
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilica gel-immobilised chiral 1,2-benzenedisulfonimide: a Brønsted acid heterogeneous catalyst for enantioselective multicomponent Passerini reaction†
Achille Antenucci ab,
Francesco Marraa and
Stefano Dughera*a
ab,
Francesco Marraa and
Stefano Dughera*a
aDipartimnto di Chimica, Università di Torino, C.so Massimo d'Azeglio 48, 10125 Torino, Italy
bNIS Interdepartmental Centre, Reference Centre for INSTM, Università di Torino, via Gioacchino Quarello 15/A, 10135 Torino, Italy
First published on 29th July 2021
Abstract
A chiral heterogeneous catalyst derivative of (−)-4,5-dimethyl-3,6-bis(1-naphthyl)-1,2-benzenedisulfonimide is proven here to be efficient in a three-component asymmetric Passerini protocol, carried out in a deep eutectic solvent. Reaction conditions are mild and green, while enantioselectivity is excellent. The catalyst was easily recovered and reused with no decrease in its catalytic activity.
Introduction
Organocatalysis has become a highly dynamic research area.1 From the various groups of possible catalysts, Brønsted acids stand out as they present a range of benefits, including a lack of sensitivity to moisture and oxygen, ready availability, low cost and low toxicity.2 The importance of catalysis also highlights the fact that the ninth principle (use of catalysts should be preferred wherever possible) of Green Chemistry3 is devoted exclusively to this issue. In light of these, one of the key challenges for organic synthesis is to design and develop new and efficient catalysts that are active under mild and sustainably conditions.4 In order to develop increasingly sustainable and eco-compatible procedures, a widespread practice in catalysis is to convert a successful homogeneous organocatalyst into an heterogeneous catalytic system.5By using efficient heterogeneous catalysts, many processes can be carried out under milder conditions, significantly simplifying the usually cost-intensive and energy-consuming removal of the catalyst after reaction.6 Moreover, such catalysts create the possibility of their effective and simple recycling. For these reasons, heterogeneous Brønsted acid organocatalysts can be used effectively as a toolbox for Green Chemistry.7
The preparation of enantiopure chemicals has been one of the most rapid growth areas in chemistry over the last decade with heterogeneous catalysis being recognised as providing new possibilities.7 A plethora of different strategies has been developed to obtain enantioselectivity through heterogeneous catalysis: the use of chiral species to modify the solid surface,8 homogeneous formation of a chiral complex before reaction at the solid surface9 and the grafting of a chiral catalytic complex on a solid support, namely the heterogenised homogeneous catalyst.10
Our previous studies have seen 1,2-benzenedisulfonimide (1) and its chiral derivatives (−)-2 and (−)-3 used as excellent, versatile, safe and eco-friendly homogeneous Brønsted acid catalysts (Fig. 1).11a–f
 | ||
| Fig. 1 1,2-Benzenedisulfonimide (1) and its chiral derivative (−)-2 and (−)-3. | ||
More generally, chiral disulfonimides have received recently significant attention as robust and effective catalysts in asymmetric synthesis.12 In fact, they are not only more acidic11g than phosphoric acids but they also have real C2 symmetry which facilitates, especially in the presence of bulky substituents close to the acid site, an increase of stereochemical communication between the catalyst and the substrate.12a Furthermore, three chiral derivatives of disulfonimides namely adducts 4, 5 and 6 (Fig. 2) are commercially available from Sigma-Aldrich.
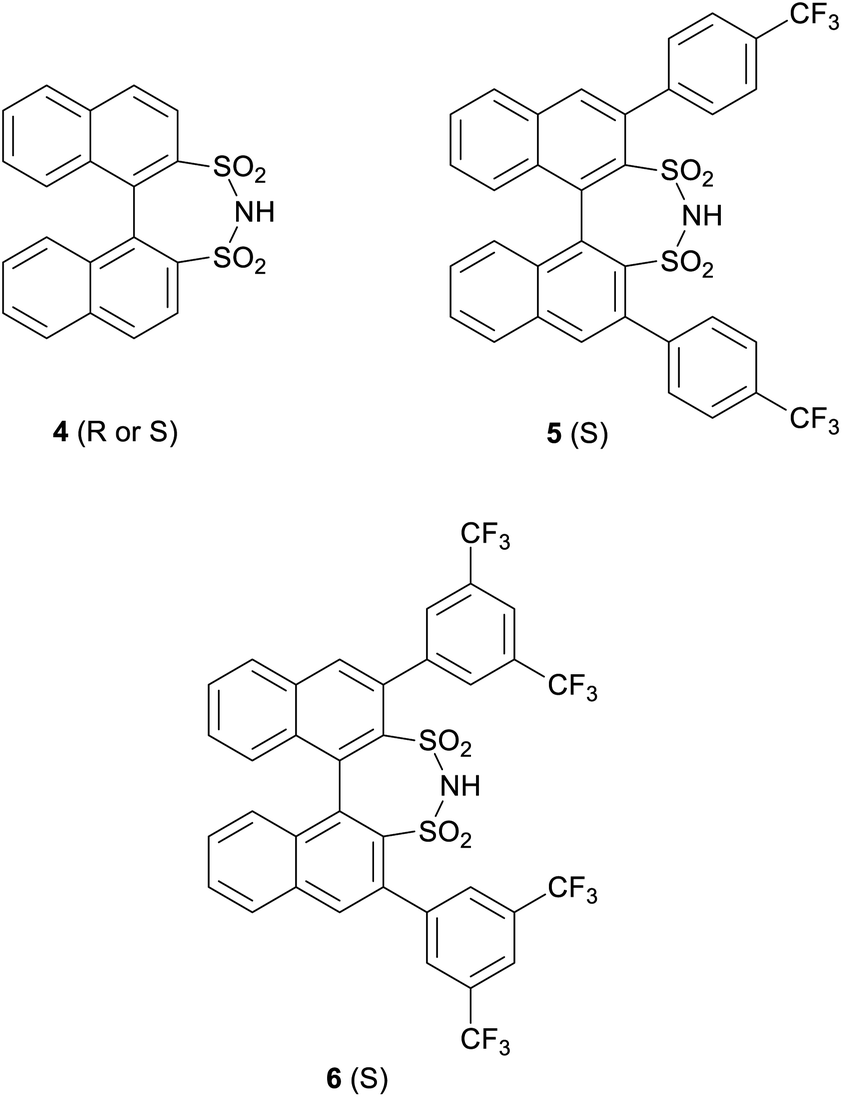 | ||
| Fig. 2 Commercial disulfonimides. | ||
In continuation of our research, we anchored, through an amide bond, an appropriately functionalised derivative of 1, namely 1,2-benzenedisulfonimide-4-yl propionic acid (8), to a 3-aminopropyl functionalized silica gel support (7) thus obtaining a new specie having increased robustness and recyclability with respect to (1), furthermore paving the way for its successful employment in heterogeneous catalysis (9; Scheme 1).13
 | ||
| Scheme 1 Synthesis of heterogeneous catalyst 9. | ||
