
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfonic acid-functionalized PCP(Cr) catalysts with Cr3+ and –SO3H sites for 5-ethoxymethylfurfural production from glucose†
Luxin Zhang *,
Yuting Liu,
Ruijun Sun and
Simin Yi
*,
Yuting Liu,
Ruijun Sun and
Simin Yi
College of Environmental and Municipal Engineering, Shaanxi Key Laboratory of Environmental Engineering, Key Lab of Northwest Water Resource, Environment and Ecology, MOE, Xi'an University of Architecture and Technology, Xi'an 710055, P. R. China. E-mail: zhangluxinxx@126.com; Fax: +86 29 82205652; Tel: +86 29 82205652
First published on 19th October 2021
Abstract
5-Ethoxymethylfurfural (EMF) has been identified as a potential biofuel and fuel additive, for which the production from glucose (the most abundant and inexpensive monosaccharide) in a one-step process would be highly desirable. Here, the synthesis of sulfonic acid-functionalized porous coordination polymers (PCPs) and their application as catalysts for EMF synthesis are reported. PCP(Cr)-BA (PCP material with Cr3+ ions and H2BDC-SO3H linkers) and PCP(Cr)-NA (PCP material with Cr3+ ions and H2NDC(SO3H)2 linkers) materials containing both Cr3+ sites and Brønsted-acidic –SO3H sites were prepared. The morphology, pore structure, acidity, chemical composition, and thermal stability of the two functionalized PCP(Cr) catalysts were analyzed by systematic characterization. The catalysts featured a porous morphology and dual Cr3+ and –SO3H sites, which enabled the cascade conversion of glucose to EMF. PCP(Cr)-BA exhibited higher performance than PCP(Cr)-NA with an EMF yield of 23.1% in the conversion of glucose at 140 °C after 22 h in an ethanol/water system. In addition, the as-prepared catalyst exhibited a high stability in the current catalytic system for EMF production from glucose with a constant catalytic activity in a four-run recycling test without an intermediate regeneration step.
1. Introduction
The increasing demand for energy and useful chemicals in modern society has led to the accelerated depletion of non-renewable fossil resources.1,2 With fossil fuel resources diminishing and CO2-emission concerns rising, there has been considerable interest in the production of multipurpose materials, fuels, and chemicals from renewable resources to create a carbon-neutral valorization cycle.3–6 5-Hydroxymethylfurfural (HMF) and its derivatives are interesting building blocks or platform chemicals that can be utilized as both biofuels and precursors for various products and therefore have the potential to replace petrochemicals and fossil fuels. Among HMF derivatives, 5-ethoxymethylfurfural (EMF), which can be obtained by ethanolysis of raw bioresources and renewable biomass-derived carbohydrates, has recently attracted considerable attention. EMF, as a potential biofuel alternative, is an oily liquid at room temperature.7 EMF has a high energy density and exhibits many excellent chemical properties. For example, when used as a fuel additive, the amounts of exhaust gas, smoke, nitrogen oxides, and sulfur oxides are reduced because of its high oxidation stability, while it delivers a superior combustion performance when used as a fuel. Consequently, EMF has emerged as a potential excellent fuel additive and new biofuel.8,9 In addition, EMF can also be used as a flavoring agent for foods and wines.In recent years, EMF production from HMF or commercial carbohydrates via catalytic routes has been reported.10–12 Acidic catalysts are mainly applied in the synthesis of EMF from these feedstocks, and both homogeneous and heterogeneous catalysts are widely investigated for these reactions.13,14 EMF can be prepared from HMF by etherification reaction in high yields (>80%) with catalysts including mineral acids (e.g., sulfuric acid), heteropolyacids (e.g., phosphotungstic acid (H3PW12O40)15) or other solid acids (e.g., MOF-based heteropolyacid materials),16 tungstophosphoric acid supported on mesoporous niobium oxophosphate17 or MCM-41,18 layered niobium-molybdate,19 and Ag1H2PW (partially silver exchanged phosphotungstic acid).20 When using fructose as feedstock, satisfactory EMF yields can also be achieved in the presence of a variety of homogeneous catalysts (e.g., sulfuric acid,21 ZrOCl2·8H2O,22 hydrogen sulfate ionic liquids,23 deep eutectic solvent24) or heterogeneous catalysts (e.g., sulfonic acid-functionalized solid acids,25–29 acidic nanospheres,30 carbonaceous solid acids12,31). Although the above-mentioned routes proceed easily and with high EMF yields, their large-scale applications are limited due to the high prices of fructose and HMF.
Glucose, the most abundant and least expensive available monosaccharide, has a price advantage over fructose or HMF for EMF production.32 The one-pot transformation of glucose to EMF would be an economically more feasible process, because it avoids the isolation of HMF or fructose and the subsequent purification steps. Currently, single catalysts (e.g., ionic liquids),33 Al(DS)3 (aluminum tridodecyl sulfate),34 multi-functional zeolites35) or paired catalysts (e.g., H-USY(Si/Al = 6) + Amberlyst-15,13 AlCl3·6H2O + PTSA–POM (polymer of p-toluenesulfonic acid (PTSA) and paraformaldehyde (POM))36 have been tested for EMF production from glucose. However, the reactions are more difficult, and the obtained EMF yields are lower than those from ketose-based carbohydrates. Using glucose as raw material, at least one of three continuous processes consisting of three consecutive reactions each must be promoted: (i) “isomerization of glucose to fructose” – “dehydration of fructose to HMF” – “etherification of HMF to EMF”, (ii) “ethanolysis of glucose to ethyl glucoside (EG)” – “isomerization of EG to ethyl fructoside (EF)” – “dehydration of EF to EMF”, or (iii) “isomerization of glucose to fructose” – “ethanolysis of fructose to EF” – “dehydration of EF to EMF”.
Brønsted acids are beneficial to the dehydration and etherification reactions.14,33,37 The investigations on the catalytic synthesis of EMF from glucose or real raw biomass feedstocks containing glucosyl units require as catalysts a combination of Brønsted acids and active sites which are beneficial for overcoming equilibrium limitations of the glucose-to-fructose or EG-to-EF isomerization reactions. Therefore, the design of a bifunctional acidic catalyst with both Brønsted acid sites and catalytic sites conducive to isomerization is reasonable for achieving the “one-pot” catalytic conversion of glucose to EMF. Other real biomass or waste materials containing glucose sub-units, such as starch,7 corn stover,38 food waste,39 or sewage sludge4 have been explored as alternative renewable feedstocks for producing EMF catalyzed by homogeneous catalytic systems, but these systems encounter limitations such as equipment corrosion, catalyst separability, and recycling problems. Therefore, the development of a stable, recyclable, and efficient heterogeneous solid catalyst is required.
Porous coordination polymers (PCPs) are porous crystalline materials, which are formed by self-assembly of metal ions/clusters and organic ligands, and are characterized by an adjustable and modified pore structure and the presence of functional groups.40,41 In recent decades, PCP materials have widespread applications because of their special structure. As a new type of solid catalysts, PCPs show broad development prospects in the field of catalytic applications. In the present work, sulfonic acid-functionalized heterogeneous PCP(Cr) catalysts, with both Cr3+ and –SO3H sites on the surface, were synthesized and used for the direct production of EMF from glucose. The Cr3+ and –SO3H sites on the catalytic materials were expected to promote the catalytic conversion of glucose to EMF, while the synergistic effect between porosity and acidity would be conducive to enhancing catalytic performance. The structures of the catalysts were systematically characterized, the relationship of catalytic activity and physicochemical properties in the synthesis of EMF were investigated and analyzed. Furthermore, the role of co-solvent, the possible reaction pathway, the reuse and recycle of the catalyst were examined.
2. Experimental
2.1 Materials
All chemicals were used as received without further purification. Glucose (≥99%), 2-sulfoterephthalic acid monosodium salt (H2BDC-SO3Na, ≥98%) was purchased from Shanghai Aladdin Industrial Inc. 4,8-Disulfonyl-2,6-naphthalenedicarboxylic acid (H2NDC(SO3H)2, ≥98%) was purchased from Chemsoon Chemical Co., Ltd. (Shanghai, China). CrCl3·6H2O, Cr(NO3)3·9H2O, N,N-dimethylformamide (DMF), hydrochloric acid, hydrofluoric acid, and other chemicals were supplied by Tianjin Kermel Chemical Co., Ltd. (Tianjin, China).2.2 Catalyst preparation
CrCl3·6H2O (10 mmol), H2BDC-SO3Na (5 mmol), hydrochloric acid (20 mmol), deionized water (50 mL) were mixed in a 100 mL Teflon-lined stainless-steel autoclave and then heated at 210 °C for 24 h. After the reaction system was cooled down to room temperature, the formed green powder was collected by centrifugation and washed three times with water. Then, after washing three times each with DMF and methanol, the product was dried under vacuum at 80 °C for 12 h. The obtained powder was denoted as PCP(Cr)-BA.Cr(NO3)3·9H2O (1 mmol), H2NDC(SO3H)2 (1 mmol), HF (∼47 wt%, 110 μL, 2.6 × 10−3 mmol) and DMF (6 mL) were added into a 100 mL Teflon-lined stainless-steel autoclave. The autoclave was heated at 190 °C for 24 h, the following washing and drying procedure was similar to that of PCP(Cr)-BA, the obtained product was denoted as PCP(Cr)-NA.
2.3 Catalyst characterization
The surface morphologies of the catalysts and elemental mappings (Cr, S) were observed using a JSM-6510LV scanning electron microscope (SEM) from JEOL Ltd., Japan. X-ray photoelectron spectroscopy (XPS) patterns of samples were determined with a monochromatic Al Kα X-rays from Thermo Scientific ESCALAB 250Xi. Fourier transform infrared (FTIR) spectra were recorded on a Nicolet NEXUS-6700 FTIR spectrometer from Thermo Fisher Scientific Inc., USA, in the wave number range of 400–4000 cm−1. Thermogravimetric (TG) analysis was conducted under nitrogen flow by using a TGA/SDTA851 thermal analyzer from Mettler-Toledo International Inc., USA. A sample was placed in a sample pan and heated from room temperature to 350 °C at a heating rate of 10 °C min−1. The N2 physisorption isotherms were recorded by an ASAP 2460 system surface analyzer from Micromeritics Instruments Corp., USA, at 77 K applying the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) method. The Cr and S contents in PCP(Cr)-BA and PCP(Cr)-NA were measured by ICP-AES (Agilent 725) analysis. The crystalline structures of the two catalysts were analyzed on a Bruker METALJET D8 Venture powder X-ray diffraction (XRD) with Cu Kα radiation (λ = 0.154 nm).3. Results and discussion
3.1 Characterization of the PCP(Cr)-BA and PCP(Cr)-NA catalyst
The microstructures of PCP(Cr)-NA and PCP(Cr)-BA are clearly visible in Fig. 1. The two catalysts exhibited an entirely different morphology. For PCP(Cr)-BA, spherical microstructures were successfully fabricated through the hydrothermal process, and numerous nanofibers were present on the surface of these microspheres. PCP(Cr)-NA exhibited an irregular block-particle morphology, in which many particles with different diameters were formed during the synthesis process. PCP(Cr)-BA and PCP(Cr)-NA showed homogeneous dispersions of the Cr and S elements as observed by elemental mapping. | ||
| Fig. 1 SEM and elemental mapping of PCP(Cr)-BA and PCP(Cr)-NA. (a)–(f) PCP(Cr)-BA, (g)–(l) PCP(Cr)-NA. | ||
The porosity characteristics of the catalysts can be revealed by the shape of the N2 adsorption/desorption isotherm (Fig. 2). BET surface area and BJH pore volume were determined as 21 m2 g−1 and 0.04 cm3 g−1 and as 92 m2 g−1 and 0.46 cm3 g−1 for PCP(Cr)-BA and PCP(Cr)-NA, respectively. The pore size and pore volume of PCP(Cr)-NA were larger than those of PCP(Cr)-BA. At a relative pressure of 0.4 < P/P0 < 0.98, a steep increase was observed. The morphological and textural property measurements and analysis confirmed the presence of porous structures in these PCP(Cr) catalysts. As evident from the data of ICP-AES (Table 1), PCP(Cr)-BA contained higher amounts of S and Cr than PCP(Cr)-NA.
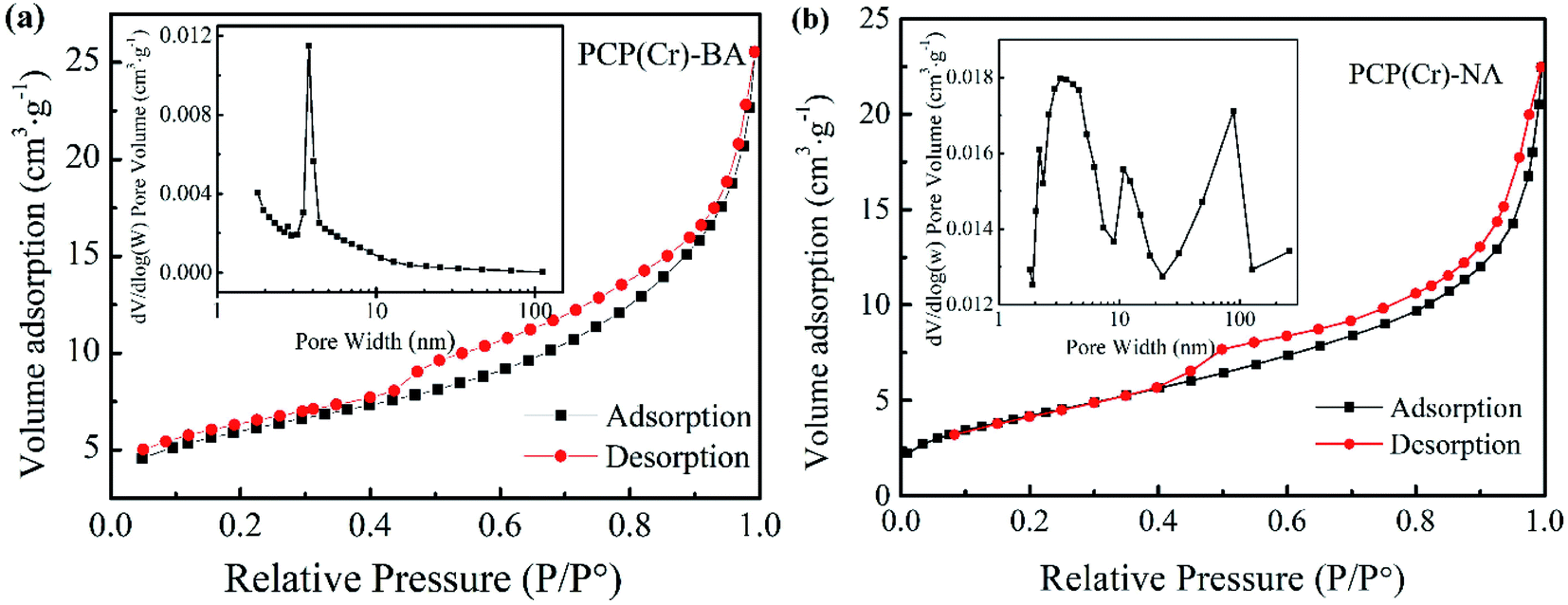 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms and pore size distribution curves of PCP(Cr)-BA and PCP(Cr)-NA. | ||
| Catalyst | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Smicro (m2 g−1) | Vmicro (cm3 g−1) | Pore size (nm) | S content (wt%) | Cr content (wt%) |
|---|---|---|---|---|---|---|---|
| PCP(Cr)-BA | 21 | 0.04 | 9 | 0.0006 | 7 | 9.3 | 8.9 |
| PCP(Cr)-NA | 92 | 0.46 | 18 | 0.07 | 27 | 6.7 | 8.6 |
The XPS results for PCP(Cr)-BA and PCP(Cr)-NA are displayed in Fig. 3. According to Fig. 3(a), both catalysts contained Cr, O, C, and S elements. In Fig. 3(b), the binding energy peaks at 577 eV and 587 eV could be assigned to Cr 2p3/2 and Cr 2p1/2. The S 2p binding energy peaks of PCP(Cr)-BA and PCP(Cr)-NA in Fig. 3(c) were in similar positions at around 167 eV, indicating all the S species were S6+.
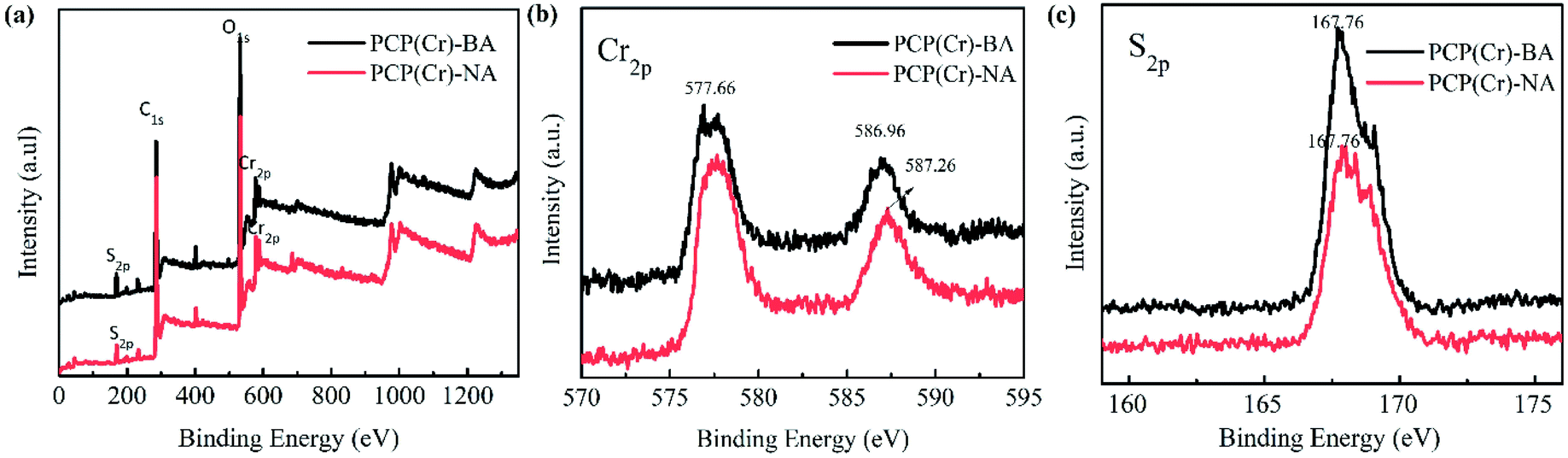 | ||
| Fig. 3 XPS patterns of PCP(Cr)-BA and PCP(Cr)-NA. | ||
The chemical structure of PCP(Cr)-BA and PCP(Cr)-NA was characterized by FTIR analysis (Fig. 4). For PCP(Cr)-BA, the band at 1629 cm−1 was assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, the bands at 1044 and 1185 cm−1 could be assigned to OSO symmetric stretching vibrations in –SO3H, and the band at 1449 cm−1 was assigned to OSO asymmetric stretching vibrations in –SO3H. For PCP(Cr)-NA, the peak at 1035 cm−1 in the IR spectra can be assigned to the symmetric stretching vibration of –SO3−-groups.42 The band at 1186 cm−1 could be assigned to OSO symmetric stretching vibrations. The peaks at 1423 cm−1 and 1641 cm−1 were the absorption peaks of carboxyl groups and were assigned to the asymmetric stretching vibration and symmetric stretching vibration of –COOH, respectively. These results indicated that the sulfonic acid groups were retained in the catalyst surface after solvothermal reaction at high temperature.
O stretching vibrations, the bands at 1044 and 1185 cm−1 could be assigned to OSO symmetric stretching vibrations in –SO3H, and the band at 1449 cm−1 was assigned to OSO asymmetric stretching vibrations in –SO3H. For PCP(Cr)-NA, the peak at 1035 cm−1 in the IR spectra can be assigned to the symmetric stretching vibration of –SO3−-groups.42 The band at 1186 cm−1 could be assigned to OSO symmetric stretching vibrations. The peaks at 1423 cm−1 and 1641 cm−1 were the absorption peaks of carboxyl groups and were assigned to the asymmetric stretching vibration and symmetric stretching vibration of –COOH, respectively. These results indicated that the sulfonic acid groups were retained in the catalyst surface after solvothermal reaction at high temperature.
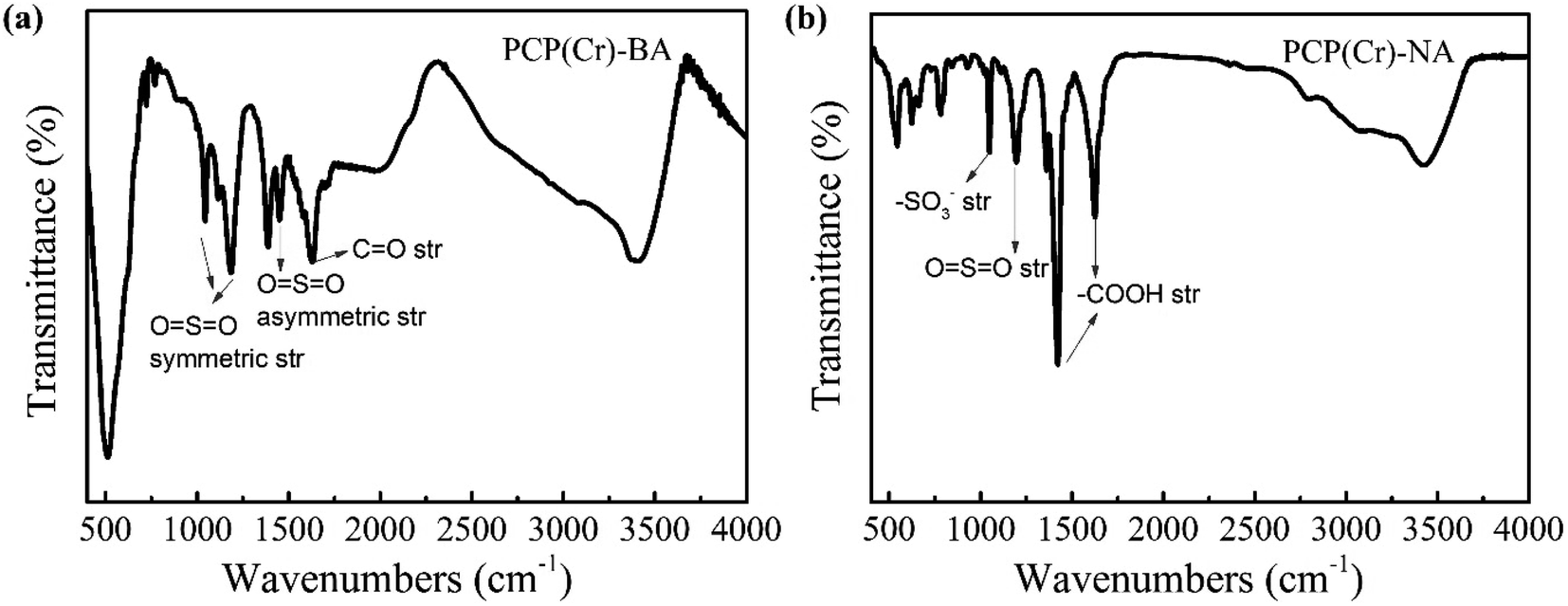 | ||
| Fig. 4 FTIR spectra of PCP(Cr)-BA and PCP(Cr)-NA. | ||
To clarify the crystal structure, the XRD patterns of PCP(Cr)-NA and PCP(Cr)-BA were provided, as shown in Fig. 5. The crystal structures of PCP(Cr)-NA and PCP(Cr)-BA were totally different. For PCP(Cr)-BA, there were obvious diffraction peaks at 22.4°, 23.6°, 29.3°, 39.2° and 46.3°. For PCP(Cr)-NA, the diffraction peaks were at 9°, 14.2°, 15.6°, 17.9° and 22.6°. PCP(Cr)-NA exhibited larger particle size than PCP(Cr)-BA, the particle size for PCP(Cr)-BA and PCP(Cr)-NA were 21.6 and 55 nm, respectively.
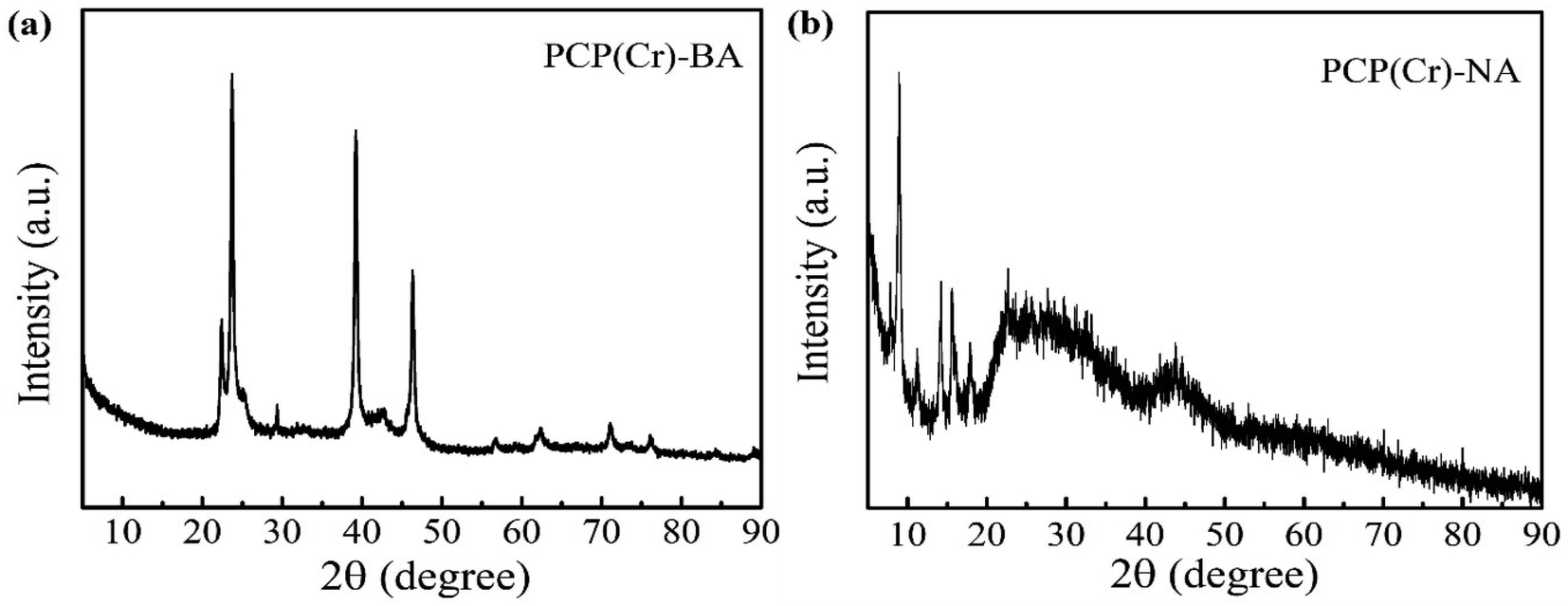 | ||
| Fig. 5 XRD patterns of PCP(Cr)-BA and PCP(Cr)-NA. | ||
The thermal decomposition properties of the PCP(Cr)-BA and PCP(Cr)-NA catalysts were investigated by TG analysis, and the results are shown in Fig. 6. The mass loss of catalyst below 150 °C was assigned to the loss of free water molecules in the pores of PCP(Cr)-BA and PCP(Cr)-NA. PCP(Cr)-BA is more stable than PCP(Cr)-NA and retained about 93% of its mass at 350 °C.
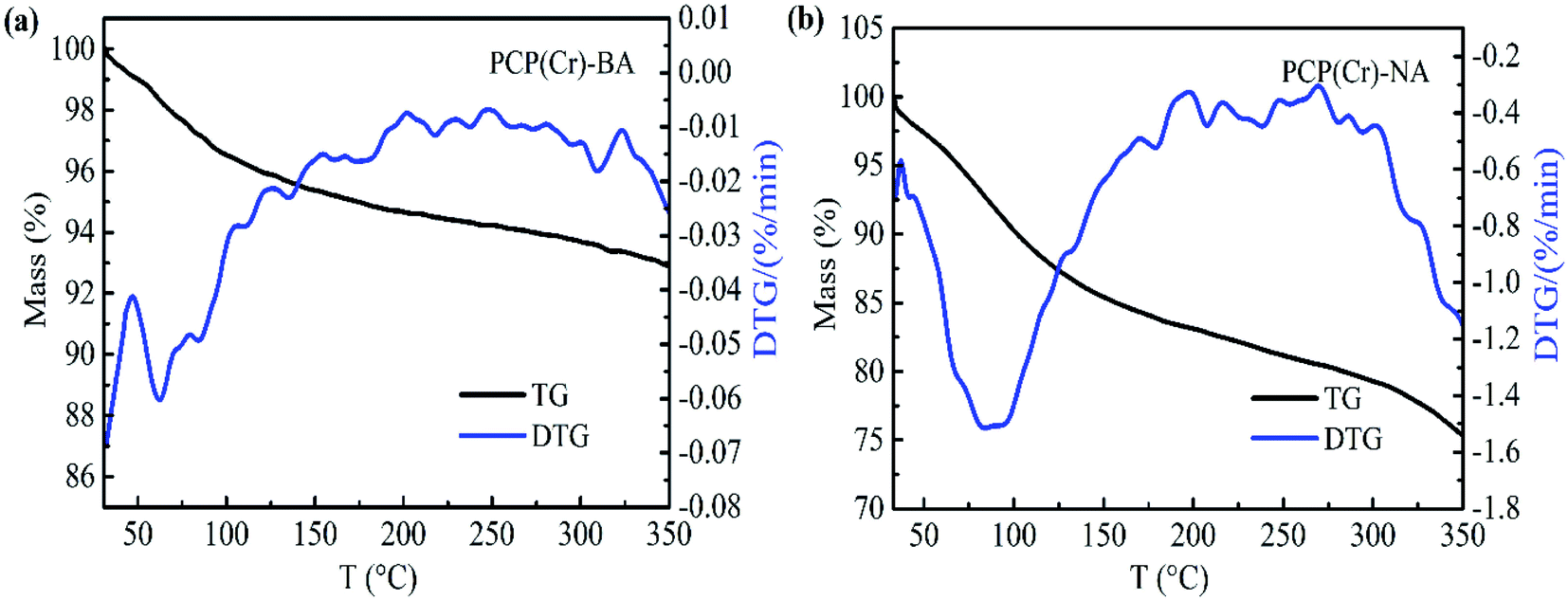 | ||
| Fig. 6 TG profiles of PCP(Cr)-BA and PCP(Cr)-NA. | ||
3.2 Catalytic properties of PCP(Cr)-BA and PCP(Cr)-NA catalyst
Reaction temperature and time are key factors in the study of catalytic performance and optimal reaction conditions. To investigate the effects of temperature and reaction time, catalytic experiments were carried out by employing PCP(Cr)-NA at different temperatures (130 °C to 150 °C) and for different reaction times (4 to 28 h) in ethanol/water system. Referring to Fig. 7, the yield of EMF progressively increased with the gradual increase of time between 4 and 24 h, while prolonging the reaction time after reaching the peak would lead to a decrease in EMF yield. The tested temperatures had a small effect on glucose conversion, because the glucose conversion reached 100% after 4 h for different temperatures.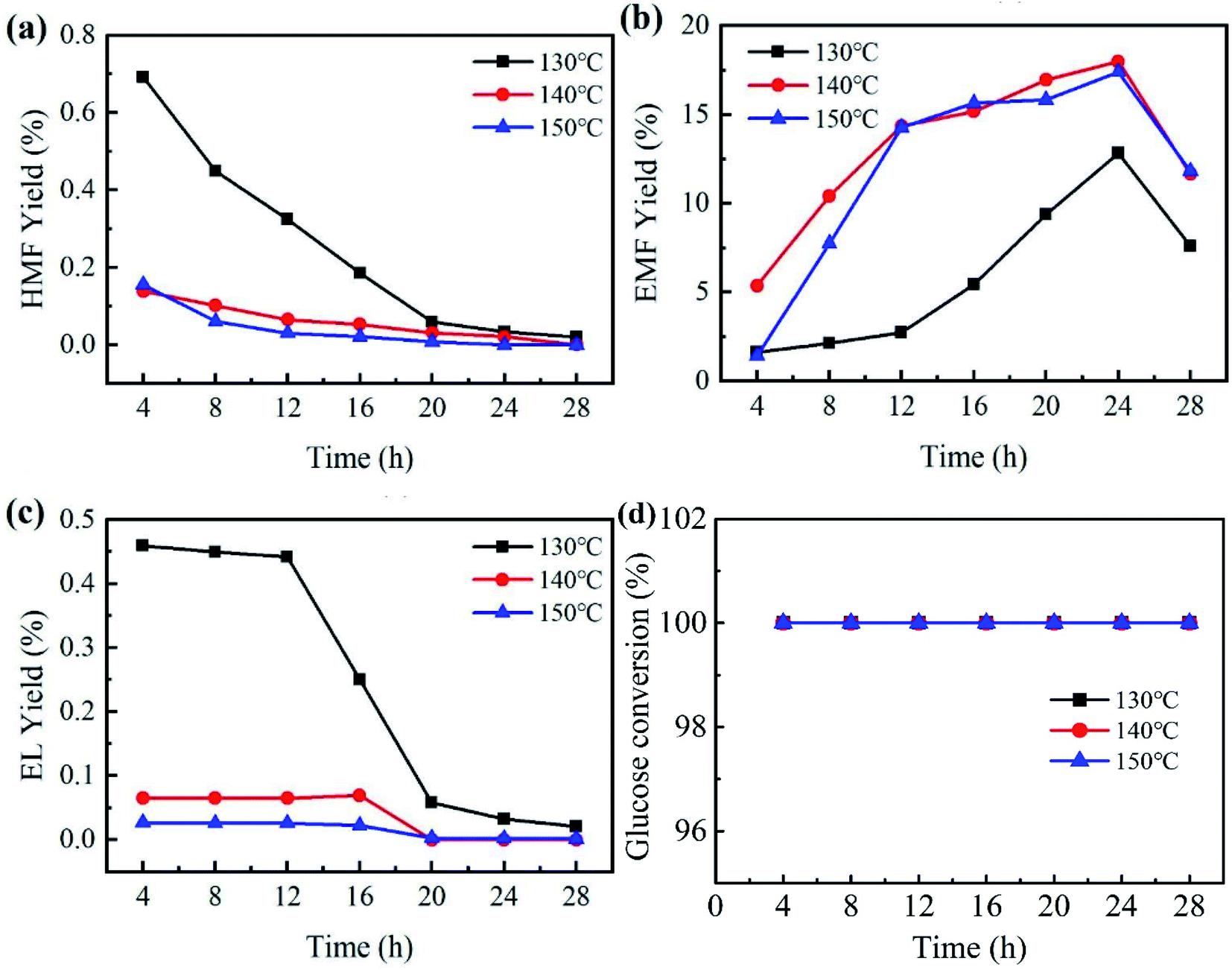 | ||
| Fig. 7 Effect of temperature and time on EMF production from glucose catalyzed by PCP(Cr)-NA. Reaction conditions: 20 mg glucose, 10 mg PCP(Cr)-NA, ethanol + H2O = 1.9 + 0.1 mL. | ||
An EMF yield of 12.8% was obtained after 24 h at 130 °C. As the temperature rose to 140 °C, the yield of EMF increased to 18% after the same reaction time. Raising the temperature accelerated the conversion efficiency of glucose, which might be explained by atoms being more likely to donate or receive electrons at higher temperatures, leading to higher reaction rates. However, when the reaction temperature was further raised to 150 °C for 24 h, the EMF yield decreased to 17.4%. The reason for this phenomenon may be that the high temperature led to further transformation of EMF to other unwanted by-products, which resulted in the decrease in EMF yield.
In addition to the catalyst, the solvent system has a noticeable effect on the solubility of raw materials, product yields, as well as the stability of key intermediates.43 Since the introduction of a co-solvent may change the properties of the solvent system and then affect reaction selectivity, the effect of co-solvent on EMF production catalyzed by PCP(Cr)-BA and PCP(Cr)-NA was investigated by using ethanol/DMSO, ethanol/H2O, or pure ethanol as the reaction medium.
As depicted in Fig. 8, the co-existence of Cr3+ and –SO3H sites in a single PCP(Cr) catalyst enabled “one-pot” cascading and concatenating reactions for glucose to EMF. Generally, PCP(Cr)-BA exhibited better catalytic activity than PCP(Cr)-NA in the EMF production from glucose, and the presence of water as co-solvent improved the reaction efficiency remarkably. Without the addition of co-solvent, 6.9% yield of EMF was obtained with PCP(Cr)-NA as catalyst in 24 h at 140 °C, while 15.7% yield was achieved in the presence of PCP(Cr)-BA with a reaction time of 22 h. When 5 vol% water was introduced into the reaction system, the yield of EMF increased for both catalysts. When using PCP(Cr)-BA as catalyst, the yield of EMF increased noticeably from 15.7% to 21.3%, and the yield of HMF (the main intermediate product) increased from 0.5% to 5% at 140 °C in 22 h.
 | ||
| Fig. 8 Effect of reaction time and co-solvent on the ethanolysis of glucose catalyzed by PCP(Cr)-BA or PCP(Cr)-NA at 140 °C. Reaction conditions: 20 mg glucose, 10 mg catalyst, 2 mL ethanol, ethanol + H2O = 1.9 + 0.1 mL, ethanol + DMSO = 1.9 + 0.1 mL. | ||
When DMSO (5 vol%) was introduced into the reaction system with the PCP(Cr)-BA catalyst, the yield of HMF increased remarkably, but the yield of EMF decreased greatly. Different from the results with PCP(Cr)-BA, the addition of DMSO increased EMF yield with PCP(Cr)-NA as catalyst compared with pure ethanol reaction system. Notably, the yield of ethyl levulinate (EL) increased by adding DMSO under PCP(Cr)-NA catalysis, however, EL yield remained in low level for PCP(Cr)-BA catalysis with DMSO or water as co-solvent. The results above indicated that catalyst property and solvent have synergistic catalytic effects on the reactions for ethanolysis of glucose. The addition of DMSO caused different yield increase or decrease for EMF, HMF and EL over different catalysts, may be because the “solvent effect” was corelated to catalyst pore size. As shown in Fig. 2 and Table 1, the pore volume and pore size of PCP(Cr)-NA were larger than those of PCP(Cr)-BA, the cooperation of pore dimension and solvent characteristics resulted in differences for products distribution.
In Fig. 8, glucose conversion of 100% being obtained for all the solvents. Furthermore, both EMF and HMF yields increased in the presence of the two catalysts at the beginning of the reaction, and as time progressed the yields decreased gradually, whereas the EL yield continuously increased with reaction time. EL was proved to be formed not only from the rehydration of EMF, but another feasible alternative route is the production of levulinic acid (LA) via HMF followed by its esterification to EL.44–46 Long reaction times were not favorable for EMF production due to the instability of EMF and easy conversion to by-products.
These two catalysts are materials with dual Cr3+ and –SO3H sites. The metal sites and organic ligands of PCP materials can function as catalytically active centers. Both PCP(Cr)-BA and PCP(Cr)-NA contained Cr3+ sites with coordinative unsaturation, with the potential to catalyze glucose-to-EG isomerization. On the other hand, the sulfonic acid functional groups, H2BDC-SO3H, or H2NDC(SO3H)2 were dispersed on the surfaces of PCP(Cr)-BA or PCP(Cr)-NA to introduce catalytically active centers with Brønsted acidity. The Cr3+ sites in the PCP(Cr)-B/NA catalysts served as active sites for the isomerization step in the cascade reaction from glucose to EMF, and Brønsted-acidic sulfonic acid group sites catalyzed the subsequent dehydration reactions (fructose to HMF, EF to EMF) and etherification reaction (HMF to EMF). PCP(Cr)-BA was more active than PCP(Cr)-NA, mainly because PCP(Cr)-BA contained more Cr3+ sites and –SO3H sites than PCP(Cr)-NA (Table 1). The efficiency of PCP(Cr)-BA and PCP(Cr)-NA in catalyzing glucose to 5-EMF was more noticeably affected by the contents of Cr3+ sites and –SO3H sites than textual property. The 5-EMF yield was not strongly correlated with high BET. PCP(Cr)-BA demonstrated higher activity than that of PCP(Cr)-NA, whereas the overall BET surface area, pore volume, and pore size of PCP(Cr)-BA was significantly lower than that of PCP(Cr)-NA.
To verify the superiority of the synergistic effect between Cr3+ and –SO3H active sites, the catalytic performance of catalyst with single Cr3+ or single –SO3H group was tested. Commercial MIL-101(Cr) with single Cr3+ site and commercial UIO-66-SO3H with single –SO3H group were used as catalyst for the comparative experiments. As can be seen from entries 1 and 2 in Table 2, UIO-66-SO3H afforded 0.7% EMF yield, while no EMF was detected in the presence of MIL-101(Cr). These observations indicated that the combination of Cr3+ and –SO3H sites was essential for EMF production from glucose. Furthermore, in bifunctional catalyst, the proximity of the two active sites may play a crucial role on the catalytic performance. Therefore, comparative tests were carried out by (i) adding single MIL-101(Cr) and single UIO-66-SO3H (Table 2, entry 3); (ii) simple powder mixing between MIL-101(Cr) and UIO-66-SO3H particles (Table 2, entry 4); and (iii) grinding a powder mixture of MIL-101(Cr) and UIO-66-SO3H in an agate mortar (Table 2, entry 5). The results showed that the closer distance between Cr3+ and –SO3H group, the higher EMF yield, which further verified the superiority of the synergistic effect between Cr3+ and –SO3H group active sites.
| Entry | Catalyst | Catalyst loading/mg | Reaction conditions | EMF yield/% |
|---|---|---|---|---|
| a Reaction conditions: 20 mg glucose, ethanol + H2O = 1.9 + 0.1 mL.b Single MIL-101(Cr) and single + UIO-66-SO3H were added to the reaction.c Simple powder mixing between single MIL-101(Cr) and single UIO-66-SO3H particles.d Grinding a powder mixture of MIL-101(Cr) and UIO-66-SO3H in an agate mortar. | ||||
| 1 | MIL-101(Cr) | 10 | 140 °C, 22 h | — |
| 2 | UIO-66-SO3H | 10 | 140 °C, 22 h | 0.7 |
| 3b | MIL-101(Cr)+ UIO-66-SO3H | 10 + 10 | 140 °C, 22 h | 13.4 |
| 4c | MIL-101(Cr)+ UIO-66-SO3H | 10 + 10 | 140 °C, 22 h | 14.5 |
| 5d | MIL-101(Cr)+ UIO-66-SO3H | 10 + 10 | 140 °C, 22 h | 15.7 |
3.3 Effect of glucose concentration and catalyst concentration
Catalyst and feedstock loading affected the products distribution significantly. As listed in Table 2 for initial glucose loading of 20 mg, the yield of EMF reached the maximum value of 23.1% with PCP(Cr)-BA catalyst loading of 10 mg (Table 3, entry 1), while increasing the catalyst loading to 15 mg or decreasing it to 5 mg exhibited a remarkable negative impact on the EMF yield in the reaction system. When increasing the PCP(Cr)-NA catalyst loading from 5 mg to 20 mg, the EMF yields first increased and then decreased with a maximum value of 18% at 10 mg PCP(Cr)-NA (Table 3, entry 5), while the yield of EL constantly increased with the increase of catalyst amount. Furthermore, at 140 °C and 10 mg PCP(Cr)-NA, the EMF yield varied with different initial glucose concentration (Table 3, entry 5, 8, 9). At initial glucose amounts of 10 mg and 30 mg, the EMF yields reached 5.7% and 11.7%, respectively. Higher glucose concentration promoted side reactions and resulted in a decrease in EMF yield. A lower EMF yield was observed with a glucose amount of 5 mg compared to 10 mg, mainly because the lower glucose concentration resulted in excess catalytic sites, which were more conducive to catalyzing side reactions.| Entry | Catalyst | Catalyst loading (mg) | Reaction time (h) | Glucose conversion (%) | HMF yield (%) | EMF yield (%) | EL yield (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: ethanol + H2O = 1.9 + 0.1 mL, 20 mg glucose, 140 °C.b 10 mg glucose.c 30 mg glucose. | |||||||
| 1 | PCP(Cr)-BA | 10 | 22 | 100 | 5.0 | 23.1 | 0.1 |
| 2 | PCP(Cr)-BA | 5 | 22 | 100 | 1.6 | 17.9 | — |
| 3 | PCP(Cr)-BA | 15 | 22 | 100 | 4.2 | 21.1 | 0.2 |
| 4 | PCP(Cr)-NA | 5 | 24 | 100 | 1.7 | 3.7 | — |
| 5 | PCP(Cr)-NA | 10 | 24 | 100 | 0.02 | 18 | 0.1 |
| 6 | PCP(Cr)-NA | 15 | 24 | 100 | 0.1 | 15.6 | 0.2 |
| 7 | PCP(Cr)-NA | 20 | 24 | 100 | 0.02 | 5 | 0.5 |
| 8b | PCP(Cr)-NA | 10 | 24 | 100 | 1.8 | 5.7 | ND |
| 9c | PCP(Cr)-NA | 10 | 24 | 100 | 0.06 | 11.7 | 0.1 |
3.4 Catalyst recycling experiments
The recycling evaluation experiments of the PCP(Cr)-BA catalyst were carried out by conducting the synthesis of EMF from glucose in ethanol/H2O under the same conditions as with the fresh catalyst. After each reuse reaction run, the PCP(Cr)-BA catalyst was separated by simple filtration. The obtained yields of the main products, i.e., EMF, EL, and HMF, were displayed in Fig. 9, which indicate an approximately constant yield over four runs. Furthermore, we conducted ICP-AES analysis for the solvents after reactions, and no Cr or S was detected, indicating the heterogeneous nature of the reaction. Consequently, PCP(Cr)-BA can be considered as a PCP material with a good thermal stability in ethanol/H2O reaction environment and with a good catalytic activity in the recycling tests for EMF production from glucose. Therefore, PCP(Cr)-BA possesses a great potential application in the catalytic conversion of glucose to biofuel EMF.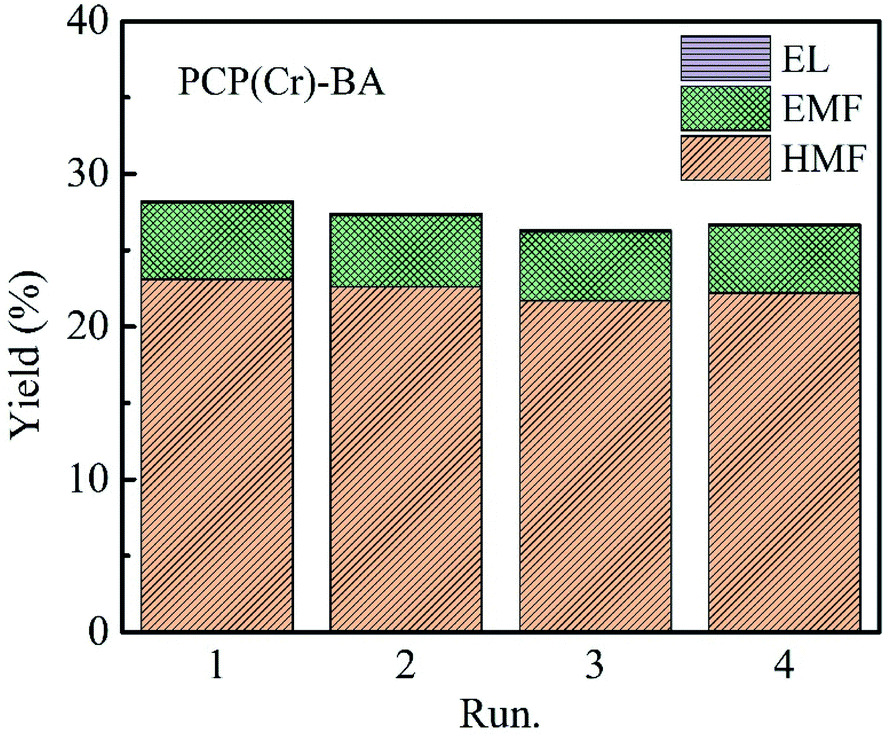 | ||
| Fig. 9 Recycle experiments of glucose conversion into EMF catalyzed by PCP(Cr)-BA. Reaction conditions: 20 mg glucose, 10 mg PCP(Cr)-BA, ethanol + H2O = 1.9 + 0.1 mL, 22 h, 140 °C. | ||
3.5 Discussion of the reaction networks for the production of EMF
The PCP(Cr)-BA catalyst featured a porous structure and both Cr3+ and –SO3H groups, which were conducive to EMF production from glucose via multi-step reactions in ethanol/co-solvent reaction systems. EMF was also detected with EG, EF, fructose or HMF as raw materials (Table 4). Based on the literatures47–49 and the determination of products in the applied reaction system under different conditions, the co-existence of three main reaction pathways can be inferred, as shown in Fig. 10; (i) glucose–EG–EF–EMF; (ii) glucose–fructose–HMF–EMF; and (iii) glucose–fructose–EF–EMF. The reaction proceeded via the isomerization reaction of glucose to fructose (and EG to EF) over Cr3+ sites on PCP(Cr)-BA, and then the dehydration reaction of fructose to HMF (and EF to EMF), followed by the etherification reaction of HMF and ethanol to EMF over the Brønsted-acidic sulfonic acid group sites on PCP(Cr)-BA catalyst.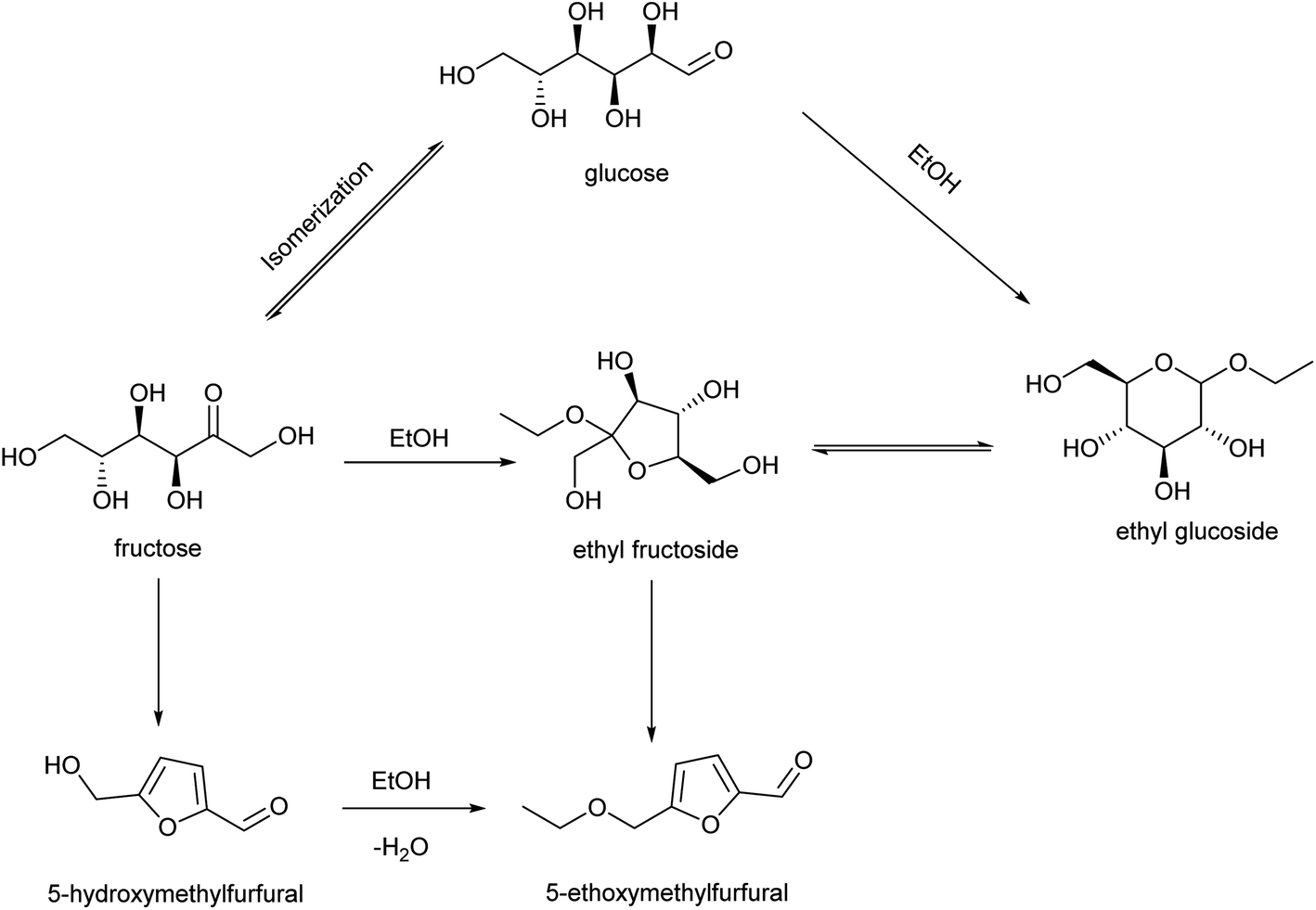 | ||
| Fig. 10 Reaction routes for EMF production from glucose catalyzed by PCP(Cr)-BA. | ||
4. Conclusion
In this work, a facile method to synthesize sulfonic acid-functionalized PCPs via solvothermal-induced self-assembly was demonstrated. The prepared PCP(Cr)-BA and PCP(Cr)-NA catalysts contained both Cr3+ and Brønsted-acidic –SO3H sites in a single PCP(Cr) material. Because of their acidity features, these materials were explored as catalysts for EMF production from glucose. The amounts of Cr3+ and –SO3H sites significantly contributed to the catalytic performance. The presence of water in the reaction medium notably improved the catalytic efficiency, enhancing EMF yields in the reactions catalyzed by PCP(Cr)-BA or PCP(Cr)-NA. It was found that PCP(Cr)-BA could convert glucose to EMF with the highest yield of 23% in an ethanol/H2O system. Cr3+ sites were identified as the major catalytic species for the isomerization steps of the cascade reactions from glucose to EMF, while –SO3H groups on the catalyst surface were beneficial for the dehydration and etherification reaction steps for synthesizing EMF. In recyclability studies consisting of a four-run recycling test, the possible reuse of the PCP(Cr)-BA catalyst was demonstrated without significant changes in product yields, confirming good recyclability and stability of the catalyst. This work provides a novel strategy to biofuel EMF synthesis and valorization of carbohydrates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21607119), Key Research and Development Plan of Shaanxi Province-Industrial Innovation Chain Project (2020ZDLNY06-08), Young Talents Support Program of Colleges and Universities Association for Science and Technology of Shaanxi Province (20190420), Special Funds of the Education Department of Shaanxi Province (19JK0475), Innovative Talents Promotion Plan-Science and Technology Innovation Group of Shaanxi Province (2019TD-025), Special Program in Natural Science of Xi'an University of Architecture and Technology (ZR19020) and for the financial support.References
- M. Y. Lui, C. Y. Y. Wong, A. W.-T. Choi, Y. F. Mui, L. Qi and I. T. Horváth, ACS Sustain. Chem. Eng., 2019, 7, 17799–17807 CrossRef CAS.
- B. S. Rao, D. D. Lakshmi, P. K. Kumari, P. Rajitha and N. Lingaiah, Sustain. Energy Fuels, 2020, 4, 3428–3437 RSC.
- G. Gómez Millán, S. Hellsten, J. Llorca, R. Luque, H. Sixta and A. M. Balu, ChemCatChem, 2019, 11, 2022–2042 CrossRef.
- L. di Bitonto, V. Locaputo, V. D'Ambrosio and C. Pastore, Appl. Energy, 2020, 259, 114163 CrossRef CAS.
- L. Tian, L. Zhang, Y. Liu, Y. He and J. Xiang, J. Clean. Prod., 2020, 268, 122296 CrossRef CAS.
- Q. Q. Mei, X. J. Shen, H. Z. Liu and B. X. Han, Chin. Chem. Lett., 2019, 30, 15–24 CrossRef CAS.
- Y. Chen, L. Peng, J. Zhang and L. He, Catal. Commun., 2020, 137, 105947 CrossRef CAS.
- D. Li, Y. Liu, S. Wu and J. Zhuang, Mod. Chem. Ind., 2020, 40, 58–62 Search PubMed.
- X. Liu and R. Wang, in Biomass, Biofuels, Biochemicals, Elsevier, 2020, pp. 355–375 Search PubMed.
- S. Alipour, H. Omidvarborna and D. S. Kim, Renew. Sustain. Energy Rev., 2017, 71, 908–926 CrossRef CAS.
- Y. Mori, Y. Katayama, T. Shikata and N. Kasuya, Res. Chem. Intermed., 2020, 46, 609–620 CrossRef CAS.
- T. Chen, L. Peng, X. Yu and L. He, Fuel, 2018, 219, 344–352 CrossRef CAS.
- H. Li, S. Saravanamurugan, S. Yang and A. Riisager, Green Chem., 2016, 18, 726–734 RSC.
- H. Li, K. S. Govind, R. Kotni, S. Shunmugavel, A. Riisager and S. Yang, Energy Convers. Manag., 2014, 88, 1245–1251 CrossRef CAS.
- M. Zuo, K. Le, Y. Feng, C. Xiong, Z. Li, X. Zeng, X. Tang, Y. Sun and L. Lin, Ind. Crop. Prod., 2018, 112, 18–23 CrossRef CAS.
- Z. Wang and Q. Chen, Green Chem., 2016, 18, 5884–5889 RSC.
- K. P. Krishna, R. B. Srinivasa, L. D. Dhana, S. P. N. Ruthwik, C. Sumana and N. Lingaiah, Catal. Today, 2019, 325, 53–60 CrossRef.
- A. Liu, Z. Zhang, Z. Fang, B. Liu and K. Huang, Ind. Eng. Chem., 2014, 20, 1977–1984 CrossRef CAS.
- F. Yang, J. Tang, R. Ou, Z. Guo, S. Gao, Y. Wang, X. Wang, L. Chen and A. Yuan, Appl. Catal. B Environ., 2019, 256, 117786 CrossRef CAS.
- Y. Ren, B. Liu, Z. Zhang and J. Lin, J. Ind. Eng. Chem., 2015, 21, 1127–1131 CrossRef CAS.
- T. Flannelly, S. Dooley and J. J. Leahy, Energy Fuels, 2015, 29, 7554–7565 CrossRef CAS.
- H. Liu, X. Tang, W. Hao, X. Zeng, Y. Sun, T. Lei and L. Lin, J. Energy Chem., 2018, 27, 375–380 CrossRef.
- H. Guo, X. Qi, Y. Hiraga, T. M. Aida and R. L. Smith, Chem. Eng. J., 2017, 314, 508–514 CrossRef CAS.
- A. B. Gawade and G. D. Yadav, Biomass Bioenergy, 2018, 117, 38–43 CrossRef CAS.
- J. Zhang, K. Dong, W. Luo and H. Guan, Fuel, 2018, 234, 664–673 CrossRef CAS.
- L. Zhang, Y. Zhu, L. Tian, Y. He and F. Deng, Fuel Process. Technol., 2019, 193, 39–47 CrossRef CAS.
- Y. Wen, Z. Yu, K. Li, H. Guo, Y. Dai and L. Yan, Green Energy Environ., 2018, 3, 384–391 CrossRef.
- J. Dai, Z. Liu, Y. Hu, S. Liu, L. Chen, T. Qi, H. Yang, L. Zhu and C. Hu, Catal. Sci. Technol., 2019, 9, 483–492 RSC.
- C. Wang, K. Li and P. Ji, J. Chem. Technol. Biotechnol., 2021, 96, 163–171 CrossRef CAS.
- Z. Wang, H. Li, C. Fang, W. Zhao, T. Yang and S. Yang, Energy Technol., 2017, 5, 2046–2054 CrossRef CAS.
- Y. Y. Bai, S. Su, S. Wang, B. Wang, R. C. Sun, G. Song and L. P. Xiao, Energy Technol., 2018, 6, 1951–1958 CrossRef CAS.
- M. Zuo, W. Jia, Y. Feng, X. Zeng, X. Tang, Y. Sun and L. Lin, Renew. Energy, 2021, 164, 23–33 CrossRef CAS.
- H. Guo, A. Duereh, Y. Hiraga, X. Qi and R. L. Smith, Energy Fuels, 2018, 32, 8411–8419 CrossRef CAS.
- Y. Mori, Y. Katayama, T. Shikata and N. Kasuya, Res. Chem. Intermed., 2020, 46, 609–620 CrossRef CAS.
- Y. Bai, L. Wei, M. Yang, H. Chen, S. Holdren, G. Zhu, D. T. Tran, C. Yao, R. Sun, Y. Pan and D. Liu, J. Mater. Chem. A, 2018, 6, 7693–7705 RSC.
- H. Xin, T. Zhang, W. Li, M. Su, S. Li, Q. Shao and L. Ma, RSC Adv., 2017, 7, 41546–41551 RSC.
- B. Xiang, Y. Wang, T. Qi, H.-Q. Yang and C.-W. Hu, J. Catal., 2017, 352, 586–598 CrossRef CAS.
- B. Chen, G. Xu, C. Chang, Z. Zheng, D. Wang, S. Zhang, K. Li and C. Zou, Energy Fuels, 2019, 33, 4310–4321 CrossRef CAS.
- L. di Bitonto, G. Antonopoulou, C. Braguglia, C. Campanale, A. Gallipoli, G. Lyberatos, I. Ntaikou and C. Pastore, Bioresour. Technol., 2018, 266, 297–305 CrossRef CAS PubMed.
- M. S. Yao, J. J. Zheng, A. Q. Wu, G. Xu, S. S. Nagarkar, G. Zhang, M. Tsujimoto, S. Sakaki, S. Horike, K. Otake and S. Kitagawa, Angew. Chem., Int. Ed., 2020, 59, 172–176 CrossRef CAS PubMed.
- J.-S. M. Lee, K.-i. Otake and S. Kitagawa, Coord. Chem. Rev., 2020, 421, 213447 CrossRef CAS.
- C. R. Li, S. L. Li and X. M. Zhang, Cryst. Growth Des., 2016, 9, 1702–1707 CrossRef.
- S. Gajula, K. Inthumathi, S. R. Arumugam and K. Srinivasan, ACS Sustain. Chem. Eng., 2017, 5, 5373–5381 CrossRef CAS.
- J. A. Melero, G. Morales, J. Iglesias, M. Paniagua, B. Hernández and S. Penedo, Appl. Catal. Gen., 2013, 466, 116–122 CrossRef CAS.
- Z. Wang, Z. Li, T. Lei, M. Yang, T. Qi, L. Lin, X. Xin, A. Ajayebi, Y. Yang, X. He and X. Yan, Appl. Energy, 2016, 183, 170–181 CrossRef CAS.
- H. Guo, A. Duereh, Y. Su, E. J. M. Hensen, X. Qi and R. L. Smith, Appl. Catal. B Environ., 2020, 264, 118509 CrossRef CAS.
- G. Bharath, K. Rambabu, A. Hai, P. P. Morajkar, A. V. Salkar, S. W. Hasan, P. L. Show and F. Banat, Int. Energy Res., 2021, 45, 1–12 CrossRef.
- F. W. Dini, H. Helmiyati and Y. K. Krisnandi, Bull. Chem. React. Eng. Catal., 2021, 16, 320–330 CrossRef.
- P. K. Kumari, B. S. Rao, D. Mallesh and N. Lingaiah, J. Mol. Catal., 2021, 508, 111607 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05103b |
| This journal is © The Royal Society of Chemistry 2021 |