
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConvergent synthesis of 2-thioether-substituted (N)-methanocarba-adenosines as purine receptor agonists†
R. Rama Suresh a,
Russell B. Poeb,
Baorui Linc,
Kexin Lvc,
Ryan G. Campbella,
Zhan-Guo Gaoa,
Theodore E. Listonb,
Kiran S. Totia and
Kenneth A. Jacobson*a
a,
Russell B. Poeb,
Baorui Linc,
Kexin Lvc,
Ryan G. Campbella,
Zhan-Guo Gaoa,
Theodore E. Listonb,
Kiran S. Totia and
Kenneth A. Jacobson*a
aMolecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bldg. 8A, Rm. B1A-19, Bethesda, MD 20892-0810, USA. E-mail: kennethj@niddk.nih.gov; Fax: +1-301-480-8422; Tel: +1-301-496-9024
bAstrocyte Pharmaceuticals, Cambridge, MA 02142, USA
cWuXi Apptec (Tianjin) Co., Ltd, No. 168 Nanhai Road, TEDA, Tianjin, China
First published on 11th August 2021
Abstract
A linear route has been used to prepare (N)-methanocarba-nucleoside derivatives, which serve as purine receptor ligands having a pre-established, receptor-preferred conformation. To introduce this rigid ribose substitute, a Mitsunobu reaction of a [3.1.0]bicyclohexane 5′-trityl intermediate 3 with a nucleobase is typically followed by functional group modifications. We herein report an efficient scalable convergent synthesis for 2-substituted (N)-methanocarba-adenosines, which were demonstrated to bind to the A3 adenosine receptor. The adenine moiety was pre-functionalized with 2-thioethers and other groups before coupling to the bicyclic precursor (3) as a key step to facilitate a high yield Mitsunobu product. This new approach provided the (N)-methanocarba-adenosines in moderate to good yield, which effectively increased the overall yield compared to a linear synthesis and conserved a key intermediate 3 (a product of nine sequential steps). The generality of this convergent synthesis, which is suitable as an optimized preclinical synthetic route, was demonstrated with various 2-thioether and 2-methoxy substituents.
Introduction
Nucleoside derivatives are used widely in a therapeutic capacity in cancer, infectious diseases and other conditions.1,2 One approach to increase the specificity of action of nucleosides and nucleotides is to constrain the ribose ring in a preformed conformation that is complementary to the requirements at a target biopolymer, such as an enzyme or receptor protein. For example, the introduction of a North (N)-methanocarba ([3.1.0]bicyclohexane) ring system in place of the tetrahydrofuryl group of native ribose lowers the energy barrier in binding at a biological target resulting in increased affinity and selectivity,3–6 e.g. of nucleosides at the A3 adenosine receptor (AR) or of nucleotides at the P2Y1 receptor (P2Y1R). Substitution at the adenine C2 position with secondary amines, ethers, thioethers or alkynes is of particular interest in biological studies at purinergic receptors. For example, adenosine 2-thioethers in the native ribose series displayed enhanced AR affinity, and an adenine 2-methylthio group is a favored substitution in various P2 receptor ligands.7,8 Also of note are 2-methylthio nucleotide derivatives that act as potent P2YR agonists, including selective P2Y1R agonist MRS2365 1 (Ki 0.4 nM), which is a (N)-methanocarba analogue of 2-methylthioadenosine 5′-diphosphate.6 Similarly, among AR ligands the potent A3AR agonist MRS3611 2 (Ki 1.5 nM) is an (N)-methanocarba analogue having a 2-methylthio substitution (Chart 1).5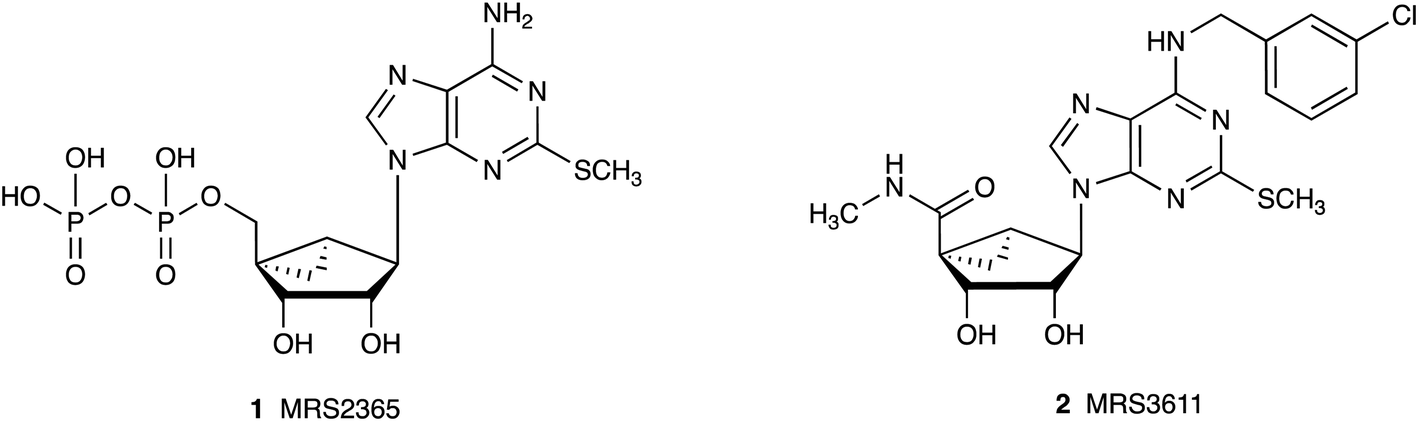 | ||
| Chart 1 Structures of potent and selective purinergic agonists (1, P2Y1R; 2, A3AR) that combine a (N)-methanocarba modification of ribose with an adenine 2-thioether. | ||
Although (N)-methanocarba nucleosides have broad applications as ligands for various G protein-coupled receptors (GPCRs) and enzymatic targets,9,10 the conventional synthetic routes involve many linear steps and the overall final yield is typically <1% from readily available starting materials such as D-ribose.9,11–14 Thus, it is of interest to identify more efficient synthetic approaches that might be adaptable to pharmaceutical development. Here, we compared a linear synthesis of 2-methylthio-(N)-methanocarba-adenosine (MRS4322, an A3AR agonist with cerebroprotective efficacy)15,16 with a convergent approach that is designed to increase overall yield and to optimally use the precious [3.1.0]bicyclohexane intermediate. We investigated the generality and scalability of the convergent route to the synthesis of (N)-methanocarba-adenosine derivatives having other C2 position substitution.
Results and discussion
Linear route
Typically, a linear route (Scheme 1, from key intermediate 3 via 5–7) is used to prepare appropriately functionalized (N)-methanocarba-adenosine derivatives, including those containing a 2-alkylthio group, e.g. 2-methylthio derivative 8a.9,12,13 The final 2-alkylthioadenine nucleosides and related nucleotides are designed to activate purine receptors.5,6 The synthesis features a protected bicyclic intermediate, shown here as a 5′-trityl protected derivative 3, which is a precursor of the pseudoribose moiety. Most commonly, a tert-butyldimethylsilyl (TBDMS) ether protection of the 5′-hydroxyl group of the intermediate analogous to 3 has been used.13 However, this precursor and subsequent nucleoside intermediates are sticky materials, which are difficult to handle for large scale production in process chemistry.13 Here, we have used a 5′-trityl protecting group of the corresponding bicyclic intermediates, following the routes of Choi et al. and Michel and Strazewski,12,14 which provided easily crystallizable intermediates leading to 3 in nine steps from D-ribose. As shown in Scheme 1, the nucleoside derivative 5 was obtained in a relatively low yield (42%) from the intermediate 3 by a Mitsunobu reaction. We initially prepared the AR agonist 8a (MRS4322) on a scale of 137 g with an overall yield of 1.0% after thirteen steps starting from D-ribose (7.0 kg), including 28.1% for the last four steps (Scheme 1), using this linear route with 5′-trityl protection.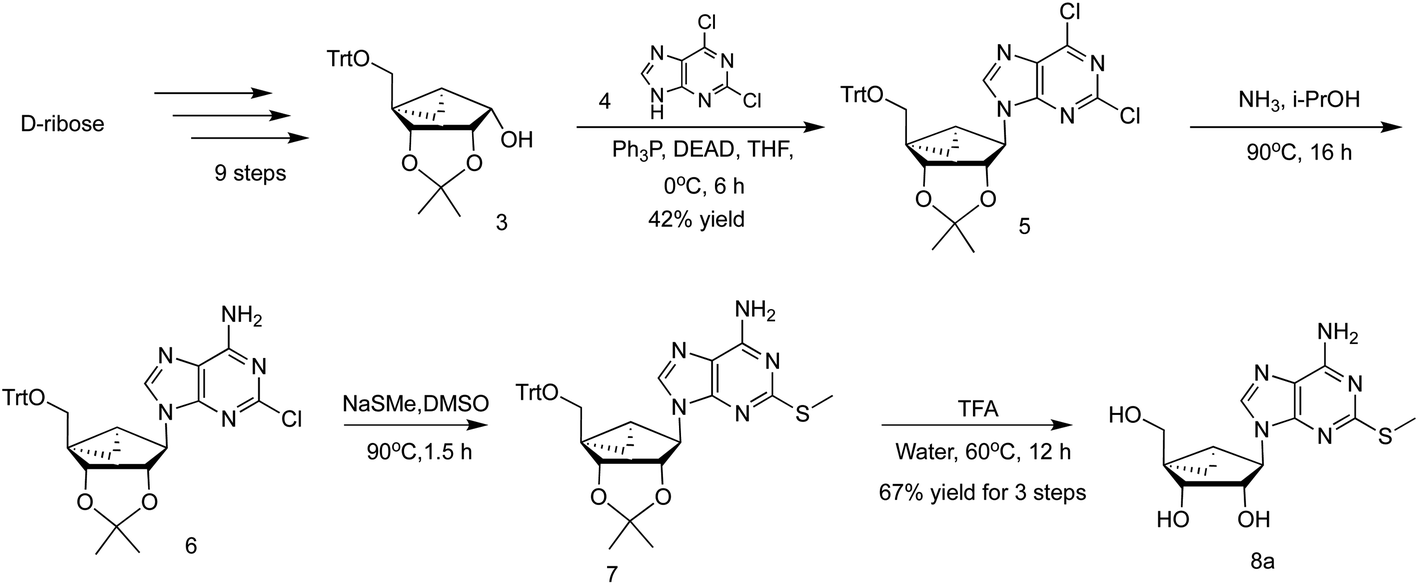 | ||
| Scheme 1 Linear route for preparation of 2-substituted (N)-methanocarba adenosine derivatives, shown here for 2-MeS-adenine analogue 8a, similar to published synthetic schemes.6,13 The intermediate 3 was prepared according to published procedures with minor modification.12,14 More synthetic details concerning the synthesis of bicyclic intermediate 3 are found in ESI (Scheme S1†). | ||
Moreover, the Mitsunobu reaction of 2,6-dichloropurine 4 with alcohols can lead to the undesired N7-regioisomer.17 There are a few reports that also mentioned the N9-alkylation of 6-chloro-2-NH-Boc adenine derivatives in good yields, using various alcohols via a Mitsunobu reaction.18,19 Therefore, we explored new methods that could successfully transform the key precursor 3 into the desired nucleoside N9-regioisomer with a 2-alkylthioadenine nucleobase.
Adenine pre-functionalization for alternative routes
As an alternative synthetic approach, rather than coupling a bicyclic intermediate (e.g. 3), either in the ribo (2′-OH) or 2′-deoxy (N)-methanocarba series, to a reactive (6-chloro or 2,6-dichloro) purine nucleobase precursor (e.g. 4),5,6,11 we pre-functionalized the purine in a manner that would facilitate high yields in a subsequent Mitsunobu step. Selective protection of the exocyclic primary amine was necessary, because earlier attempts to couple various adenine derivatives containing a free 6-NH2 through a Mitsunobu reaction failed.11 It remains unclear if this was due to the formation of phosphazane complexes20,21 with the betaine intermediate, or to the poor solubility of the 6-NH2-purine.The N6-Boc protecting group was considered for 2-chloroadenine 9 (Scheme S2†). With the goal of improving the synthetic route to adenine 2-thioether derivatives, we first prepared N6,N6-di-Boc-2-chloro-adenine 11 in two steps via tri-Boc intermediate 10, based on a literature report.14 The di-Boc protected adenine derivative obtained was used in a Mitsunobu reaction with the alcohol (3). However, it was found that only ∼10% of the coupling product 12 was observed by 1H-NMR in contrast to the quantitative synthesis of similar kind of analog under identical conditions that was reported by Michel et al.14 Other approaches to protection of the exocyclic amine of 9 were explored (e.g. 14, 15, Scheme S3, ESI†).22
Convergent route
The need for an efficient and scalable synthesis led us to seek a convergent route that envisaged the use of N6-protected forms of 2-substituted adenine derivatives 16 (Scheme 2A) as precursors for a subsequent Mitsunobu reaction to obtain the corresponding (N)-methanocarba nucleoside derivative 8a and its S-alkyl homologues. In this context, we first synthesized 2-methylthioadenine 16a in high yield (94%) by treating 2-Cl-adenine 9 with sodium methylthiolate at elevated temperature.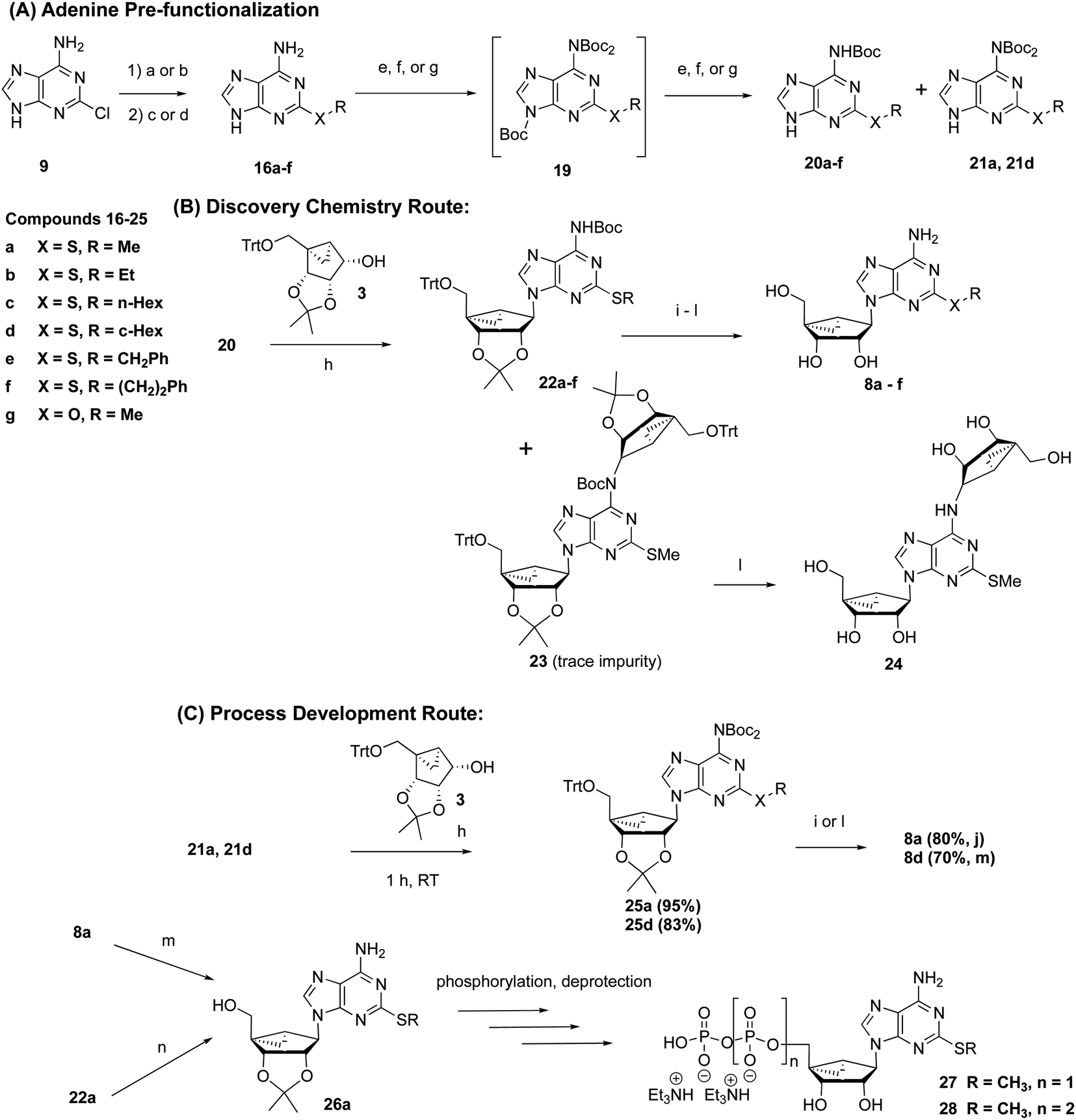 | ||
Scheme 2 General reaction scheme using the convergent route for preparation of 2-thioether-substituted (N)-methanocarba adenosine derivatives (8a–g) and nucleotides (27 and 28, Scheme S5†). The key differences from Scheme 1 are the pre-installation of a 2-thioether (A) and use of a Boc-protected exocyclic amine on adenine (B) prior to the Mitsunobu coupling with the bicyclic pseudoribose. Part (C) shows the scalable process route. An “a” designation after the compound numbers refers to R = Me. Other substituents are shown in Table S1.† Reaction conditions and yields: [a] (94%) (i) aq. NaSMe (3.0 equiv.), 140 °C, autoclave, 16 h; (ii) 6 N HCl to pH = 7–8; [b] (67%) (i) NaSMe (2.5 equiv.), DMF, 110 °C, 16 h; (ii) 6 N HCl, 60 °C, 2 h; (iii) 23% aqueous NH4OH; [c] (40–99%) RSH (5 equiv.), Cs2CO3 (3.0–3.5 equiv.), DMF, 140 °C, 1 day; [d] (75%) NaOMe (20 equiv.), MeOH, 150 °C, 4 days; [e] (28–69%) (i) Boc2O (4.0 equiv.), DMAP (0.2 equiv.), THF; (ii) aq. 10% NaOH, MeOH, 5–6 h; [f] (94%) (i) Boc2O (4.0 equiv.), DMAP (0.2 equiv.), THF; (ii) aq. NH4OH (23%), THF, 6 h; [g] (23–45%) (i) Boc2O (4.0 equiv.), DMAP (0.2 equiv.), THF; (ii) sat. NaHCO3, MeOH–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 60 °C, 5–16 h; [h] (67–94%) 20 or 21 (1.1–1.2 equiv.), alcohol 3 (1.0 equiv.) PPh3 (1.5–2.0 equiv.), DIAD (1.5–2.0 equiv.), THF (∼0.1 M), 1–2 h; [i] (59–70%) (i) aq. 4 N HCl or 4 N HCl (g)/MeOH, 35 °C, 16 h; (ii) Na2CO3, MeOH/H2O. [j] (88%) 1 N HCl in H2O, 50 °C, 18 h; (ii) Amberlite resin-93, MeOH, 16 h; [k] (46–75%) aq. 4 N HCl in MeOH or EtOH, 35 °C, 16 h; (ii) Amberlite resin-93, MeOH, 16 h; [l] (45%) aq. TFA in MeOH 50 °C, 17 h; (ii) Amberlite resin-93, MeOH, 16 h; [m] (variable yield, see ESI†) anhyd. acetone, 2,2-dimethoxypropane, p-TSA, room temperature, 18 h. [n] (58%) anhyd. acetone-TFA (1:2), room temperature, 3 h or anhyd. ZnBr2, DCM, 10–20 min. :1), 60 °C, 5–16 h; [h] (67–94%) 20 or 21 (1.1–1.2 equiv.), alcohol 3 (1.0 equiv.) PPh3 (1.5–2.0 equiv.), DIAD (1.5–2.0 equiv.), THF (∼0.1 M), 1–2 h; [i] (59–70%) (i) aq. 4 N HCl or 4 N HCl (g)/MeOH, 35 °C, 16 h; (ii) Na2CO3, MeOH/H2O. [j] (88%) 1 N HCl in H2O, 50 °C, 18 h; (ii) Amberlite resin-93, MeOH, 16 h; [k] (46–75%) aq. 4 N HCl in MeOH or EtOH, 35 °C, 16 h; (ii) Amberlite resin-93, MeOH, 16 h; [l] (45%) aq. TFA in MeOH 50 °C, 17 h; (ii) Amberlite resin-93, MeOH, 16 h; [m] (variable yield, see ESI†) anhyd. acetone, 2,2-dimethoxypropane, p-TSA, room temperature, 18 h. [n] (58%) anhyd. acetone-TFA (1:2), room temperature, 3 h or anhyd. ZnBr2, DCM, 10–20 min. | ||
Consequently, we were curious to know the relative reactivity of N6-Boc-protected 2-MeS-adenine 20a with the alcohol 3 in a Mitsunobu reaction (Scheme 2B). The 2-thioether 16a was first Boc-protected with excess Boc-anhydride to yield a mixture of N-tert-butoxycarbonyladenine intermediates, 19a (N6,N6,N9-tri-tert-butoxycarbonyladenine) and 21a (N6,N6-di-tert-butoxycarbonyladenine). The corresponding tri-Boc derivative 19a formed during the reaction was largely cleaved in mild basic conditions to the N6-mono-Boc derivative 20a. The di-Boc intermediate 21a was found to be less stable than mono-Boc 20a, as it gradually decomposed, even upon long-term storage as a solid at room temperature, to mono-Boc 20a as indicated by TLC. The phthaloyl group (e.g. 17, Scheme S4, ESI†) was also considered for the amino-protection of 16a, but Boc protection was more successful.22,23
The Mitsunobu reaction with either mono-Boc 20a or di-Boc 21a nucleobase (Schemes 2B and C) with a 5′-O-trityl bicyclic intermediate 3 proceeded in high yield (94%) to provide only N9-regioisomers 22a (Table S1†) or 25a, respectively. Following acidic deprotection of 22a or 25a, nucleoside 8a was obtained, and this step to remove three protecting groups simultaneously proceeded in high yield (88% at 50 °C). The isolated mono-Boc intermediate 20a contained a small amount of unprotected 16a as an impurity, which was problematic for the purity of subsequent steps leading to 8a. The presence of a small amount of the di-Boc compound 21a in the mono-Boc intermediate 20a was not detrimental during the Mitsunobu reaction, because its Mitsunobu product (25a) was later deprotected to yield the same product 8a. However, we preferred the di-Boc approach (Scheme 2C) for the scalable process development, as the mono-Boc route (Scheme 2B) produced a bis-alkylated impurity (24) via bis-adduct (23), which was inseparable from the desired product. The purging of 24 was unsuccessful by crystallization after global deprotection since the product (8a) and 24 have the same polarity characteristics [(24), analytical HPLC: retention time 6.89, 466 (m/z). Retention time for MRS4322 (8a): 6.64, 324 (m/z)].
For subsequent 5′-phosphorylation, compound 8a could be reprotected with a 2′,3′-isopropylidene group to provide 26a, which was then phosphorylated (and subsequently deprotected) to yield high potency P2Y1R agonists, e.g. 27 and 28.6 Although our published method for the synthesis of 27 used benzoyl peroxide as oxidant in the phosphitylation reaction,6 we found that use of H2O2 resulted in less undesired thioether oxidation. Alternatively, the trityl group and Boc protection of 22a were removed simultaneously using ZnBr2 to yield 26a directly.24
The overall yield of 8a on a scale of 520 g from 3 using Scheme 2B was 60%. The outstanding advantage of the convergent route is that it spares the precious intermediate 3, which itself required nine steps from D-ribose to prepare. We calculate that the linear synthesis required 540 g of 3 per 100 g of 8a by the linear route compared to 230 g of 3 per 100 g of 8a for the convergent route. Thus, the molar ratio of the key precursor 3 in the convergent route was reduced by 57% compared to the linear synthesis.
To test the applicability of this approach to other 2-thioether substituents, 2-Cl-adenine (9) was treated with various alkyl thiols, aryl–alkyl thiols, and the corresponding sodium salt to provide 2-thioethers of general formula 16b–16f,25 selected based upon the commercial availability of the thiols (Scheme 2). The 2-thioethers were then mono-Boc-protected (20b–20f), as with 2-methylthioadenine, and subjected to a Mitsunobu reaction with the alcohol (3) to produce the desired products 22b–22f in excellent yields (Table S1†). For the selective deprotection of tri-Boc intermediate (19a), we screened several conditions for the scale-up process, and we found that basic conditions consisting of aq. sodium hydroxide (NaOH) in MeOH gave 20a in good yield (62% from 16a). The yields varied with this combination for other examples (20f, 20g and 21d). For example, the yield of 20f with aq. NaOH in MeOH was 28%. However, we admit that these conditions were not optimized, as our principal objective was to obtain the corresponding product (20) in a reasonable or modest yield for examining the Mitsunobu reaction. Thus, only the conditions using sodium bicarbonate or aq. ammonia, which resulted in satisfactory yields, were shown in the Table S1.† In the case of cyclohexylthio derivative 8d, the isolation of pure product was difficult from the corresponding mono-Boc adduct 22d even by HPLC. However, we were able to obtain homogeneous 8d via the di-Boc intermediate 25d (Scheme 2C).
Additionally, to evaluate the generality of our methodology, we prepared 2-methoxyadenine (16g), which was subjected to the same reaction sequence with similar results as the 2-thioethers (Scheme 2). A Mitsunobu reaction of 2-MeO-adenine (16g) with the alcohol 3 proceeded well in moderate yield (67%). It is noteworthy that we achieved improved yields since the reactivity of N9 of the N6-mono-Boc-adenine derivatives 20 towards alkylation depends upon the substituent present at the C2 position. The 2-thioether derivatives typically resulted in 77–95% yield in the Mitsunobu reaction (Table S1†). The low reactivity of 2-halo-substituted adenine (e.g. Cl and I,13 Scheme S2, ESI†) is probably due to decreased electron density of the purine ring. However, the convergent synthetic approach has fewer synthetic steps after the Mitsunobu reaction than the linear synthesis to produce 12 (Scheme S2, ESI†) and the corresponding deprotected product. Thus, this convergent approach is still sparing of the precious intermediate alcohol 3. Activating/electron donating groups such as S-alkyl and O-alkyl likely increased the nucleophilic character of the purine nitrogen atoms to enhance yields. Thus, use of a 2-alkyloxy adenine precursor instead of 2-alkylthio is expected to be suitable for this synthetic approach.
Pharmacological evaluation
The 2-substituted (N)-methanocarba-adenosine analogues 8a–8g were evaluated in a standard radiolabeled A3AR agonist binding assay (Table 1). All of the analogues exceeded the affinity of the parent 8a, containing a 2-methylthio substituent. The highest affinity was observed for the S-benzyl derivative 8e, with a Ki value of 49.8 nM, i.e. 30-fold higher affinity than 8a. A3AR agonists are under development for treatment of ischemic and inflammatory conditions.16,26,27 We note that 2-alkylthio modifications of adenosine have already been well explored for receptor affinity in the ribose series,30 but not in the (N)-methanocarba series. The receptor affinity of 2-alkyloxo-ether modified adenosine analogues was also studied in detail.31,32| Compound | Ki (nM) |
|---|---|
| a Binding in membranes of HEK293 cells stably expressing hA3AR,16 using [125I]N6-(4-amino-3-iodobenzyl)-adenosine-5′-N-methyluronamide ([125I]I-AB-MECA, 0.1 nM) as radioligand.b As reported in Liston et al.16 | |
| 8a | 1490 ± 410b |
| 8b | 970 ± 60 |
| 8c | 548 ± 44 |
| 8d | 1080 ± 90 |
| 8e | 49.8 ± 5.5 |
| 8f | 291 ± 89 |
| 8g | 1140 ± 84 |
Conclusions
We conclude that the most effective route is to install a 2-thioether or 2-ether group on the adenine precursor prior to N6 protection and subsequent Mitsunobu coupling to the pseudoribose ((N)-methanocarba) moiety. Thus, we have identified a di-Boc-protected 2-methylthioadenine intermediate 21a that facilitates an efficient convergent synthetic route to known purine receptor ligands and is applicable to exploration of novel SAR of (N)-methanocarba derivatives at adenosine and P2Y receptors. This convergent synthesis of 2-substituted (N)-methanocarba-adenosines effectively increased the overall yield of the sequence compared to the previously reported linear synthesis. In particular, the quantity of the precious intermediate alcohol 3 needed for the convergent reaction scheme was reduced (by 57% for 8a) compared to the linear route. In order to optimize the convergent route, we compared various adenine amine protecting groups and found that N6-di-Boc protection was the most versatile and provided the most easily purified intermediates. The thioether extended analogues prepared by the convergent route displayed enhanced receptor affinity. In summary, we have developed a facile convergent route, suitable for preclinical development, for the synthesis of (N)-methanocarba-nucleoside derivatives. This route allows for a variety of 2-substitutions to aid in structure activity studies. These bicyclic nucleoside derivatives serve as purine receptor ligands with a pre-established receptor-preferred conformation and display efficacy in various disease models.Experimental section
Chemical synthesis
2-Chloroadenine (9) was purchased from Ark Pharm (Arlington Heights, IL, USA). All other reagents were from Sigma-Aldrich (St. Louis, MO, USA). 1H-NMR spectra were obtained with a Bruker 400 MHz spectrometer in CDCl3 (7.26 ppm), CD3OD (HOD = 4.87 ppm) or in (CD3)2SO (1H = 2.50 ppm and 13C = 39.52 ppm). The chemical shifts are expressed as ppm downfield and coupling constants (J) are given in Hz. TLC analysis was carried out on glass sheets precoated with silica gel F254 (0.2 mm) from Sigma-Aldrich. The purity of final nucleoside derivatives was checked using a Hewlett–Packard 1100 HPLC equipped with an Eclipse 5 μm XDB-C18 analytical column (50 mm × 4.6 mm; Agilent Technologies Inc., Palo Alto, CA). Mobile phase: linear gradient solvent system, 10 mM TEAA (triethylammonium acetate):CH3CN from 95:5 to 0:100 in 20 min; the flow rate was 1.0 mL min−1. Peaks were detected by UV absorption with a diode array detector at 230, 254, and 280 nm. All derivatives tested for biological activity showed >95% purity in the HPLC systems. Low-resolution mass spectrometry was performed with a JEOL SX102 spectrometer with 6 kV Xe atoms following desorption from a glycerol matrix or on an Agilent LC/MS 1100 MSD, with an Atlantis C18 column (Waters Corp., Milford, MA, USA). High resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine, unless noted. Mass accuracies were observed.
General procedure for the synthesis of adenine 2-thioether derivatives (16a–16f)
1H NMR (400 MHz, DMSO-d6) δ 12.76 (s, 1H), 7.97 (s, 1H), 7.18 (s, 2H), 2.44 (s, 3H).
Yield: 67%; 3.80 g of 16a from 5.30 g of 2-chloroadenine (9).
Large scale yield for step 1 leading to 520 g 8a, using conditions (a) 94%; 500 g of 16a from 500 g of 2-chloroadenine (9). See ESI.†
Compounds 16b–16gc were prepared by this method with slight modifications.
2-(Ethylthio)-9H-purin-6-amine, 16b. Compound 16b was prepared from 2-chloroadenine (510 mg, 3.0 mmol) (9) and 10.0 equiv. of ethanethiol in DMF (∼0.4 M). Yield: 99%; 580 mg.
1H NMR (400 MHz, DMSO-d6) δ 12.75 (s, 1H), 7.94 (s, 1H), 7.17 (s, 2H), 3.04 (q, J = 7.3 Hz, 2H), 1.30 (t, J = 7.3 Hz, 3H).
13C NMR (100 MHz, DMSO-d6) δ 164.38, 155.28, 152.65, 139.47, 115.54, 25.08, 15.43.
HRMS (ESI) m/z: [M + H]+ calculated for C7H10N532S: 196.0657; found 196.0655.
2-(Hexylthio)-9H-purin-6-amine, 16c. Compound 16c was prepared from 2-chloroadenine (340 mg, 2.0 mmol) (9) and 5.0 equiv. of n-hexanethiol in DMF (∼0.4 M). Yield: 40%; 200 mg.
1H NMR (400 MHz, DMSO-d6) δ 12.65 (s, 1H), 7.96 (s, 1H), 7.15 (s, 2H), 3.05 (t, J = 7.2 Hz, 2H), 1.63 (p, J = 7.3 Hz, 2H), 1.39 (t, J = 7.5 Hz, 2H), 1.33–1.13 (m, 4H), 0.92–0.76 (m, 3H).
13C NMR (100 MHz, DMSO) δ 163.18, 154.87, 151.86, 138.22, 115.25, 30.85, 29.93, 29.07, 27.99, 22.00, 13.85.
HRMS (ESI) m/z: [M + H]+ calculated for C11H17N5S: 182.0930; found 182.0936.
2-(Cyclohexylthio)-9H-purin-6-amine, 16d. Compound 16d was prepared from 2-chloroadenine (340 mg, 2.0 mmol) (9) and 6.0 equiv. of n-hexanethiol in DMF (∼0.4 M). Yield: 60%; 298 mg.
1H NMR (400 MHz, DMSO-d6) δ 7.95 (s, 1H), 7.13 (s, 2H), 3.70 (h, J = 4.4, 3.9 Hz, 1H), 2.05 (dd, J = 9.9, 4.7 Hz, 2H), 1.70 (dt, J = 10.0, 4.7 Hz, 2H), 1.58 (d, J = 12.4 Hz, 1H), 1.39 (q, J = 8.3, 6.4 Hz, 4H), 1.29–1.18 (m, 1H).
13C NMR (100 MHz, DMSO) δ 162.94, 154.92, 151.96, 138.30, 115.33, 42.13, 32.84, 25.60, 25.29.
HRMS (ESI) m/z: [M + H]+ calculated for C11H16N532S: found 250.1128.
2-(Benzylthio)-9H-purin-6-amine, 16e. Compound 16e was prepared from 2-chloroadenine (510 mg, 3.0 mmol) (9) and 5.0 equiv. of benzyl mercaptan in DMF (∼0.4 M). Yield: 80%; 620 mg.
1H NMR (400 MHz, DMSO-d6) δ 12.76 (s, 1H), 7.98 (s, 1H), 7.49–7.37 (m, 2H), 7.36–7.12 (m, 5H), 4.35 (s, 2H).
13C NMR (100 MHz, DMSO) δ 162.69, 155.39, 150.91, 138.67, 137.91, 128.90, 128.27, 126.77, 116.37, 34.16.
HRMS (ESI) m/z: [M + H]+ calculated for C12H12N532S: 258.0813; found 258.0804.
2-(Phenethylthio)-9H-purin-6-amine, 16f. Compound 16f was prepared from 2-chloroadenine (510 mg, 3.0 mmol) (9) and 5.0 equiv. of 2-phenylethanethiol in (∼0.4 M). Yield: 87%; 710 mg.
1H NMR (400 MHz, DMSO-d6) δ 7.98 (d, J = 1.9 Hz, 1H), 7.31 (d, J = 4.4 Hz, 4H), 7.21 (dt, J = 8.0, 4.0 Hz, 3H), 3.29 (dd, J = 9.2, 6.3 Hz, 2H), 2.96 (dd, J = 9.1, 6.4 Hz, 2H).
13C NMR (100 MHz, DMSO) δ 162.79, 155.00, 152.10, 140.72, 138.50, 128.61, 128.30, 126.15, 115.45, 35.40, 31.50.
HRMS (ESI) m/z: [M + H]+ calculated for C13H14N532S: 272.0970; found 272.0968.
2-Methoxy-9H-purin-6-amine, 16g. Compound 16g was prepared according to a published report.29 Yield: 75% (710 mg) from (510 mg, 3.0 mmol) (9) and 20 equiv. of NaOMe in MeOH (∼0.3 M) for 4 days at 150 °C in a sealed tube.
1H NMR (400 MHz, DMSO-d6) δ 12.57 (s, 1H), 7.90 (s, 1H), 7.10 (s, 2H), 3.78 (s, 3H).
HRMS (ESI) m/z: [M + H]+ calculated for C6H8N5O: 166.0729; found 166.0728.
Synthesis of mono-Boc-2-MeS-adenine derivatives
1H-NMR (400 MHz, chloroform-d) δ 11.31 (s, 1H), 8.19 (s, 1H), 7.75 (s, 1H), 2.62 (s, 3H), 1.54 (s, 9H).
13C NMR (100 MHz, DMSO) δ 162.79, 155.00, 152.10, 140.72, 138.50, 128.61, 128.30, 126.15, 115.45, 35.40, 31.50.
ESMS calculated for C22H24N5O7: (M + H) 470.2, found 470.2.
Large scale yield for step 3 leading to 520 g 8a, using conditions (e) 62%, 180 g of 20a from 500 g of 19 using conditions [e]. See ESI.†
For compounds 20b–20g, 0.560–1.17 mmol of the 16b–16g was used; all other amounts were as given for the preparation of 8a.
Typically, to a stirred solution of the compound 16b (195 mg, 1.0 mmol, 1.0 equiv.) in THF (5 mL, ∼0.2 M) were added Boc2O (875 mg, 4.0 mmol, 4.0 equiv.) and DMAP (24 mg, 0.2 equiv.) and the mixture was stirred overnight at room temperature. The solvent (THF) was removed under reduced pressure by rotary evaporation and the crude was dissolved in ethyl acetate (EtOAc, 15 mL). The solution was washed with water (2 × 20 mL), separated the EtOAc layer, and concentrated to afford the crude oil, which was used directly for the next step without further purification. The obtained crude oil was dissolved in MeOH (3 mL) and added aq. 10% NaOH (3 mL). The reaction mixture was stirred for 5 h at room temperature. The reaction mixture was diluted with EtOAc (20 mL) and neutralized with 10% solution of sodium dihydrogen phosphate (NaH2PO4·H2O) until pH becomes 7–7.5. The phases were separated, and the aqueous phase was extracted with EtOAc (2 × 20 mL). The combined EtOAc layer was dried over Na2SO4 and filtered. The filtrate was concentrated by rotary evaporation to obtain an oily residue, which was purified by silica column to get a white solid. Eluent: 30–50% EtOAc in hexane. Yield: 58%; 170 mg.
1H NMR (400 MHz, chloroform-d) δ 8.30 (s, 1H), 8.27 (s, 1H), 3.15 (q, J = 7.4 Hz, 2H), 1.46 (s, 9H), 1.32 (t, J = 7.3 Hz, 4H).
13C NMR (100 MHz, chloroform-d) δ 164.55, 162.42, 152.63, 144.34, 143.74, 109.95, 83.24, 28.03, 25.26, 14.56.
HRMS (ESI) m/z: [M + H]+ calculated for C12H18N5O232S: 296.1185; found 296.1181.
:1, ∼0.2 M) under room temperature for 5 h. Yield: 60%; 130 mg.1H NMR (400 MHz, chloroform-d) δ 8.22 (s, 1H), 8.03 (s, 1H), 3.20 (t, J = 7.3 Hz, 2H), 1.70 (q, J = 7.4 Hz, 2H), 1.50 (s, 9H), 1.41 (t, J = 7.8 Hz, 2H), 1.31–1.19 (m, 4H), 0.84 (t, J = 8.0 Hz, 3H).
13C NMR (100 MHz, chloroform-d) δ 164.96, 163.26, 152.81, 143.70, 143.56, 109.35, 83.66, 31.51, 31.12, 29.25, 28.69, 28.13, 22.62, 14.09.
ESMS calculated for C16H25N5O2S: (M + H) 352.2; found 352.2.
To a stirred solution of the compound 16d (100 mg, 0.401 mmol, 1.0 equiv.) in THF (2 mL, ∼0.2 M) were added Boc2O (350 mg, 4.0 mmol, 4.0 equiv.) and DMAP (10 mg, 0.2 equiv.). The reaction mixture was stirred overnight at room temperature. The solvent (THF) was removed under reduced pressure by rotary evaporation and the crude was dissolved in ethyl acetate (EtOAc, 15 mL). The solution was washed with water (2 × 20 mL), separated the EtOAc layer, and concentrated to afford the crude product, which was used directly for the next step without further purification. The obtained crude product was dissolved in THF (5 mL) and added 10% NH4OH (5 mL). The reaction mixture was stirred for 40 h at room temperature. The reaction mixture was neutralized with 4.0 N HCl solution until pH 7.0–7.5. The phases were separated, and the aqueous phase was extracted with EtOAc (2 × 20 mL). The combined EtOAc layer was dried over Na2SO4 and filtered. The filtrate was concentrated by rotary evaporation to obtain an oily residue, which was purified by silica column to get a white solid. Eluent: 30–40% EtOAc in hexane. Yield: 94%; 170 mg.
1H NMR (400 MHz, chloroform-d) δ 11.33 (s, 1H), 8.37 (s, 1H), 3.95–3.75 (m, 1H), 2.13–2.08 (m, 2H), 1.75–1.70 (m, 2H), 1.59–1.50 (m, 1H), 1.50–1.34 (m, 22H), 1.28–1.21 (m, 1H).
13C NMR (100 MHz, chloroform-d) δ 164.79, 157.62, 150.07, 147.32, 143.27, 119.86, 84.42, 43.74, 32.90, 27.73, 25.92, 25.70.
ESMS calculated for C21H32N5O4S: (M + H) 450.2; found 450.3.
:1, ∼0.1 M) under room temperature for 6 h. Yield of 20e: 69%; 107 mg from 200 mg of 21e.1H NMR (400 MHz, chloroform-d) δ 8.35 (s, 1H), 8.11 (s, 1H), 7.48–7.27 (m, 5H), 4.66 (s, 2H), 1.68 (d, J = 1.3 Hz, 9H).
13C NMR (100 MHz, chloroform-d) δ 164.44, 161.95, 152.63, 144.20, 143.49, 137.71, 129.30, 128.57, 127.24, 109.67, 84.00, 35.65, 28.17.
HRMS (ESI) m/z: [M + H]+ calculated for C17H20N5O2S: 358.1338; found 357.1340.
:1, ∼0.2 M) under room temperature for 5 h. Yield: 28%; 58 mg.Using the conditions [g] on a (326 mg, 1.201 mmol) scale. Step (i) in THF (6 mL, 0.2 M), and step (ii) sat. NaHCO3, MeOH:H2O (6 mL, 1:1, 0.2 M), 60 °C, 12 h; yield of 20f: 36%, 160 mg; yield of 21f: 131 mg, 23%.
1H NMR (400 MHz, chloroform-d) δ 8.23 (s, 1H), 7.34–7.25 (m, 4H), 7.21 (tt, J = 5.2, 3.4 Hz, 1H), 3.53–3.44 (m, 2H), 3.13–3.04 (m, 2H), 1.56 (s, 9H).
13C NMR (100 MHz, chloroform-d) δ 164.44, 162.67, 152.71, 144.03, 143.57, 140.72, 128.89, 128.53, 126.42, 109.67, 83.91, 35.86, 32.55, 28.19.
HRMS (ESI) m/z: [M + H]+ calculated for C18H22N5O232S: 372.1494; found 372.1490.
:H2O (6 mL, 1:1, 0.2 M), 60 °C, 12 h. Yield: 41%; 132 mg.1H NMR (400 MHz, DMSO-d6) δ 11.89 (s, 1H), 10.57 (s, 1H), 8.24 (s, 1H), 3.87 (s, 3H), 1.51 (s, 9H).
13C NMR (100 MHz, DMSO) δ 163.94, 161.94, 160.95, 152.76, 145.66, 108.73, 81.33, 54.25, 27.91.
HRMS (ESI) m/z: [M + H]+ calculated for C11H15N5O2H+: 266.1253; found 266.1252.
Mitsunobu reaction of 20a–20c, 20e–20g, and 21d with 5′-O-trityl bicyclic intermediate 3; typical procedure for 22a–22c, 22e–22g, and 25d
In a 25 mL round bottom flask, mono-Boc-2-MeS-adenine (20a) (300 mg, 1.07 mmol, 1.5 equiv.), alcohol 3 (315 mg, 0.711 mmol, 1.0 equiv.), and triphenylphosphine (PPh3) [373 mg, 1.42 mmol, 2.0 equiv.] were added. The contents were co-evaporated with dry toluene (3 × 5 mL) and the residue dried under vacuum for 3 h. The mixture was dissolved in dry THF (10 mL) and DIAD (280 μL, 1.42 mmol, 2.0 equiv.) was added drop wisely via syringe at room temperature under nitrogen atmosphere. The reaction mixture was stirred for 1–2 h at room temperature and monitored by TLC. The solvent (THF) was removed under reduced pressure by rotary evaporation, and the crude product was purified by silica gel column chromatography to afford the product (22a) along with di-(isopropyloxycarbonyl)hydrazine byproduct (DIAD-H2) [UV inactive; ∼13% based on the C–H proton (septet) integration of isopropoxy group by 1H-NMR], which could be observed as a yellow spot after developing the TLC with p-anisaldehyde stain. Eluent: 15–30% EtOAc in hexane. TLC: Rf ∼ 0.3 (30% EtOAc in hexane). Corrected yield by 1H-NMR: 94%, 470 mg. Purity: 87% by 1H-NMR.Large scale yield for step 4 leading to 520 g 8a: 1.8 kg, crude product was obtained as a foamy solid from 20a (769 g, 2.73 mol, 1.10 eq.) and compound 3 (1.10 kg, 2.49 mol, 1.00 eq.). See ESI.†
1H NMR (400 MHz, chloroform-d) δ 8.14 (s, 1H), 7.81–7.70 (m, 1H) 7.78 (s, 1H), 7.43–7.37 (m, 6H), 7.33–7.13 (m, 9H), 5.38–5.29 (m, 1H), 5.07 (d, J = 3.2 Hz, 1H), 4.63 (dd, J = 7.2, 1.6 Hz, 1H), 3.76 (d, J = 10.0 Hz, 1H), 3.04 (d, J = 10.0 Hz, 1H), 2.56 (s, 3H), 1.55 (s, 9H), 1.53 (s, 3H), 1.25 (s, 4H), 1.16 (t, J = 5.0 Hz, 1H), 0.94–0.85 (m, 1H).
13C NMR (100 MHz, chloroform-d) δ 166.67, 151.72, 149.75, 149.28, 143.84, 139.86, 128.76, 128.06, 127.29, 119.27, 112.49, 88.85, 87.01, 82.17, 81.81, 64.82, 59.02, 37.39, 30.71, 28.33, 27.91, 26.09, 24.47, 22.08, 21.97, 14.86, 13.19.
ESMS (ESI) m/z: [M + H]+ calculated for C40H44N5O5S: 706.3; found 706.3.
Compounds 22a–22c, 22e–22g, and 25d
1H NMR (400 MHz, chloroform-d) δ 8.12 (s, 1H), 7.77 (s, 1H), 7.38 (m, 6H), 7.23 (m, 9H), 5.29 (d, J = 6.9 Hz, 1H), 5.04 (d, J = 3.6 Hz, 1H), 4.60 (d, J = 7.0 Hz, 1H), 3.73 (dd, J = 10.2, 3.5 Hz, 1H), 3.14 (ddd, J = 14.0, 8.8, 5.2 Hz, 2H), 3.03 (dd, J = 10.2, 3.4 Hz, 1H), 1.53 (s, 9H), 1.50 (s, 3H), 1.36 (t, J = 7.3 Hz, 3H), 1.23 (m, 4H), 1.14 (m, 1H), 0.91–0.86 (m, 1H). Product was contaminated with hydrazine impurity (∼9%).
13C NMR (100 MHz, chloroform-d) δ 166.27, 151.69, 149.68, 149.28, 143.81, 139.66, 128.71, 128.00, 127.24, 119.12, 112.42, 88.78, 86.98, 82.08, 81.75, 64.82, 59.00, 37.32, 30.54, 28.27, 26.04, 25.76, 24.40, 14.52, 13.15.
HRMS (ESI) m/z: [M + H]+ calculated for C41H46N5O532S: 720.3220; found 720.3212.
1H NMR (400 MHz, chloroform-d) δ 8.16 (s, 1H), 7.88 (s, 1H), 7.44–7.42 (m, 6H), 7.35–7.21 (m, 9H), 5.35–5.33 (m, 1H), 5.08 (s, 1H), 4.63 (dd, J = 7.1, 1.5 Hz, 1H), 3.79 (d, J = 10.0 Hz, 1H), 3.25–3.12 (m, 2H), 3.06 (d, J = 10.0 Hz, 1H), 1.56 (s, 9H), 1.54 (s, 4H), 1.48–1.45 (m, 2H), 1.33 (q, J = 3.7 Hz, 4H), 1.28 (s, 2H), 1.26 (s, 3H), 1.18 (t, J = 5.0 Hz, 1H), 0.92–0.89 (m, 4H).
Product was contaminated with hydrazine impurity (∼26%).
13C NMR (100 MHz, chloroform-d) δ 166.35, 151.68, 149.66, 149.26, 143.76, 139.62, 128.66, 127.96, 127.18, 119.23, 112.35, 88.74, 86.93, 81.92, 81.65, 64.78, 58.83, 37.26, 31.50, 31.44, 30.50, 29.36, 28.66, 28.22, 25.98, 24.36, 22.64, 21.98, 14.11, 13.08.
ESMS calculated for calculated for C45H53N5O5S: 775.4; found 775.4.
1H NMR (400 MHz, chloroform-d) δ 8.24 (s, 1H), 7.44–7.32 (m, 5H), 7.28–7.10 (m, 10H), 5.29 (d, J = 7.0 Hz, 1H), 5.04 (s, 1H), 4.60 (dd, J = 7.1, 1.5 Hz, 1H), 3.78 (dd, J = 10.1, 7.0 Hz, 2H), 3.04 (d, J = 10.0 Hz, 1H), 2.10–2.07 (m, 2H), 1.77–1.67 (m, 2H), 1.61 (m, 1H), 1.51 (s, 4H), 1.45 (s, 22H), 1.26–1.17 (m, 4H), 1.17–1.11 (m, 1H), 0.90–0.86 (m, 1H). Product was contaminated with hydrazine impurity (∼10%).
13C NMR (100 MHz, chloroform-d) δ 165.35, 153.49, 150.56, 150.01, 143.77, 141.85, 128.65, 127.94, 127.15, 125.73, 112.32, 88.66, 86.97, 83.64, 81.73, 64.66, 59.11, 43.88, 37.34, 32.94, 32.88, 30.26, 27.86, 25.97, 24.33, 21.74, 14.14, 12.96, 10.93.
ESMS calculated for C50H60N5O7S: 874.4; found 874.5.
1H NMR (400 MHz, chloroform-d) δ 8.18 (s, 1H), 7.85 (s, 1H), 7.50–7.45 (m, 2H), 7.43–7.36 (m, 6H), 7.33–7.19 (m, 13H), 5.31 (d, J = 7.1 Hz, 1H), 5.10 (s, 1H), 4.60 (d, J = 7.1 Hz, 1H), 4.45 (s, 2H), 3.80 (d, J = 10.2 Hz, 1H), 3.04 (d, J = 10.1 Hz, 1H), 1.57 (s, 9H), 1.54 (s, 3H), 1.29–1.25 (m, 4H), 1.18 (t, J = 5.1 Hz, 1H), 0.94–0.87 (m, 1H). Product was contaminated with hydrazine impurity (∼10%).
13C NMR (100 MHz, chloroform-d) δ 165.56, 151.61, 149.66, 149.28, 143.79, 139.76, 138.22, 129.36, 128.68, 128.32, 127.97, 127.91, 127.20, 126.95, 119.35, 112.39, 88.82, 86.95, 82.08, 81.73, 64.82, 59.04, 37.33, 35.85, 30.49, 28.23, 26.02, 24.41, 21.99, 13.11.
HRMS (ESI) m/z: [M + H]+ calculated for C46H48N5O532S: 782.3376; found 782.3384.
1H NMR (400 MHz, chloroform-d) δ 8.19 (d, J = 1.4 Hz, 1H), 7.77 (s, 1H), 7.43–7.41 (m, 5H), 7.38–7.36 (m, 2H), 7.31–7.21 (m, 13H), 5.33 (d, J = 7.1 Hz, 1H), 5.14 (s, 1H), 4.61–4.59 (m, 1H), 3.83 (d, J = 10.1 Hz, 1H), 3.40 (qt, J = 13.6, 6.6 Hz, 2H), 3.07 (t, J = 7.9 Hz, 2H), 3.00 (d, J = 10.1 Hz, 1H), 1.57 (s, 9H), 1.54 (s, 3H), 1.33 (d, J = 6.3 Hz, 1H), 1.23 (s, 3H), 1.19 (t, J = 5.1 Hz, 1H), 0.89 (dd, J = 9.4, 5.7 Hz, 1H). Product was contaminated with hydrazine impurity (∼38%).
13C NMR (100 MHz, chloroform-d) δ 165.90, 151.67, 149.57, 149.38, 143.77, 140.94, 139.54, 128.91, 128.67, 128.37, 127.97, 127.20, 126.24, 112.38, 88.82, 86.98, 81.94, 81.55, 64.85, 58.75, 37.32, 36.37, 32.92, 30.42, 28.22, 26.01, 24.37, 13.14.
HRMS (ESI) m/z: [M + H]+ calculated for C47H50N5O532S: 796.3533; found 796.3527.
1H NMR (400 MHz, chloroform-d) δ 8.08 (s, 1H), 7.87 (s, 1H), 7.44–7.35 (m, 5H), 7.31–7.17 (m, 10H), 5.33 (d, J = 7.1 Hz, 1H), 5.02 (s, 1H), 4.66 (dd, J = 7.3, 1.5 Hz, 1H), 3.91 (s, 3H), 3.74 (d, J = 9.9 Hz, 1H), 3.07 (d, J = 9.9 Hz, 1H), 1.55 (s, 9H), 1.53 (s, 3H), 1.27 (s, 1H), 1.25 (s, 3H), 1.16 (t, J = 5.0 Hz, 1H), 0.96–0.90 (m, 1H). Product was contaminated with hydrazine impurity (∼12%).
13C NMR (100 MHz, chloroform-d) δ 162.29, 152.35, 150.88, 149.50, 143.80, 139.51, 128.69, 127.97, 127.23, 117.82, 112.40, 88.80, 86.91, 82.05, 81.91, 64.75, 59.16, 55.06, 37.34, 30.54, 28.23, 26.05, 24.40, 13.15.
HRMS (ESI) m/z: [M + H]+ calculated for C40H44N5O6: 690.3292; found 690.3297.
Compounds 8a–8f
Conditions (j): 1.0 N HCl (5.0 mL) was added to a sealed tube containing the compound (22a) (70 mg, 0.1 mmol). The suspension was stirred at 50 °C for 18 h. The solvent was removed, and the crude was co-evaporated with toluene (2 × 5 mL). The residue was dissolved in MeOH (5.0 mL) and treated with 1 mL of Amberlite resin-93 (∼1.5 g, 1.2 mmol), which was previously washed with MeOH (3 × 3 mL). The reaction mixture was stirred for 16 h. The MeOH solution was filtered, concentrated, and the crude was purified by silica gel column chromatography to get 8a. Eluent: 5% of 10% aq. NH4OH (23%) in MeOH, then 5–15% MeOH in DCM. Yield: 32 mg, 88%.Large scale yield for step 5 leading to 520 g 8a, using conditions (i).
Six individual runs (each with 315 g of 22a, 446 mmol, 1.00 eq.) were carried out to get a combined 520 g of 8a as a white solid, 1.61 mol, 60% yield for 2 steps. See ESI.†
13C NMR (100 MHz, DMSO-d6). HRMS (ESI) m/z: [M + H]+ calculated for C8H11N3O2H+: 182.0930; found 182.0936.
Compounds 8b–8f were prepared using various acidic conditions.
Aq. 4.0 N HCl in MeOH (2.0 mL) was added to a 25 mL round-bottomed flask containing the compound (22b) (30 mg, 0.042 mmol). The solution was stirred at 35 °C for 16 h. The solvent was removed, and the crude was co-evaporated with ethanol (2 × 5 mL). The residue was dissolved in MeOH (5.0 mL) and treated with 1 mL of Amberlite resin-93 (1.2 mmol), which was previously washed with MeOH (3 × 3 mL). The reaction mixture was stirred for 16 h. The MeOH solution was filtered, concentrated, and the crude was purified by silica gel column chromatography to get 8b. Eluent: 10–20% MeOH in DCM. Yield: 46%; 6.0 mg.
1H NMR (600 MHz, methanol-d4) δ 8.39 (s, 1H), 4.83 (s, 1H), 4.74 (dd, J = 6.8, 1.7 Hz, 1H), 4.24 (d, J = 11.6 Hz, 1H), 3.87 (dt, J = 6.7, 1.4 Hz, 1H), 3.31 (d, J = 11.7 Hz, 1H), 3.24–3.10 (m, 2H), 1.60 (ddd, J = 8.8, 3.9, 1.5 Hz, 1H), 1.53 (dd, J = 5.2, 4.0 Hz, 1H), 1.38 (t, J = 7.3 Hz, 3H), 0.74 (ddd, J = 8.7, 5.2, 1.7 Hz, 1H).
13C NMR (151 MHz, methanol-d4) δ 166.68, 156.78, 151.34, 139.72, 117.57, 77.75, 72.20, 64.43, 62.94, 37.82, 26.22, 24.54, 15.28, 12.30.
HRMS (ESI) m/z: [M + H]+ calculated for C14H20N5O332S: 338.1287; found 338.1293.
Aq. 4.0 N HCl (1.0 mL) was added to a 25 mL cylindrical sealed tube containing the compound (22c) (28 mg, 0.431 mmol) in EtOH (3 mL). The solution was stirred at 35 °C for 16 h. The solvent was removed, and the crude was co-evaporated with ethanol (2 × 5 mL). The residue was dissolved in MeOH (5.0 mL) and treated with 1 mL of Amberlite resin-93 (1.2 mmol), which was previously washed with MeOH (3 × 3 mL). The reaction mixture was stirred for 1 h. The MeOH solution was filtered, concentrated, and the crude was purified by preparative HPLC to get the pure 8c. Yield: 8.0 mg, 56%.
1H NMR (400 MHz, methanol-d4) δ 8.39 (s, 1H), 4.85 (s, 1H), 4.76 (d, J = 6.7 Hz, 1H), 4.25 (d, J = 11.6 Hz, 1H), 3.89 (d, J = 6.6 Hz, 1H), 3.36–3.28 (m, 1H), 3.25 (dt, J = 14.1, 7.3 Hz, 1H), 3.13 (dt, J = 13.6, 7.3 Hz, 1H), 1.75 (p, J = 7.2 Hz, 2H), 1.61 (dd, J = 9.0, 3.9 Hz, 1H), 1.54 (t, J = 4.7 Hz, 1H), 1.53–1.41 (m, 2H), 1.36 (h, J = 4.0 Hz, 4H), 0.97–0.89 (m, 3H), 0.79–0.71 (m, 1H). Compound was purified by HPLC and was contaminated with triethylammonium acetate buffer (∼17% based on NCH2 protons of the buffer at 3.03).
Prep. HPLC method: Phenomenex Luna 5 μm C18(2) 100 A, LC column (250 × 21.2 mm). Linear gradient solvent system: ACN:10 mM TEAA from 40:80 to 80:20 in 40 minutes. Rt 43.32 min.
13C NMR (100 MHz, methanol-d4) δ 157.36, 147.25, 141.87, 130.23, 108.12, 68.35, 62.77, 54.98, 53.48, 28.39, 23.15, 22.49, 21.29, 20.20, 15.15, 14.19, 4.90, 2.83.
HRMS (ESI) m/z: [M + H]+ calculated for C18H26N5O332S: 394.1913; found 394.1920.
Aq. 4.0 N HCl in MeOH (2.0 mL) was added to a 25 mL round-bottomed flask containing the compound (25d) (96 mg, 0.1098 mmol). The solution was stirred at 35 °C for 16 h. The solvent was removed, and the crude was co-evaporated with ethanol (2 × 5 mL). The residue was dissolved in MeOH (3.0 mL) and added aq. Na2CO3 (2.2 M) until the pH of the reaction mixture becomes ∼8. The suspension was stirred at room temperature for 20 minutes. The solution was filtered and washed the precipitate with EtOH (2 × 5 mL). The combined solution was concentrated, and the crude was purified by silica gel column chromatography to get 8d. Eluent: 10% MeOH in DCM. Yield: 70%; 30.0 mg.
1H NMR (400 MHz, methanol-d4) δ 8.39 (s, 1H), 4.81 (s, 1H), 4.75 (dd, J = 6.7, 1.6 Hz, 1H), 4.25 (d, J = 11.6 Hz, 1H), 3.89 (d, J = 6.8 Hz, 2H), 3.36–3.27 (m, 1H), 2.16–2.10 (m, 2H), 1.81–1.78 (m, 2H), 1.68–1.58 (m, 2H), 1.54–1.47 (m, 5H), 1.36–1.27 (m, 2H), 0.75 (ddd, J = 8.8, 5.1, 1.7 Hz, 1H).
13C NMR (100 MHz, methanol-d4) δ 166.58, 156.78, 151.31, 139.72, 117.58, 77.79, 72.24, 64.47, 63.05, 44.73, 37.86, 34.50, 34.22, 27.16, 27.11, 26.97, 24.55, 12.32.
ESMS calculated for C18H26N5O3S: 392.2; found 392.2.
Aq. 4.0 N HCl in MeOH (2.0 mL) was added to a 25 mL round-bottomed flask containing the compound (22e) (89 mg, 0.1184 mmol). The solution was stirred at 35 °C for 16 h. The solvent was removed, and the crude was co-evaporated with ethanol (2 × 5 mL). The residue was dissolved in MeOH (5.0 mL) and treated with 1 mL of Amberlite resin-93 (1.2 mmol), which was previously washed with MeOH (3 × 3 mL). The reaction mixture was stirred for 6 h. The MeOH solution was filtered, concentrated, and the crude was purified by silica gel column chromatography to get 8e. Eluent: 5–10% MeOH in DCM. The product was crystallized on trituration with ethanol to get pure 8e. Yield: 63%; 30.0 mg.
1H NMR (600 MHz, methanol-d4) δ 8.40 (s, 1H), 7.47–7.39 (m, 2H), 7.26 (t, J = 7.7 Hz, 2H), 7.19–7.14 (m, 1H), 4.74 (dd, J = 6.6, 1.7 Hz, 1H), 4.49–4.34 (m, 2H), 4.23 (d, J = 11.6 Hz, 1H), 3.88 (dt, J = 6.5, 1.4 Hz, 1H), 3.29 (d, J = 11.7 Hz, 1H), 1.59 (ddd, J = 8.8, 3.9, 1.5 Hz, 1H), 1.54 (dd, J = 5.2, 3.9 Hz, 1H), 0.74 (ddd, J = 8.7, 5.2, 1.7 Hz, 1H). One proton peak obscured by solvent peaks at 4.87.
13C NMR (151 MHz, methanol-d4) δ 166.14, 156.73, 151.27, 139.90, 139.75, 130.19, 129.35, 127.95, 117.67, 77.76, 72.18, 64.40, 62.89, 37.80, 36.33, 24.56, 12.33.
HRMS (ESI) m/z: [M + H]+ calculated for C8H11N3O2H+: 182.0930; found 182.0936.
Aq. 4.0 N HCl in MeOH (2.0 mL) was added to a 25 mL round-bottomed flask containing the compound (22f) (92 mg, 0.116 mmol). The solution was stirred at 35 °C for 16 h. The solvent was removed, and the crude was co-evaporated with toluene (2 × 5 mL). The residue was dissolved in MeOH (5.0 mL) and treated with 1 mL of Amberlite resin-93 (1.2 mmol), which was previously washed with MeOH (3 × 3 mL). The reaction mixture was stirred for 16 h. The MeOH solution was filtered, concentrated, and the crude was purified by silica gel column chromatography to get 8f. Eluent: 5–10% MeOH in DCM. The product was crystallized on trituration with ethanol to get pure 8f. Yield: 75%; 36.0 mg.
1H NMR (500 MHz, methanol-d4) δ 8.38 (s, 1H), 7.30–7.28 (m, 2H), 7.25–7.22 (m, 2H), 7.15–7.12 (m, 1H), 4.73 (dd, J = 6.6, 1.6 Hz, 1H), 4.23 (d, J = 11.6 Hz, 1H), 3.86 (d, J = 6.6 Hz, 1H), 3.39 (ddd, J = 13.3, 8.7, 7.0 Hz, 1H), 3.33–3.22 (m, 2H), 3.00 (t, J = 7.3 Hz, 2H), 1.58 (ddd, J = 8.7, 3.9, 1.4 Hz, 1H), 1.54–1.52 (m, 1H), 0.72 (ddd, J = 8.6, 5.1, 1.7 Hz, 1H). One proton peak obscured by solvent peaks at 4.87.
13C NMR (126 MHz, methanol-d4) δ 166.41, 156.74, 151.30, 142.11, 139.62, 129.85, 129.39, 127.22, 77.74, 72.18, 64.38, 62.81, 37.81, 37.38, 33.60, 24.64, 12.30.
HRMS (ESI) m/z: [M + H]+ calculated for C20H24N5O332S: 414.1600; found 414.1607.
10% TFA in water (1.0 mL) was added to a 25 mL cylindrical sealed tube containing the compound (22g) (25 mg, 0.0362 mmol) in MeOH (5 mL). The solution was stirred at 50 °C for 17 h. The solvent was removed, and the crude was co-evaporated with ethanol (2 × 5 mL). The residue was dissolved in MeOH (5.0 mL) and treated with 1 mL of Amberlite resin-93 (1.2 mmol), which was previously washed with MeOH (3 × 3 mL). The reaction mixture was stirred for 1 h. The MeOH solution was filtered, concentrated, and the crude was the crude was purified by silica gel column chromatography to get 8e. Eluent: 15–20% MeOH in DCM. Yield: 45%; 5.0 mg.
1H NMR (400 MHz, chloroform-d) δ 8.32 (d, J = 5.8 Hz, 1H), 4.82–4.71 (m, 2H), 4.25 (dd, J = 11.8, 5.6 Hz, 1H), 3.96 (dd, J = 4.9, 2.7 Hz, 3H), 3.89 (d, J = 6.3 Hz, 1H), 3.30 (dd, J = 11.5, 5.5 Hz, 1H), 1.60 (p, J = 4.4 Hz, 1H), 1.51 (p, J = 4.7, 4.0 Hz, 1H), 0.80–0.68 (m, 1H).
13C NMR (100 MHz, methanol-d4) δ 163.78, 158.14, 152.21, 139.68, 116.31, 77.82, 72.24, 64.48, 63.28, 55.18, 37.93, 24.57, 12.22.
HRMS (ESI) m/z: [M + H]+ calculated for C13H18N5O4: 308.1359; found 308.1353.
Conflicts of interest
T. E. L. and R. B. P. are employees of Astrocyte Pharmaceuticals Inc. The other authors have no competing interest to declare.Acknowledgements
Support from the Intramural Research Program of NIDDK, National Institutes of Health (ZIADK031117) and Astrocyte Pharmaceuticals (NIH CRADA DK15-0856) is acknowledged. We thank Robert O'Connor (NIDDK) for assistance with NMR.References
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef CAS.
- K. A. Jacobson, D. K. Tosh, K. S. Toti and A. Ciancetta, Polypharmacology of conformationally locked methanocarba nucleosides, Drug Discovery Today, 2017, 22, 1782–1791 CrossRef CAS PubMed.
- V. E. Marquez, M. A. Siddiqui, A. Ezzitouni, P. L. Russ, J. Wang, R. W. Wagner and M. D. Matteucci, Nucleosides with a twist. Can fixed forms of sugar ring pucker influence biological activity in nucleosides and oligonucleotides?, J. Med. Chem., 1996, 39, 3739–3747 CrossRef CAS.
- V. E. Marquez, The properties of locked methanocarba nucleosides in biochemistry, biotechnology and medicine, in Modified Nucleosides in Biochemistry Biotechnology and Medicine, ed. P. Herdewijn, Wiley-VCH, 2008, pp. 307–341 Search PubMed.
- S. Tchilibon, B. V. Joshi, S. K. Kim, H. T. Duong, Z. G. Gao and K. A. Jacobson, (N)-Methanocarba 2, N6-disubstituted adenine nucleosides as highly potent and selective A3 adenosine receptor agonists, J. Med. Chem., 2005, 48, 1745–1758 CrossRef CAS PubMed.
- R. G. Ravi, H. S. Kim, J. Servos, H. Zimmermann, K. Lee, S. Maddileti, J. L. Boyer, T. K. Harden and K. A. Jacobson, Adenine nucleotides analogues locked in a Northern methanocarba conformation: enhanced stability and potency as P2Y1 receptor agonists, J. Med. Chem., 2002, 45, 2090–2100 CrossRef CAS.
- R. Volpini, S. Costanzi, C. Lambertucci, F. R. Portino, S. Taffi, S. Vittori, K.-N. Klotz and G. Cristalli, Adenosine receptor agonists: synthesis and binding affinity of 2-(aryl)alkylthioadenosine derivatives, ARKIVOC, 2004, 5, 301–311 Search PubMed.
- A. El-Tayeb, J. Iqbal, A. Behrenswerth, M. Romio, M. Schneider, H. Zimmermann, J. Schrader and C. E. Müller, Nucleoside-5′-monophosphates as prodrugs of adenosine A2A receptor agonists activated by ecto-5′-nucleotidase, J. Med. Chem., 2009, 52, 7669–7677 CrossRef CAS PubMed.
- V. E. Marquez, P. Russ, R. Alonso, M. A. Siddiqui, S. Hernandez, C. George, M. C. Nicklaus, F. Dai and H. Ford, Synthesis of conformationally restricted carbocyclic nucleosides: the role of the O(4)-atom in the key hydration step of adenosine deaminase, Helv. Chim. Acta, 1999, 82, 2119–2129 CrossRef CAS.
- K. A. Jacobson, D. K. Tosh, K. S. Toti and A. Ciancetta, Polypharmacology of conformationally locked methanocarba nucleosides, Drug Discovery Today, 2017, 22, 1782–1791 CrossRef CAS PubMed.
- B. V. Joshi, A. Melman, R. L. Mackman and K. A. Jacobson, Synthesis of ethyl (1S,2R,3S,4S,5S)-2,3-O-(isopropylidene)-4-hydroxy-bicyclo[3.1.0]hexane-carboxylate from L-ribose: a versatile chiral synthon for preparation of adenosine and P2 receptor ligands, Nucleosides, Nucleotides Nucleic Acids, 2008, 27, 279–291 CrossRef CAS.
- Y. Choi, H. R. Moon, Y. Yoshimura and V. E. Marquez, Recent advances in the synthesis of conformationally locked nucleosides and their success in probing the critical question of conformational preferences by their biological targets, Nucleosides, Nucleotides Nucleic Acids, 2003, 22, 547–557 CrossRef CAS.
- D. K. Tosh, J. Padia, D. Salvemini and K. A. Jacobson, Efficient, large-scale synthesis and preclinical studies of MRS5698, a highly selective A3 adenosine receptor agonist that protects against chronic neuropathic pain, Purinergic Signalling, 2015, 11, 371–387 CrossRef CAS PubMed.
- B. Y. Michel and P. Strazewski, Total syntheses of a conformationally locked north-type methanocarba puromycin analogue and a dinucleotide derivative, Chem.–Eur. J., 2009, 15, 6244–6257 CrossRef CAS PubMed.
- W. S. Korinek, J. D. Lechleiter and T. E. Liston, Compounds and Methods for Treating Neurological and Cardiovascular Conditions, US Pat., 2018021363A1, U.S. Patent and Trademark Office, Washington, DC, Jan. 25, 2018 Search PubMed.
- T. E. Liston, S. Hinz, C. E. Müller, D. M. Holstein, J. Wendling, R. J. Melton, M. Campbell, W. S. Korinek, R. R. Suresh, D. Sethre-Hofstad, Z. G. Gao, D. K. Tosh, K. A. Jacobson and J. D. Lechleiter, Nucleotide P2Y1 receptor agonists are in vitro and in vivo prodrugs of A1/A3 adenosine receptor agonists: implications for roles of P2Y1 and A1/A3 receptors in health and disease, Purinergic Signalling, 2020, 16, 543–559 CrossRef CAS PubMed.
- W. Lu, S. Sengupta, J. L. Petersen, N. G. Akhmedov and X. Shi, Mitsunobu coupling of nucleobases and alcohols: an efficient, practical synthesis for novel nonsugar carbon nucleosides, J. Org. Chem., 2007, 72, 5012–5015 CrossRef CAS PubMed.
- S. Fletcher, V. M. Shahani and P. T. Gunning, Facile and efficient access to 2,6,9-tri-substituted purines through sequential N9, N2 Mitsunobu reactions, Tetrahedron Lett., 2009, 50, 4258–4261 CrossRef CAS.
- S. Fletcher, V. M. Shahani, A. J. Lough and P. T. Gunning, Concise access to N9-mono-, N2-mono- and N2,N9-di-substituted guanines via efficient Mitsunobu reactions, Tetrahedron, 2010, 66, 4621–4632 CrossRef CAS.
- K. Toti, D. Osborne, A. Ciancetta, D. Boison and K. A. Jacobson, South (S)- and North (N)-methanocarba-7-deazaadenosine analogues as inhibitors of human adenosine kinase, J. Med. Chem., 2016, 59, 6860–6877 CrossRef CAS.
- R. H. C. Yu, B. H. Brown, R. P. Polniaszek, B. R. Graetz, K. Sujino, D. Tran, A. S. Triman, K. M. Kent and S. Pfeiffer, Processes and intermediates for preparing anti-HIV agents, US Pat., 20180079773, WO 2012/159047 Al, 2018 Search PubMed.
- J. W. Arico, A. K. Calhoun, K. J. Salandria and L. W. McLaughlin, Tetramethylsuccinimide as a directing/protecting group in purine glycosylations, Org. Lett., 2010, 12, 120–122 CrossRef CAS.
- P. Y. Reddy, S. Kondo, T. Toru and Y. Ueno, Lewis acid and hexamethyldisilazane-promoted efficient synthesis of N-alkyl- and N-arylimide derivatives, J. Org. Chem., 1997, 62, 2652–2654 CrossRef CAS.
- V. Kohli, H. Blocker and H. Koster, The triphenylmethyl(trityl) group and its uses in nucleotide chemistry, Tetrahedron Lett., 1980, 21, 2683–2686 CrossRef CAS.
- G. Breault, C. J. Eyermann and B. Geng, Derivatives of adenine and 8-aza-adenine and uses thereof-796, WO2009034386A1, 2009.
- K. A. Jacobson, S. Merighi, K. Varani, P. A. Borea, S. Baraldi, M. A. Tabrizi, R. Romagnoli, P. G. Baraldi, A. Ciancetta, D. K. Tosh, Z. G. Gao and S. Gessi, A3 adenosine receptors as modulators of inflammation: from medicinal chemistry to therapy, Med. Res. Rev., 2018, 38, 1031–1072 CrossRef CAS PubMed.
- D. K. J. E. von Lubitz, R. C.-S. Lin, P. Popik, M. F. Carter and K. A. Jacobson, Adenosine A3 receptor stimulation and cerebral ischemia, Eur. J. Pharmacol., 1994, 263, 59–67 CrossRef CAS PubMed.
- M. Söderström, E. Zamaratski and L. R. Odell, BF3·SMe2 for thiomethylation, nitro reduction and tandem reduction/SMe insertion of nitrogen heterocycles, Eur. J. Org. Chem., 2019, 5402–5408 CrossRef.
- W. Bergmann and M. F. Stempien, Marine products. XLIII. Nucleosides of sponges. 5. Synthesis of spongosine, J. Org. Chem., 1957, 22, 1575–1577 CrossRef CAS.
- A. El-Tayeb, S. Michael, A. Abdelrahman, A. Behrenswerth, S. Gollos, K. Nieber and C. E. Müller, Development of polar adenosine A2A receptor agonists for inflammatory bowel disease: synergism with A2B antagonists, ACS Med. Chem. Lett., 2011, 2, 890–895 CrossRef CAS.
- Z. G. Gao, L. K. Mamedova, P. Chen and K. A. Jacobson, 2-Substituted adenosine derivatives: affinity and efficacy at four subtypes of human adenosine receptors, Biochem. Pharmacol., 2004, 68, 1985–1993 CrossRef CAS PubMed.
- M. Higginbottom, E. D. Savory, G. A. Brown, V. A. Horgan and E. J. Chapman, Adenosine derivatives for the treatment of pain, WO 2008/000745 A2, 2009.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental detail, 1H and 13C NMR spectra. See DOI: 10.1039/d1ra05096f |
| This journal is © The Royal Society of Chemistry 2021 |