Result and discussion
Preparation of the catalyst 11
Encouraged by above results, in order to transform homogeneous chiral catalysts (−)-3 into an efficient heterogeneous catalytic system, our first goal was to immobilize (−)-3 by means a strong covalent bond, namely an amide bond, on 3-aminopropyl functionalized silica gel (7; 1 mmol g−1 NH2 loading), selected as the solid support because of its commercial availability, chemical inertness and low toxicity. Moreover, it displays excellent chemical stability, high thermal and mechanical robustness. In order to form an amide bond, we needed to presence of two carboxyl groups on the central benzene ring. This objective was easily achieved by oxidising the methyl groups of (−) 3 with KMnO4 in a basic environment (Scheme 2), obtaining in this way (−)-4,5-dicarboxy-3,6-bis(1-naphthyl)-1,2-benzenedisulfonimide (10). As reported in our previous paper,13 the condensation between NH2 and COOH groups was promoted by N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC; Scheme 2). The material obtained was isolated via filtration over Büchner funnel and was washed firstly with dilute hydrochloric acid in order to fully restore the acid function of the sulfonimide and then with acetone in order to remove any possible organic residue. | ||
| Scheme 2 Synthesis of chiral heterogeneous catalyst 11. | ||
The analysis of the IR spectrum of this solid, compared with that of 10 and with that of 7, confirmed that the anchoring occurred with the formation of an amide bond (Fig. 3). In fact, In the IR spectrum of 11, two bands were evident at 1633 and 1542 cm−1. The band at 1633 cm−1 is due to the CO stretching vibration, while the band at 1542 cm−1 is due to NH bending vibration of secondary amides. The weak band observed at 1595 cm−1 is probably associated to NH bending vibration of sulfonimide NH.
 | ||
| Fig. 3 IR spectra of 7, 10 and 11. | ||
Once 11 was obtained, we decided to disfruit it as an heterogeneous chiral catalyst in order to develop a green asymmetric protocol for Passerini reaction (Scheme 3).14
 | ||
| Scheme 3 Passerini reaction. | ||
Use of the catalyst 11 in asymmetric Passerini reactions
Multicomponent reactions (MCRs) are chemical transformations in which more than two reagents afford a new product which incorporates all of the atoms of the reagents, with an efficient one-step and high atom economy approach to building up complex molecules in compliance to the first principle of green chemistry.15 In light of this, MCRs are a valid tool for achieving the goal of sustainable and eco-compatible chemistry. The Passerini reaction, first described about a century ago, is one of the oldest MCRs and it is the first isocyanide-based MCR.14 It consists in one-pot, three component reaction of carboxylic acids 12 with aldehydes 13 (or ketones) and isocyanides 14, offering an inexpensive and rapid way to generate a wide library of α-acyloxy carboxamides 15. These compounds are present in the structures of many natural products, such as pharmacologically active depsipeptides and can lead to potentially bioactive peptidomimetics compounds.16 As a stereocenter is formed, the development of enantioselective protocols has become a major goal. Although the literature highlights a number of examples of diastereoselective Passerini reactions17 or asymmetric Passerini-like reactions,18 only few examples are reported for asymmetric classic three-component passserini reactions.19 These include, notably, a study by X.-Y. Liu, B. Tan et al.,19d involving the use of BINOL-based chiral phosphoric acids that efficiently catalyze the reaction, allowing for excellent enantiomeric excesses to be obtained. It must be stressed that this is the only example in which a homogeneous chiral Brønsted acid catalyst was applied in Passerini reactions. In light of this, an asymmetric and green version of classic Passerini reactions with a broad scope to application still remains a major goal to be achieved to date.Optimization of the reaction conditions
Passerini reactions are usually carried out in common organic aprotic solvents.14 However, in the last years, several optimizations have been implemented in order to minimise the ecological impact of this reaction, carrying out it in green solvents such ionic liquids,20 water21 or deep eutectic solvents (DESs).22 DESs23 are considered to be a non-toxic, environmentally benign, sustainable and easily accessible alternative to conventional organic solvents. They not only possess the advantages of ionic liquids, such as designability, chemical stability, and negligible vapour pressure, but they also provide many other advantages, such as tunability, low-cost and easily sourced raw materials, as well as green and simple synthesis without using other organic solvents. Therefore, DESs appear to be a good alternative to volatile organic solvents, and it can definitely be said that DESs have every chance of becoming “the reaction media of the century”.23b In fact, since 2016,24a DESs have been profitably used as solvents for reactions carried out in the presence of organic catalysts.24b However, despite these advantages, the employment of DESs in asymmetric organocatalyzed reactions remains very scarce24c but, given their promising features, it may be expected that many applications in these areas will appear in the coming years.On this basis, a model reaction between benzoic acid (12a), benzaldehyde (13a), and tert-butyl isocyanide (14a) was studied (Table 1) at room temperature without any catalyst in several common organic solvents including DCM (Table 1; entry 1), toluene (Table 1; entry 2) THF (Table 1, entry 3), DMSO (Table 1; entry 4), EtOH (Table 1; entry 5) and in H2O (Table 1; entry 6). It was not possible to carry out the reaction in neat conditions at room temperature (Table 1; entry 7). Furthermore, two different kinds of DESs were used, formed by a glycerol and choline chloride (GL/CC; ratio 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; Table 1, entries 9 and 10) and by urea and choline chloride (U/CC; 2:1 ratio; Table 1, entries 11 and 12) respectively. It is evident that U/CC DES is a good solvent for this reaction, providing yields of 15a comparable to those obtained with toluene and DCM, though the reactions carried out in DCM or toluene are much faster than those carried out in DESs.
:1; Table 1, entries 9 and 10) and by urea and choline chloride (U/CC; 2:1 ratio; Table 1, entries 11 and 12) respectively. It is evident that U/CC DES is a good solvent for this reaction, providing yields of 15a comparable to those obtained with toluene and DCM, though the reactions carried out in DCM or toluene are much faster than those carried out in DESs.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp (°C) | Time (h) | Yield (%) of 15aa,b |
| a Yields refer to pure and isolated 15a.b The reaction was carried out with 1 mmol of 12a, 13a and 14a.c After 36 h the reaction was not complete. | ||||
| 1 | DCM | Rt | 16 | 90 |
| 2 | Toluene | Rt | 18 | 85 |
| 3 | THF | Rt | 36 | —c |
| 4 | DMSO | Rt | 36 | —c |
| 5 | EtOH | Rt | 36 | —c |
| 6 | H2O | Rt | 36 | 87 |
| 7 | Neat | Rt | 36 | —c |
| 8 | Neat | 60 | 16 | 45 |
| 9 | Glycerol/choline chloride 4:1 |
Rt | 26 | —c |
| 10 | Glycerol/choline chloride 4:1 |
60 | 24 | 77 |
| 11 | Urea/choline chloride 2:1 |
Rt | 36 | 87 |
| 12 | Urea/choline chloride 2:1 |
60 | 14 | 88 |

At the end of the reaction carried out in U/CC DES (Table 1; entry 11), after adding H2O a white solid precipitated, which was separated by filtration and washed with small amounts of diethyl ether. In this way, pure 15a was obtained without further purification. After this optimization, choosing U/CC DES as the privileged solvent, the scope of this reaction was extended using different carboxylic acids 12, aldehydes 13 and isocyanides 14. The results of these reactions are showed in a Table included in ESI† (Page 6). A number of aromatic 12a–d or heteroaromatic carboxylic acids 12g and aromatic aldehydes 13a–e containing both electron-donating and electron-withdrawing groups were used and afforded excellent yields of 15. Acids 12e–f and aldehydes 13f–g were also applied successfully in this reaction. Finally, it must be stressed that the reactions proceeded effectively with both aromatic 14b and aliphatic isocyanides 14a. On the contrary, the reaction of acetophenone (13h) could not reach completion.
Whit these optimized conditions in hand, we chose four kinds of solvents, namely DCM, toluene, H2O and DES urea/choline chloride and we carried out the reactions in the presence of heterogeneous chiral catalysts 11. We successfully gained good enantiomeric excesses in the reaction carried out with both DCM or toluene (Table 2; entries 1 and 2) and U/CC DES (Table 2; entry 4) at room temperature. It is interesting to note that the reaction times are significantly lower than the reactions carried out in the absence of a catalyst.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Temp (°C) | Time (h) | Yield (%) of 15aa,b | eec (%) |
| a Yields refer to pure and isolated 15a.b The reaction was carried out with 1 mmol of 12a, 13a, 14a and 5 mol% of catalyst 11.c Ee was determined by chiral analyses on HPLC.d The same results were obtained using 10 mol% of catalyst 11. | |||||
| 1 | DCM | Rt | 8 | 87 | 94.0 |
| 2 | Toluene | Rt | 10 | 90 | 94.2 |
| 3 | H2O | Rt | 24 | 88 | 65.2 |
| 4 | U/CC 2:1 |
Rt | 20 | 87 | 94.8d |
| 5 | U/CC 2:1 |
60 | 12 | 90 | 22.3 |

Then, using U/CC at room temperature as favorite and green solvent, we expanded the reaction to the same terms previously used, obtaining approximately the same yields as the reactions carried out in the absence of catalyst and excellent enantiomeric excesses, always higher than 92% (Table 3). The heterogeneous catalyst 11 was almost completely recovered by easy filtration on a Büchner funnel and reused four consecutive runs. The results are listed in Table 4, where it can be seen that the yields of 15a and the enantioselectivity were consistently good over the various runs.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 in RCOOH 12 | R2 in RCHO 13 | R3 in RNC 14 | Time (h) | 15 | Yielda (%) | Eeb (%) |
| a Yields refer to pure and isolated 15. Times and yields refer to the reactions carried out with 1 mmol of 12a, 13a, 14a with chiral catalyst 11 (5% mol).b Enantiomeric excess was determined by chiral analyses on HPLC (ee).c The same results were obtained using 10 mol% of catalyst 11. | |||||||
| 1 | Ph; 12a | Ph; 13a | t-Bu; 14a | 20 | 15a | 87c | 94.8 |
| 2 | Ph; 12a | 3-NO2C6H4; 13b | t-Bu; 14a | 24 | 15b | 93 | 96.4 |
| 3 | Ph; 12a | 4-MeOC6H4; 13c | t-Bu; 14a | 20 | 15c | 85 | 96.8 |
| 4 | Ph; 12a | 4-ClC6H4; 13d | t-Bu; 14a | 22 | 15d | 93 | 96.9 |
| 5 | Ph; 12a | 4-MeC6H4; 13e | t-Bu; 14a | 24 | 15e | 87 | 97.6 |
| 6 | Ph; 12a | PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH; 13f CH; 13f |
t-Bu; 14a | 28 | 15f | 86 | 92.7 |
| 7 | Ph; 12a | i-Pr; 13g | t-Bu; 14a | 20 | 15g | 87 | 95.5 |
| 8 | 4-NO2C6H4; 12b | Ph; 13a | t-Bu; 14a | 24 | 15h | 84 | 95.6 |
| 9 | 4-MeOC6H4; 12c | Ph; 13a | t-Bu; 14a | 30 | 15i | 87 | 95.9 |
| 10 | 4-ClC6H4; 12d | Ph; 13a | t-Bu; 14a | 20 | 15j | 90 | 96.8 |
| 11 | Me; 12e | Ph; 13a | t-Bu; 14a | 20 | 15k | 86 | 96.5 |
| 12 | PhCH2; 12f | Ph; 13a | t-Bu; 14a | 22 | 15l | 87 | 93.1 |
| 13 | 3-Pyridyl; 12g | 4-ClC6H4; 13d | t-Bu; 14a | 24 | 15m | 78 | 96.5 |
| 14 | Ph; 12a | Ph; 13a | 2-Naphthyl; 14b | 20 | 15n | 87 | 98.4 |
| 15 | Ph; 12a | 4-ClC6H4; 13d | 2-Naphthyl; 14b | 20 | 15o | 83 | 95.3 |
| 16 | Me; 12e | 3-NO2C6H4; 13b | 2-Naphthyl; 14b | 24 | 15p | 84 | 97.6 |
| 17 | Me; 12e | i-Pr; 13g | 2-Naphthyl; 14b | 22 | 15q | 86 | 96.1 |

| Entry | Time (h) | Yield (%) of 15aa,b | Recovery of 11 | ee (%) in 15a |
|---|---|---|---|---|
| a Yields refer to the pure and isolated product.b The reaction was performed at room temperature with 1 mmol of 12a, 13a, 14a and 5 mol% of 11.c Recovered 11 was used as a catalyst in entry 2.d Recovered 11 was used as a catalyst in entry 3.e Recovered 11 was used as a catalyst in entry 4.f Recovered 11 was used as a catalyst in entry 5. | ||||
| 1 | 24 | 87 | 0.06 gc | 94.8 |
| 2 | 24 | 85 | 0.06 gd | 95.0 |
| 3 | 24 | 87 | 0.06 ge | 93.2 |
| 4 | 28 | 88 | 0.05 gf | 93.1 |
| 5 | 28 | 84 | 0.05 g | 94.0 |
Furthermore, also the solvent system U/CC was easily recovered at the end of the reactions (for details see Experimental) and was used in three consecutive runs, without observing a decrease in the yield of 15a and in the enantioselectivity of the reaction.
Mechanism of the reaction
Although Passerini's reaction has been known for nearly a century, some uncertainties remain regarding its exact mechanism.25 It has been hypothesized that the reaction can proceed either via an “open chain” mechanism (where the key intermediates is a nitrilium ion) or via a “concerted mechanism” (where an intermediate nitrilium ion is not formed; Scheme 4). In polar solvents such as methanol or water, the reaction should proceed with an “open chain mechanism”26 via protonation of the carbonyl component followed by nucleophilic addition of the isocyanide and carboxylate residue, respectively. The resultant intermediate undergoes acyl group transfer to give the α-acyloxyamide. In nonpolar solvents, a “concerted mechanism” should be favored: a trimolecular reaction between the isocyanide, the carboxylic acid, and the carbonyl in a sequence of nucleophilic additions has been proposed, without the formation of a real nitrilium intermediate. The transition state is depicted as a 5-membered ring. The last step (Mumm rearrangement) includes the acyl migration to the neighbouring hydroxyl group to produce the desired ester. | ||
| Scheme 4 Proposed mechanisms for Passerini reaction. | ||
In the already mentioned elegant paper by Liu and Tan,15d an efficient enantioselective Passerini reaction was developed in the presence of a chiral phosphoric acid catalyst, deriving from BINOL. The authors proposed an interesting mechanism in which, inspired by List's previous studies,27 a key intermediate, such as heterodimer, could form between the chiral phosphoric acid catalyst and carboxylic acid, creating a chiral environment suitable for asymmetric induction of the reaction (Fig. 4).
 | ||
| Fig. 4 Key intermediates for chiral phosphoric acid catalysed Passerini reaction. | ||
Regarding the Passerini reactions carried out in the presence of 11, we believe that two possible mechanisms can be hypothesized. In the first and, in particular, in the reactions carried out with DCM and toluene (apolar or not very polar solvents), heterogeneous catalyst 11 could act similarly to phosphoric acid. In fact, if the structure of sulfonimides is compared with that of the phosphoric acids, it is clear that both have acidic and basic sites. Indeed, if the phosphoric acid can be considered a bidentate system, sulfonimides, thanks to the presence of the SO2 group, can be considered a multidentate system that can form five hydrogen bonds (Fig. 5).28
 | ||
| Fig. 5 Disulfonimide 11 as multidentate system. | ||
Moreover, the high acidity of catalyst 11 (higher than that of a phosphoric acid) could, on the one hand, facilitate the formation of the heterodimer and, on the other hand, make the carboxylic acid more nucleophilic, and therefore more reactive. Indeed, the reaction times carried out in the presence of 11 (8 h. Table 3; entry 1) were significantly lower than those carried out without 11 (16 h. Table 1; entry 1). Therefore, thanks to the formation of the heterodimer 16 and its interaction with aldehyde 13, a chiral fashion is created, facilitating the enantioselective attack of the nucleophile isocyanate 14, with the formation of a nitrilium intermediate 17. It then undergoes nucleophilic attack by the acid 12; finally, Mumm rearrangement of the imidate 18 leads to the formation of the final product 15 (Scheme 5). In a recent review of Passerini reaction mechanism, Morokuma demonstrated, using DFT calculations, that the nitrilium intermediate is stable in solution and the rate of the reactions is significantly higher in low-polar solvents (e.g. DCM) not because the nitrilium ion is not formed, as is commonly believed, but because a network of hydrogen bonds makes its formation more difficult.24 This could be confirmed by our experimental data: the reactions carried out in DCM (8 h. Table 3; entry 1) are much faster than the reactions carried out in DES (20 h. Table 2; entry 1). In his mechanism hypothesis, Morokuma believes that the Mumm rearrangement can be catalyzed by a second carboxylic acid molecule. Therefore, it might be is possible that the acid catalyst also plays a role in facilitating Mumm rearrangement.
 | ||
| Scheme 5 Proposed mechanism of Passerini reaction in DCM catalysed by 11. | ||
On the other hand, it is dubious that the reaction occurs the same way in DES. The polarity of U/CC DES,29 far superior to that of DCM or toluene, should favour an “open chain” mechanism. Therefore, we assume that, in DES, the reaction could occur precisely with this mechanism (Scheme 6). We think that the activation of aldehyde is due to the action of 11, mainly by means of the acid site of the sulfonimide (NH group), but also with those belonging to the silica support. The weak basicity of U/CC DES30 should not hinder this activation After its formation, 19 undergoes the enantioselective attack of 14 with the formation of the intermediate 20 which, in turn, undergoes the nucleophilic attack of the OH of 12. The intermediate 18 is obtained and finally the latter, by means of Mumm rearrangement, gives the product 15. Assuming the above mechanism, this reaction may therefore be counted as an asymmetric counteranion-directed catalysis (ACDC), according to list classification.31
 | ||
| Scheme 6 Proposed mechanism of Passerini reaction in DES catalysed by 11. | ||
Conclusions
In this paper, a sustainable version of asymmetric three component Passerini reaction has been proposed. In our conditions, we are able to achieve important green benefits, including the use of a DES as a solvent at room temperature, the use of a novel and safe heterogeneous catalyst, its complete recovery and reuse, stoichiometric reagent ratio and mild reaction conditions. High yields of target products (17 examples) and excellent enantiomeric excess (17 examples; average 96%) have been obtained; furthermore, they are obtained in adequate purity, making further chromatographic purification unnecessary. It must be stressed that the use of chiral disulfonimides as acid catalysts in asymmetric synthesis, thanks to the important advantages they provide, is now widely consolidated. However, the synthetic protocols for obtaining such catalysts can be quite complex; this is proven by the fact that the cost of the commercially available disulfonimides is very high. Therefore, a disulfonimide which acts as a heterogeneous catalyst and which can be used several times without losing its catalytic activity, certainly represents an added value for this family of catalysts.Experimental
General remarks
Analytical grade reagents were used and reactions were monitored by GC, GC-MS. Column chromatography were performed on Merck silica gel 60 (70–230 mesh ASTM) and GF 254, respectively. Petroleum ether (PE) refers to the fraction boiling in the range 40–70 °C. Mass spectra were recorded on an HP 5989B mass selective detector connected to an HP 5890 GC with a cross-linked methyl silicone capillary column. HRMS analyses were performed on an Orbitrap Fusion instrument. HPLC analyses were performed on HPLC Waters 1525 connected to a column Chiralpack IG-SFC. 1H NMR and 13C NMR spectra were recorded on a Jeol ECZR spectrometer at 600 and 150 MHz respectively. IR spectra were recorded on a IR PerkinElmer UATR-two spectrometer. Alternatively, IR spectra of 7, 10 and 11 were recorded in the form of self-supporting pellets (using the powders either as such or in KBr dispersion) by means of a Bruker IFS Vector 22 spectrophotometer (resolution 4 cm−1), equipped with a MCT cryodetector. For the determination of optical rotations, a Jasco P-2000 polarimeter was used. (−)-4,5-Dimethyl-3,6-bis(1-naphthyl)-1,2-benzenedisulfonimide (3) was synthesized as previously reported by us.11d Urea/choline chloride DES were prepared as reported in the literature.32 All reagents were purchased from Sigma-Aldrich or Alfa-Aesar. Structures and purity of all the products obtained in this research were confirmed by their spectral (NMR, MS, IR) data. Yields and enantiomeric excesses of the pure (GC, GC-MS and NMR) and isolated 15 are reported in Table 2. Satisfactory microanalyses were obtained for new compounds.Synthesis of (−)-4,5-dicarboxy-3,6-bis(1-naphthyl)-1,2-benzenedisulfonimide 10
To a solution of (−)-4,5-Dimethyl-3,6-bis(1-naphthyl)-1,2-benzenedisulfonimide (3, 1 mmol, 0.50 g) in H2O (15 mL) were added KMnO4 (26 mmol, 4.11 g) and Na2CO3 (18 mmol, 1.91 g). The resulting mixture was heated at 100 °C for 24 h. In order to remove KMnO4, the mixture was filtered on a Büchner funnel and the purple filtrate was decoloured with charcoal. H2O was evaporated under reduced pressure. The crude residue, dissolved in H2O was passed through a Dowex (HCR-W2) column (H2O), affording pure 10 (0.56 g, 100% yield) as a grey waxy solid.[α]22D-25.6 (c 0.15 in CH2Cl2). Found: C, 60.48; H, 3.41; N 2.38. Calc. for C28H17NO8S2: C, 60.10; H, 3.06; N, 2.50. νmax/cm−1 3012 (OH), 1695 (CO). δH (600 MHz, D2O) 7.85–7.49 (m, 6H), 7.47–7.34 (m, 8H). δC (150 MHz, D2O) 167.9, 136.2, 133.7, 133.5, 132.0, 129.6, 128.5, 128.3, 127.8, 127.8, 126.4, 126.2, 125.9. HRMS (ESI, m/z): 560.05 (M + H+).
4,5-Dicarboxy-3,6-bis(1-naphthyl)-1,2-benzenedisulfonimide immobilized on 3-aminopropyl functionalized silica gel (11)
Adduct 10 (0.5 mmol, 0.28 g) and N-(3-dimethylaminopropyl)-N-ethylcarbodiimmide hydrochloride (EDC, 1.5 mmol, 0.28 g) were added to a stirred suspension of 3-aminopropyl functionalized silica gel 7 (1 g; 1 mmol g−1 NH2) in H2O (5 mL). The reaction mixture was vigorously stirred at r.t. for 5 h. The resulting white solid was filtered over Büchner funnel. Firstly was washed with 10 mL of 2 N HCl (in order to fully restore the sulfonimide acidity) and then with 10 mL of acetone (in order to remove eventual organic residues arising from 11 and catalyst); lastly, it was dried in oven at 70 °C overnight. After this treatment, 1.10 g of 11 was obtained (87% yield). Aqueous and acetone washings were collected and evaporated under reduced pressure. No traces of possible unreacted 10 were detected. In a collateral proof 2 (1 g) and 10 (0.5 mmol, 0.28 g) were reacted without EDC. IR spectrum of the obtained solid (0.99 g), included in ESI.† (Page 5), was markely different from that of 11 and the characteristic bands of the amide bond were not present.11 as a heterogeneous catalyst in Passerini reactions. General procedure
Chiral heterogeneous catalyst 11 (1; 5 mol%, 0.06 g equivalent to 0.05 mmol of immobilized acid) was added at room temperature to a stirring mixture of aldehyde 13 (1 mmol) and carboxylic acid 12 (1 mmol) in urea/choline chloride (U/CC) deep eutectic solvent system (5 mL). After 10 min, isocyanide 14 (1 mmol) was added. The mixtures were stirred at room temperature for the times listed in Table 3 (yield in bracket and in blue), until the GC and GC-MS analyses showed the complete disappearance of starting compounds and the complete formation of α-acyloxyamide 15. In order to recover the catalyst, the reaction mixture was filtered on a Hirsch funnel and recovered solid was washed with of H2O. Recovered catalyst was dried in oven at 70 °C. After this treatment the recovered catalyst (0.06 g, 100% of recovery) was reused in other four consecutive catalytic runs as reported in Table 4. The yield and the enantiomeric excess of 15a and the recovery of 11 were always consistently good. Cold H2O (5 mL) was added to the filtrate, under vigorous stirring. A white solid formed and was recovered by filtration on a Hirsch funnel. It was washed with additional cold H2O and diethyl ether (2 mL each). Virtually pure (GC, GC-MS, 1H NMR, 13C NMR) 15 was obtained. The liquid residue was collected and, in order to remove H2O and diethyl ether, was evaporated under reduced pressure. The recovered U/CC solvent system showed NMR and IR spectra almost identical to the initial one (ESI†). Physical and spectral data of adducts 15 are reported in ESI.† After the above work-up, enantiomeric excesses were measured. However, in order to verify the accuracy of these measures, the crude residue of 15a, obtained after extraction with DCM, was chromatographed on a short column (eluent: EP–EtOEt, 4:1) and then enantiomeric excess was determined. The values were identical (96.4% on HPLC).
Author contributions
AA was responsible of the chiral analyses; FM took care of the synthesis of the heterogeneous chiral catalyst. SD coordinated the research. From the experimental point of view he performed the Passerini reactions with the chiral catalyst and he took care of the writing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Ministero dell'Università e della Ricerca and by University of Torino.References
- (a) Organocatalysis, ed, M. Reetz, B. List, S. Jaroch, H. Weinmann Springer, Dordrecht, 2008 Search PubMed; (b) V. da Gama Oliveira, M. F. do Carmo Cardoso and L. da Silva Magalhães Forezi, Catalysts, 2018, 8, 605–634 CrossRef; (c) S.-H. Xiang and B. Tan, Nat. Comm., 2020, 11, 3786–3791 CrossRef CAS.
- (a) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999–1010 CrossRef CAS; (b) T. Akiyama, Stronger Brønsted acids, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS; (c) D. Kampen, C. M. Reisinger and B. List, Top. Curr. Chem., 2010, 29, 395–456 Search PubMed; (d) J. Merad, C. Lalli, G. Bernadat, J. Maury and G. Masson, Chem. –Eur. J., 2018, 24, 3925–3943 CrossRef CAS PubMed.
- (a) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice. Oxford University Press, Oxford, 1998 Search PubMed; (b) V. K. Ahluwalia and M. Kidwai M. Basic Principles of Green Chemistry: New Trends in Green Chemistry, Springer, Dordrecht, 2004 Search PubMed; (c) R. A. Sheldon, I. Arends and U. Hanefelds, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007 CrossRef.
- (a) D. Kristofikova, V. Modrocka, M. Meciarova and R. Sebesta, ChemSusChem, 2020, 13, 2828–2858 CrossRef CAS PubMed; (b) A. Antenucci, S. Dughera and P. Renzi, ChemSusChem, 2021 DOI:10.1002/cssc.202100573.
- (a) R. T. Baker, S. Kobayashi and W. Leitner, Adv. Synth. Catal., 2006, 348, 1337–1340 CrossRef CAS; (b) F. Cozzi, Adv. Synth. Catal., 2006, 348, 1367–1390 CrossRef CAS; (c) A. Corma and H. Garcia, Adv. Synth. Catal., 2006, 348, 1391–1412 CrossRef CAS; (d) M. Benaglia, New J. Chem., 2006, 30, 1525–1533 RSC; (e) S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed; (f) A. E. Fernandes, A. M. Jonas and O. Riant, Tetrahedron, 2014, 70, 1709–1731 CrossRef CAS.
- (a) C. M. Friend and B. Xu, Acc. Chem. Res., 2017, 50, 517–521 CrossRef CAS PubMed; (b) B. Torok, C. Schaefer and A. Kokel, Heterogeneous Catalysis in Sustainable Synthesis, Elsevier, 2021 Search PubMed.
- (a) A. K. Mutyala and N. T. Patil, Org. Chem. Front., 2014, 1, 582–586 RSC; (b) C. B. Watson, A. Kuechle and D. E. Bergbreiter, Green Chem., 2021, 23, 1266–1273 RSC.
- S. J. Jenkins, Chirality at Solid Surfaces, Wiley-VCH, Weinheim, 2018 Search PubMed.
- G. Szollosi, Catal. Sci. Technol., 2018, 8, 389–422 RSC.
- Heterogenized Homogeneous Catalysts for Fine Chemicals Production, ed. P. Barbaro and F. Liguori, Springer, Dordrecht, 2010 Search PubMed.
- (a) M. Barbero and S. Dughera, Targets in Heterocycl. Syst., 2019, 23, 178–200 CAS; (b) M. Barbero, S. Bazzi, S. Cadamuro, L. Di Bari, S. Dughera, G. Ghigo, D. Padula and S. Tabasso, Tetrahedron, 2011, 67, 5789–5797 CrossRef CAS; (c) M. Barbero, S. Cadamuro, S. Dughera and G. Ghigo, Org. Biomol. Chem., 2012, 10, 4058–4068 RSC; (d) M. Barbero, S. Cadamuro, S. Dughera and R. Torregrossa, Org. Biomol. Chem., 2014, 12, 3902–3911 RSC; (e) M. Barbero, S. Cadamuro and S. Dughera, Tetrahedron Asymm, 2015, 26, 1180–1188 CrossRef CAS; (f) M. Barbero, S. Cadamuro and S. Dughera, Green Chem., 2017, 19, 1529–1535 RSC; (g) M. Barbero, S. Berto, S. Cadamuro, P. G. Daniele, S. Dughera and G. Ghigo, Tetrahedron, 2013, 69, 3212–3217 CrossRef CAS.
- (a) T. James, M. van Gemmeren and B. List, Chem. Rev., 2015, 115, 9388–9409 CrossRef CAS PubMed; (b) M. C. Benda and S. France, Org. Biomol. Chem., 2020, 18, 7485–7513 RSC.
- M. Barbero, G. Cerrato, E. Laurenti, S. Zanol and S. Dughera, ChemistrySelect, 2017, 2, 3178–3183 CrossRef CAS.
- (a) A. Domling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef CAS; (b) A. Varadi, T. C. Palmer, R. Notis Dardashti and S. Majumdar, Molecules, 2016, 21, 19 CrossRef PubMed.
- (a) C. de Graaff, E. Ruijter and R. V. A. Orru, Chem. Soc. Rev., 2012, 41, 3969–4009 RSC; (b) R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC; (c) Multicomponent Reactions, ed, T. J. J. Müller. Thieme, Stuttgart, vol. 2, 2014 Search PubMed; (d) S. Zhi, X. Ma and W. Zhang, Org. Biomol. Chem., 2019, 17, 7632–7650 RSC; (e) P. S. Gonçalves Nunes, H. D. Almeida Vidal and A. G. Corrêa, Org. Biomol. Chem., 2020, 18, 7751–7773 RSC.
- A. R. Kazemizadeh and A. Ramazani, Curr. Org. Chem., 2012, 16, 418–450 CrossRef CAS.
- (a) L. Moni, L. Banfi, A. Basso, E. Martino and R. Riva, Org. Lett., 2016, 18, 1638–1641 CrossRef CAS PubMed; (b) C. Lambruschini, L. Moni and L. Banfi, Eur. J. Org. Chem., 2020, 3766–3778 CrossRef CAS.
- (a) S. Denmark and Y. Fan, J. Am. Chem. Soc., 2003, 125, 7825–7827 CrossRef CAS PubMed; (b) S. Denmark and Y. Fan, J. Org. Chem., 2005, 70, 9667–9676 CrossRef CAS PubMed; (c) S.-X. Wang, M.-X. Wang, D.-X. Wang and J. Zhu, Org. Lett., 2007, 9, 3615–3618 CrossRef CAS PubMed; (d) T. Yue, M.-X. Wang, D.-X. Wang, G. Masson and J. Zhu, J. Org. Chem., 2009, 74, 8396–8399 CrossRef CAS PubMed; (e) H. Mihara, Y. Xu, N. E. Shepherd, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 8384–8385 CrossRef CAS PubMed; (f) X. Zeng, K. Ye, M. Lu, P. J. Chua, B. Tan and G. Zhong, Org. Lett., 2010, 12, 2414–2417 CrossRef CAS PubMed; (g) Q. Wang, D.-X. Wang, M.-X. Wang and J. Zhu, Acc. Chem. Res., 2018, 51, 1290–1300 CrossRef CAS PubMed.
- (a) U. Kusebauch, B. Beck, K. Messer, E. Herdtweck and D. Alexander, Org. Lett., 2003, 5, 4021–4024 CrossRef CAS PubMed; (b) P. R. Andreana, C. C. Liu and S. L. Schreiber, Org. Lett., 2004, 5, 4231–4233 CrossRef PubMed; (c) S.-X. Wang, M.-X. Wang, D.-X. Wang and J. Zhu, Angew. Chem., Int. Ed., 2008, 47, 388–391 CrossRef CAS PubMed; (d) J. Zhang, S.-X. Lin, D.-J. Cheng, X.-Y. Liu and B. Tan, J. Am. Chem. Soc., 2015, 137, 14039–14042 CrossRef CAS PubMed.
- F. Shirini, K. Rad-Moghadam and S. Akbari-Dadamahaleh, Green solvents II, Application of Ionic Liquids in Multicomponents Reactions, Springer, Dordrecht, 2012., pp. 289−334 Search PubMed.
- M. C. Pirrung and K. D. Sarma, J. Am. Chem. Soc., 2004, 126, 444–445 CrossRef CAS PubMed.
- A. Shaabani, R. Afshari and S. E. Hooshmand, Res. Chem. Intermed., 2016, 42, 5607–5616 CrossRef CAS.
- (a) E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed; (b) D. A. Alonso, A. Baeza, R. Chinchilla, G. Guillena, I. M. Pastor and D. J. Ramón, Eur. J. Org. Chem., 2016, 612–632 CrossRef CAS; (c) B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed.
- (a) E. Massolo, S. Palmieri, M. Benaglia, V. Capriati and F. M. Perna, Green Chem., 2016, 18, 792–797 RSC; (b) N. Fanjul-Moisterin and V. del Amo, Tetrahedron, 2021, 84, 131967 CrossRef; (c) D. D. Alonso, S.-J. Burlingham, R. Chinchilla, G. Guillena, D. J. Ramon and M. Tiecco, Eur. J. Org. Chem., 2021, 1–8 Search PubMed.
- R. Ramozzi and K. Morokuma, J. Org. Chem., 2015, 80, 5652–5657 CrossRef CAS PubMed.
- J. Tarana, A. Ramazani, S. W. Joo, K. Slepokura and T. Lis, Helv. Chim. Acta, 2014, 97, 1088–1096 CrossRef.
- M. R. Monaco, B. Poladura, M. Diaz de Los Bernardos, M. Leutzsch, R. Goddard and B. List, Angew. Chem., Int. Ed., 2014, 53, 7063–7067 CrossRef CAS PubMed.
- A. Galvan, A. B. Gonzalez-Pÿrez, R. Alvarez, A. R. de Lera, F. J. Fañanas and F. Rodriguez, Angew. Chem., Int. Ed., 2016, 55, 3428–3432 CrossRef CAS PubMed.
- A. Valvi, J. Dutta and S. Tiwari, J. Phys. Chem. B, 2017, 121, 11356–11366 CrossRef CAS PubMed.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jerome, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/d1ra05297g |
| This journal is © The Royal Society of Chemistry 2021 |