
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeuroinflammatory inhibitors from Gardneria nutans Siebold & Zuccarini†
Ying-Ying Si ab,
Wei-Wei Wanga,
Qing-Mei Fenga,
Zhen-Zhu Zhaoab,
Gui-Min Xueab,
Yan-Jun Sunab,
Wei-Sheng Fengab,
Jun-Im Young*c and
Xian-Shi Wang*acd
ab,
Wei-Wei Wanga,
Qing-Mei Fenga,
Zhen-Zhu Zhaoab,
Gui-Min Xueab,
Yan-Jun Sunab,
Wei-Sheng Fengab,
Jun-Im Young*c and
Xian-Shi Wang*acd
aCollege of Pharmacy, Henan University of Chinese Medicine, Zhengzhou 450046, China
bThe Engineering and Technology Center for Chinese Medicine Development of Henan Province, Zhengzhou 450046, China
cCollege of Pharmacy, Chonnam National University, Gwangju 500-757 61186, South Korea. E-mail: imyoungjun@jnu.ac.kr
dSchool of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou 325035, China. E-mail: xianshifree@126.com
First published on 9th August 2021
Abstract
Two new monoterpene indole alkaloid glycosides nutanoside A–B (1–2), two new phenolic glycoside esters nutanester A–B (6–7), together with five known compounds (3–5, 8–9) were isolated from the ethanol extract of Gardneria nutans Siebold & Zuccarini. Their structures were established on the basis of extensive spectroscopic analysis and TDDFT/ECD calculations. Compounds 1 and 2 are two rare monoterpene indole alkaloids with the glucosyl moiety located at C-12 and represent the first two examples of enantiomer of ajmaline type monoterpene indole alkaloids. Compounds 3, 4 and 6 displayed significant inhibitory effects on NO production in over-activated BV2 microglial cells, with the IC50 values of 2.29, 6.36, and 8.78 μM, respectively. Compounds 1, 5, 7 could significantly inhibit the mRNA expression of inflammatory factors TNF-α and IL-6 induced by LPS in BV2 microglial cells at the effective concentration. Moreover, compound 3 exhibited stronger cytotoxicities against U87 and HCT116 cell lines than taxol with IC50 values of 10.58 and 14.60 μM, respectively.
Introduction
Neuroinflammation is considered to be one of the main causes of neurodegenerative diseases like Alzheimer's disease (AD) and Parkinson's disease (PD),1,2 and microglia cells play an important role in the occurrence and development of neuroinflammation.3 Microglia cells are the permanent immune cells of central nervous system, which have endogenous immune defense function.4 Under normal physiological conditions, microglia cells are in a resting or dormant state and provide nutritional support for neurons. In the brain of AD patients, microglia cells can be activated by Aβ and then clear them, thus playing a role in the repair and protection of nervous system.5 However, over-activation of microglia cells leads to the release of large amounts of inflammatory factors, including nitric oxide (NO), tumor necrosis factor (TNF-α), interleukin (IL-6, IL-1β), reactive oxygen species (ROS) and so on. The continuous and a mass of release of these neurotoxic factors will directly damage neurons, or further activate microglia cells to form a vicious cycle, and eventually lead to the gradual degenerative changes or apoptosis of neurons and the occurrence of neurodegenerative diseases such as AD.6,7 Therefore, inhibition of over-activation of microglia cells has become an effective way to inhibit neuroinflammation. The lack of effective drugs to treat neuroinflammation has led researchers to turn their attention to natural products.In our screening of natural herbals for therapy of neuroinflammation, the ethanol extract of Gardneria nutans Siebold & Zuccarini was selected as a good candidate. G. nutans is derived from the genus Gardneria in the Loganiaceae family. The genus Gardneria is endemic to Asia and mainly distributed in the southeast of Asia. The plants of this genus are usually used as folk medicines in China, such as G. multiflora and G. angustifolia.8,9 Studies on the chemical constituents and pharmacological activities of this genus have revealed that monoterpene indole alkaloids were responsible for diverse bioactivities exhibited by plants of the Gardneria genus, including antitumor, sympathetic block, central nervous excitation, dilate blood vessels, reduce blood pressure and nerve conduction10–14 activities and so on. G. nutans is a member of the genus, but there are few reports on its chemical constituents and biological activities. Thus, investigation on the ethanol extract of G. nutans was conducted, which led to the isolation of five monoterpene indole alkaloids (1–5) and four phenolic glucosides (6–9) (Fig. 1). Among the isolated compounds, four compounds (1–2, 6–7) were previously undescribed and others were known. The anti-neuroinflammation activities and cytotoxicities against U87 and HCT116 cell lines were also investigated. Herein, the isolation, structural elucidation, anti-neuroinflammation activities, and cytotoxicities of these components are discussed.
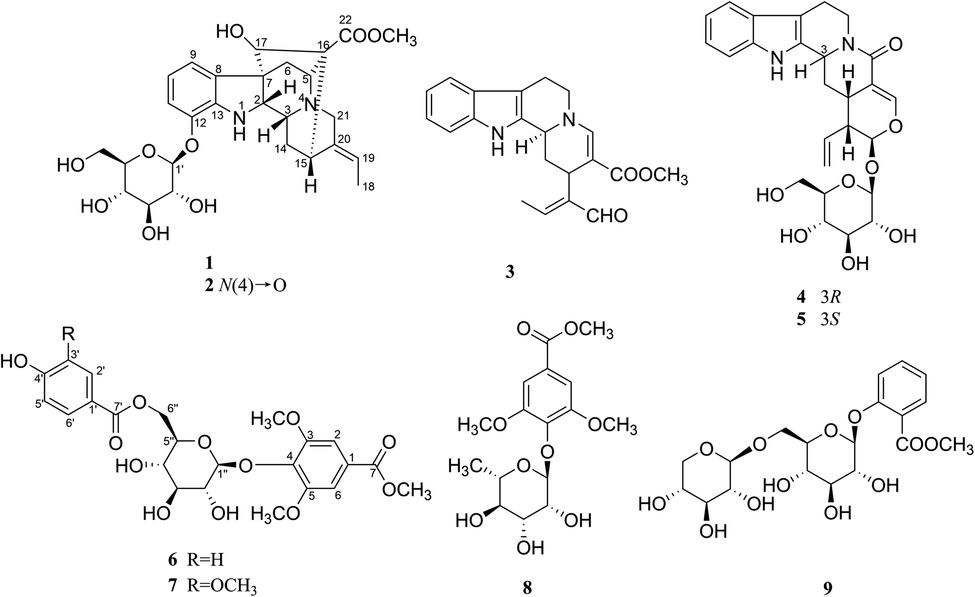 | ||
| Fig. 1 Structures of compounds 1–9. | ||
Results and discussion
Nutanoside A (1) was obtained as a white acicular crystal. The molecular formula was determined as C27H34N2O9 on the basis of the HR-ESI-MS peak at m/z 531.2359 [M + H]+ (C27H35N2O9, calcd for 531.2343), corresponding to 12 indices of hydrogen deficiency. The 1H NMR and 13C NMR data (Table 1) displayed signals characteristic of a 1,2,3-trisubstituted phenyl group provided by signals at δH 6.84 (1H, d, J = 7.6 Hz), 6.75 (1H, t, J = 7.6 Hz), 6.56 (1H, d, J = 7.6 Hz), and δC 143.0 (C-12), 141.7 (C-13), 131.9 (C-8), 119.6 (C-9), 118.2 (C-10), 114.3 (C-11); a methoxy at δH 3.53 (3H, s) and δC 50.9 (22-OCH3); a methyl at δH 1.52 (3H, d, J = 6.6 Hz) and δC 12.5 (C-18); a carbonyl group at δC 172.7 (C-22); a trisubstituted double bond at δH 5.14 (1H, q, J = 6.6 Hz) and δC 114.2 (C-19), 139.0 (C-20); and a glucosyl group at δH 4.67 (1H, d, J = 7.3 Hz) and δC 101.7 (C-1′), 73.4 (C-2′), 77.1 (C-3′), 69.9 (C-4′), 76.0 (C-5′), 60.9 (C-6′). Comparison of its 1D NMR data with those of quebrachidine15 indicated that they have a similar monoterpenoid indole alkaloid skeleton, except for the presence of a glucosyl group in compound 1. The attachment of the glucosyl group to C-12 was confirmed by the correlations from H-1′ (δH 4.67) to C-12 (δC 143.0) in HMBC spectrum (Fig. 2), and the configuration was confirmed to be β-D by the large coupling constant (7.3 Hz) of the anomeric proton and acid hydrolysis as described in the Experimental section. The NOESY correlations of H-2 with H-5 (Fig. 3) suggested the same β-orientations of H-2 and H-5. The polycyclic rigid framework formed by the connections of C15–C16(C5)–C17–C7 determined the same β-orientations of H-15, H-3 and H-5. The relative configuration at C-17 can be deduced from the observed NOESY correlation signals between H-15 and H-17, which indicated the orientation of H-17 is β. The reciprocal NOESY correlations observed for H-15/H-18 and H-19/H-21 established the geometry configuration of the 19,20-double bond as E. Then, theoretical calculations were carried out to determine the absolute configuration of 1. The ECD spectra of (2R, 3R, 5S, 7R, 15S, 16R, 17S)-1a and its enantiomer (2S, 3S, 5R, 7S, 15R, 16S, 17R)-1b were calculated at B3LYP/6-31+G** level on the base of TDDFT method (Fig. 4). Comparison of the calculated ECD spectra data with that of experimental data of compound 1 indicated the absolute configuration of compound 1 was assigned to be (2R, 3R, 5S, 7R, 15S, 16R, 17S).| No. | Compound 1 | Compound 2 | ||
|---|---|---|---|---|
| δHa J (Hz) | δCb | δHa J (Hz) | δCb | |
| a Recorded at 500 MHz in DMSO-d6.b Recorded at 125 MHz in DMSO-d6. | ||||
| 1 | 5.33 d, 3.2 | 6.29 s | ||
| 2 | 3.51 d, 4.2 | 68.9 | 4.01 br. s | 63.4 |
| 3 | 3.34 m | 53.8 | 3.68 br. s | 70.9 |
| 4 | ||||
| 5 | 3.22 m | 61.0 | 3.72 d, 3.5 | 74.7 |
| 6 | 2.40 dd, 10.9, 4.8 | 36.0 | 2.31 d, 11.9 | 31.2 |
| 1.47 d, 10.9 | 2.24 m | |||
| 7 | 57.7 | 57.0 | ||
| 8 | 131.9 | 129.7 | ||
| 9 | 6.75 d, 7.6 | 119.6 | 6.66 d, 7.7 | 118.9 |
| 10 | 6.56 t, 7.6 | 118.2 | 6.53 t, 7.7 | 118.6 |
| 11 | 6.84 d, 7.6 | 114.3 | 6.77 d, 7.7 | 112.7 |
| 12 | 143.0 | 143.4 | ||
| 13 | 141.7 | 141.2 | ||
| 14 | 2.46 dd, 13.8, 4.9 | 22.1 | 2.56 dd, 13.2, 4.1 | 21.8 |
| 1.36 dd, 13.8, 9.9 | 1.89 t, 11.8 | |||
| 15 | 3.35 m | 30.0 | 3.49 d, 4.1 | 29.0 |
| 16 | 59.9 | 60.5 | ||
| 17 | 4.08 d, 6.6 | 73.2 | 3.93 s | 72.7 |
| 18 | 1.52 d, 6.6, 3H | 12.5 | 1.57 d, 6.2, 3H | 12.3 |
| 19 | 5.14 q, 6.6 | 114.2 | 5.33 q, 6.2 | 117.8 |
| 20 | 139.0 | 131.3 | ||
| 21 | 3.28 m | 54.8 | 4.22 d, 15.0 | 69.7 |
| 3.25 m | 3.65 m | |||
| 22 | 172.7 | 170.7 | ||
| 1′ | 4.67 d, 7.3 | 101.7 | 4.73 d, 7.3 | 100.6 |
| 2′ | 3.23 m | 73.4 | 3.23 m | 73.4 |
| 3′ | 3.29 m | 77.1 | 3.32 m | 76.8 |
| 4′ | 3.14 m | 69.9 | 3.15 m | 70.0 |
| 5′ | 3.26 m | 76.0 | 3.30 m | 76.0 |
| 6′ | 3.72 m | 60.9 | 3.71 m | 60.9 |
| 3.46 m | 3.45 m | |||
| –OCH3 | 3.53 s, 3H | 50.9 | 3.60 s, 3H | 51.7 |
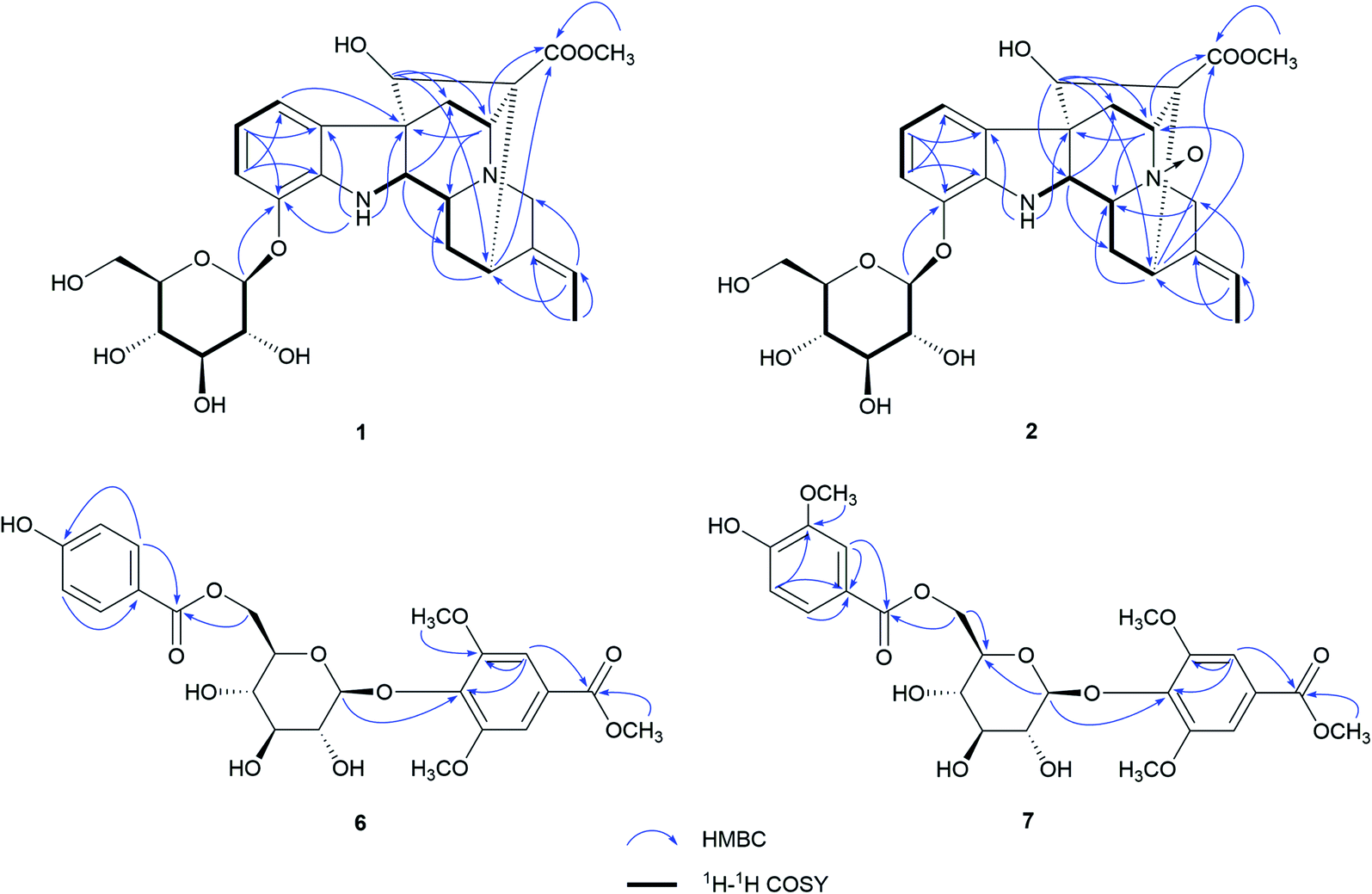 | ||
| Fig. 2 Key HMBC and 1H–1H COSY correlations of 1, 2, 6, and 7. | ||
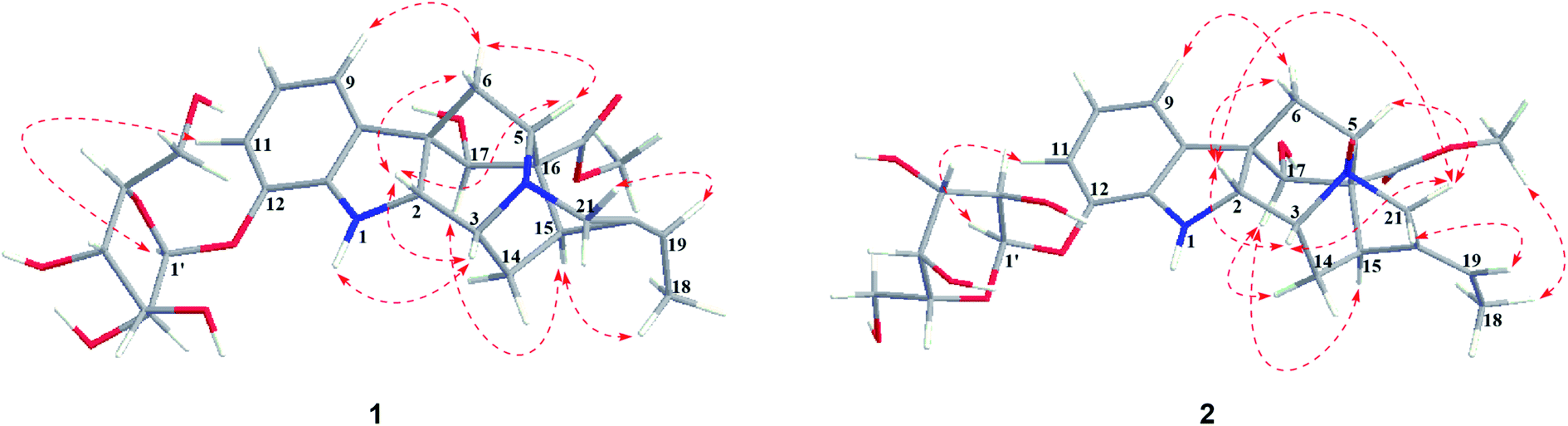 | ||
| Fig. 3 Key NOESY correlations of 1–2. | ||
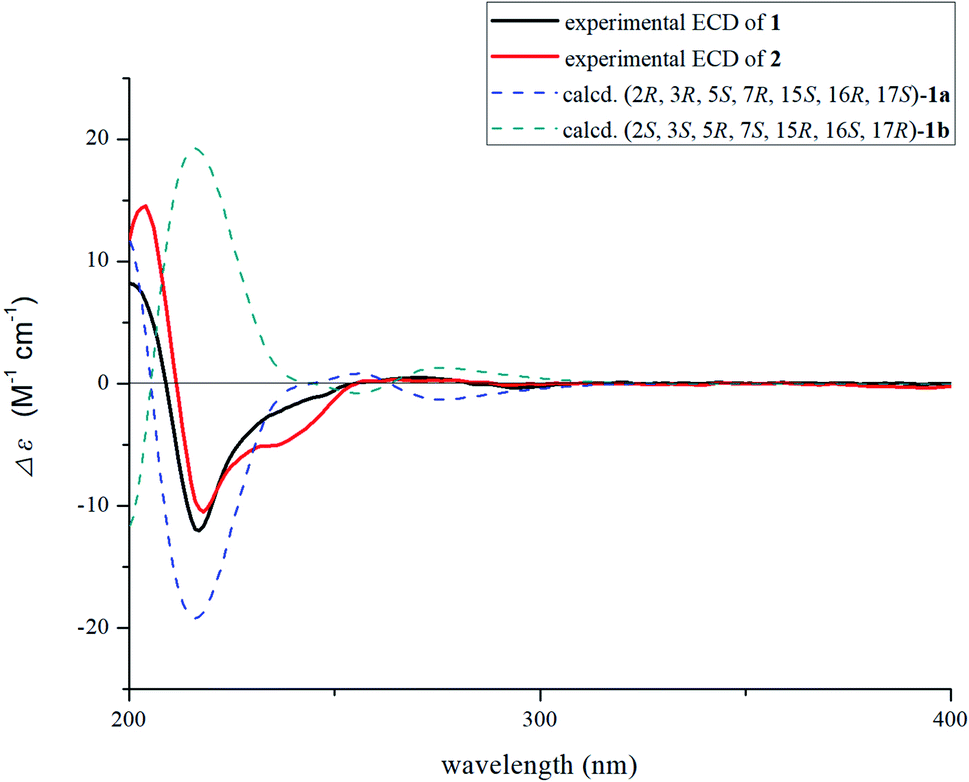 | ||
| Fig. 4 Calculated ECD spectrum of compound 1 and experimental ECD curves of 1–2. | ||
Nutanoside B (2) was obtained as a yellowish oil. It had a molecular formula of C27H34N2O10 by the HR-ESI-MS peak at m/z 547.2296 [M + H]+ (C27H35N2O10, calcd for 547.2292), which was 16 mass units higher than that of compound 1. The NMR spectra data (Table 1) were similar to those of 1, except for the characteristic downfield shifts of the carbon resonances for C-3, C-5, and C-21. Accordingly, the planar structure of compound 2 was identified as the N(4)-oxide of compound 1. The relative configuration was secured by 2D NMR data, including HSQC, HMBC, 1H–1H COSY, and NOESY. The NOESY correlations (Fig. 3) of H-21α with H-2, H-3 and H-5; H-15 with H-17 indicated the same relative configuration at these chiral centers compared with compound 1. Similarly, the NOESY correlations observed for H-15/H-18 and H-19/H-21 established the geometry configuration of the 19,20-double bond as E. In addition, HMBC correlations (Fig. 2) from H-1′ (δH 4.73) to C-12 (δC 143.4) assigned the C-12 attachment of the glucosyl moiety, and the coupling constants (7.3 Hz) of anomeric protons and acid hydrolysis established the sugar units as β-D-glucose. The absolute configuration of 2 was established as (2R, 3R, 5S, 7R, 15S, 16R, 17S), which was same as 1, according to their highly similarity on ECD spectrum (Fig. 4). Generally, the glucosyl moiety of monoterpene indole alkaloids are located at monoterpene moiety and occasionally at C-10 or C-11 of the indole skeleton. Compounds 1 and 2 are two rare monoterpene indole alkaloids with the glucosyl moiety located at C-12. Moreover, compounds 1 and 2 represent the first two examples of enantiomer of ajmaline type monoterpene indole alkaloids.
Nutanester A (6) was obtained as a yellowish amorphous powder. The molecular formula of 6 was determined as C23H26O12 on the basis of the quasi-molecular ion peak at m/z 517.1334 [M + Na]+ (C23H26O12Na, calcd for 517.1322) in its HR-ESI-MS spectrum. The 1H NMR and 13C NMR spectrum (Table 2) showed the presence of a 1,3,4,5-tetrasubstituted benzene ring by two magnetically equivalent aromatic proton signals at [δH 7.14 (2H, s) and δC 124.7 (C-1), 106.8 (C-2, 6), 152.6 (C-3, 5), 137.9 (C-4)], with two magnetically equivalent methoxy by signals at δH 3.73 (6H, s) and δC 56.2; a 1,4-disubstituted benzene ring at [δH 7.55 (2H, d, J = 8.5 Hz), 6.76 (2H, d, J = 8.5 Hz); δC 120.2 (C-1′), 131.3 (C-2′, 6′), 115.1 (C-3′, 5′), 162.0 (C-4′)]; a glucosyl group at [δH 5.10 (1H, d, J = 6.5 Hz, H-1′′); δC 101.6 (C-1′′), 73.9 (C-2′′), 76.5 (C-3′′), 70.4 (C-4′′), 74.1 (C-5′′), 63.5 (C-6′′)]; two carbonyl groups at δC 165.8 (C-7) and 165.2 (C-7′); and a methoxy at δH 3.85 (3H, s) and δC 52.2. The connection of the glucose unit at C-4 was confirmed by the HMBC correlation from H-1′′ to C-4 (Fig. 2). The p-hydroxy benzoyl was shown to be involved in an ester linkage at the C-6′′ position by HMBC correlations observed from H-6′′ to C-7′. Then, the large coupling constants (6.5 Hz) of anomeric protons, acid hydrolysis and alkaline methanolysis established the sugar units as β-D-glucose. Accordingly, compound 6 was identified as 4-[6-O-(p-hydroxy benzoyl)-β-D-glucopyranoside]-3,5-dimethoxy methyl benzoate, named as nutanester A.
| No. | Compound 6 | Compound 7 | ||
|---|---|---|---|---|
| δHa J (Hz) | δCb | δHa J (Hz) | δCb | |
| a Recorded at 500 MHz in DMSO-d6.b Recorded at 125 MHz in DMSO-d6. | ||||
| 1 | 124.7 | 124.7 | ||
| 2 | 7.14 s | 106.8 | 7.13 s | 106.8 |
| 3 | 152.6 | 152.5 | ||
| 4 | 137.9 | 138.0 | ||
| 5 | 152.6 | 152.5 | ||
| 6 | 7.14 s | 106.8 | 7.13 s | 106.8 |
| 7 | 165.8 | 165.7 | ||
| 1′ | 120.2 | 119.6 | ||
| 2′ | 7.55 d, 8.5 | 131.3 | 7.22 br. s | 112.4 |
| 3′ | 6.76 d, 8.5 | 115.1 | 147.5 | |
| 4′ | 162.0 | 142.3 | ||
| 5′ | 6.76 d, 8.5 | 115.1 | 6.71 d, 8.1 | 115.1 |
| 6′ | 7.55 d, 8.5 | 131.3 | 7.17 br. d, 8.1 | 123.5 |
| 7′ | 165.2 | 165.4 | ||
| 1′′ | 5.10 d, 6.5 | 101.6 | 5.11 d, 6.0 | 101.7 |
| 2′′ | 3.27 m | 73.9 | 3.28 m | 74.3 |
| 3′′ | 3.26 m | 76.5 | 3.27 m | 76.5 |
| 4′′ | 3.22 m | 70.4 | 3.21 m | 70.5 |
| 5′′ | 3.38 m | 74.1 | 3.40 m | 74.1 |
| 6′′ | 4.43 d, 12.1 | 63.5 | 4.43 m | 63.6 |
| 4.09 dd, 12.1, 7.3 | 4.10 m | |||
| 3,5-OCH3 | 3.73 s, 6H | 56.2 | 3.73 s, 6H | 56.2 |
| 7-OCH3 | 3.85 s, 3H | 52.2 | 3.84 s, 3H | 52.2 |
| 3′-OCH3 | 3.71 s, 3H | 55.5 | ||
Nutanester B (7) was obtained as a yellowish amorphous powder. The HR-ESI-MS spectrum exhibited a quasi-molecular ion peak at m/z 547.1429 [M + Na]+ (C24H28O13Na, calcd for 547.1428), allowing the molecular formula of 7 to be assigned as C24H28O13, which was 30 mass units higher than that of compound 6. The 1H NMR and 13C NMR spectra data (Table 2) were similar to those of 6, except for the presence of an additional methoxy at [δH 3.71 (3H, s); δC 55.5]. Meanwhile, the aromatic proton signals for 1,4-disubstituted benzene ring in compound 6 were substituted by a set of ABX coupling proton signals at [δH 7.22 (1H, br. s), 6.71 (1H, d, J = 8.1 Hz), and 7.17 (1H, br. d, J = 8.1 Hz)] in compound 7. Thus, compound 7 was identified as the 3′-methoxy substituted analogue of compound 6, which was further confirmed by 2D NMR spectrum (Fig. 2).
The five known compounds, isovallesiachotamine (3),16 vincosamide (4),17 strictosamide (5),17 methyl syringate 4-O-α-L-rhamnoside (8),18 methyl salicylate 2-O-β-D-xylosyl (1→6) β-D-glucopyranoside (9),19 were identified by analysis of physicochemical properties and comparison of spectral data with those reported in literatures.
Biological activities
The anti-inflammatory activities of the isolated compounds 1–8 were assayed in BV2 microglial cells by monitoring LPS-induced NO production. Beforehand, cell viabilities of compounds 1–8 on BV2 microglial cells were tested by MTT assay in order to avoid the possible effect of reduced viability on NO release. Results (Fig. 5) showed that compounds 3, 4, 8 exhibited cytotoxicity at 10 μM, and compounds 2, 7 were toxic to the cells at 100 μM, which may affect their inhibitory effect on LPS-induced NO release. That is to say, such effect of these compounds may mainly due to cell death and not to their inhibitory effect on LPS-induced NO release. Taking this into consideration, concentrations that did not affect the cell viability were selected to test for anti-inflammatory activities of compounds 1–8 in BV2 microglial cells. Among them, 1 μM was the maximum test concentration for compounds 3, 4, 8 and 10 μM for compounds 1–2, 5–7. As shown in Table 3, compounds 3, 4 and 6 displayed significant inhibitory effects on NO production in over-activated BV2 microglial cells, with the IC50 values of 2.29, 6.36, and 8.78 μM, respectively. In addition, compounds 2, 5 and 8 also showed moderate inhibitory effects with IC50 values ranging from 14.41 to 47.30 μM.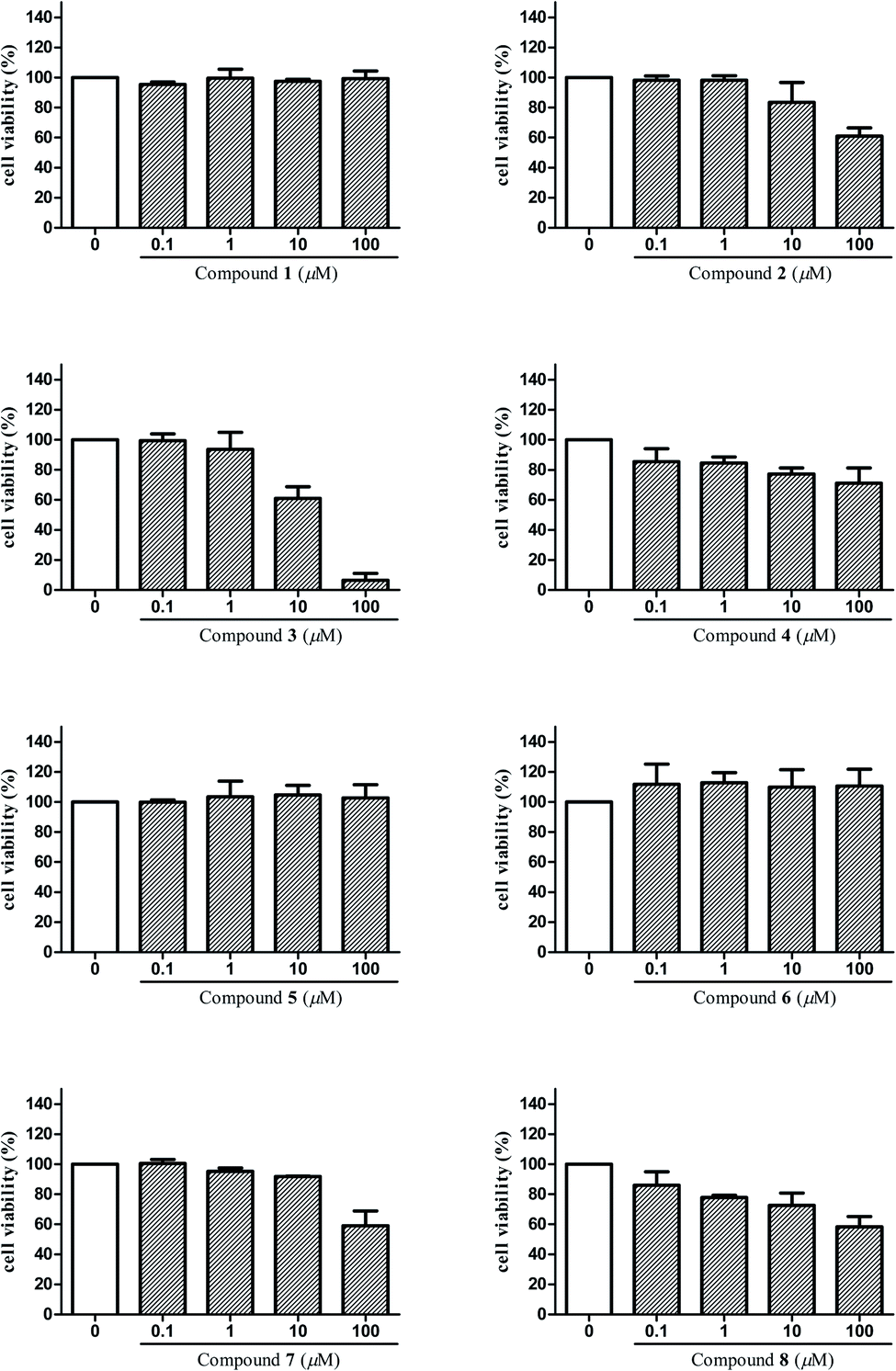 | ||
| Fig. 5 Cell viability assay of compounds 1–8 in BV2 microglial cells. | ||
| Compounds | IC50 (μM) | Compounds | IC50 (μM) |
|---|---|---|---|
| 1 | 74.43 ± 1.34 | 5 | 15.18 ± 0.81 |
| 2 | 47.30 ± 0.69 | 6 | 8.78 ± 1.57 |
| 3 | 2.29 ± 0.08 | 7 | >100 |
| 4 | 6.36 ± 0.72 | 8 | 14.41 ± 0.03 |
Then, quantitative real-time PCR (qRT-PCR) was also used to further investigate the anti-neuroinflammatory activities of compounds 1–8. The results (Fig. 6) revealed that compounds 1, 7 (0.1, 1 and 10 μM), and compound 5 (10 μM) could significantly inhibit the mRNA expression of inflammatory factors TNF-α induced by LPS in BV2 microglial cells. Compounds 5 (1 and 10 μM) and 7 (10 μM) could also greatly inhibit the mRNA expression of inflammatory factors IL-6. In addition, compounds 1, 3, 6 and 8 also exhibited a moderate inhibitory effect on the inflammatory factors TNF-α or IL-6 at 0.3–10 μM.
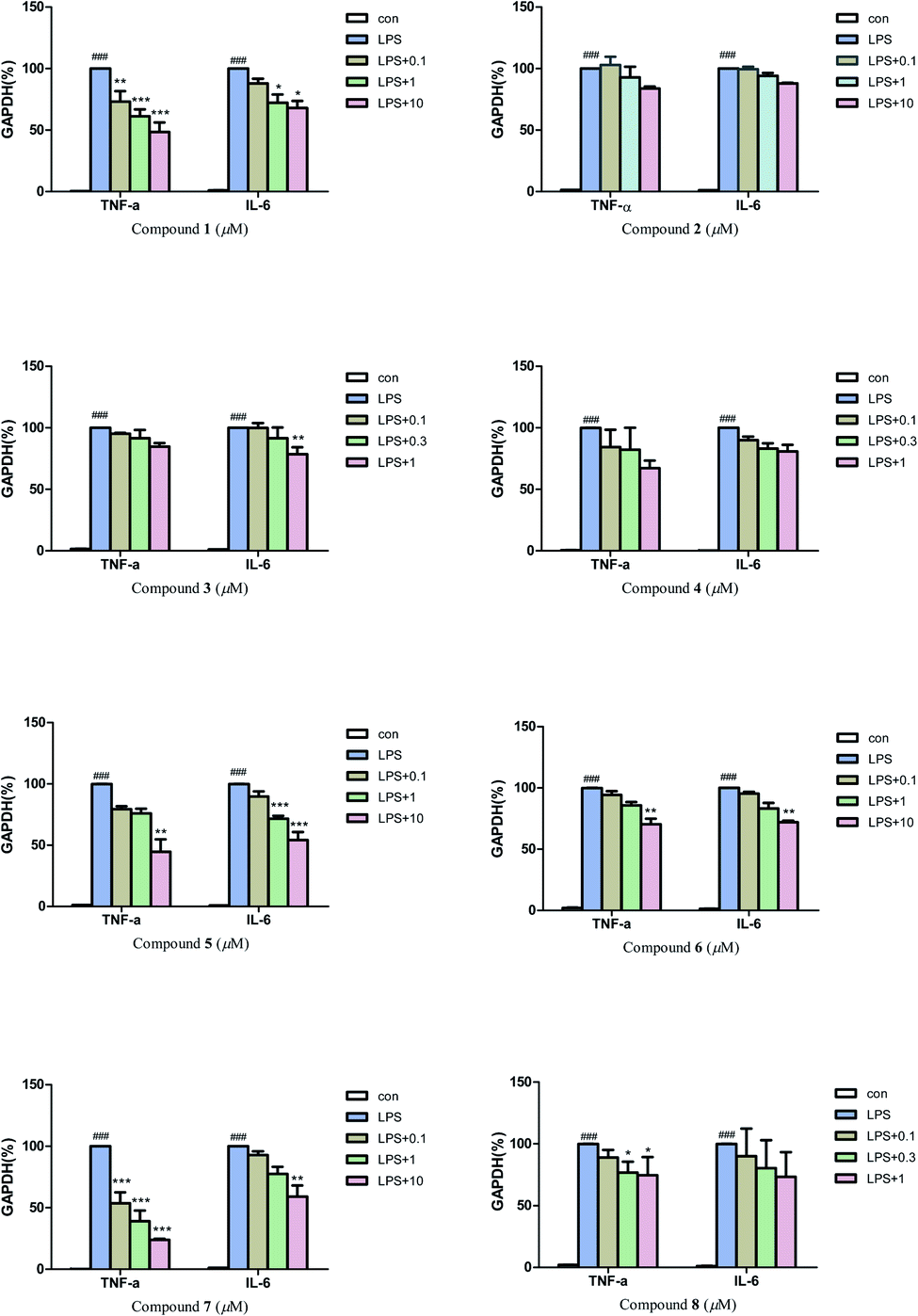 | ||
| Fig. 6 Effect of compounds 1–8 on LPS-induced mRNA expressions of TNF-α, and IL-6 in BV-2 microglial cells. Each bar represents the means ± SD of three independent experiments. Significance: *P < 0.05, **p < 0.01, ***p < 0.001, compared to LPS group; ###P < 0.001, compared to control group. | ||
Considering the anti-neuroinflammatory activities and structural characteristics of the isolated compounds, a brief structure–activity relationship (SAR) could be concluded as follows: by comparing compound 4 with 5, we found that the 3R configuration could cause an increase of NO inhibitory activity but attenuated the inhibitory effect on inflammatory factors TNF-α and IL-6 in over-activated BV2 microglial cells. A similar situation occurred between compounds 6 and 7, the presence of 3′-OCH3 significantly reduced the NO inhibitory activity, while greatly enhanced the inhibitory activities on inflammatory factors TNF-α and IL-6. Moreover, the presence of coordination bond at N-4 in compound 2 (compared with 1) could weaken the inhibitory effect on inflammatory factors TNF-α and IL-6 in BV2 microglial cells.
Furthermore, considering the effect of some compounds on the survival rate of microglia cells, the cytotoxic activities on two human cancer cell lines (U87 and HCT116) of compounds 3, 4 and 8 were investigated by MTT method. As shown in Table 4, compound 3 exhibited stronger cytotoxic activities against U87 and HCT116 cell lines than taxol, which was used as the positive control, with IC50 values of 10.58 and 14.60 μM, respectively. Compound 4 showed selective inhibitory effect on HCT116 cells with IC50 values of 49.21 μM.
Experimental method
General experimental procedures
CD spectrum was tested using Chirascanq CD spectrometer (Applied Photophysics) at room temperature. Optical rotation was analyzed using the APIV (Rudolph Research Analytical). NMR spectra were performed on Bruker Avance III 500 MHz spectrometer (Bruker, Karlsruhe, Germany) using TMS as a standard (500 MHz for 1H NMR and 125 MHz for 13C NMR). Bruker maXis HD mass spectrometer was used to collect HR ESI-TOF MS data in m/z (rel.%). MPLC was performed on Agela ODS flash column (C-18, 120 g, 40–60 μm, Tianjin, China), and the FL-H050G MPLC system (Tianjin, China). HPLC separation was carried out on a semi-preparative YMC-pack ODS-A column (250 × 10 mm, YMC-pack, Kyoto, Japan) equipped with Saipuruisi LC-52 instrument with a UV200 detector (Beijing, China). Silica gel GF254 for TLC and silica gel (200–300 mesh) for column chromatography were obtained from Qingdao Haiyang Chemical Co., Ltd. (Qingdao, China). D-Glucose and L-glucose (SigmaAldrich, St. Louis, MO, USA) were used to determine the absolute configuration of sugar moiety. Sephadex LH-20 for column chromatography was obtained from Pharmacia (Piscataway, NJ, USA).Plant material
The whole plant of Gardneria nutans Siebold & Zuccarini was collected from Guizhou province, China, in October, 2018, and identified by Prof. Cheng-Ming Dong (Henan University of Chinese Medicine). A voucher specimen (no. 201810) was deposited in the Research Department of Natural Product Chemistry, School of Pharmacy, Henan University of Chinese Medicine.Extraction and isolation
The dried whole plant of G. nutans (17.5 kg) were extracted with 95% ethanol under reflux for three times (2 h/time). After concentration and combination, the crude extract was dispersed in 0.5% HCl and extracted with isopyknic EtOAc for three times. The acidic water layer was basified with 10% ammonia solution to pH 9–10 and extracted with EtOAc to give an alkaloidal extract (17 g). The alkaloidal extract was subjected to MPLC on ODS column, eluting gradually with MeOH–H2O (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80, 40:60, 60:40, 80:20, and 100:0, v/v) to afford five fractions Fr. A–E. Fr. B was applied to a silica gel column and eluted with a CH2Cl2–MeOH gradient (from 100:0 to 0:100) to afford 11 fractions Fr. B1–B11. Fr. B9 was further purified by semi-preparative HPLC eluted with 32% MeOH–H2O to yield compound 1 (20 mg, tR = 64 min). Fr. B10 was chromatographed over pre-HPLC (MeOH–H2O, 22:78, v/v) to yield compound 2 (12.5 mg, tR = 32 min). Fr. B4 was further purified by pre-HPLC (MeOH–H2O, 40:60, v/v) to afford compounds 6 (2.1 mg, tR = 57 min), 7 (1.5 mg, tR = 50 min), and 8 (4.6 mg, tR = 38 min). Fr. B7 was applied to Sephadex LH-20 with MeOH eluent to obtain ten fractions Fr. B7-1–B7-10, and then compound 9 (13.4 mg, tR = 21 min) was obtained from Fr. B7-4 by pre-HPLC (MeOH–H2O, 35:65, v/v). Fr. C was subjected to Sephadex LH-20 (MeOH) to give four fractions Fr. C1–C4. Fr. C3 was chromatographed on a silica gel column eluting with CH2Cl2–MeOH (20:1, 13:1, 1:0) to give 16 subfractions Fr. C3-1–C3-16. Compound 4 (3.6 mg, tR = 42 min) was obtained from Fr. C3-5 by pre-HPLC (48% MeOH), and compound 5 (2.8 mg, tR = 23 min) was yielded from Fr. C3-12 by pre-HPLC (57% MeOH). Compound 3 (41 mg) was purified by Sephadex LH-20 (MeOH) and recrystallization from Fr. D.
:80, 40:60, 60:40, 80:20, and 100:0, v/v) to afford five fractions Fr. A–E. Fr. B was applied to a silica gel column and eluted with a CH2Cl2–MeOH gradient (from 100:0 to 0:100) to afford 11 fractions Fr. B1–B11. Fr. B9 was further purified by semi-preparative HPLC eluted with 32% MeOH–H2O to yield compound 1 (20 mg, tR = 64 min). Fr. B10 was chromatographed over pre-HPLC (MeOH–H2O, 22:78, v/v) to yield compound 2 (12.5 mg, tR = 32 min). Fr. B4 was further purified by pre-HPLC (MeOH–H2O, 40:60, v/v) to afford compounds 6 (2.1 mg, tR = 57 min), 7 (1.5 mg, tR = 50 min), and 8 (4.6 mg, tR = 38 min). Fr. B7 was applied to Sephadex LH-20 with MeOH eluent to obtain ten fractions Fr. B7-1–B7-10, and then compound 9 (13.4 mg, tR = 21 min) was obtained from Fr. B7-4 by pre-HPLC (MeOH–H2O, 35:65, v/v). Fr. C was subjected to Sephadex LH-20 (MeOH) to give four fractions Fr. C1–C4. Fr. C3 was chromatographed on a silica gel column eluting with CH2Cl2–MeOH (20:1, 13:1, 1:0) to give 16 subfractions Fr. C3-1–C3-16. Compound 4 (3.6 mg, tR = 42 min) was obtained from Fr. C3-5 by pre-HPLC (48% MeOH), and compound 5 (2.8 mg, tR = 23 min) was yielded from Fr. C3-12 by pre-HPLC (57% MeOH). Compound 3 (41 mg) was purified by Sephadex LH-20 (MeOH) and recrystallization from Fr. D.
Nutanoside A (1), white acicular crystal; [α]25D-68.28 (c 0.07, MeOH); IR νmax 3369, 1645, 1463, 1297, 1249, 1207, 1017 cm−1; 1H NMR (500 MHz, DMSO-d6) and 13C NMR (125 MHz, DMSO-d6) see Table 1; HR-ESI-MS m/z 531.2359 [M + H]+ (calcd for C27H35N2O9, 531.2343).
Nutanoside B (2), yellowish oil; [α]25D-13.10 (c 0.07, MeOH); IR νmax 3314, 2923, 1731, 1593, 1482, 1458, 1230, 1244, 1206, 1175, 1076, 1042, 879, 742 cm−1; 1H NMR (500 MHz, DMSO-d6) and 13C NMR (125 MHz, DMSO-d6) see Table 1; HR-ESI-MS m/z 547.2296 [M + H]+ (calcd for C27H35N2O10, 547.2292).
Nutanester A (6), yellowish amorphous powder; IR νmax 3424, 1698, 1640, 1608, 1502, 1462, 1341, 1277, 1126, 1066, 1018, 768, 699 cm−1; 1H NMR (500 MHz, DMSO-d6) and 13C NMR (125 MHz, DMSO-d6) see Table 2; HR-ESI-MS m/z 517.1334 [M + Na]+ (calcd for C23H26O12Na, 517.1322).
Nutanester B (7), yellowish amorphous powder; IR νmax 3486, 2920, 1649, 1258, 775, 725, 701 cm−1; 1H NMR (500 MHz, DMSO-d6) and 13C NMR (125 MHz, DMSO-d6) see Table 2; HR-ESI-MS m/z 547.1429 [M + Na]+ (calcd for C24H28O13Na, 547.1428).
Absolute configurations determination of sugar moieties
Compounds 1–2 (each 1.0 mg) were separately treated with 2 M HCl (2 mL) for 3 h at 85 °C. Each reaction product was dissolved in H2O after evaporation and then extracted with CHCl3 for three times. After being concentrated to dryness, the aqueous layer was subjected to HPLC on a chiral-phase CHIRALPAK AD-H column (250 × 4.6 mm) eluting with n-hexane: EtOH:TFA (750:250:0.25, v/v/v, 0.5 mL min−1), and equipped with an evaporative light scattering detector (ELSD). D-Glucose was detected in the acid hydrolysates of compounds 1–2 according to their same retention times with those of known D-glucose (tR = 18.3 min) in the same condition.
Compounds 6–7 (each 1.0 mg) were treated with 6% NaOMe in MeOH (2 mL) at room temperature for 2 h. The reaction mixture was dissolved in H2O after evaporation and then extracted with CHCl3 for three times. The aqueous layer was dried and then D-glucose was determined by using acid hydrolysis method described above.
Anti-inflammatory assay in BV2 cells
Cell viability was detected by MTT method. Cells were seeded in 96-well plates (3 × 105 cells per mL) and incubated for 24 h. After treatment with tested compounds for 24 h, cells were incubated with MTT (0.25 mg mL−1) for 4 h at 37 °C. The culture medium was removed, and then the crystals were dissolved in DMSO. The cell viability rate was measured by optical densities at 570 nm with a microplate reader.The nitrite concentration was measured in the supernatant of cells by the Griess reagent system according to protocol. Cells were seeded into 96-well plates and treated with tested compounds in presence of LPS (100 ng mL−1) for 24 h. 50 μL of culture supernatant fluids were mixed with 50 μL Griess reagent at room temperature, and then the absorbance at 540 nm was determined after 15 min.
Quantitative real-time PCR
BV-2 microglial cells were seeded in 96-well plates (4 × 105 cells per mL) and incubated for 24 h. After treatment with tested compounds for 1 h, cells were incubated with LPS (100 ng mL−1) for 24 h. Total RNA was extracted with Trizol (Invitrogen) according to the manufacturer's protocol, and qRT-PCR assays were performed with the CFX Connect™ real-time PCR detection system (Bio-Rad) using total RNA and the GoTaq one-step real-time PCR kit with SYBR green.Cytotoxicity assay
A MTT assay was used to determine the cytotoxicity of each compound against two cultured human cancer cell lines. The cell lines used were U87 (human glioma cell line) and HCT116 (human colon cancer cell line). In brief, cells suspensions were plated in 96-well microtiter plates (2.5 × 104 cells per mL) and incubated for 24 h at 37 °C under 5% CO2. After treatment with tested compounds for 72 h, cells were incubated with MTT (50 μL, 0.4%) for 4 h at 37 °C. Then, the MTT was removed from the wells and the formazan crystals were dissolved in DMSO (200 μL) for 10 min with shaking. The plate was read immediately on a microtiter plate reader at a wavelength of 570 nm to record the optical density. The IC50 value was defined as the concentration of the control in the MTT assay. Taxol was used as a positive control.ECD computation method
Conformational search of compound 1 was carried out by Spartan software using the MMFF94S force field. Then, the structural optimization of target conformations was performed by DFT theory at the B3LYP/6-31G* level. TDDFT ECD calculations for these optimized conformers were carried out at the B3LYP/6-31+G* level with a CPCM model in MeOH solvent.20Conclusions
In this research, a phytochemical investigation on the ethanol extract of G. nutans was carried out and led to the isolation of two new monoterpene indole alkaloid glycosides nutanoside A–B (1–2), two new phenolic glycoside esters nutanester A–B (6–7) and five known compounds (3–5, 8–9). Compounds 1 and 2 are two rare monoterpene indole alkaloids with the glucosyl moiety located at C-12. Moreover, compounds 1 and 2 represent the first two examples of enantiomer of ajmaline type monoterpene indole alkaloids. The results of anti-neuroinflammatory activities showed that compounds 3, 4 and 6 displayed significant inhibitory effects on NO production in over-activated BV2 microglial cells, with the IC50 values of 2.29, 6.36, and 8.78 μM, respectively, and compounds 1, 5, 7 could significantly inhibit the mRNA expression of inflammatory factors TNF-α and IL-6 induced by LPS in BV2 microglial cells at the effective concentration, which provide evidence for compounds 1, 3–7 to be used as leading compounds for medicine design for neurodegenerative diseases. In addition, compound 3 exhibited stronger cytotoxicities against U87 and HCT116 cell lines than taxol, which indicated that compound 3 could be potential anti-tumor drug candidates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research project was financially supported by the 2018 doctoral research start-up foundation of Henan University of Chinese Medicine (BSJJ2018-02), and innovation training program for college students of Henan University of Chinese Medicine (No. 202110471034).Notes and references
- F. D. Zhu, Y. J. Hu, L. Yu, X. G. Zhou, J. M. Wu, Y. Tang, D. L. Qin, Q. Z. Fan and A. G. Wu, Front. Pharmacol., 2021, 12, 683935 CrossRef CAS PubMed.
- S. Koppula, R. Alluri and S. R. Kopalli, EXCLI J., 2021, 20, 835–850 Search PubMed.
- M. Guo, Y. Hao, Y. Feng, H. Li, Y. Mao, Q. Dong and M. Cui, Front. Mol. Neurosci., 2021, 14, 630808 CrossRef.
- M. Colonna and O. Butovsky, Annu. Rev. Immunol., 2017, 35, 441–468 CrossRef CAS.
- D. V. Hansen, J. E. Hanson and M. Sheng, J. Cell Biol., 2018, 217, 459–472 CrossRef CAS PubMed.
- Z. Fan, D. J. Brooks, A. Okello and P. Edison, Brain, 2017, 140, 792–803 Search PubMed.
- J. D. Alibhai, A. B. Diack and J. C. Manson, FASEB J., 2018, 32, 5766–5777 CrossRef CAS.
- W. X. Yang, Y. F. Chen, J. Yang, T. Huang, L. L. Wu, N. Xiao and Y. H. Zhang, Fitoterapia, 2018, 124, 8–11 CrossRef CAS PubMed.
- J. H. Jiang, Y. M. Zhang, Y. Zhang, G. M. Yang and Y. G. Chen, Yunnan Chem. Technol., 2012, 39, 32–35 CAS.
- X. H. Zhong, L. Xiao, Q. Wang, B. J. Zhang, M. F. Bao, H. X. Cai and L. Peng, Phytochem. Lett., 2014, 10, 55–59 CrossRef CAS.
- T. Feng, X. N. Li, B. H. Zhang, Y. Li, X. H. Cai, Y. P. Liu and X. D. Luo, Bioorg. Med. Chem. Lett., 2013, 23, 5563–5565 CrossRef CAS.
- W. X. Yang, T. Huang, J. X. Zhang, J. H. Liu, X. J. Hao and Y. H. Zhang, Chin. Pharm. J., 2016, 51, 1113–1115 CAS.
- G. H. Xie, L. Ma, Z. P. Zhang and L. H. Hu, Chin. J. Nat. Med., 2007, 5, 255–258 CAS.
- M. Harada and Y. Ozaki, Chem. Pharm. Bull., 1978, 26, 48–52 CrossRef CAS.
- M. R. Yagudaev, Chem. Nat. Compd., 1982, 18, 693–696 CrossRef.
- F. Bahadori, G. Topçu, M. Boğa, A. Türkekul, U. Kolak and M. Kartal, Nat. Prod. Commun., 2012, 7, 731–734 CrossRef CAS.
- Z. Z. Zhang, H. N. ElSohly, M. R. Jacob, D. S. Pasco, L. A. Walker and A. M. Clark, J. Nat. Prod., 2001, 64, 1001–1005 CrossRef CAS.
- Y. L. Wang, Y. J. Li, A. M. Wang, X. He, S. G. Liao and Y. Y. Lan, J. Asian Nat. Prod. Res., 2010, 12, 765–769 CrossRef CAS.
- J. Li, F. Li, Y. Y. Lu, Y. J. Zhang and X. S. Huang, Guangxi Sci., 2006, 13, 217–218 CAS , 225.
- Y. Y. Si, M. X. Tang, S. Lin, G. Chen, H. M. Hua, J. Bai, Y. B. Wang, H. F. Wang and Y. H. Pei, J. Asian Nat. Prod. Res., 2019, 21, 528–534 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The 1D & 2D NMR, and HR-ESI-MS data of compounds 1–9. See DOI: 10.1039/d1ra05204g |
| This journal is © The Royal Society of Chemistry 2021 |