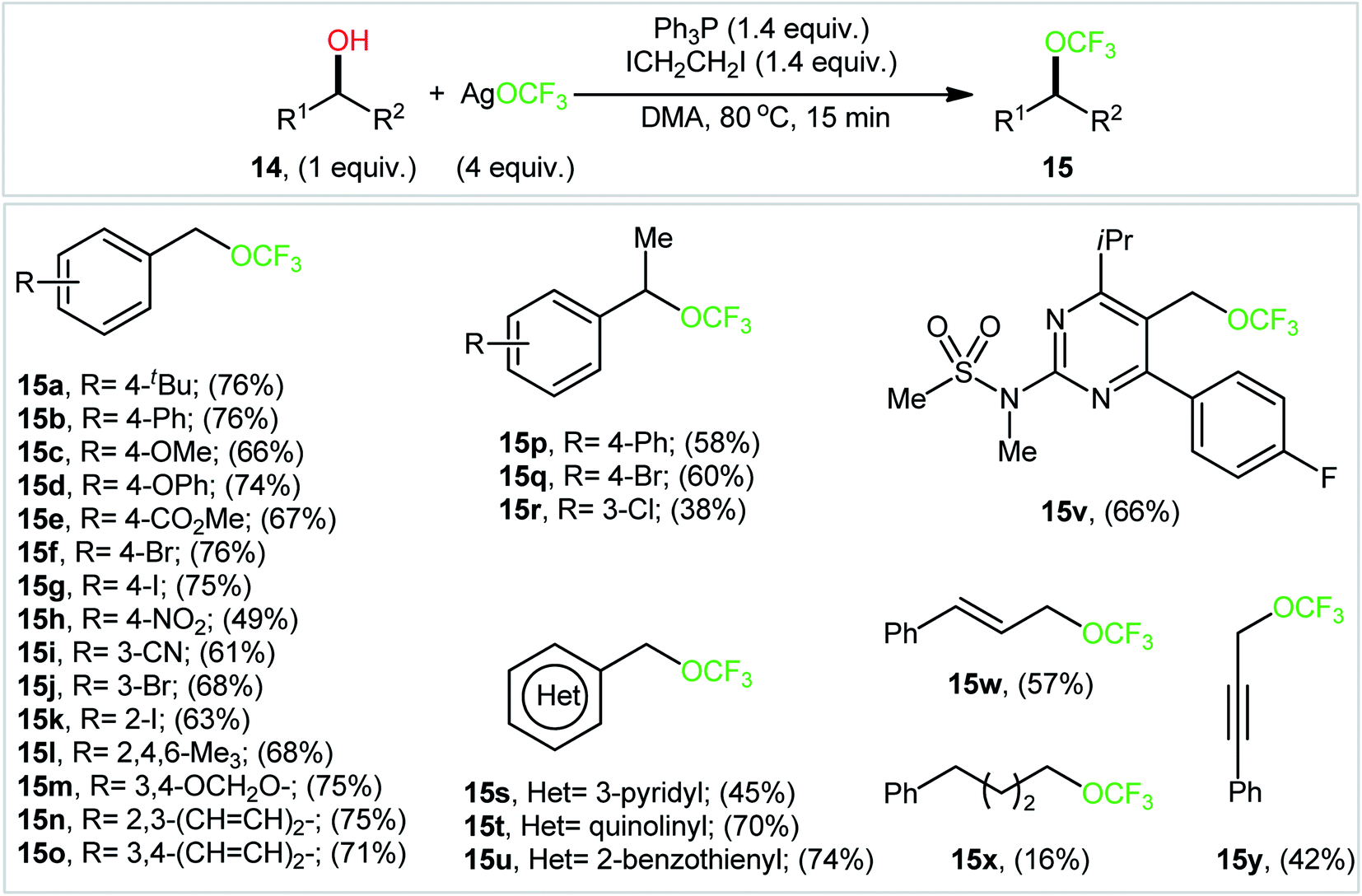 | ||
| Scheme 8 Ph3P/ICH2CH2I-promoted dehydroxylative trifluoromethoxylation of aliphatic alcohols 14 using AgOCF3 as a nucleophilic trifluoromethoxylating reagent. | ||
Mechanistically (Scheme 9), the reaction may be initiated by the reaction of Ph3P with ICH2CH2I to give diiodophosphonium salt A, which upon coordination with the reaction solvent DMF, furnishes complex B. Subsequently, substitution of an alcohol 14 with a DMF molecule in complex B yields complex C, and after nucleophilic attack by trifluoromethoxy anion, generated from AgOCF3 by precipitating AgI, affords the observed alkyl trifluoromethyl ether 15. In another possibility, a sequential P–O bond formation and C–O bond cleavage process converts complex B into a triphenylphosphine oxide (Ph3PO) and the Vilsmeier–Haack-type intermediate D. Later, nucleophilic substitution of alcohol 14 with intermediate D leads to the formation of intermediate E, which after nucleophilic trifluoromethoxylation with CF3O−, provides the final product 15.
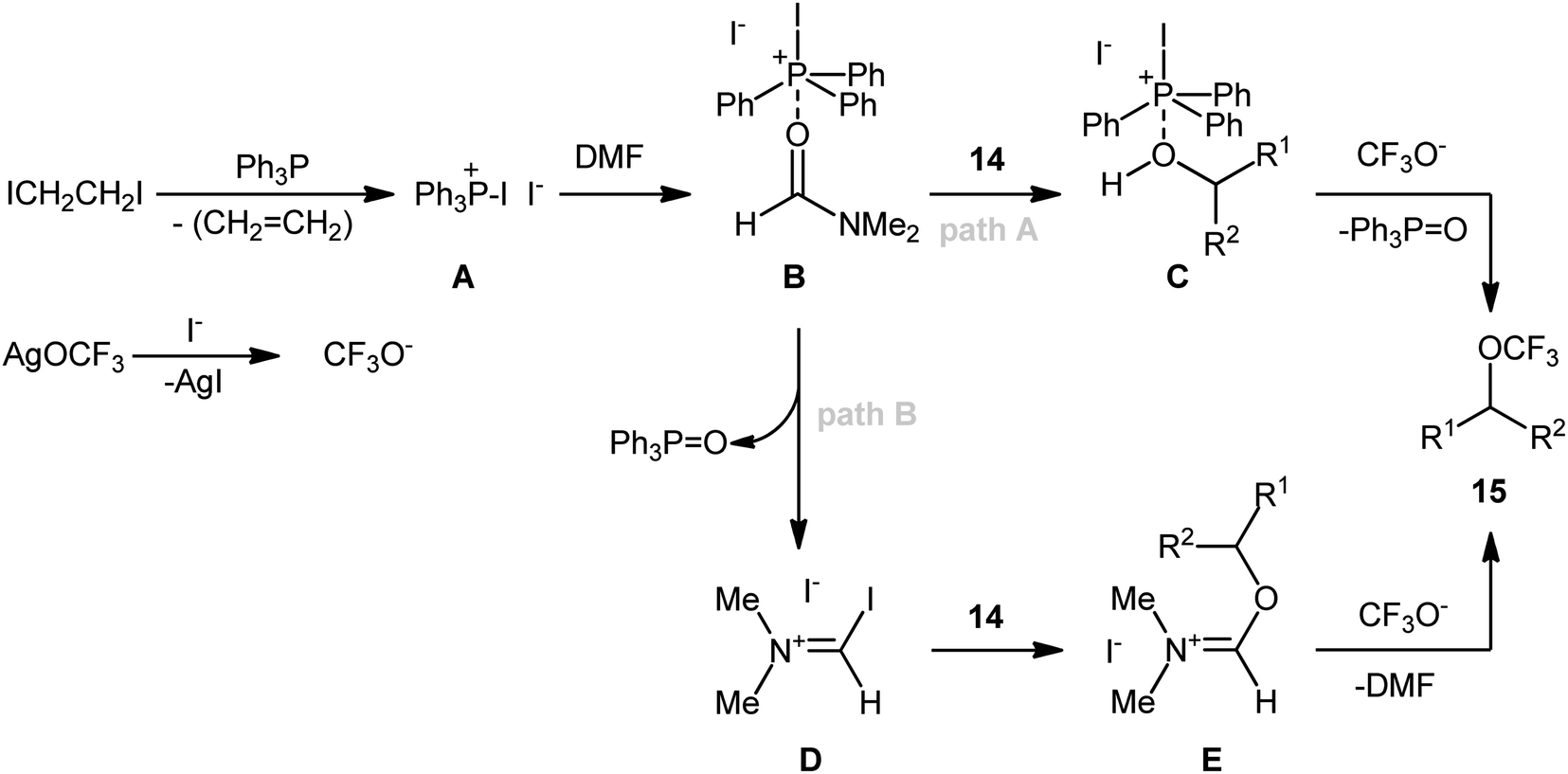 | ||
| Scheme 9 Mechanistic proposal for the formation of trifluoromethyl ethers 15. | ||
4. Dehydroxylative trifluoromethylthiolation
The first report of the direct dehydroxylative trifluoromethylthiolation of alcohols was published by Rueping and co-workers in 2014,26 who disclosed that the treatment of benzylic alcohols 16 with CuSCF3 as a stable and readily available SCF3 source and BF3·Et2O as a Lewis acid additive under an air atmosphere resulted in the formation trifluoromethylthioethers 17 in moderate to quantitative yields within 30 min (Scheme 10). All three types of alcohols (primary, secondary, and tertiary) were applicable to this reaction, and in all cases, the corresponding trifluoromethylthioethers were selectively obtained. However, due to the strong acidic conditions, poor functional group tolerance occurs with this method. It should be mentioned that the presence of BF3·Et2O was essential for the success of this C–S bond formation reaction. No product was detected with the lack of this additive, even at elevated temperatures.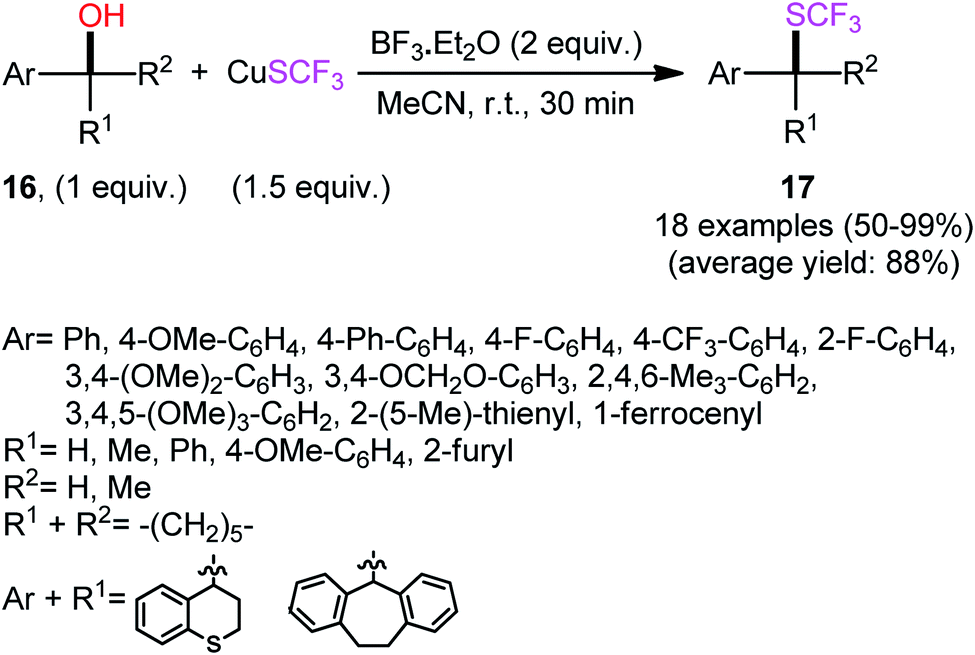 | ||
| Scheme 10 Lewis acid-mediated dehydroxylative trifluoromethylthiolation of benzylic alcohols 16 with CuSCF3, as reported by Rueping. | ||
Other Brønsted or Lewis acids such as MsOH, TsOH, TFA, Sc(OTf)3, Bi(OTf)3, and In(OTf)3 were also tested and proved to be completely ineffective. The identical reaction conditions were also applied for trifluoromethylthiolation of allylic alcohols to give the corresponding allylic trifluoromethyl thioethers in good to excellent yields (9 examples, 73–96% yield) and high regioselectivities, in which regardless of the substitution pattern, conjugated aryl/olefin products were predominantly formed in the case of aryl-substituted allyl alcohols. Mechanistic investigations indicated that the reaction may occur via an SN1-type process, as evidenced by the formation of racemic products from enantiopure alcohols.
Immediately after, Qing and collaborators developed a similar dehydroxytrifluoromethylthiolation of alkyl alcohols 18 with AgSCF3, employing a large excess of the mild reagent n-Bu4NI as activator and toluene as solvent (Scheme 11).27 The reaction was shown to be quite general, and a diverse range of primary aliphatic, benzylic, allylic, and propargylic alcohols participated in the trifluoromethylthiolation. Moreover, secondary alcohols also accomplished production of the corresponding products albeit the addition of a very large amount of another activator, KI (8 equiv.), and elevated reaction temperature (120 °C) were needed to prevent the competitive elimination reaction. However, tertiary alcohols were not suitable substrates for this transformation.
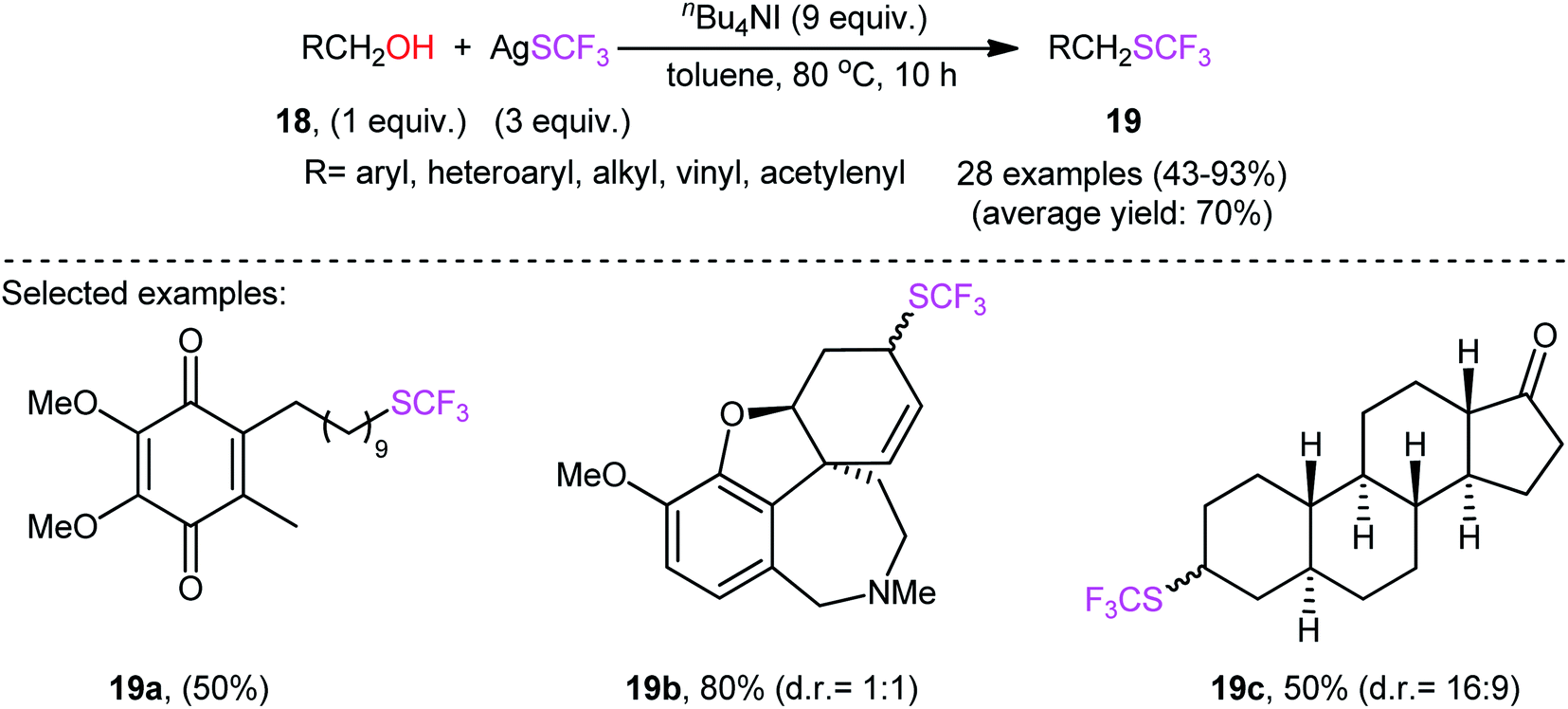 | ||
| Scheme 11 Qing's synthesis of trifluoromethylthioethers 19. | ||
Interestingly, several biologically active alcohols such as idebenone 18a (an anti-Alzheimer's drug), galantamine 18b (an anti-Alzheimer's and anti-dementia drug), and epiandrosterone 18c (a steroid hormone) also responded to the reaction. Notably, the authors observed that changing the ratio of AgSCF3/n-Bu4NI from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 to 1:1 led to the selective formation of alkyl fluorides instead of the expected alkyl trifluoromethylthioethers. A plausible mechanism based on previous studies is outlined in Scheme 12.
:3 to 1:1 led to the selective formation of alkyl fluorides instead of the expected alkyl trifluoromethylthioethers. A plausible mechanism based on previous studies is outlined in Scheme 12.
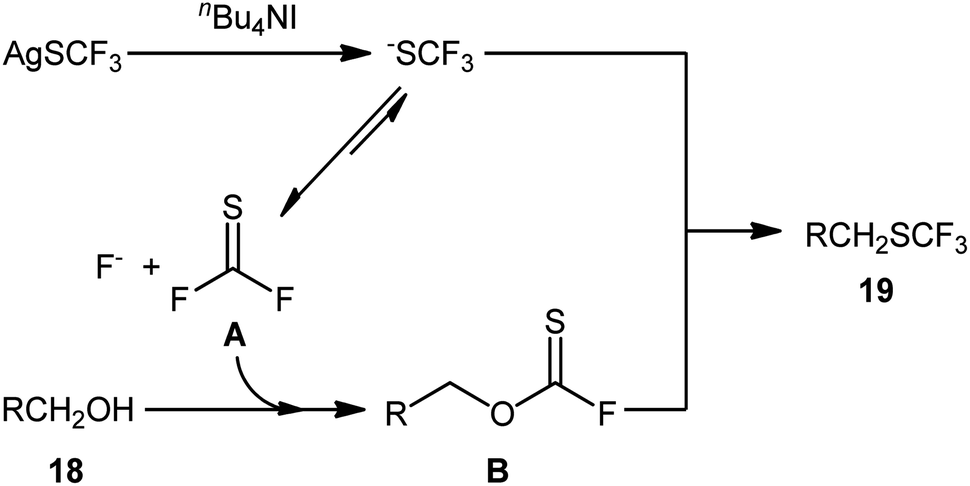 | ||
| Scheme 12 Proposed reaction mechanism for the synthesis of trifluoromethylthioethers 19 starting from alcohols 18 with AgSCF3. | ||
In their subsequent studies, this research group extended the scope of their methodology to enols.28 Thus, a library of (Z)-ethyl 2-(aryl)-3-hydroxyacrylates 20 were reacted with AgSCF3, n-Bu4NI, and KI in toluene at 120 °C leading to the respective α-aryl-β-(trifluoromethylthio)acrylates 21 in moderate to excellent yields and satisfactory stereoselectivities in favor of the (E)-products (Scheme 13). Under the same conditions, they also executed the direct dehydroxytrifluoromethylthiolation of a small series of 3-(hydroxymethylene)indolin-2-one derivatives 22, offering a decent yield of the desired 3-(((trifluoromethyl)thio)methylene)indolin-2-ones 23 (Scheme 14). Interestingly, two electron-deficient phenols were also tested and gave products in satisfactory yields. To the best of our knowledge, this is the first and only reported example of dehydroxylative trifluoromethylthiolation of C(sp2)–OH bonds.
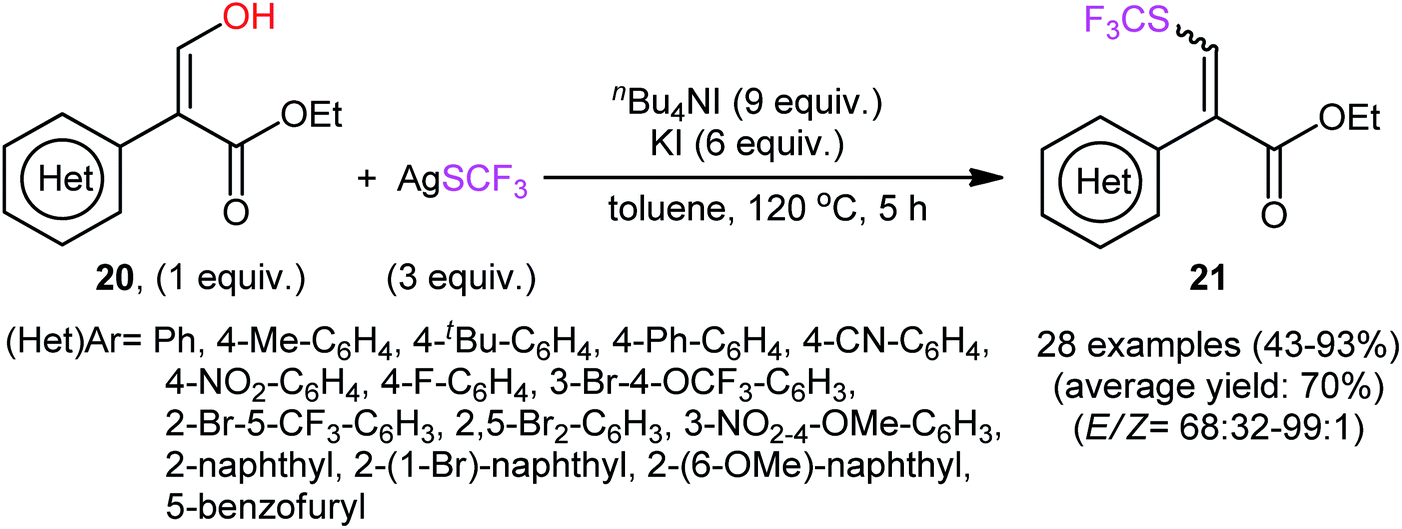 | ||
| Scheme 13 Qing's synthesis of α-aryl-β-(trifluoromethylthio)acrylates 21. | ||
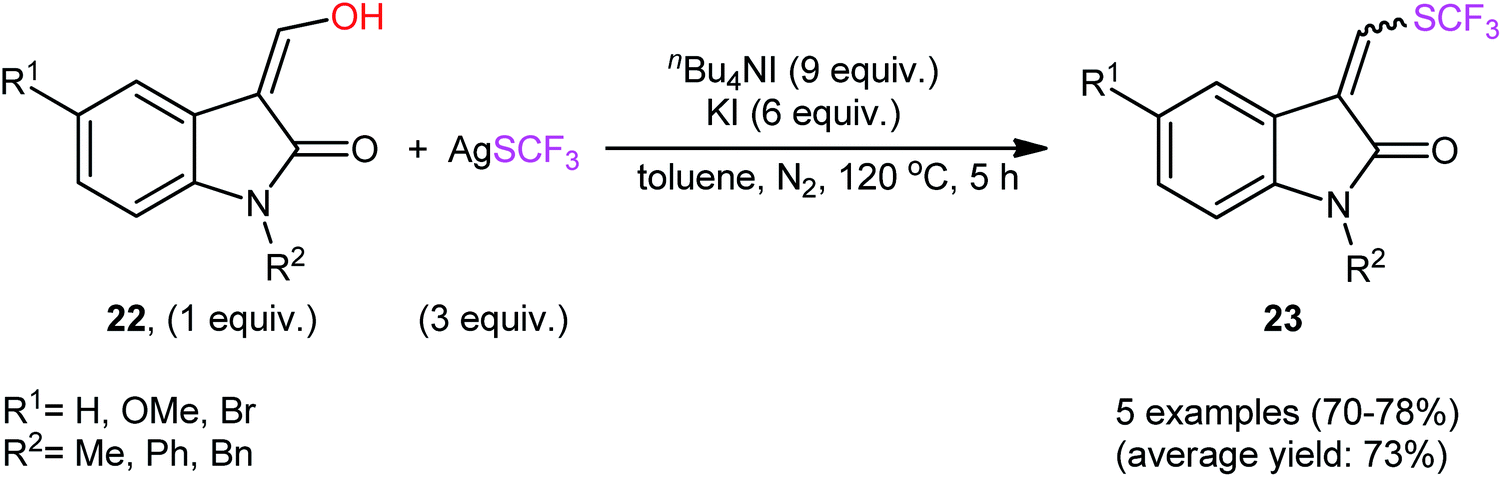 | ||
| Scheme 14 Synthesis of 3-(((trifluoromethyl)thio)methylene)indolin-2-ones 23 via nBu4NI-mediated dehydroxytrifluoromethylthiolation of 3-(hydroxymethylene)indolin-2-one derivatives 22 with AgSCF3. | ||
At the outset of 2016, Billard's research group devised an elegant metal-free method for dehydroxylative trifluoromethylthiolation of alkyl alcohols 24 using the second-generation trifluoromethanesulfenamide reagent 25 as an SCF3 source and n-Bu4NI as an activator in refluxing acetone to afford the corresponding alkyl trifluoromethylthioethers 26 in an efficient manner (Scheme 15a).29 The experiments demonstrated that the outcome of this reaction was not highly sensitive to the electronic nature of substrates, and therefore benzylic alcohols with either neutral, electron-donating, or electron-withdrawing substituents gave the trifluoromethylthiolated products in relatively similar yields. However, the steric effect was very strong (92% yield for benzyl alcohol to 46% for α-methylbenzyl alcohol). Unfortunately, the applicability of tertiary alcohols as starting materials was not explored in this study.
 | ||
| Scheme 15 (a) Metal-free dehydroxylative trifluoromethylthiolation of alkyl alcohols 24 with trifluoromethanesulfenamide 25; (b) solvent-free microwave-assisted synthesis of alkyl trifluoromethylthioethers 28 from alkyl alcohols 27 and AgSCF3. | ||
A presumptive mechanism for this dehydroxylative trifluoromethylthiolation reaction is represented in Scheme 16. Subsequently, a straightforward and greener approach for the synthesis of alkyl trifluoromethylthioethers 28 by the reaction between alkyl alcohols 27 and 1-n-butyl-3-methylimidazolium trifluoromethylthiolate ([bmim][SCF3]) generated in situ from [bmim][I] and AgSCF3 was reported by Pégot, Magnier, and co-workers.30 The reactions were implemented under microwave irradiation and solvent-free conditions, tolerated primary, secondary, as well as tertiary alcohols, and rapidly provided the desired products in poor to excellent yields (Scheme 15b). Recycling tests indicated that the ionic liquid can be reused in several consecutive trials without significant loss of its activity (from 96% in the first run to 96% in the fifth run).
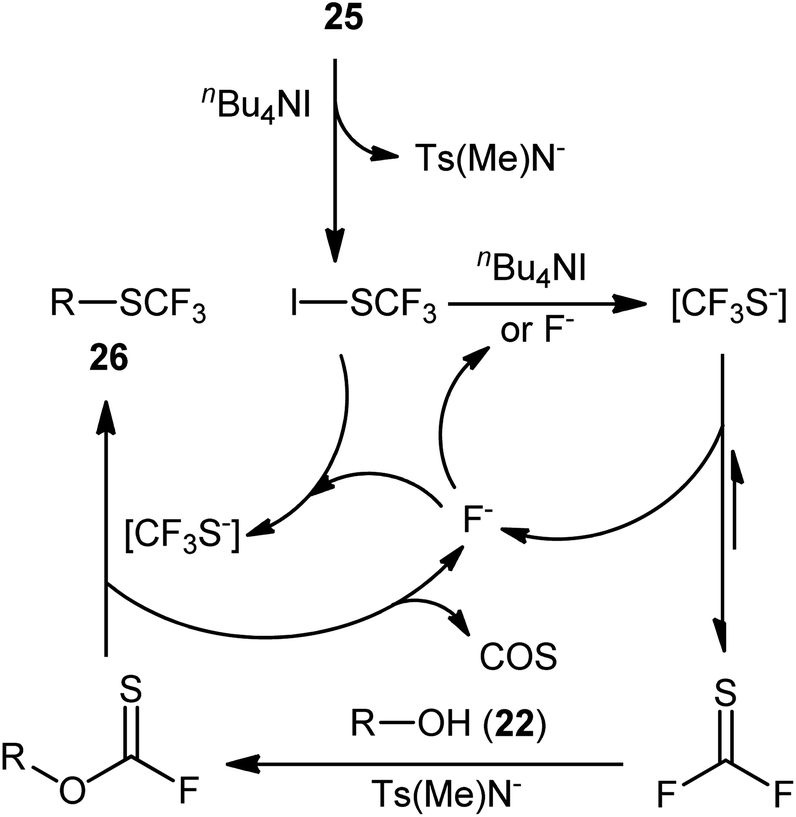 | ||
| Scheme 16 Proposed pathway of alkyl trifluoromethylthioethers 25 formation. | ||
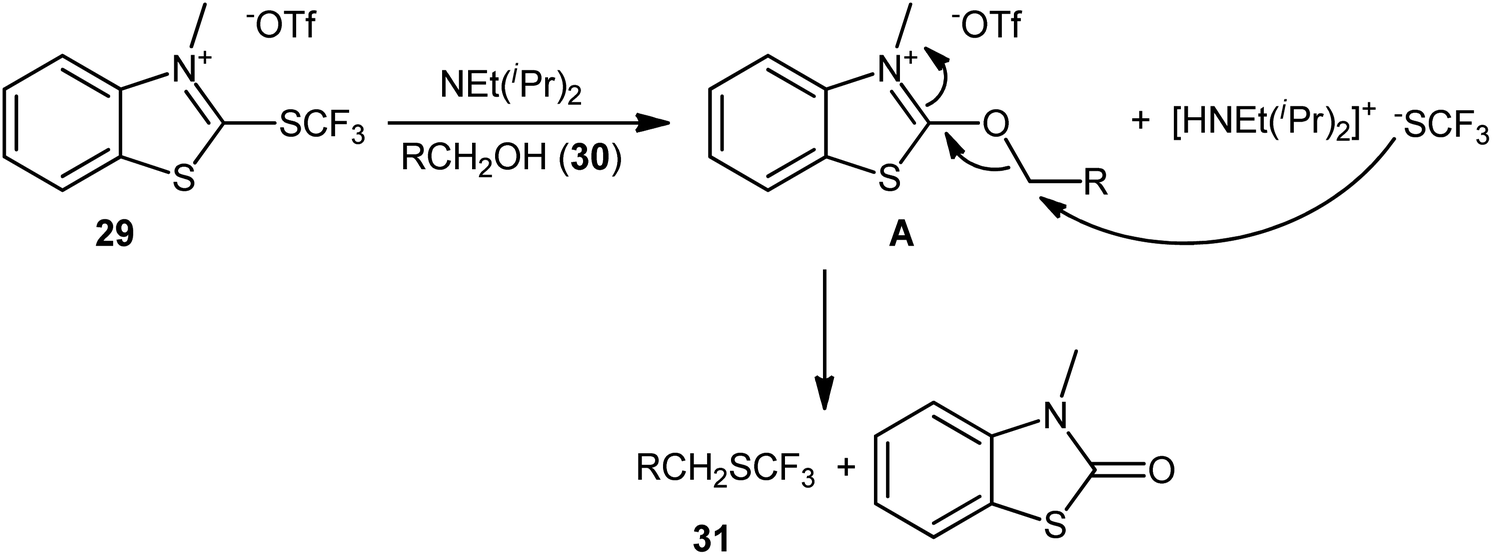
In 2019, Hopkinson and co-workers designed and synthesized a new bench-stable 2-trifluoromethylthio-substituted benzothiazolium salt (BT-SCF3; 29) through visible-light-induced, Ir-catalyzed trifluoromethylthiolation of inexpensive 2-mercaptobenzothiazole disulfide (MBTS) with the Langlois reagent (NaSO2CF3) and subsequent reaction of the generated 2-SCF3-substituted benzothiazole with MeOTf in DCM at room temperature (Scheme 17a).31 The activity of this purely organic trifluoromethylthiolating reagent has been evaluated in the dehydroxylative trifluoromethylthiolation of a broad set of alkyl/benzyl/propargyl alcohols 30 in the presence of Hünig's base (NEt(iPr)2) in MeCN. Moderate to almost quantitative yields of the target trifluoromethyl thioethers 31 were obtained within 1–2 h at room temperature (Scheme 17b). The reaction exhibited satisfactory tolerance for an array of catalytically reactive functional groups (e.g., F, Cl, Br, I, CF3, CO2Me, SMe, NO2), and thus, promised further elaboration of the end products. It is worthwhile to note that the authors nicely adapted this approach to the direct construction of SeCF3-substituted compounds from alcohols by developing a similar trifluoromethylselenyl-substituted benzothiazolium salt (BT-SeCF3).
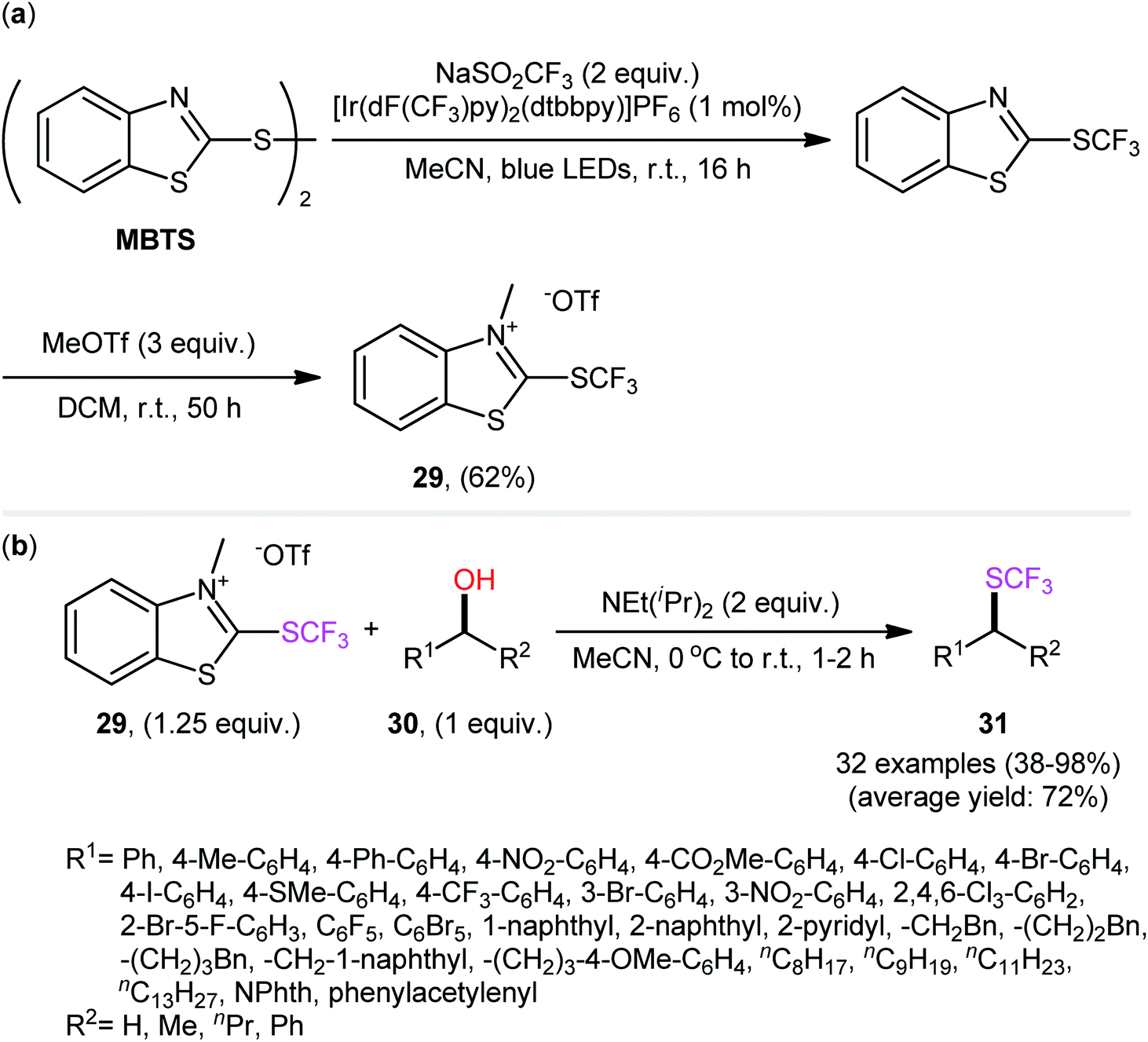 | ||
| Scheme 17 Hünig's base-mediated dehydroxylative trifluoromethylthiolation of alkyl/benzyl/propargyl alcohols 30 with benzothiazolium reagent 29. | ||
As for the mechanism, the authors speculated that the reaction most likely proceeds through the formation of key electrophilic 2-alkoxybenzothiazolium species A via nucleophilic attack of the alcohol 30 in the presence of NEt(iPr)2 at the C2-position of the BT-SCF3 reagent 29 and subsequent nucleophilic substitution reaction with in situ generated –SCF3 anion (Scheme 18). Guided by the same principle, a similar dehydroxylative functionalization strategy was applied by the same research group towards the synthesis of various perfluoroalkyl thioethers32 and thioesters.33
 | ||
| Scheme 18 Mechanism proposed to explain the formation of trifluoromethylthioethers 31. | ||
In a recent report, Wu, Xiao, and colleagues accomplished the direct conversion of alcohols 32 into the corresponding trifluoromethyl thioethers 33 using AgSCF3 as a source of F3CS group and the Ph3P/ICH2CH2I/n-Bu4NI combination as an activation system in a 2:1 mixture of DMF and MeCN.22 The reaction was compatible with a variety of functionalized benzylic alcohols, as well as heterobenzylic alcohols such as hydroxymethyl pyridines, quinolines, thiazoles, benzothiols, even simple allylic and propargylic alcohols (Scheme 19). As for alkyl alcohols, the corresponding products were obtained in poor yields. In this case, when Ph2PCHCH2 was used instead of Ph3P, the product yields were significantly increased. The plausible mechanism for this transformation is analogous to the one depicted in Scheme 5. It should be mentioned that this mechanism is tentative and lacks experimental evidence.
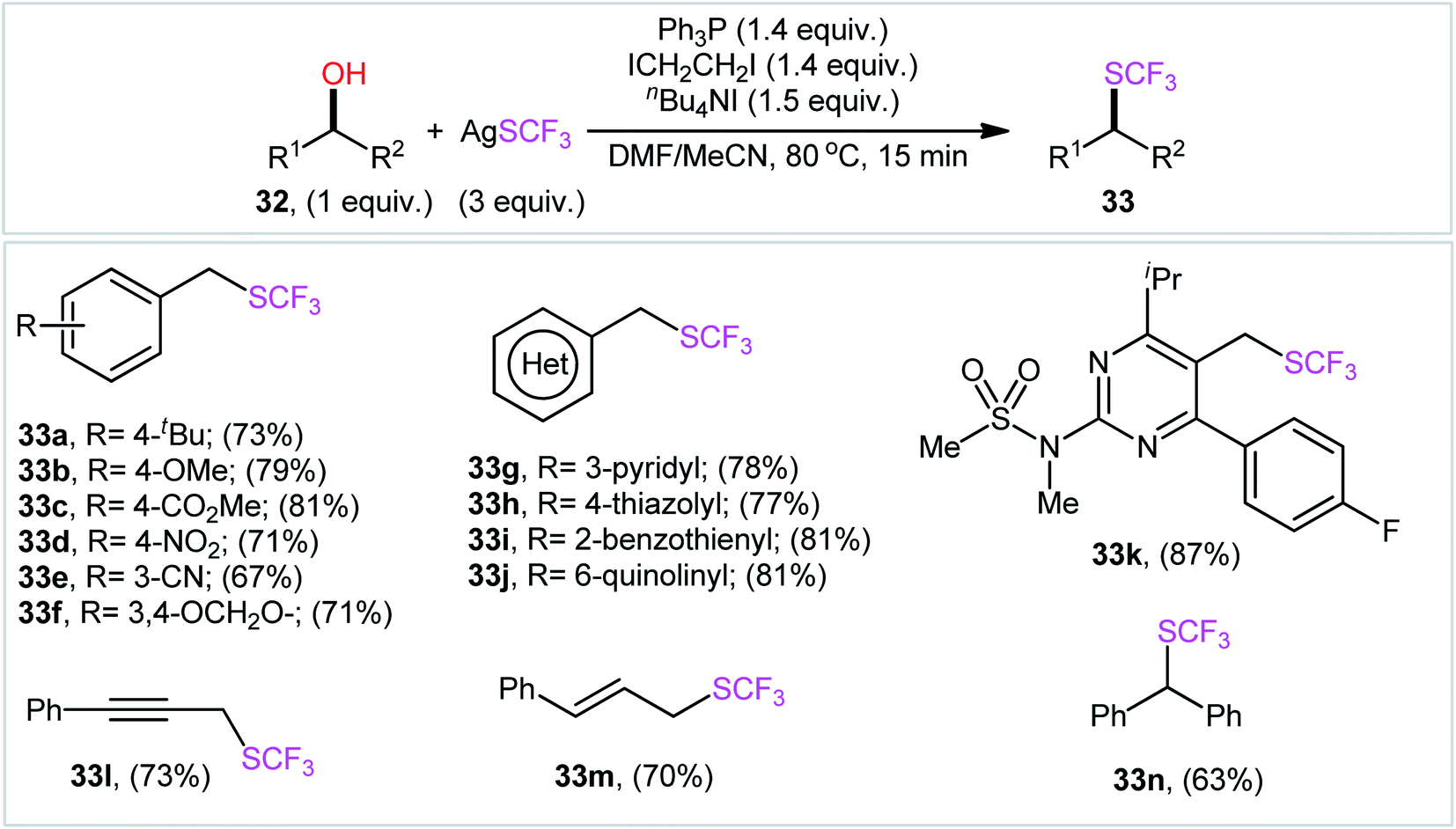 | ||
| Scheme 19 Ph3P/ICH2CH2I/n-Bu4NI-mediated direct conversion of alcohols 32 into the corresponding trifluoromethyl thioethers 33 using AgSCF3. | ||
5. Dehydroxylative trifluoromethylselenylation
Compared to trifluoromethylation, trifluoromethoxylation, and trifluoromethylthiolation, the direct trifluoromethylselenylation of alcohols is relatively less explored, although there has been much recent attention on the development of novel and efficient methodologies for the synthesis of SeCF3-substituted compounds.34 In 2019, in the same study describing the deoxytrifluoromethylthiolation of alcohols with the benzothiazolium salt BT-SCF3, Hopkinson's research team reported the successful NEt(iPr)2-promoted preparation of trifluoromethylselenoalkanes 36 from the respective alcohols 34 employing 2-trifluoromethylseleno-substituted benzothiazolium salt (BT-SeCF3; 35) (Scheme 20).31 Notably, BT-SeCF3 was synthesized via the same general strategy used to prepare BT-SCF3.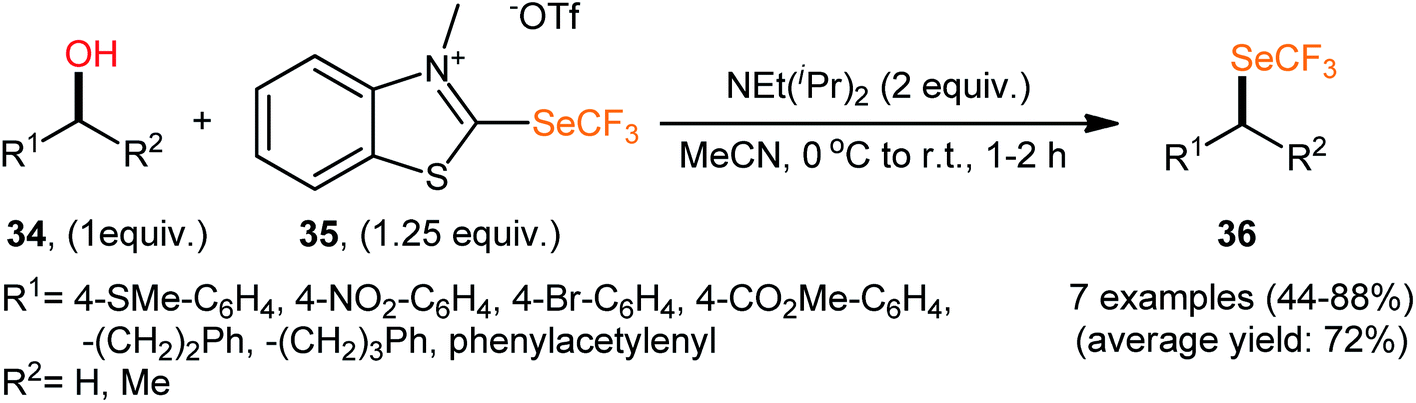 | ||
| Scheme 20 Hopkinson's synthesis of trifluoromethylselenoalkanes 36. | ||
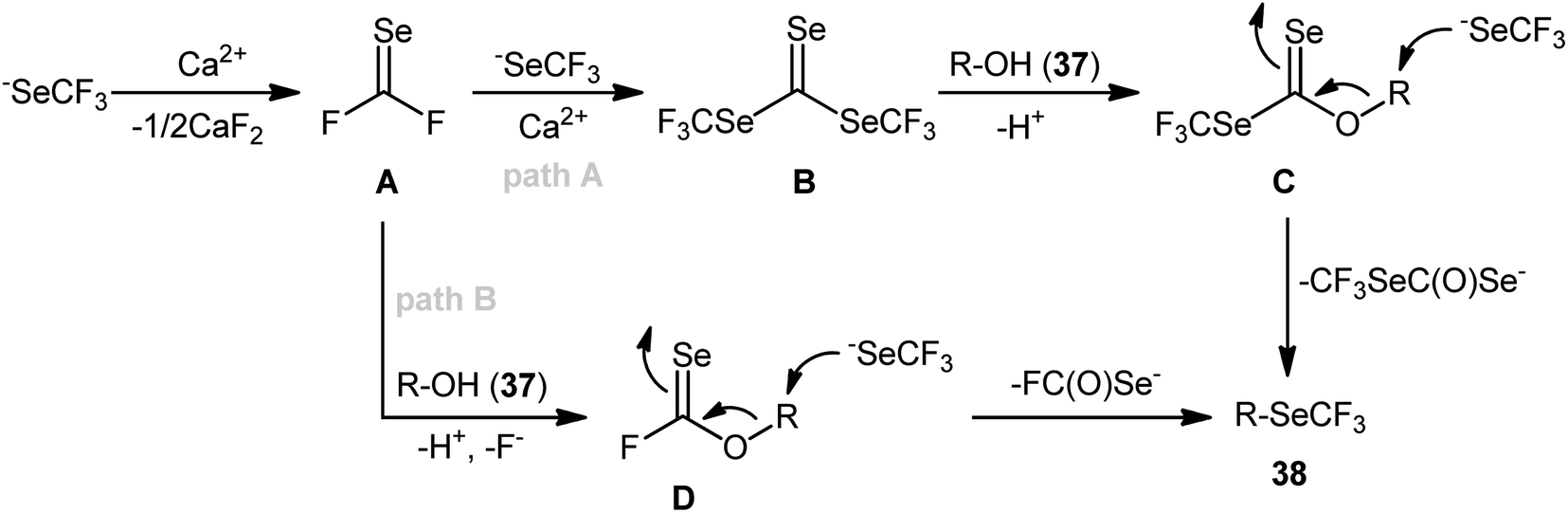
In 2020, using [Me4N][SeCF3] salt as a stable, non-volatile, and readily accessible source of nucleophilic SeCF3, Zhang and colleagues engineered the direct dehydroxylative trifluoromethylselenylation of alcohols 37 for the synthesis of valuable alkyl trifluoromethyl selenoethers 38 under catalyst-free conditions.35 By employing 3-phenylpropyl alcohol as the model reactant, several additives such as CaF2, CaCl2, CaBr2, Ca(OTf)2, Ca(C2O4), CaSO4, HCl, and LiI were carefully screened. Among them, excellent results were obtained for this transformation with CaCl2, whereas MeCN was found to be the most effective solvent among the other common organic solvents tested (DMA, DMF, NMP, DMSO, MeCN, DCM, toluene, and 1,4-dioxane).36 Evaluation of the substrate scope clearly demonstrated that the reaction was tolerant to a variety of primary and secondary aliphatic alcohols (Scheme 21). However, tertiary alcohols provided complicated mixtures. In order to elucidate the mechanism of the reaction, the authors performed several control experiments, such as GC-MS analyses, 19F NMR studies, and others.37
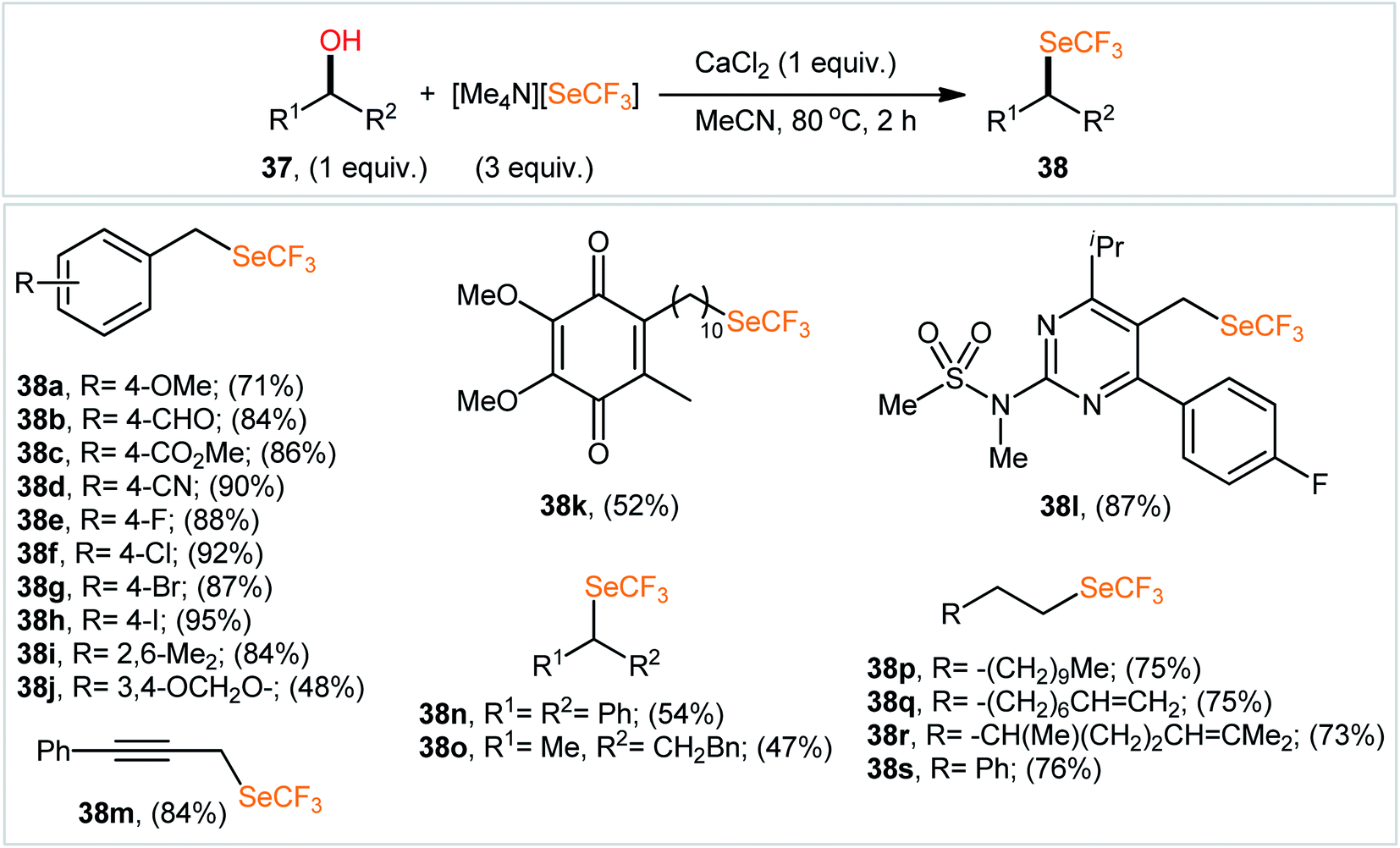 | ||
| Scheme 21 Selected examples of CaCl2-promoted dehydroxylative trifluoromethylselenylation of alcohols 37 with [Me4N][SeCF3]. | ||
From these results, the authors proposed two possible pathways for this transformation. The first pathway (Scheme 22, path A) starts with the formation of carbonoselenoic difluoride intermediate A through the decomposition of −SeCF3 with CaCl2 as a fluoride scavenger, which after reaction with another two equivalents of −SeCF3 in the presence of Ca2+ cations provides bis(trifluoromethyl)-carbonotriselenoate B (this intermediate was detected by 19F NMR and GC-MS analyses). Subsequently, the nucleophilic substitution of key intermediate B by alcohol 37 leads to a carbonoselenoate C. Finally, the nucleophilic attack of intermediate C with −SeCF3 anion provides the target trifluoromethylselenylated product 38. The key steps of the second possible route (Scheme 22, path B) are the generation of O-alkyl carbonofluoridoselenoate D via straightforward reaction of intermediate A with alcohol 37 and its nucleophilic substitution by −SeCF3 to form the observed product 38. According to the authors, pathway B is not likely the major process in the Ca-mediated dehydroxy trifluoromethylselenylation,38 especially when using a large excess of [Me4N][SeCF3].
 | ||
| Scheme 22 Mechanistic proposal for the reaction in Scheme 21. | ||
6. Conclusion
Because of their unique physicochemical and biological properties, there has been broad interest in fluorinated compounds in various research fields such as pharmaceuticals, medical imaging, agrochemicals, and materials science. Among all fluorine-containing functionalities, the trifluoromethyl (CF3), trifluoromethoxyl (OCF3), trifluoromethylthiol (SCF3), and trifluoromethylselenyl (SeCF3) groups are becoming increasingly prominent in new drugs due to their strong electron-withdrawing nature and high lipophilic properties. Therefore, the high demand for biologically active compounds has stimulated significant efforts to develop efficient methods for the installation of these groups into organic molecules. As shown in this Mini-Review, recently direct dehydroxylative trifluoromethylation, trifluoromethoxylation, trifluoromethylthiolation, and trifluoromethylselenylation of alcohols have emerged as efficient new methods for the construction of CF3/OCF3/SCF3/SeCF3-functionalized compounds from inexpensive and readily available starting materials without the need for time-consuming pre-functionalization steps.As illustrated, various aliphatic and benzylic alcohols were applicable in these reactions. However, aromatic alcohols were mainly unsuitable substrates. Therefore, many more studies are needed to develop efficient procedures that allow trifluoro-methylation, -methoxylation, -methylthiolation, and -methylselenylation reactions of aromatic alcohols. Moreover, there are insufficient reported examples for some reactions such as trifluoromethylselenylations, and thus, additional study is necessary to determine the scope and limitations of these reactions.
Conflicts of interest
There are no conflicts to declare.References
- (a) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS; (b) J. Cheng, Z. Tan, Y. Xing, Z. Shen, Y. Zhang, L. Liu and S. Liu, J. Mater. Chem. A, 2021, 9, 5787–5795 RSC; (c) H. Wang, J. Cui, Y. Zhao, Z. Li and J. Wang, Green Chem., 2021, 23, 405–411 RSC; (d) H. Wang, T. Song, Z. Li, J. Qiu, Y. Zhao, H. Zhang and J. Wang, ACS Appl. Mater. Interface, 2021, 13, 25918–25925 CrossRef CAS PubMed; (e) Z. Li, Y. Shi, A. Zhu, Y. Zhao, H. Wang, B. P. Binks and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 3928–3933 CrossRef CAS PubMed.
- (a) C. Hansch, A. Leo, S. H. Unger, K. H. Kim, D. Nikaitani and E. J. Lien, J. Med. Chem., 1973, 16, 1207–1216 CrossRef CAS PubMed; (b) X. Wang, Z. Feng, B. Xiao, J. Zhao, H. Ma, Y. Tian and L. Tan, Green Chem., 2020, 22, 6157–6169 RSC; (c) L. Zhang, M. Zhang, S. You, D. Ma, J. Zhao and Z. Chen, Sci. Total Environ., 2021, 780, 146505 CrossRef CAS PubMed; (d) L. Zhang, J. Zheng, S. Tian, H. Zhang, X. Guan, S. Zhu and Z. Li, J. Environ. Sci., 2020, 91, 212–221 CrossRef PubMed; (e) X. Wang, P. Gao, Y. Liu, H. Li and F. Lu, Bioinformatics, 2020, 15, 493–502 CAS; (f) S. Sun, L. Xu, Q. Zou, G. Wang and J. Gorodkin, Bioinformatics, 2021, 37, 1319–1321 CrossRef CAS PubMed.
- (a) S. M. Vrouenraets, F. W. Wit, J. v. Tongeren and J. M. Lange, Expert. Opin. Pharmacother., 2007, 8, 851–871 CrossRef CAS PubMed; (b) H. M. Bryson, B. Fulton and P. Benfield, Drugs, 1996, 52, 549–563 CrossRef CAS PubMed; (c) M. Kirby, H. Carageorgiou-Markomihalakis and P. Turner, Br. J. Clin. Pharmacol., 1975, 2, 541–542 CrossRef CAS PubMed.
- Y. Wang, Z. Ye, H. Zhang and Z. Yuan, Adv. Synth. Catal., 2021, 363, 1835–1854 CrossRef CAS.
- (a) G. Yan, K. Qiu and M. Guo, Org. Chem. Front., 2021, 8, 3915–3942 RSC; (b) K. K. Goh, A. Sinha, C. Fraser and R. D. Young, RSC Adv., 2016, 6, 42708–42712 RSC; (c) R. K. Belter, J. Fluorine Chem., 2010, 131, 1302–1307 CrossRef CAS; (d) L. V. Sokolenko, R. K. Orlova, A. A. Filatov, Y. L. Yagupolskii, E. Magnier, B. Pégot and P. Diter, Molecules, 2019, 24, 1249 CrossRef PubMed; (e) L. Xu, J. Cheng and M. L. Trudell, J. Org. Chem., 2003, 68, 5388–5391 CrossRef CAS PubMed; (f) F. Brüning, C. R. Pitts, J. Kalim, D. Bornemann, C. Ghiazza, J. de Montmollin, N. Trapp, T. Billard and A. Togni, Angew. Chem., Int. Ed., 2019, 58, 18937–18941 CrossRef PubMed.
- (a) H. Liu, Z. Gu and X. Jiang, Adv. Synth. Catal., 2013, 355, 617–626 CrossRef CAS; (b) T. Koike and M. Akita, J. Fluorine Chem., 2014, 167, 30–36 CrossRef CAS; (c) P. Gao, X. R. Song, X. Y. Liu and Y. M. Liang, Chem.–Eur. J., 2015, 21, 7648–7661 CrossRef CAS PubMed.
- (a) X. Zhang and P. Tang, Sci. China: Chem., 2019, 62, 525–532 CrossRef CAS; (b) B. Manteau, P. Genix, L. Brelot, J. P. Vors, S. Pazenok, F. Giornal, C. Leuenberger and F. R. Leroux, Eur. J. Org. Chem., 2010, 6043–6066 CrossRef CAS.
- (a) F. Toulgoat, S. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415–2428 CrossRef CAS; (b) H. Chachignon and D. Cahard, Chin. J. Chem., 2016, 34, 445–454 CrossRef CAS; (c) A. L. Barthelemy, E. Magnier and G. Dagousset, Synthesis, 2018, 50, 4765–4776 CrossRef CAS; (d) X. Guo-Liang, J. Shang-Hua, S. Xiangdong and F. Behmagham, J. Fluorine Chem., 2020, 109524 CrossRef; (e) A. Tlili and T. Billard, Angew. Chem., Int. Ed., 2013, 52, 6818–6819 CrossRef CAS PubMed.
- (a) C. Zhang, J. Chin. Chem. Soc., 2017, 64, 457–463 CrossRef CAS; (b) Y. Wang, Z. Ye, H. Zhang and Z. Yuan, Adv. Synth. Catal., 2021, 363, 1835–1854 CrossRef CAS; (c) A. Tlili, E. Ismalaj, Q. Glenadel, C. Ghiazza and T. Billard, Chem.–Eur. J., 2018, 24, 3659–3670 CrossRef CAS PubMed; (d) C. Ghiazza, T. Billard and A. Tlili, Chem.–Eur. J., 2019, 25, 6482–6495 CrossRef CAS PubMed; (e) C. Ghiazza and A. Tlili, Beilstein J. Org. Chem., 2020, 16, 305–316 CrossRef CAS PubMed.
- W. L. Hu, X. G. Hu and L. Hunter, Synthesis, 2017, 49, 4917–4930 CrossRef CAS.
- 1 (a) S. Arshadi, E. Vessally, L. Edjlali, R. Hosseinzadeh-Khanmiri and E. Ghorbani-Kalhor, Beilstein J. Org. Chem., 2017, 13, 625–638 CrossRef CAS PubMed; (b) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 1–32 CrossRef PubMed; (c) F. A. H. Nasab, L. Z. Fekri, A. Monfared, A. Hosseinian and E. Vessally, RSC Adv., 2018, 8, 18456–18469 RSC; (d) A. Hosseinian, P. D. K. Nezhad, S. Ahmadi, Z. Rahmani and A. Monfared, J. Sulfur Chem., 2019, 40, 88–112 CrossRef CAS; (e) M. Hamzehloo, A. Hosseinian, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Fluorine Chem., 2019, 224, 52–60 CrossRef CAS; (f) M. R. J. Sarvestani, N. Mert, P. Charehjou and E. Vessally, J. Chem. Lett., 2020, 1, 93–102 Search PubMed; (g) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 9–18 Search PubMed; (h) S. Majedi, L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 25–31 Search PubMed.
- Z. Liu, A. Ebadi, M. Toughani, N. Mert and E. Vessally, RSC Adv., 2020, 10, 37299–37313 RSC.
- B. Azizi, M. R. P. Heravi, Z. Hossaini, A. Ebadi and E. Vessally, RSC Adv., 2021, 11, 13138–13151 RSC.
- S. Ahmadi, A. Hosseinian, P. D. Kheirollahi Nezhad, A. Monfared and E. Vessally, Iran. J. Chem. Chem. Eng., 2019, 38, 1–19 CAS.
- E. Vessally, S. Mohammadi, M. Abdoli, A. Hosseinian and P. Ojaghloo, Iran. J. Chem. Chem. Eng., 2020, 39, 11–19 CAS.
- X. Ma, Z. Kexin, W. Yonggang, A. G. Ebadi and M. Toughani, Iran. J. Chem. Chem. Eng., 2021, 40, 1364–1374 Search PubMed.
- R. T. Kareem, B. Azizi, M. Asnaashariisfahani, A. Ebadi and E. Vessally, RSC Adv., 2021, 11, 14941–14955 RSC.
- (a) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 1–19 CrossRef PubMed; (b) W. Peng, E. Vessally, S. Arshadi, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 1–22 CrossRef CAS PubMed; (c) Y. Yang, D. Zhang and E. Vessally, Top. Curr. Chem., 2020, 378, 1–32 CrossRef PubMed; (d) Z. He, D. Wu and E. Vessally, Top. Curr. Chem., 2020, 378, 1–30 CrossRef PubMed; (e) L. Feng, X. Li, B. Liu and E. Vessally, J. CO2 Util., 2020, 40, 101220 CrossRef CAS; (f) W. Xu, A. G. Ebadi, M. Toughani and E. Vessally, J. CO2 Util., 2020, 43, 101358 CrossRef; (g) A. Bakhtiary, M. R. P. Heravi, A. Hassanpour, I. Amini and E. Vessally, RSC Adv., 2020, 11, 470–483 RSC.
- C. Zhang, X. Liu, C. Liu and X. Luo, J. Kans. Entomol. Soc., 2021, 93, 267–281 Search PubMed.
- F. de Azambuja, S. M. Lovrien, P. Ross, B. R. Ambler and R. A. Altman, J. Org. Chem., 2019, 84, 2061–2071 CrossRef CAS PubMed.
- C. Han, L. M. Alabanza, S. M. Kelly and D. L. Orsi, Org. Process Res. Dev., 2019, 23, 1695–1702 CrossRef CAS.
- W. Zhang, J. H. Lin, W. Wu, Y. C. Cao and J. C. Xiao, Chin. J. Chem., 2020, 38, 169–172 CrossRef CAS.
- X. Jiang, Z. Deng and P. Tang, Angew. Chem., Int. Ed., 2018, 57, 292–295 CrossRef CAS PubMed.
- X. Jiang and P. Tang, Chin. J. Chem., 2021, 39, 255–264 CrossRef CAS.
- W. Zhang, J. Chen, J. H. Lin, J. C. Xiao and Y. C. Gu, iScience, 2018, 5, 110–117 CrossRef CAS PubMed.
- X. Ji, C. Hou, M. Shi, Y. Yan and Y. Liu, Food Rev. Int., 2020 DOI:10.1080/87559129.2020.1771363.
- X. Ji, B. Peng, H. Ding, B. Cui, H. Nie and Y. Yan, Food Rev. Int., 2021 DOI:10.1080/87559129.2021.1904973.
- Y. L. Liu, X. H. Xu and F. L. Qing, Tetrahedron, 2018, 74, 5827–5832 CrossRef CAS.
- Q. Glenadel, A. Tlili and T. Billard, Eur. J. Org. Chem., 2016, 1955–1957 CrossRef CAS.
- E. Anselmi, C. Simon, J. Marrot, P. Bernardelli, L. Schio, B. Pégot and E. Magnier, Eur. J. Org. Chem., 2017, 6319–6326 CrossRef CAS.
- S. Dix, M. Jakob and M. N. Hopkinson, Chem.–Eur. J., 2019, 25, 7635–7639 CrossRef CAS PubMed.
- A. Ariamajd, N. J. Gerwien, B. Schwabe, S. Dix and M. N. Hopkinson, Beilstein J. Org. Chem., 2021, 17, 83–88 CrossRef CAS PubMed.
- M. Tironi, L. M. Maas, A. Garg, S. Dix, J. P. Götze and M. N. Hopkinson, Org. Lett., 2020, 22, 8925–8930 CrossRef CAS PubMed.
- X. H. Yang, D. Chang, R. Zhao and L. Shi, Asian J. Org. Chem., 2021, 10, 61–73 CrossRef CAS.
- S. Wu, T. H. Jiang and C. P. Zhang, Org. Lett., 2020, 22, 6016–6020 CrossRef CAS PubMed.
- V. Amani, M. Zakeri and R. Ahmadi, Iran. J. Chem. Chem. Eng., 2020, 39, 113–122 Search PubMed.
- S. J. S. J. Tabatabaei Rezaei, H. Khorramabadi, A. Hesami, A. Ramazani, V. Amani and R. Ahmadi, Ind. Eng. Chem. Res., 2017, 56, 12256–12266 CrossRef CAS.
- R. Ahmadi and M. R. R. Jalali Sarvestani, Russ. J. Phys. Chem. B, 2020, 14, 198–208 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |