
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-assembled nanotubes with controlled curvature radius using a hydrogen bond regulation strategy†
Lai-Cheng Zhou‡
,
Yun-Han Yang‡,
Ran He‡,
Yang Qin and
Ling Zhang *
*
PCFM Lab, GDHPRC Lab, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, China. E-mail: ceszl@mail.sysu.edu.cn
First published on 22nd October 2021
Abstract
The design of co-organized nanotube systems with controlled curvature radius that are realized by tilt modulation of co-assembled molecules, induced by the strength of non-covalent interactions in aqueous media, remains a significant challenge. Here, we report success in utilizing a hydrogen bond regulation strategy to stimulate molecular tilt for the formation of nanotubes with controlled curvature radius based on the co-assembly of two kinds of achiral cationic building blocks in aqueous solution. Computations and electron microscopy experiments suggest that the nanotube curvature radius drastically decreases as the tilt angle θ of co-assembled molecules increases with an increase of hydrogen bond strength. Interestingly, a slight change in the co-assembled molecular tilt causes a drastic change in the nanotube curvature radius.
1. Introduction
Tubular and helical nanostructures with controllable curvature radius from the co-assembly of achiral molecules in aqueous solution are attracting attention owing to the need to improve miniaturization and device performance in the emerging fields of bio-nanotechnology and related nanotechnologies, and microchip and microelectronics applications.1,2 Understanding how the strength of non-covalent interactions affects nanotube curvature radius at the molecular level would facilitate a more efficient and systematic approach to generating rationalized tubular libraries. Tubular and helical structures with different curvature radius seem to be rather common in nature. A key feature of nature's tubular and helical structures via complex multifunctional co-assembly is that tubular and helical structures with different curvature radius can play a crucial role in a vast number of physical, chemical, and biological process in aqueous environment.3 Thus it is very important to have an overall understanding of the co-assembly process of tubular and helical nanostructures with controllable curvature radius in aqueous solution for materials science and biology.Over the past years, the growing number of synthesized monomers has contributed significantly to the development of general design rules to facilitate the nanotubes with the change of the diameters for supramolecular assembly in water.4–9 However, to further guide their rational design, it is necessary to understand factors that influence the strengths of competing noncovalent interactions between co-assembled monomers on the molecular tilt for controlled nanotube curvature radius. Here, we demonstrate the impact of hydrogen bonding strength on the molecular tilt for controlled curvature radius of co-assembled nanotubes in aqueous solution.
We recently found a CO2-triggered co-assembled nanotube system with controlled chirality through a CO2-bubble-induced vortex.10 For the co-assembled nanotubes, the π–π interactions between the aromatic rings of azobenzene and intermolecular hydrogen bonding interactions (N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) between co-assembled cations play very important roles for forming chiral helical assemblies, resulting in the emergence and amplification of supramolecular chirality. Herein, we trigger co-assemblies to control their nanotube curvature radius using a hydrogen bond regulation strategy. To achieve this, exquisite molecular design of hydrogen bond in the molecular structure need to be localized so as to adjust co-assembled aromatic molecular tilt. To date, although there are several reports on the supramolecular curvature regulation based on molecular self-assembly through altering the packing mode of aromatic molecules,11–14 controlled nanotube curvature radius based on the co-assembled molecular tilt induced by the hydrogen bond strength in aqueous solution is rather scarce and remains a significant challenge.
C) between co-assembled cations play very important roles for forming chiral helical assemblies, resulting in the emergence and amplification of supramolecular chirality. Herein, we trigger co-assemblies to control their nanotube curvature radius using a hydrogen bond regulation strategy. To achieve this, exquisite molecular design of hydrogen bond in the molecular structure need to be localized so as to adjust co-assembled aromatic molecular tilt. To date, although there are several reports on the supramolecular curvature regulation based on molecular self-assembly through altering the packing mode of aromatic molecules,11–14 controlled nanotube curvature radius based on the co-assembled molecular tilt induced by the hydrogen bond strength in aqueous solution is rather scarce and remains a significant challenge.
2. Results and discussion
In this paper, to construct the models of hydrogen bond strength regulation, we have selected 2-(3-dimethylamino-propylamino)-N-ethyl-acetamide for introducing N,N-dimethylamino species by the activation of carboxylate groups to form carbodiimide adducts to the azobenzene molecule (Fig. 1a; see Fig. S1† for molecular structure of compound 2, as well as for that of compound 3 based on 1-ethyl-3-(3-dimethylaminoisopropyl) carbodiimide (EDC),10 as hydrogen-bond donors of molecular coassembly). To investigate co-assembled tubular nanostructures with controlled curvature radius via the hydrogen bond regulation, we chose compound 1 as the hydrogen-bond acceptor of molecular coassembly, which is made up of a hydrophobic tail containing alkyl and azobenzene functionalities attached through an ethylene glycol hydrophilic linker to N,N-dimethylamino head groups (Fig. 1a). Ureas are excellent hydrogen-bond donors,15 and are geometrically compatible with the hydrogen-bond acceptor capability of compound 1.16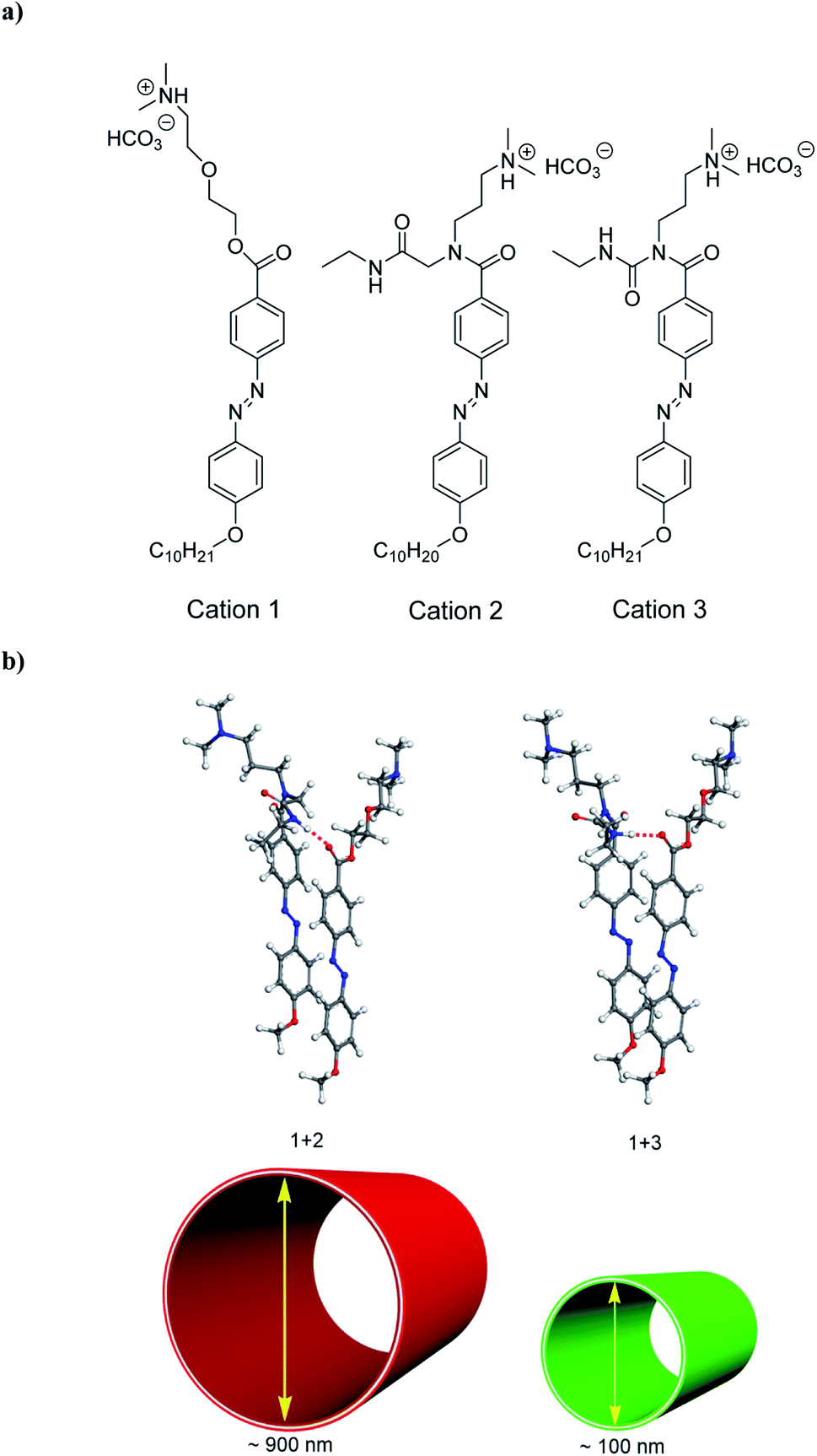 | ||
| Fig. 1 (a) Molecular structures of cations 1, 2 and 3. (b) Schematic representation of the formation of CO2-triggered co-organized nanotubes of 1 + 2 and 1 + 3 systems with controllable curvature radius by tilt modulation of co-assembled molecules induced by the hydrogen bond regulation. | ||
Compound 2 could be dissolved in CH2Cl2 at room temperature, but the compound was suspended in water, being consistent with those of compounds 1 and 3.10 When CO2 gas was bubbled through the aqueous solution of compounds 1 + 2 system with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, they could be dissolved in water to form a viscous, yellow-orange solution. By introducing CO2 into the solution, the N,N-dimethylamino species were gradually protonated by the acidic gaseous medium (Fig. 1a), leading to the formation of cations 1 + 2 system, and a successive variation of the hydrophilic–hydrophobic balance and a better dissolution in water.
:1, they could be dissolved in water to form a viscous, yellow-orange solution. By introducing CO2 into the solution, the N,N-dimethylamino species were gradually protonated by the acidic gaseous medium (Fig. 1a), leading to the formation of cations 1 + 2 system, and a successive variation of the hydrophilic–hydrophobic balance and a better dissolution in water.
Interestingly, cations 1 + 2 system (protonated monomers) could format co-assembled tubular structures with the different curvature radius in aqueous solution, compared with cations 1 + 3 system (Fig. 1b). Representative cryogenic transmission electron microscopy (cryo-TEM) images showed the CO2-triggered co-assembly of high aspect ratio nanotubes in aqueous solution, which were straight and discrete. In comparison with the inner diameter of ∼100 nm for cations 1 + 3 system,10 the nanotubes had a uniform inner diameter of ∼900 nm for cations 1 + 2 system (Fig. 2a). Considering the molecular structure of cations 1 + 2 system, the wall thickness of the nanotube of cations 1 + 2 system (∼25 nm, indicated by the red arrows in Fig. 2b) suggests a multilayer structure, being different from a bilayer structure of cations 1 + 3 system.10 For a control experiment with the presence of only cation 1 or 2 or 3, no tubular and helical nanostructures could be detected in the TEM observations, indicating that molecular interactions between cations 1 and 2 system (or cations 1 and 3 system) may play very important roles for co-assembled nanotubes.
 | ||
| Fig. 2 (a) Cryo-TEM images of co-assembled nanotubes of cations 1 + 2 system generated by exposure to CO2 under aqueous conditions. (b) Summed cryo-TEM images of a cross-section of the co-assembled nanotubes of cations 1 + 2 system. Dark regions are multilayer walls. Red bar indicates an approximate thickness of ∼25 nm. | ||
Another observation was to monitor the formation of co-assembled nanotubes of cations 1 + 2 system based on the change in electronic absorption spectra in water over CO2 aeration time (Fig. 3). For cations 1 + 2 system, a weak signal was observed before CO2 treatment, and the absorption increased in intensity upon bubbling CO2, and a further increase of intensity was observed upon prolonged CO2 aeration time. For a comparison of the spectra of the aggregates, the UV/vis spectra of the protonated monomer (cation 2) and the non-protonated monomer (compound 2) in water as control experiments are also displayed in Fig. S2a.† A weak signal of compound 2 before CO2 treatment and the increase of absorption band intensity upon CO2 stimulation were also observed, being consistent with those of the aggregates in Fig. 3.
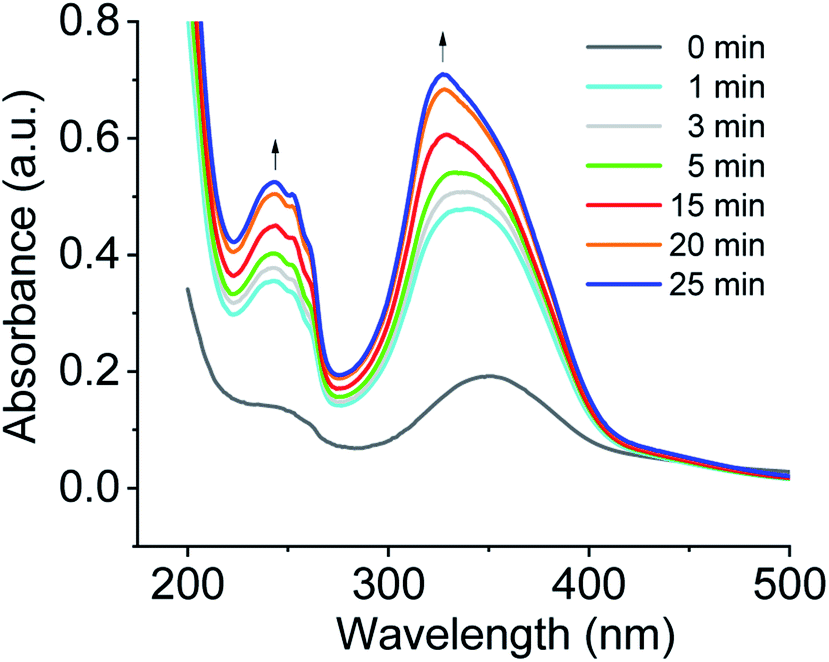 | ||
| Fig. 3 Monitoring the formation of co-assembled nanotubes of cations 1 + 2 system based on the change in electronic absorption spectra in water over CO2 aeration time at room temperature. | ||
Exciton coupling of azobenzene-based chromophores results from the interaction of their transition dipole moments in space to promote the alignment of molecules and the formation of a well-organized bilayer. In DMSO solution, an absorption maxima of the non-protonated and protonated monomer 2 appeared at ∼360 nm, characteristic of π–π* transition of trans-azobenzene derivatives (see Fig. S2b†).17 For the co-assembled bilayer structure of cations 1 + 2 system formed upon CO2 stimulation in water, the absorption maximum of trans azobenzene blue shifted to ∼327 nm (see Fig. S3†) relative to cations 1 + 2 system (λmax = 362 nm) in DMSO solution. This hypsochromic shift is indicative of an increase in the π–π stacking of the azobenzenes within the tubules in water.18 In comparison with cations 1 + 2 system, this further blue shift of cations 1 + 3 system is characteristic of an increase in the intermolecular electronic coupling between the stacked azobenzene-based chromophores of cations 1 + 3 system.19 It was also found that no absorption peaks appear at ∼450 nm which is ascribable to the n–π* transition of the cis isomer, suggesting the absence of cis-azobenzene during the co-assembly. When CO2 gas was bubbled through the aqueous solution of compounds 1 + 2 system under illumination, any assembly morphologies could not be observed by TEM, indicating that cis-azobenzene of the system could not form any assembly morphologies.
In addition, we employed Fourier-transform infrared spectroscopy (FTIR) to provide direct evidences on hydrogen bonding between OC bonds of monomer 1 and N–H bonds of monomer 2 for 1 + 2 nanotube system by observing the spectral absorption region (1692–1758 cm−1) of free carbonyl groups (Fig. 4). Generally, the infrared absorption spectra of the carbonyl groups might be shifted toward a lower frequency upon the formation of the hydrogen bonding (N–H⋯OC).20 Infrared spectroscopy of monomer 1 displayed that 1714 cm−1 is the peak of the vibration associated with the free carbonyl group (Fig. 4a), while the carbonyl absorption peak of the amide group of monomer 2 in Fig. 4b is approximately located at 1638 cm−1. Fig. 4c showed that the original ν(CO) band at 1714 cm−1 observed almost disappears for 1 + 2 co-assembled nanotube system, suggesting the formation of favorable intermolecular hydrogen bonding between OC bonds of monomer 1 and N–H bonds of monomer 2 for 1 + 2 nanotube system. Thus, we attribute the formation of the co-assembled nanotubular structures to the crystalline domain formation of the hydrophobic moieties in water, to π-stacking of the azobenzene moieties, to intermolecular hydrogen bonds (N–H⋯OC) between cations 1 and 2 and to electrostatic screening by condensed HCO3− counterions.
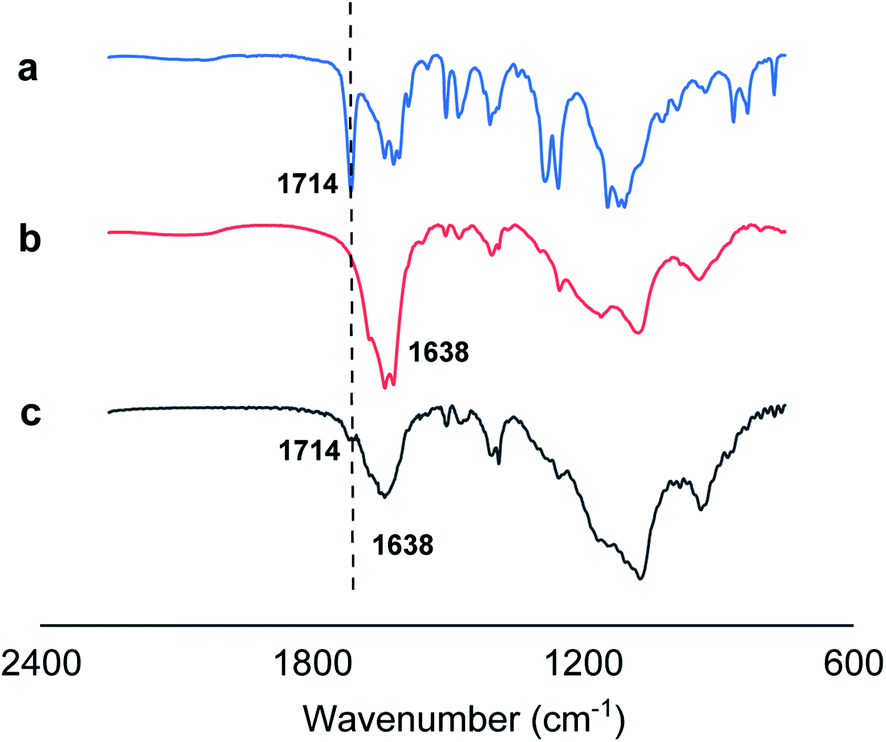 | ||
| Fig. 4 IR spectra (2250 to 750 cm−1) of monomer 1 (a), monomer 2 (b), and 1 + 2 nanotube system (c). | ||
Cryo-TEM measurements showed helical nanoribbons during the course of the nanotube formation, which indicated the helical stacking of the azobenzene units of cations 1 + 2 system (and cations 1 + 3 system) on a molecular level. As shown in Fig. 5, cations 1 + 2 system (or cations 1 + 3 system) could predominantly produce helical ribbons with the pitch angle ϕ 45° (or ϕ 55°) by cryo-TEM observations, respectively. The results show that the pitch angle ϕ of the helical ribbon could be regulated by hydrogen bond, indicating the alternation of the co-assembled molecular tilt by hydrogen bond regulation.
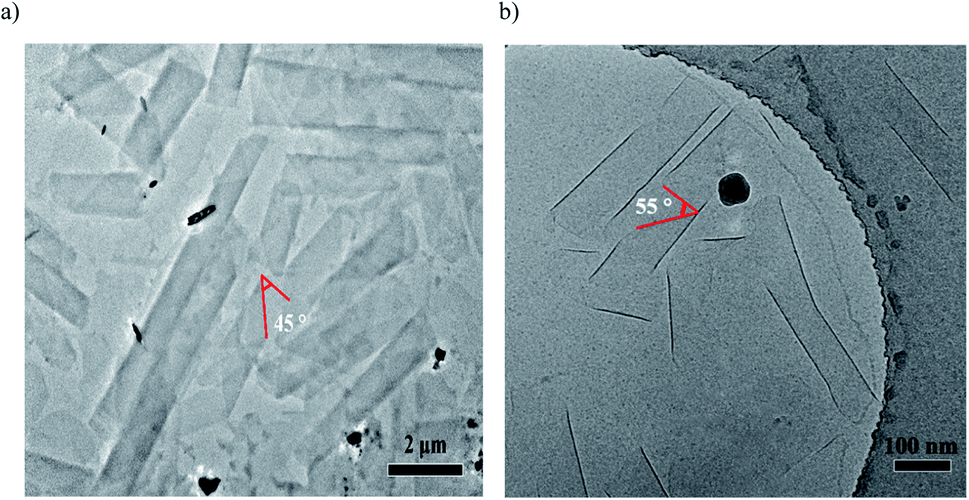 | ||
| Fig. 5 Cryo-TEM images of co-assembled helical ribbons with the pitch angle for (a) cations 1 + 2 system and (b) cations 1 + 3 system10 generated by exposure to CO2 under aqueous conditions. | ||
Theoretical calculations were applied to further clarify the co-assembled molecular tilt for the controlled nanotube curvature radius of 1 + 2 and 1 + 3 systems based on the hydrogen bond regulation strategy in water phase. The energy-minimized structures of all the building blocks (1, 2 and 3) were obtained by the density functional theory (DFT) at B3LYP/6-31++G (d, p) level. And the energy-minimized geometries of co-assembled structures (1 + 2 and 1 + 3 systems) were obtained by dihedral scan at M06-2X/6-31G (d) level. The polarizable continuum model (PCM) with water as solvent was invoked, see the ESI† for more comprehensive calculations details.
The interaction energies of the co-assemblies (1 + 2 and 1 + 3 systems) were obtained after scanning of the dihedral angle, and the related energetic features are listed in Table S1.† For better comparison with experiments, we analyze the calculated results in aqueous phase. It is noteworthy that the interaction energies of 1 + 2 and 1 + 3 systems are all less than zero, indicating that the formation of the co-assemblies are thermodynamically favorable and stable. The most stable co-assembled conformations of 1 + 2 and 1 + 3 systems occur at 13°, and −19°, respectively. Structures of the co-assemblies with the strongest interaction energy in Fig. 6 show the hydrogen bond distances and the tilt angles. In the two co-assembled systems (1 + 2 and 1 + 3 systems), the N–H⋯OC distances are 2.057 Å and 1.903 Å, respectively. Furthermore, the tilt angle θ of co-assembled molecules in a membrane were 18.041° and 21.737°, respectively. Both the N–H⋯OC hydrogen bond strength and the tilt angle θ of co-assembled molecules showed the same trend, i.e. 1 + 2 < 1 + 3, indicating that the tilt angle θ of co-assembled molecules increases with an increase of hydrogen bond strength. Furthermore, according to the blue shift observed in the above absorption spectra of cations 1 + 2 and 1 + 3 systems in water, we believe that the observed further blue shift of cations 1 + 3 system is caused by greater π–π overlap of adjacent azobenzene molecules induced by an increase of hydrogen bond strength, which probably promotes a better alignment of the transition dipoles across co-assembled azobenzene molecules of cations 1 + 3 system. Taken together, the increase of hydrogen bond strength results in stronger excitonic coupling between the co-assembled azobenzene molecules, as well as greater π–π overlap, leading to an increased tilt angle θ of co-assembled molecules.
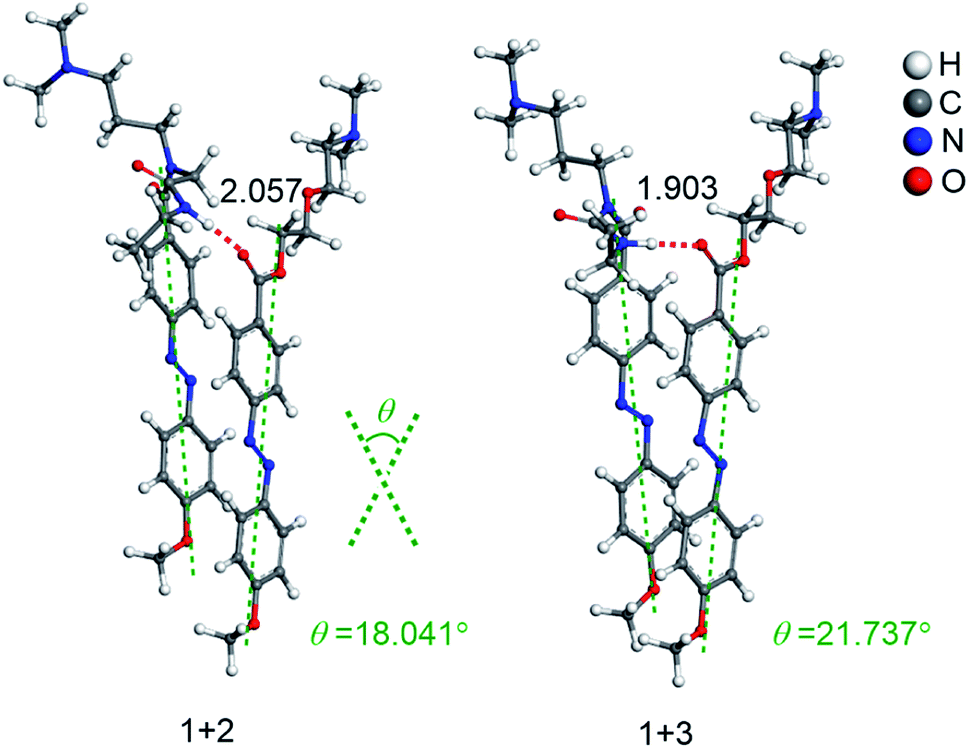 | ||
| Fig. 6 Optimized structures (in Å) of two co-assembled systems in the aqueous phase at M06-2X/6-31G (d) level of theory. | ||
The theoretical calculation results show good correlation among hydrogen bond strength, the tilt angle θ of co-assembled molecules in a membrane and the curvature radius of co-assembled nanotubes. As the tilt angle θ of co-assembled molecules increases with an increase of hydrogen bond strength, the nanotube curvature radius drastically decreases, being consistent with this prediction of these mathematical models and the elastic theories.1a,4b,21 And a slight change in the co-assembled molecular tilt causes a drastic change in the nanotube curvature radius. Computations and experiments suggest that the controlled nanotube curvature radius could be realized by regulating co-assembled molecular tilt of 1 + 2 and 1 + 3 systems with hydrogen bond strength.
3. Conclusions
We have demonstrated co-assembled tubular nanostructures with controlled curvature radius in water that are realized by co-assembled molecular tilt modulation based on a hydrogen bond regulation strategy. This is the example of how the sense of the co-assembled molecular tilt induced by hydrogen bond strength can determine the curvature radius of a co-assembled nanotube system, and such studies could provide a new strategy to regulate the nanotube curvature radius of nanostructures based on the study of co-assembled molecular tilt induced by the strength of non-covalent interactions.Author contributions
Lai-Cheng Zhou: investigation, methodology and synthesis; Yun-Han Yang: theoretical calculation, writing–original draft; Ran He: investigation; Yang Qin: investigation; Ling Zhang: conceptualization, supervision and writing–review and editing. All authors approve the publication of the manuscript in the current version.Conflicts of interest
There are no interests to declare.Acknowledgements
This research was supported by the program of Science and Technology Program of Guangzhou (Grant 201804010017), National Natural Science Foundation of China (Grant 21374137), Natural Science Foundation of Guangdong Province (Grant 2014A030313194), and Science and Technology Program of Guangdong Province (Grant 2017A050506021). The authors gratefully thank the staff of Institute of Biophysics, Chinese Academy of Sciences, Beijing, People's Republic of China, for the cryo-TEM measurements.References
- (a) T. Shimizu, M. Masuda and H. Minamikawa, Supramolecular Nanotube Architectures Based on Amphiphilic Molecules, Chem. Rev., 2005, 105, 1401–1443 CrossRef CAS PubMed; (b) T. Aida, E. W. Meijer and S. I. Stupp, Functional Supramolecular Polymers, Science, 2012, 335, 813–817 CrossRef CAS PubMed; (c) Z. Huang, S. K. Kang, M. Banno, T. Yamaguchi, D. Lee, C. Seok, E. Yashima and M. Lee, Pulsating Tubules from Noncovalent Macrocycles, Science, 2012, 337, 1521–1526 CrossRef CAS PubMed; (d) T. G. Barclay, K. Constantopoulos and J. Matisons, Nanotubes Self-assembled from Amphiphilic Molecules via Helical Intermediates, Chem. Rev., 2014, 114, 10217–10291 CrossRef CAS PubMed , and references therein..
- (a) M. Liu, L. Zhang and T. Wang, Supramolecular Chirality in Self-assembled Systems, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed; (b) E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Supramolecular Polymers in Aqueous Media, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- P. L. Luisi, The Emergence of Life: From Chemical Origins to Synthetic Biology, Cambridge University Press, New York, 2010 Search PubMed.
- (a) W. Helfrich and J. Prost, Intrinsic Bending Force in Anisotropic Membranes Made of Chiral Molecules, Phys. Rev. A, 1988, 38, 3065–3068 CrossRef CAS PubMed; (b) P. Nelson and T. Powers, Rigid Chiral Membranes, Phys. Rev. Lett., 1992, 69, 3409–3412 CrossRef CAS PubMed; (c) Z. Ou-Yang and J. Liu, Phys. Rev. A, 1991, 43, 6826–6836 CrossRef CAS PubMed; (d) J. V. Selinger and J. M. Schnur, Theory of Chiral Lipid Tubules, Phys. Rev. Lett., 1993, 71, 4091–4094 CrossRef CAS PubMed; (e) J. M. Schnur, Lipid Tubules: A Paradigm for Molecularly Engineered Structures, Science, 1993, 262, 1669–1676 CrossRef CAS PubMed.
- (a) B. N. Thomas, R. C. Corcoran, C. L. Cotant, C. M. Lindemann, J. E. Kirsch and P. J. Persichini, Phosphonate Lipid Tubules I, J. Am. Chem. Soc., 1998, 120, 12178–12186 CrossRef CAS; (b) B. N. Thomas, C. M. Lindemann, R. C. Corcoran, C. L. Cotant, J. E. Kirsch and P. J. Persichini, Phosphonate Lipid Tubules II, J. Am. Chem. Soc., 2002, 124, 1227–1233 CrossRef CAS PubMed.
- (a) M. Markowitz and A. Singh, Self-assembling Properties of 1, 2-diacyl-sn-glycero-3-phosphohydroxyethanol: a Headgroup-modified Diacetylenic Phospholipid, Langmuir, 1991, 7, 16–18 CrossRef CAS; (b) M. A. Markowitz, J. M. Schnur and A. Singh, The Influence of the Polar Headgroups of Acidic Diacetylenic Phospholipids on Tubule Formation, Microstructure Morphology and Langmuir Film Behavior, Chem. Phys. Lipids, 1992, 62, 193–204 CrossRef CAS.
- F. Gobeaux, N. Fay, C. Tarabout, C. Mériadec, F. Meneau, M. Ligeti, D.-A. Buisson, J.-C. Cintrat, K. M. H. Nguyen, L. Perrin, C. Valéry, F. Artzner and M. Paternostre, Structural Role of Counterions Adsorbed on Self-Assembled Peptide Nanotubes, J. Am. Chem. Soc., 2012, 134, 723–733 CrossRef CAS PubMed.
- (a) D. G. Rhodes and A. Singh, Structure of Polymerizable Lipid Bilayers IV: Mixtures of Long Chain Diacetylenic and Short Chain Saturated Phosphatidylcholines and Analogous Asymmetric Isomers, Chem. Phys. Lipids, 1991, 59, 215–224 CrossRef CAS PubMed; (b) M. S. Spector, A. Singh, P. B. Messersmith and J. M. Schnur, Chiral Self-assembly of Nanotubules and Ribbons from Phospholipid Mixtures, Nano Lett., 2001, 1, 375–378 CrossRef CAS; (c) A. Singh, E. M. Wong and J. M. Schnur, Toward the Rational Control of Nanoscale Structures Using Chiral Self-assembly: Diacetylenic Phosphocholines, Langmuir, 2003, 19, 1888–1898 CrossRef CAS.
- Z. Huang, S.-K. Kang, M. Banno, T. Yamaguchi, D. Lee, C. Seok, E. Yashima and M. Lee, Pulsating Tubules from Noncovalent Macrocycles, Science, 2012, 337, 1521–1526 CrossRef CAS PubMed.
- L. Zhang, L. Zhou, N. Xu and Z. Ouyang, A Carbon Dioxide Bubble-induced Vortex Triggers Co-assembly of Nanotubes with Controlled Chirality, Angew. Chem., Int. Ed., 2017, 56, 8191–8195 CrossRef CAS PubMed.
- S. Yagai, Y. Goto, X. Lin, T. Karatsu, A. Kitamura, D. Kuzuhara, H. Yamada, Y. Kikkawa, A. Saeki and S. Seki, Self-organization of Hydrogen-bonding Naphthalene Chromophores into J-type Nanorings and H-type Nanorods: Impact of Regioisomerism, Angew. Chem., 2012, 124, 6747–6751 CrossRef.
- B. Adhikari, K. Aratsu, J. Davis and S. Yagai, Photoresponsive Circular Supramolecular Polymers: A Topological Trap and Photoinduced Ring-Opening Elongation, Angew. Chem., 2019, 131, 3804–3808 CrossRef.
- B. Adhikari, Y. Yamada, M. Yamauchi, K. Wakita, X. Lin, K. Aratsu, T. Ohba, T. Karatsu, M. J. Hollamby, N. Shimizu, H. Takagi, R. Haruki, S.-I. Adachi and S. Yagai, Light-induced Unfolding and Refolding of Supramolecular Polymer Nanofibers, Nat. Commun., 2017, 8, 15254–15263 CrossRef PubMed.
- M. J. Hollamby, K. Aratsu, B. R. Pauw, S. E. Rogers, A. J. Smith, M. Yamauchi, X. Lin and S. Yagai, Simultaneous SAXS and SANS Analysis for the Detection of Toroidal Supramolecular Polymers Composed of Noncovalent Supermacrocycles in Solution, Angew. Chem., 2016, 128, 10044–10047 CrossRef.
- (a) A. G. Doyle and E. N. Jacobsen, Small-Molecule H-Bond Donors in Asymmetric Catalysis, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed; (b) N. Volz and J. Clayden, The Urea Renaissance, Angew. Chem., Int. Ed., 2011, 50, 12148–12155 CrossRef CAS PubMed.
- (a) K. A. Haushalter, J. Lau and J. D. Roberts, An NMR Investigation of the Effect of Hydrogen Bonding on the Rates of Rotation about the C-N Bonds in Urea and Thiourea, J. Am. Chem. Soc., 1996, 118, 8891–8896 CrossRef CAS; (b) E. Fan, S. A. Van Arman, S. Kincaid and A. D. Hamilton, Molecular Recognition: Hydrogen-bonding Receptors that Function in Highly Competitive Solvents, J. Am. Chem. Soc., 1993, 115, 369–370 CrossRef CAS; (c) S. T. Victorio, D. Joyal, C. H. Wu, Z. L. Wen, L. Francesca, B. S. S. G. Karthick, R. Raisa, B. Mahsa, H. Alexander, T. T. Thuat, E. Pablo, K. V. Ilja, I. W. Judy and E. K. Roxanne, Thiosquaramide-based Supramolecular Polymers: Aromaticity Gain in a Switched Mode of Self-assembly, J. Am. Chem. Soc., 2020, 142, 19907–19916 CrossRef PubMed.
- A. A. Beharry and G. A. Woolley, Azobenzene Photoswitches for Biomolecules, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- (a) H. Menzel, B. Weichart, A. Schmidt, S. Paul, W. Knoll, J. Stumpe and T. Fischer, Small-Angle X-ray Scattering and Ultraviolet-Visible Spectroscopy Studies on the Structure and Structural Changes in Langmuir-Blodgett Films of Polyglutamates with Azobenzene Moieties Tethered by Alkyl Spacers of Different Length, Langmuir, 1994, 10, 1926–1933 CrossRef CAS; (b) Q. Bo and Y. Zhao, Fluorescence from an Azobenzene-containing Diblock Copolymer Micelle in Solution, Langmuir, 2007, 23, 5746–5751 CrossRef CAS PubMed.
- F. C. Spano, The Spectral Signatures of Frenkel Polarons in H-and J-aggregates, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed.
- (a) N. Nandi and D. Vollhardt, Effect of Molecular Chirality on the Morphology of Biomimetic Langmuir Monolayers, Chem. Rev., 2003, 103, 4033 CrossRef CAS PubMed; (b) G. Q. Wang, C. X. Zhang, X. H. Guo and Z. Y. Ren, Experimental FTIR and Simulation Studies on H-bonds of Model Polyurethane in Solutions. I: In Dimethylformamide (DMF), Spectrochim. Acta, Part A, 2008, 69, 407–412 CrossRef PubMed.
- Z. Ou-Yang and J. Liu, Helical Structures of Tilted Chiral Lipid Bilayers Viewed as Cholesteric Liquid Crystals, Phys. Rev. Lett., 1990, 65, 1679 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05251a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |